
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of supramolecular encapsulation of camptothecin by cucurbit[7]uril: reduced toxicity and preserved anti-cancer activity†‡
Xue
Yang
a,
Ziyi
Wang
ab,
Yanan
Niu
a,
Xiuping
Chen
a,
Simon M. Y.
Lee
a and
Ruibing
Wang
*a
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Avenida da Universidade, Taipa, Macau, China. E-mail: rwang@umac.mo
bUndergraduate Research Program, China Pharmaceutical University, Nanjing, China
First published on 18th May 2016
Abstract
Camptothecin (CPT), a natural antineoplastic agent that was originally isolated from the bark and stem of Camptotheca acuminata, targets a wide range of cancers by inhibition of topoisomerase I during DNA replication. In this study, we investigated the use of cucurbit[7]uril (CB[7]) for molecular encapsulation of CPT and the associated benefits of such encapsulation of CPT on its anti-cancer activity, and side effects have been systemically evaluated both in vitro and in vivo. The cytotoxicity of the free CPT and complexed CPT (CPT@CB[7]) was examined by MTT assay using both a healthy liver cell line (L02) and a liver cancer cell line (Bel-7402). Interestingly, both the free CPT and CPT@CB[7] demonstrated comparable anti-cancer efficacy in vitro on the cancer cell line Bel-7402, whereas CPT@CB[7] showed obviously lower toxicity on the healthy liver cell line L02, in comparison with the free CPT. The anti-cancer/anti-angiogenic activity and non-specific toxicity of both free CPT and CPT@CB[7] were further investigated in vivo with both transgenic and wild-type zebrafish models, showing again that CPT@CB[7] exhibited reduced non-specific toxicities with well-preserved anti-angiogenic activities, when compared with the free CPT. These results demonstrate that CB[7] may work as a functional excipient for alleviation of undesired side-effects of selected organic drugs.
Introduction
Camptothecin (CPT) (Fig. 1), a natural cytotoxic alkaloid, which was isolated from the Chinese plant Camptotheca acuminate and characterized by Wall et al. in 1966, is a well-known potent anti-cancer agent against many types of human cancers.1,2 CPT targets the nuclear enzyme topoisomerase I (topo I) and forms stable conjugates via hydrogen bonds, thereby inhibiting DNA synthesis and inducing cell apoptosis.3 Although CPT has exhibited promising antitumor activity with various in vitro, ex vivo, and in vivo models,4 its clinical application has been limited by the instability of its lactone ring at physiological pH and severe side-effects that have been associated with this drug's hydrolyzed carboxylate form.5 In particular, the lactone form (Fig. 1) plays an essential role in maintaining CPT's antitumor activity, as the hydrolyzed carboxylate form (Fig. 1) has been found as the cause of severe toxicities and is less active pharmacologically against various cancer cell lines.6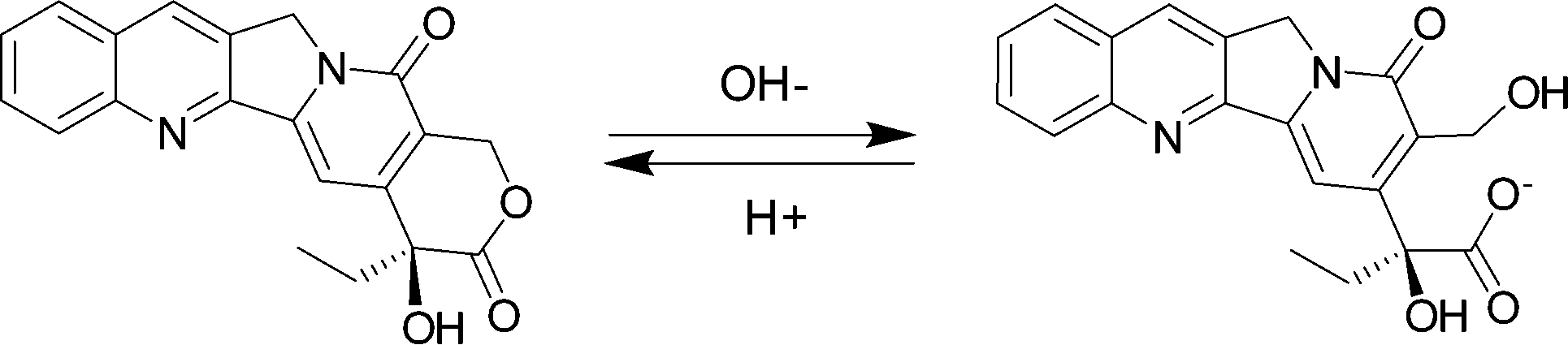 | ||
| Fig. 1 Molecular structures of lactone and carboxylate CPT. | ||
A variety of CPT derivatives have been developed to increase the lactone ring stability and to improve water-solubility. Among these efforts, topotecan and irinotecan, both approved for chemotherapy by the US Food and Drug Administration, are two successful examples of clinical applications as synthetic CPT analogues.7 However, derivatization often involves laborious multi-step synthesis and purification, which usually increases the cost of these drugs. A facile alternative strategy to overcome the stability and solubility problems of CPT is to develop a novel drug delivery system or formulation. For instance, liposome formulation and microsphere-based delivery of CPT have been attempted with demonstrated benefits.8 Similarly, Botella et al. successfully synthesized surface-modified silica nanoparticles (SNP) for tumor-targeted delivery of CPT and their biological evaluations demonstrated that the SNP/CPT had better antitumor activity than free CPT both in vitro and in vivo.9 It has also been reported that the hydrolysis of CPT in the cyclodextrin (CD) complexed form was much slower than that of free CPT and their preliminary cytotoxicity experiments showed that the anti-cancer activity of CD/CPT complexes was higher than that of free CPT.10 However, none of the these studies addressed the non-specific toxicities of CPT. Additionally, one limitation for the use of CD as a complexing agent for CPT is the weak bindings between CPT and CD (for instance, the binding constant of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 β-CD@CPT is only 266 M−1).10
:1 β-CD@CPT is only 266 M−1).10
In recent years, cucurbit[n]urils (CB[n], n = 5–8, 10, 14), a new family of pumpkin-shaped macrocyclic molecules, have emerged and earned significant attention due to their unique binding behaviours for a variety of cationic and neutral guest molecules.12,13 In comparison with CDs, CB[n]s typically have much higher binding affinities and higher selectivity towards a variety of guest molecules.14,15 Within the CB[n] family, CB[7] has attracted the most attention as a potent drug delivery carrier owing to its superior solubility in water and appropriate size that can form complexes with numerous organic and organometallic guest molecules of biomedical interest.16–18 In addition, it has been demonstrated that CB[7] exhibited little toxicity at sub-mM concentrations or even up to 1 mM concentration in in vitro cellular models.19,20 In addition, our recent study has shown acceptable biocompatibility of CB[7] with an in vivo zebrafish model.21 Recently, it has been shown that CPT and CB[7] form both 1:1 and 1:2 inclusion complexes (with binding constants K1 and K2 of 2.8 × 103 (±100) and 3.4 × 104 (±1890) M−1, respectively, for a step-wise complexation at neutral pH), with the latter complexes being the predominant species when the host is at a concentration >50 μM (Fig. 2).11 Another study carried out by Tao et al. indicated that the lactone form of CPT was significantly preserved by the presence of CB[7] or CB[8] under physiological conditions.22,23
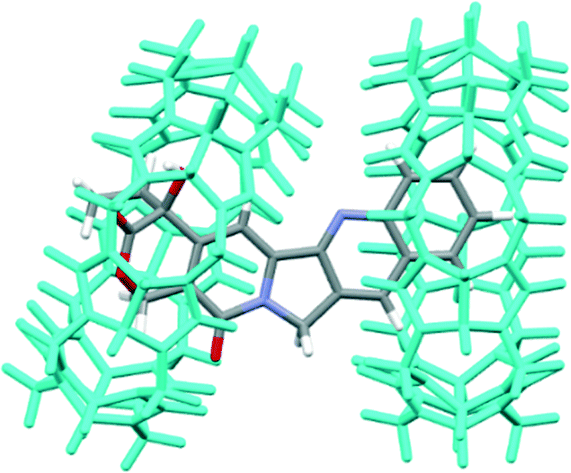 | ||
| Fig. 2 Molecular modeling of 1:2 CPT@CB[7] (re-built from courtesy data of Dr. Partha Hazra).11 | ||
Despite these discoveries and the intriguing CPT–CB[7] host–guest chemistry, the expected medical benefits as a result of improved lactone ring stability, i.e. reduced toxicity, have never been demonstrated yet with any in vitro and in vivo models. With our extensive interest and experience in the exploration of biomedical applications of CB[n],24–28 particularly the potential of CB[7] in inhibiting or alleviating side-effects of bioactive molecules,26,27 we report herein the influence of CB[7] on CPT's toxicity and antitumor/anti-angiogenic activities using both in vitro cellular models and in vivo zebrafish models. We demonstrate for the first time that supramolecular encapsulation of CPT by CB[7] may significantly reduce CPT's inherent non-specific toxicities while preserving its important antitumor/anti-angiogenic activities.
Results and discussion
In vitro cytotoxicity
The cytotoxicity of CPT and CPT@CB[7] was determined by in vitro MTT assay with two different cell lines, human hepatocarcinoma cells (Bel-7402) and healthy human liver cells (L02). As shown by Fig. 3(i), the IC50 values (upon 24 h of incubation) of CPT and CPT@CB[7] are 0.562 ± 0.417 μM and 3.562 ± 0.252 μM, respectively, suggesting that CPT@CB[7] is significantly less toxic than the free CPT in the healthy liver cell line (L02). On the other hand, as shown by Fig. 3(ii), molecular encapsulation of CPT by CB[7] yielded little effect on the cytotoxicity of CPT on the cancer cell line Bel-7402. The cellular viability of Bel-7402 cells showed approximately the same dosage-dependent behaviour against free CPT and CPT@CB[7] complexes (IC50 of ca. 100 μM). Taken together, these in vitro assay results demonstrated that CB[7]-encapsulated CPT may significantly reduce the cytotoxic effect in healthy liver cells; meanwhile, the antitumor activity of CPT may be well-maintained.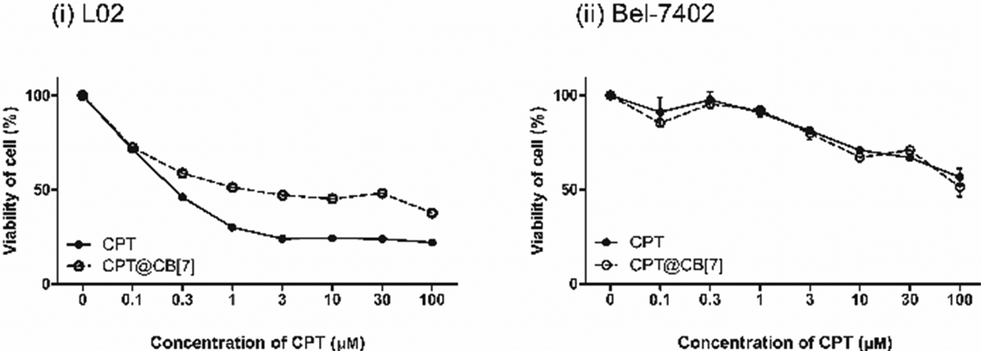 | ||
| Fig. 3 Viability (MTT assay) of L02, human healthy liver cells (i), and Bel-7402, human hepatocellular carcinoma cells (ii), incubated for 1 day with various concentrations of CPT (0–100 μM) in the absence and in the presence of CB[7] (200 μM). Each data point represents the mean ± S.E.M. from a set of experiments (n = 4). | ||
Toxicity assay with wild-type zebrafish
In order to determine the toxicity of CPT and CPT@CB[7] in vivo, we evaluated the hatching rate of wild type zebrafish treated with these reagents. No significant effect on the hatchability of zebrafish embryos was observed when they were exposed to test solutions of 0.01 to 10 nM CPT for 2 days (Fig. 4(i)). When the dosage was increased to the 100 nM level, CPT dramatically inhibited zebrafish embryonic hatching. Moreover, most of the unhatched embryos seemed to have lost vital signs, indicating that a higher concentration of CPT indeed had lethal toxic effects on the embryos rather than just retarding their development. Conversely, complexation of 100 nM CPT with CB[7] significantly improved CPT's developmental toxicity profile by showing approximately 90% hatchability. As shown in Fig. 4(ii), though the hatching rates of zebrafish embryos treated with 10 nM CPT in the absence and in the presence of CB[7] were similar, the hatched fish from the CPT@CB[7]-treated group exhibited normal morphology and physical dimensions that are comparable with those of the control group; however, the hatched fish from the free CPT-treated group showed significant morphological abnormalities. Similarly, when the CPT concentration was 100 nM, zebrafish that hatched from the CPT@CB[7]-treated group not only had a normal hatching rate, but were also morphologically healthier than the fish of the free CPT-treated group. These in vivo results demonstrated for the first time that encapsulation of CPT by CB[7] may dramatically reduce the non-specific toxicity, particularly, the fetus developmental toxicities of CPT.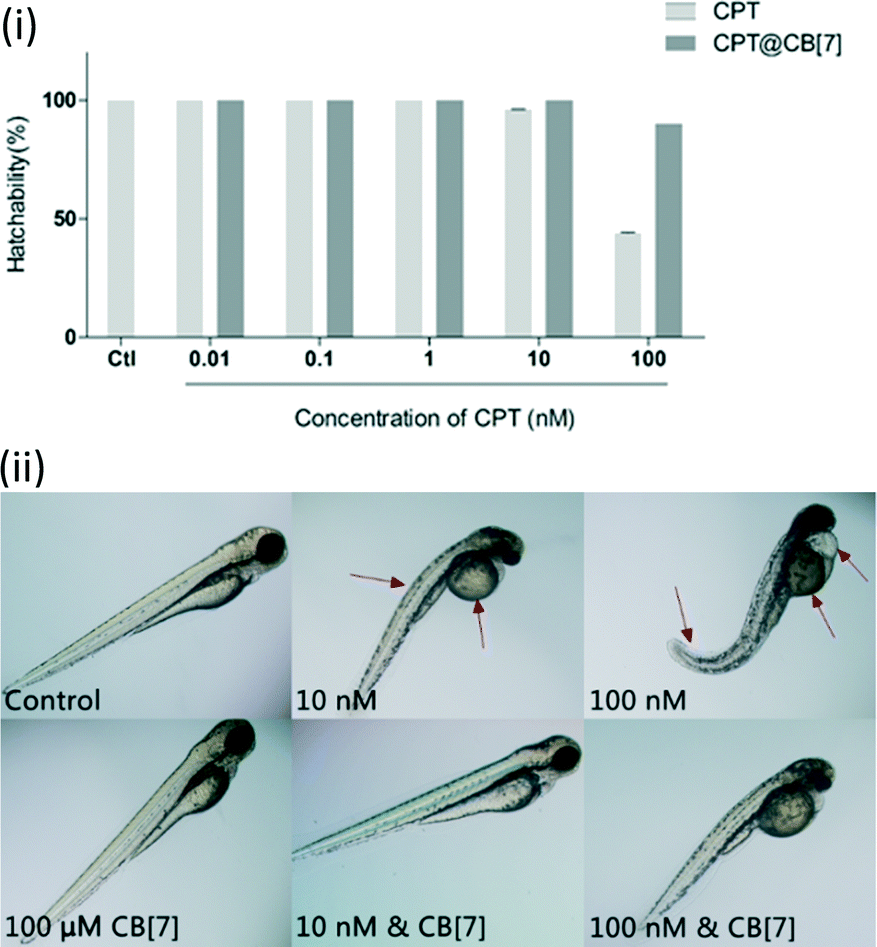 | ||
| Fig. 4 (i) Hatchability of zebrafish embryos after incubation with CPT (0.01 to 100 nM) in the absence and in the presence of CB[7] (100 μM) for 2 days. Data are presented as mean ± S.E.M. (n = 20) of three independent experiments; (ii) morphology of representative zebrafish larvae (3 dpf) after exposure to various concentrations of CPT (10 nM and 100 nM) in the absence and in the presence of CB[7] (100 μM) for 2 days. Red arrows point to the obvious abnormal morphologies. See Fig. S1 in the ESI‡ for the morphologies of fish incubated with a full range of concentrations of CPT. | ||
Anti-angiogenic activity and toxicity assay with transgenic zebrafish
Angiogenesis plays an important role in the progression of cancers.29 Numerous anti-cancer drugs possess anti-angiogenic activities.30 Notably, the responses of zebrafish to many anti-angiogenic drugs are similar to those of mammalian models, thus zebrafish has become an animal model of increasing interest for the investigation of angiogenesis.31 In particular, the Tg(fli-1:EGFP) transgenic zebrafish that express GFP in the entire vasculature make it possible to realize a fast analysis of live embryos in reaction to anti-angiogenic drugs.32 Zebrafish sub-intestinal vessels (SIVs) begin to establish angiogenesis in the developing gut by 1 day post-fertilization (dpf), forming on the dorsolateral surface of the yolk on both sides of the embryo in the shape of a basket, which allows assessment of the effects of anti-angiogenic compounds.33 It has been reported previously that CPT can inhibit angiogenesis, as demonstrated through the inhibition of the development of zebrafish SIVs.34 We therefore used the Tg(fli-1:EGFP) transgenic zebrafish model to evaluate the anti-angiogenic activity of CPT in the absence and in the presence of CB[7]. For this study, SIV angiogenesis inhibition was quantified by measurement of the total SIV length at the end of 4 dpf. In addition, the survival rate of embryos at the end of the incubation period was also examined for evaluation of the toxicity profile of CPT and CPT@CB[7] complexes.The inhibitory effect of CPT and CPT@CB[7] on SIVs in zebrafish embryos was evaluated under a fluorescence microscope for phenotypic observation after incubation for 2 days, as shown in Fig. 5(i). Both CPT and CPT@CB[7] exhibited a dosage-dependent manner in inhibition of SIV development in treated embryos. In both groups, the anti-angiogenic activity was most remarkable when CPT was at a concentration between 1 and 100 nM.
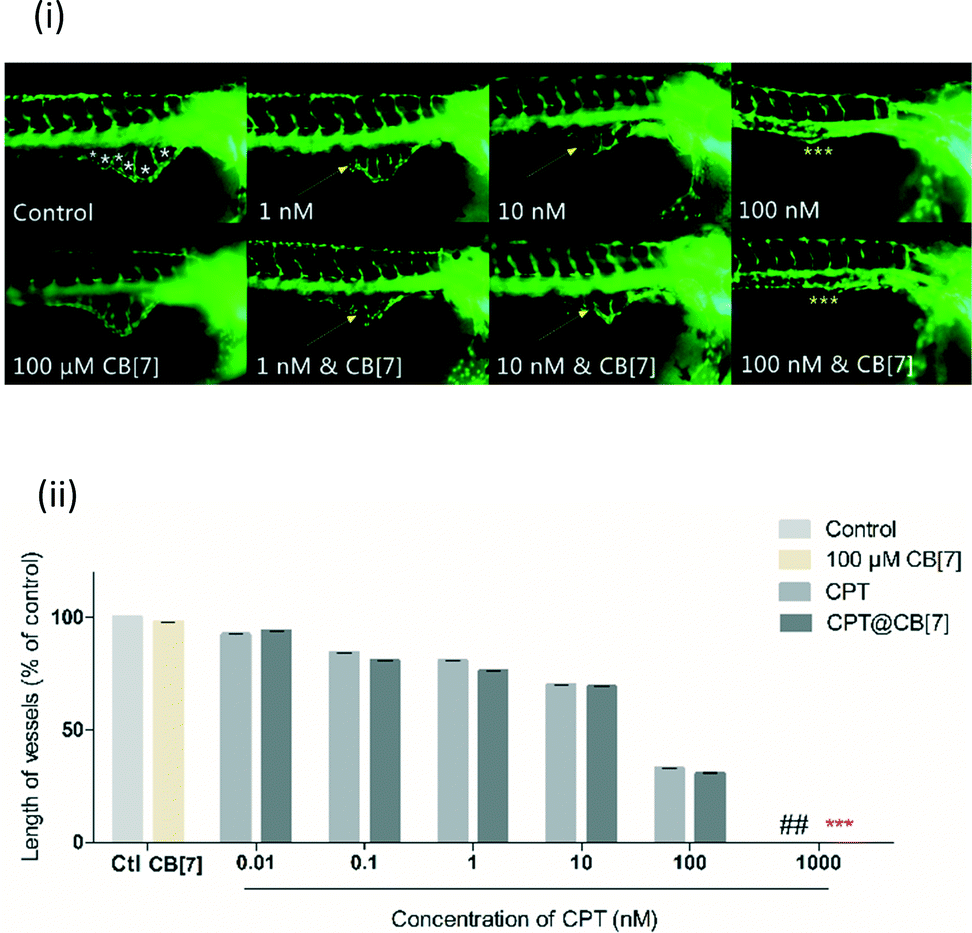 | ||
| Fig. 5 (i) Lateral view of Tg(fli-1:EGFP) zebrafish embryos at 3 dpf treated with CPT (1 to 100 nM) in the absence and in the presence of CB[7] (100 μM). Yellow arrows point to regions where SIVs were inhibited to some degree, and yellow asterisks point to regions where SIVs were completely inhibited; for zebrafish treated with a full range of concentrations of CPT, see Fig. S2;‡ (ii) quantitative measurement of SIV length of zebrafish treated with CPT (0.01 to 100 nM) in the absence and in the presence of CB[7] (100 μM). The values were expressed as a percentage of the total SIV length of treated fish to that of the blank control (% control), where the control (untreated embryos) is 100%. ## denotes 100% death of fish treated with 1000 nM CPT; *** denotes a complete inhibition of SIV growth of fish treated with 1000 nM CPT in the presence of CB[7]. Data were plotted as mean ± S.E.M. (n = 12–16) of three independent experiments. | ||
Treatment with 100 nM CPT in the absence and in the presence of CB[7] for 2 days both completely inhibited SIV formation. Conversely, there was little change in SIVs following treatment with either free or complexed CPT of less than 1 nM (Fig. S2‡). Fig. 5(ii) shows the quantitative analysis of the anti-angiogenic activity of CPT in the absence and in the presence of CB[7] by the end of treatment for 2 days. The length of SIVs, a more sensitive and quantitative parameter for evaluating vasculature viability, showed comparable anti-angiogenic effects of CPT and CPT@CB[7], which again clearly demonstrated the dosage-dependent inhibition of SIV formation in both groups. The comparable in vivo anti-angiogenic activities of CPT and CPT@CB[7] seem to be consistent with the aforementioned in vitro anti-cancer activities.
The survival rate of zebrafish by the end of incubation was examined in order to evaluate the non-specific toxicity of this drug during the in vivo anti-angiogenic activity assay. The survival rate of the CPT@CB[7]-treated group at each concentration level was generally higher than that of the free CPT-treated group (Fig. S3‡). In particular, all of the zebrafish treated with 1000 nM free CPT died by the end of the incubation period, whereas the survival rate of zebrafish treated with 1000 nM CPT in the presence of CB[7] was nearly 70%, suggesting that CB[7]-encapsulated CPT (CPT@CB[7]) has a much lower systemic toxicity to treated zebrafish. Further evaluation of gross morphological changes of these zebrafish by the end of the incubation (Fig. 6) revealed that the free CPT induced pronounced deleterious effect to the growth of embryos, in comparison with the corresponding CPT@CB[7]-treated groups and the control group. Taking therapeutic efficacy and toxicity into account, the therapeutic indexes (TI, TI = LD50/ED50) of CPT and CPT@CB[7] complexes were 0.932 and 387.78, respectively, suggesting that CPT@CB[7] complexes have a much better safety margin.
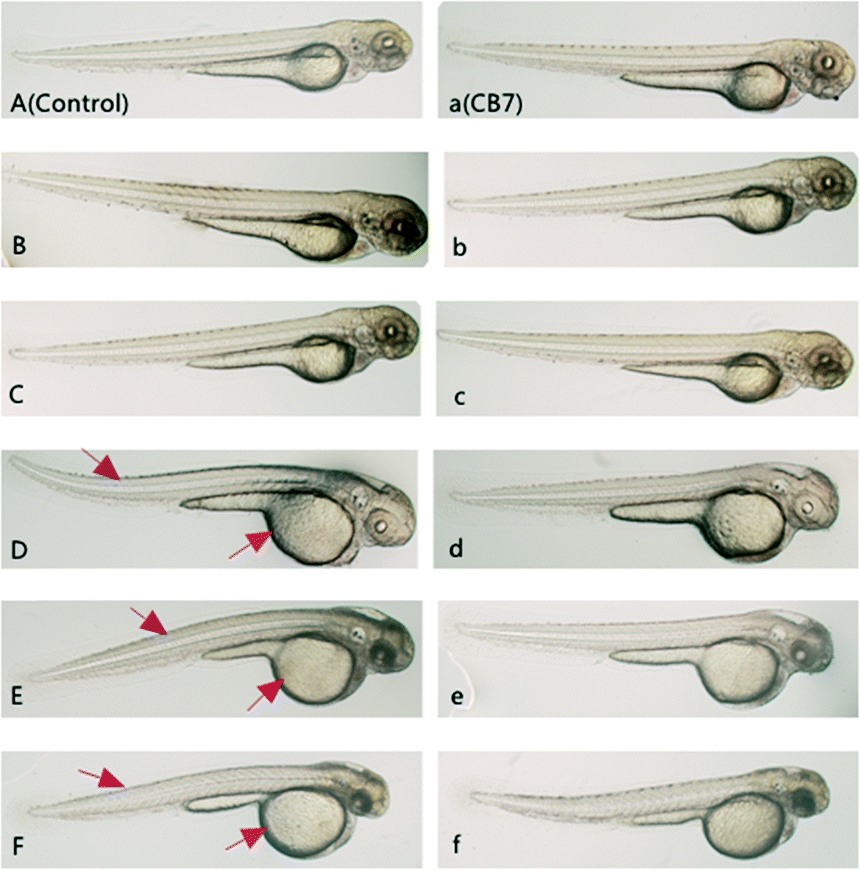 | ||
| Fig. 6 Morphological features of zebrafish embryos incubated with CPT or CPT@CB[7] for 2 days. (A) Fish of the blank control group; (a) fish treated with 100 μM CB[7]; (B–F/b–f) fish treated with various concentrations of CPT (0.1 to 100 nm) in the absence (B–F) and in the presence (b–f) of CB[7]. The red arrows point to obvious morphological abnormalities. | ||
With the in vivo Tg(fli-1:EGFP) transgenic zebrafish model, supramolecular encapsulation of CPT by CB[7] has significantly reduced its inherent non-specific systemic toxicity with well-preserved antitumor/anti-angiogenic efficacy. It is well-known that active CPT requires a lactone form; inhibition of the lactone form from hydrolysis into the carboxylate form is essential for keeping its bioactivity and inhibiting its toxic side-effects.22 The decreased toxicity of CPT upon encapsulation by CB[7] is likely attributed to a reduced hydrolysis rate of the active lactone form of CPT. Tao and co-workers have previously demonstrated that CB[7] indeed had the ability to protect CPT's active lactone form from hydrolysis in the phosphate buffer solution (pH 7.4 at 37 °C).22 When CPT is encapsulated inside the CB[7] cavity, the lactone moiety is located in the hydrophobic cavity of CB[7] (Fig. 2), thus being protected against hydrolysis. It is somewhat out of our expectation that the anticancer/anti-angiogenic activities of CPT, upon inclusion by CB[7], were not enhanced. This might be attributed to balancing effects between the increased concentration of the lactone form and a certain degree of activity inhibition caused by encapsulation.
Experimental
Ethics statement
All animal experiments were done in accord with the ethical guidelines of the Institute of Chinese Medical Sciences, University of Macau, and the protocols were approved by the Institute of Chinese Medical Sciences, University of Macau.Reagents and preparation
Camptothecin and other organic reagents including DMSO, NaCl, KCl, CaCl2, MgSO4 and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were all purchased from Sigma Aldrich (St. Louis, MO, USA). Cucurbit[7]uril was synthesized according to a literature method.35 RPMI culture solution, penicillin, streptomycin, trypsin, phosphate buffered saline (PBS) and fetal bovine serum (FBS) were supplied by GIBCO (Carlsbad, CA, USA). All of these reagents were used as received. VEGF receptor kinase inhibitor II (VRI) obtained from Calbiochem (Billerica, MA) was dissolved in DMSO and used as a positive control in the transgenic zebrafish angiogenic model. Camptothecin was dissolved in DMSO as a 10 mM stock solution and stored in the dark. The zebrafish medium was composed of 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4 (pH 7.2–7.3).Cell lines and culture
L02, a human healthy liver cell line, obtained from the Committee of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), and Bel-7402, a human hepatocellular carcinoma cell line, obtained from Shanghai Institute of Materia Medical, were cultured in RPMI 1640 media supplemented with 10% FBS and 1% penicillin–streptomycin in a humidified environment at 37 °C with 5% CO2. The medium was changed every 2 days.Cytotoxicity assay (MTT test)
Cytotoxicity of CPT and CPT@CB[7] complex were tested by colorimetric 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT) assay in vitro. Cells were plated in 96-well plates (2 × 103 per well) and were allowed to attach overnight. The test compounds were added with final doses ranging from 0.1 to 100 μM CPT (in the absence and in the presence of 200 μM CB[7]), and the cells were incubated for 24 h at 37 °C. Following incubation, 5 mg ml−1 MTT solution was added to each well and incubated for 3 h. DMSO was then added to each well to dissolve the formazan crystals. The DMSO-dissolved formazan crystals were read immediately at 540 nm with a multiplate reader (SpectraMax M5 Microplate Reader, Molecular Devices, USA).Zebrafish breeding
Zebrafish (Danio rerio) strains, including Tg(fli-1:EGFP) and wild type, were provided by the ZFIN (Oregon) for use as the in vivo animal models in our study. Zebrafish were kept at the dedicated aquarium area at the Institute of Chinese Medical Sciences, University of Macau. Maintenance and breeding were conducted in accordance with the standard procedures. Adult fish were kept in an aquaculture system with a 14 h light/10 h dark cycle under a controlled environment at a temperature of 28.5 °C. Fish were mainly fed with newly hatched brine shrimp twice daily, and occasionally fed with general tropical fish food. Mature male and female zebrafish (at a ratio of 1 male to 2 females) were moved into a dedicated aquarium tank for breeding. Embryos were collected in the morning of the second day and were subjected to a microscopic examination. Only those that developed normally were selected for subsequent culture and experiments at 28.5 °C in embryo medium. Wild type zebrafish embryos of AB background were used for the toxicity assays, whereas Tg(fli-1:EGFP) zebrafish with GFP (green fluorescent protein) specifically expressed GFP in the entire vasculature were used for anti-angiogenic assays and systemic toxicity studies.Toxicity study with wild-type zebrafish
For the toxicity study with zebrafish hatching assay, 1 day post-fertilization (dpf) wild type zebrafish embryos at the same developmental stage were randomly divided into a 24-well microplate (20 embryos per well) and incubated with 1 mL solutions of CPT at various concentrations (0, 0.01, 0.10, 1, 10, 100, 1000 nM) in the absence and in the presence of 100 μM CB[7], respectively, for 2 days. Embryos receiving 0.3% DMSO (v/v) served as a blank control group. The assay solutions in all treated groups were refreshed every 24 h and all groups were incubated at 28 °C. All experiments were repeated at least three times. After 2 days of incubation, the hatching rate was calculated through the ratio of hatched larvae/total treated embryos × 100%.Toxicity and anti-angiogenic assays with transgenic zebrafish
For anti-angiogenic assay and systemic toxicity studies, 1 dpf Tg(fli-1:EGFP) transgenic zebrafish embryos were dechorionated under a microscope by manual removal of the chorion with tweezers, and subsequently were randomly distributed into a 24-well microplate (12 to 16 fish per well), where they were exposed to 1 mL assay solutions of various concentrations of CPT (0, 0.01, 0.10, 1, 10, 100, 1000 nM) in the absence and in the presence of 100 μM CB[7], respectively, for 2 days. Embryos receiving 0.3% DMSO (v/v) served as a blank control group, and those receiving 250 nM VRI were used as a positive control group. The assay solutions in all treated groups were refreshed every day and all groups were incubated at 28 °C. All experiments were repeated at least three times.After incubation for 2 days, zebrafish embryos were removed from each well, anesthetized using 1% (w/v) tricaine and inspected for morphological changes, viability and blood vessel development using a fluorescence microscope (Olympus IX81 Motorized Inverted Microscope, Japan) equipped with a digital camera (DP controller, Soft Imaging System, Olympus, Germany). Images were analyzed with Image J and Adobe Photoshop 7.0. The length of sub-intestinal vessels (SIVs) in each zebrafish embryo was measured using software and manual point to point measurement.
Statistical analysis
All data are presented as mean ± S.E.M., from at least three independent experiments. Diagrams and statistical analysis using a one-way ANOVA test was performed with GraphPad Prism Software (GraphPad Software, La Jolla CA, USA). The statistical significance was calculated using the t-test. Meanwhile, the Mann–Whitney Test was used when comparing two treatment groups.Conclusions
In summary, we have demonstrated for the first time with both in vitro and in vivo models that supramolecular encapsulation of camptothecin by macrocyclic cucurbit[7]uril may significantly inhibit the systemic toxicities of the free drug while its antitumor/anti-angiogenic activities have been well-preserved. These results suggest that cucurbit[7]uril is a potent drug delivery carrier that may help alleviate systemic side-effects of encapsulated drugs, and it has great potential to be developed further as a new pharmaceutical excipient for future clinical applications.Acknowledgements
Macau Science and Technology Development Fund (FDCT/020/2015/A1) and University of Macau Research Fund (SRG2014-00025) are gratefully acknowledged for providing financial support. We thank Dr. Jian Ding, Shanghai Institute of Materia Medical, Chinese Academy of Sciences, for providing the Bel-7402 cell line. We also thank Dr. Partha Hazra, Department of Chemistry, Mendeleev Block, Indian Institute of Science Education and Research, for providing the molecular modeling data of CPT@CB[7], which was used to re-construct Fig. 2 in this work.Notes and references
- V. J. Venditto and E. E. Simanek, Mol. Pharmaceutics, 2010, 7, 307–349 CrossRef CAS PubMed.
- M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS.
- R. P. Hertzberg, M. J. Caranfa and S. M. Hecht, Biochemistry, 1989, 28, 4629–4638 CrossRef CAS PubMed.
- J. F. Powers, P. G. Korgaonkar, S. Fliedner, A. Giubellino, K. P. G. G. Sahagian and A. S. Tischler, PLoS One, 2014, 9, e87807 Search PubMed.
- J. Lu, C. Liu, P. Wang, M. Ghazwani, J. Xu, Y. Huang, X. Ma, P. Zhang and S. Li, Biomaterials, 2015, 62, 176–187 CrossRef CAS PubMed.
- I. Chourpa, J.-M. Millot, G. D. Sockalingum, J.-F. Riou and M. Manfait, Biochim. Biophys. Acta, 1998, 1379, 353–366 CrossRef CAS.
- K. S. Cunha, M. L. Reguly, U. Graf and H. H. Rodrigues de Andrade, Mutagenesis, 2002, 17, 141–147 CrossRef CAS PubMed.
- A. Hatefi and B. Amsden, Pharm. Res., 2002, 19, 1389–1399 CrossRef CAS.
- P. Botella, I. Abasolo, Y. Fernández, C. Muniesa, S. Miranda, M. Quesada, J. Ruiz, S. Schwartz and A. Corma, J. Controlled Release, 2011, 156, 246–257 CrossRef CAS PubMed.
- J. Kang, V. Kumar, D. Yang, P. R. Chowdhury and R. J. Hohl, Eur. J. Pharm. Sci., 2002, 15, 163–170 CrossRef CAS PubMed.
- K. Gavvala, A. Sengupta and P. Hazra, ChemPhysChem, 2013, 14, 532–542 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed.
- D. Shetty, J. K. Khedkar, K. M. Park and K. Kim, Chem. Soc. Rev., 2015, 44, 8747–8761 RSC.
- D. H. Macartney, Isr. J. Chem., 2011, 51, 600–615 CrossRef CAS.
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, Isr. J. Chem., 2011, 51, 616–624 CrossRef CAS.
- D. H. Macartney, Future Med. Chem., 2013, 5, 2075–2089 CrossRef CAS PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- R. Oun, R. S. Floriano, L. Isaacs, E. G. Rowan and N. J. Wheate, Toxicol. Res., 2014, 3, 447–455 RSC.
- H. Chen, J. Y. W. Chan, X. Yang, I. W. Wyman, D. Bardelang, D. H. Macartney, S. M. Y. Lee and R. Wang, RSC Adv., 2015, 5, 30067–30074 RSC.
- N. Dong, M. Dong, A. Zhao, Q. Zhu, Z. Tao and Y. Zhao, Sci. China: Chem., 2010, 53, 2304–2310 CrossRef CAS.
- N. Dong, S.-F. Xue, Q.-J. Zhu, Z. Tao, Y. Zhao and L.-X. Yang, Supramol. Chem., 2008, 20, 663–671 CrossRef.
- S. Li, H. Yin, G. Martinz, I. W. Wyman, D. Bardelang, D. H. Macartney and R. Wang, New J. Chem., 2016, 40, 3484–3490 RSC.
- S. Li, H. Yin, I. W. Wyman, Q. Zhang, D. H. Macartney and R. Wang, J. Org. Chem., 2016, 81, 1300–1303 CrossRef CAS PubMed.
- S. Li, H. Chen, X. Yang, D. Bardelang, I. W. Wyman, J. Wan, S. M. Lee and R. Wang, ACS Med. Chem. Lett., 2015, 6, 1174–1178 CrossRef CAS PubMed.
- H. Chen, J. Y. W. Chan, S. Li, J. J. Liu, I. W. Wyman, S. M. Y. Lee, D. H. Macartney and R. Wang, RSC Adv., 2015, 5, 63745–63752 RSC.
- X. Miao, Y. Li, I. Wyman, S. M. Y. Lee, D. H. Macartney, Y. Zheng and R. Wang, MedChemComm, 2015, 6, 1370–1374 RSC.
- J. Folkman, Nat. Med., 1995, 1, 27–30 CrossRef CAS PubMed.
- I. K. Lam, D. Alex, Y.-H. Wang, P. Liu, A.-L. Liu, G.-H. Du and S. M. Yuen Lee, Mol. Nutr. Food Res., 2012, 56, 945–956 CAS.
- U. Langheinrich, BioEssays, 2003, 25, 904–912 CrossRef CAS PubMed.
- N. D. Lawson and B. M. Weinstein, Dev. Biol., 2002, 248, 307–318 CrossRef CAS PubMed.
- G. N. Serbedzija, E. Flynn and C. E. Willett, Angiogenesis, 1999, 3, 353–359 CrossRef CAS.
- C. X. Song, S. L. Song, H. Liang and X. Liu, Adv. Mater. Res., 2013, 750–752, 1472–1475 CrossRef.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00239k |
| This journal is © The Royal Society of Chemistry 2016 |