
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Human lysosomal acid lipase inhibitor lalistat impairs Mycobacterium tuberculosis growth by targeting bacterial hydrolases†‡
J.
Lehmann§
a,
J.
Vomacka§
 a,
K.
Esser§
b,
M.
Nodwell
a,
K.
Kolbe
c,
P.
Rämer
d,
U.
Protzer
be,
N.
Reiling
*c and
S. A.
Sieber
*a
a,
K.
Esser§
b,
M.
Nodwell
a,
K.
Kolbe
c,
P.
Rämer
d,
U.
Protzer
be,
N.
Reiling
*c and
S. A.
Sieber
*a
aDepartment of Chemistry, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: stephan.sieber@tum.de
bInstitut für Virologie, Technische Universität München/Helmholtz Zentrum München, Trogerstraße 30, 81675 München, Germany
cForschungszentrum Borstel, Leibniz-Zentrum für Medizin und Biowissenschaften, FG Mikrobielle Grenzflächenbiologie, Parkallee 22, 23845 Borstel, Germany. E-mail: nreiling@fz-borstel.de
dInstitut für Medizinische Mikrobiologie, Immunologie und Hygiene, Technische Universität München, Trogerstraße 30, 81675 München, Germany
eGerman Center for Infection Research (DZIF), Munich partner site, Germany
First published on 21st July 2016
Abstract
Lalistat inhibits growth of Mycobacterium tuberculosis in bacterial culture as well as in infected macrophages. Target identification by quantitative proteomics revealed a cluster of 20 hydrolytic proteins including members of the lipase family. Lipases are essential for M. tuberculosis fatty acid production and energy storage thus representing promising antibiotic targets.
Tuberculosis (TB) is one of the world's most prevalent diseases responsible for the largest fraction of infection related casualties.1 The disease is caused by Mycobacterium tuberculosis a pathogen difficult to treat by standard antibiotics requiring therapies with a combination of drugs such as isoniazid, rifampicin, ethambutol and pyrazinamide. Moreover, in recent years a strong increase in multidrug-resistant (MDR) TB cases (resistant to at least isoniazid and rifampicin) was observed. It is estimated that around 9% of MDR TB samples were also extensively drug-resistant (XDR) TB, i.e., resistant to isoniazid and rifampin and to at least one representative of each class of the most effective second-line drugs (i.e., fluoroquinolones and the injectable drugs kanamycin, amikacin, or capreomycin).1 This augments the challenge of treatment and requires new and more efficient drug development strategies. However, these are challenged by a limited number of exploited targets and impaired uptake of small molecule drugs by the highly impermeable mycobacterial cell wall.2
M. tuberculosis is transmitted by aerosol and is initially taken up by alveolar macrophages, which phagocytose but do not kill the bacterium. Most infected individuals can immunologically control the disease. It is assumed that one-third of the world's population is latently infected. The bacteria adapt and survive in diverse environmental niches in vivo, e.g. in solid granulomas, a characteristic feature of latent TB infection.3 It is presumed that M. tuberculosis resides in these regions in a slow growing or non-replicating, phenotypically drug resistant dormant-like state, due to limited availability and supply of oxygen and nutrients.3,4 During infection experiments it has been shown that M. tuberculosis accumulates triacylglycerols (TAGs) within intracellular inclusion bodies.5–7 The hydrolysis of TAGs to free fatty acids is an essential prerequisite for M. tuberculosis growth and survival requiring diverse lipases and hydrolases.6,8–10 During infection the liberation of fatty acids represents an important energy source11 triggered by the availability of lipids in the host cell.12 Moreover, it is believed that these fatty acids are crucial for bacteria to enter and maintain the dormant state by the production of foamy lung macrophages during latent infection.9,13 Accordingly, the M. tuberculosis genome encodes numerous hydrolytic enzymes involved in lipid metabolism.14 Among those, the lip gene family consist of 24 lipid/ester hydrolases termed Lip C to Z which share a conserved active site as well as alpha/beta hydrolase fold.6,15 Specifically targeting the bacterial lipid metabolism, could represent a viable strategy to address M. tuberculosis10 and open a new opportunity to shorten the currently required long-term TB therapy, when given simultaneously with known first and second line antibiotics.16 Thus focusing on known mammalian lipase inhibitors such as lalistat (La-0)17 and orlistat18 could represent a novel therapeutic strategy inhibiting TAG metabolism. Despite their prominent role only little is known about the exact function of lipid hydrolysing enzymes in mycobacteria. Orlistat was previously shown to reduce viability and mycolic acid biosynthesis for numerous mycobacterial strains.19 In depth target analysis via activity based protein profiling (ABPP)20 with an alkynylated orlistat probe in M. bovis BCG revealed binding of multiple enzymes belonging to the Lip family. Orlistat or related hydrolase inhibitors that address the mycobacterial lipid metabolism thus represent attractive starting points for medicinal chemistry.21,22
We here show, that lalistat exhibits an inhibitory effect on M. tuberculosis reproduction and further investigated its mode of action by in situ target identification via ABPP. Several bacterial lipases were identified suggesting inhibition of TAG hydrolysis as the mode of action.
Lipases are important metabolic enzymes for many prokaryotic and eukaryotic organisms. Several compounds have been developed to inhibit eukaryotic hydrolases specifically e.g. for the treatment of obesity.23,24 The beta-lactone orlistat represents a broad lipase inhibitor acting on a wide range of triglyceride- and cholesterol ester hydrolysing mammalian enzymes.25,26 In addition, La-0 is a thiadiazole carbamate (Fig. 2A) developed as a specific inhibitor of the lysosomal acid lipase (LAL).27 This enzyme is located in cellular late endosomes hydrolysing cholesterol esters and triglycerides from incoming lipoproteins.24 Lalistat, similarly to orlistat, binds to the active site serine of lipases covalently and thereby inhibits enzyme activity.27,28 Given the structural and electronic properties of La-0 we anticipated binding to diverse lipases which may deviate from the orlistat target spectrum and complement mechanistic insights. We thus inspected if lalistat impairs viability of M. tuberculosis by several independent approaches.
First, minimal inhibitory concentrations (MICs) were determined by addition of La-0 to diluted cultures of M. tuberculosis H37Rv and utilizing the resazurin assay.29–31 The compound exhibited a MIC of 25–50 μM (Fig. S1‡) exceeding the potency of previously studied orlistat ∼30 μM.19 Currently applied standard therapeutics such as rifampicin and isoniazid exhibit MIC values in the range of 0.04 to 1.0 μM.32M. tuberculosis infection is treated by a combination of up to four different drugs; we therefore tested mixtures of La-0 and various (myco-)bacterial drugs on two-dimensional MIC plates. Remarkably, a strong effect was obtained for the combined application of lalistat and vancomycin resulting in a MIC drop of factor 4 for lalistat and 16 for vancomycin (Fig. 1A). Using the fractional inhibitory concentration index (FICI)33,34 a cooperative effect could be concluded as both molecules interfere with the composition and assembly of the cell wall. Furthermore, growth of green fluorescent protein (GFP) expressing M. tuberculosis H37Rv was monitored for 7 days in presence of either La-0 or rifampicin (control) as described by Michelucci et al.35 Interestingly, growth inhibition was observed for lalistat down to a concentration of 4 μM (Fig. 1B). Finally, the effect of La-0 on intracellular growth of M. tuberculosis in human macrophage host cells was determined. To this end we infected human monocyte derived macrophages with strain M. tuberculosis H37Rv (multiplicity of infection 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 4 h and subsequently cultured the cells for 7 days with various concentrations of La-0 and DMSO (control). Cells were then lysed and CFUs were determined as described previously.36 Again lalistat substantially reduced bacterial load by 55% compared to the untreated control suggesting that the compound can even address intracellular bacteria (Fig. 1C). These results point towards an essential mechanism addressed by lalistat and given its specificity for lipases a link to essential metabolic processes was subsequently evaluated by in situ target identification.
:1) for 4 h and subsequently cultured the cells for 7 days with various concentrations of La-0 and DMSO (control). Cells were then lysed and CFUs were determined as described previously.36 Again lalistat substantially reduced bacterial load by 55% compared to the untreated control suggesting that the compound can even address intracellular bacteria (Fig. 1C). These results point towards an essential mechanism addressed by lalistat and given its specificity for lipases a link to essential metabolic processes was subsequently evaluated by in situ target identification.
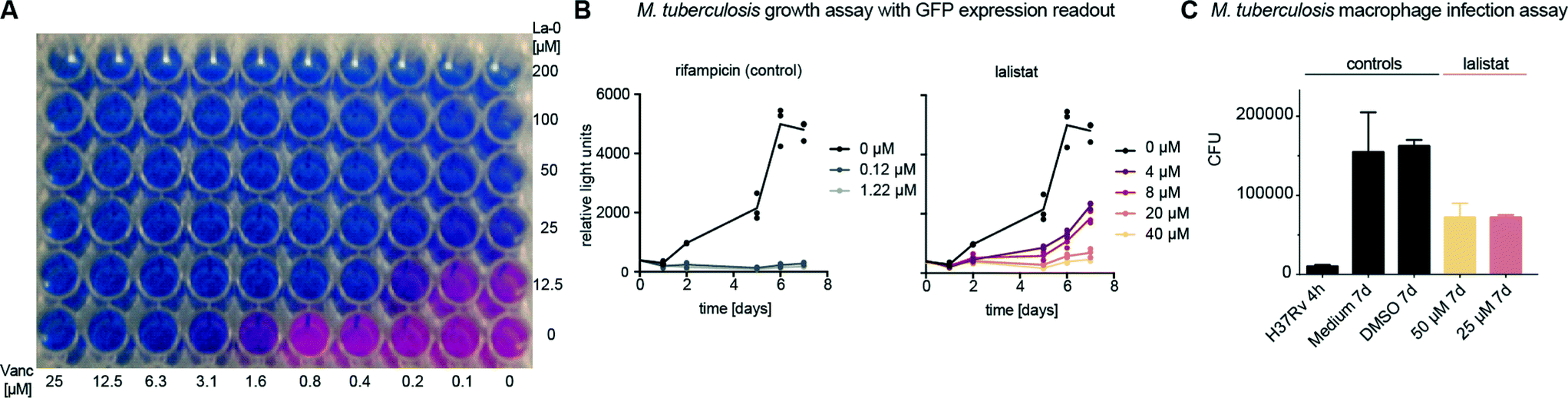 | ||
| Fig. 1 Lalistat inhibits M. tuberculosis growth. (A) Two-dimensional MIC determination. Lanes: La-0 dose-down. Rows: vancomycin dose-down. (B) Inhibition of GFP-expressing M. tuberculosis H37Rv with rifampicin (left) and lalistat (right). All experiments were performed together with the same DMSO control (identical in both graphs). (C) Colony forming units (cfu) of M. tuberculosis H37Rv recovered from human macrophages in presence and absence of inhibitors. Shown is one representative experiment out of two performed, each consisting of three (B) and two (C) technical replicates. | ||
To analyse the mode of action we identified lalistat protein targets in live M. tuberculosis cells via ABPP. For this technology a chemical probe bearing an alkyne tag (La-1) was synthesized following established protocols (Fig. 2A).27 In brief, a piperidine moiety was introduced in commercially available 3,4-dichloro-1,2,5-thiadiazole, replacing one of the chlorine substituents while the other was subsequently converted into an hydroxyl-function using potassium hydroxide (Scheme 1). The newly synthesized hydroxy-heterocycle was utilized as a common precursor for lalistat (La-0) and its alkynylated probe (La-1) using piperidyl chloroformate or 4-ethylylpiperidine chloroformate, respectively, to form the essential carbamate functionality.
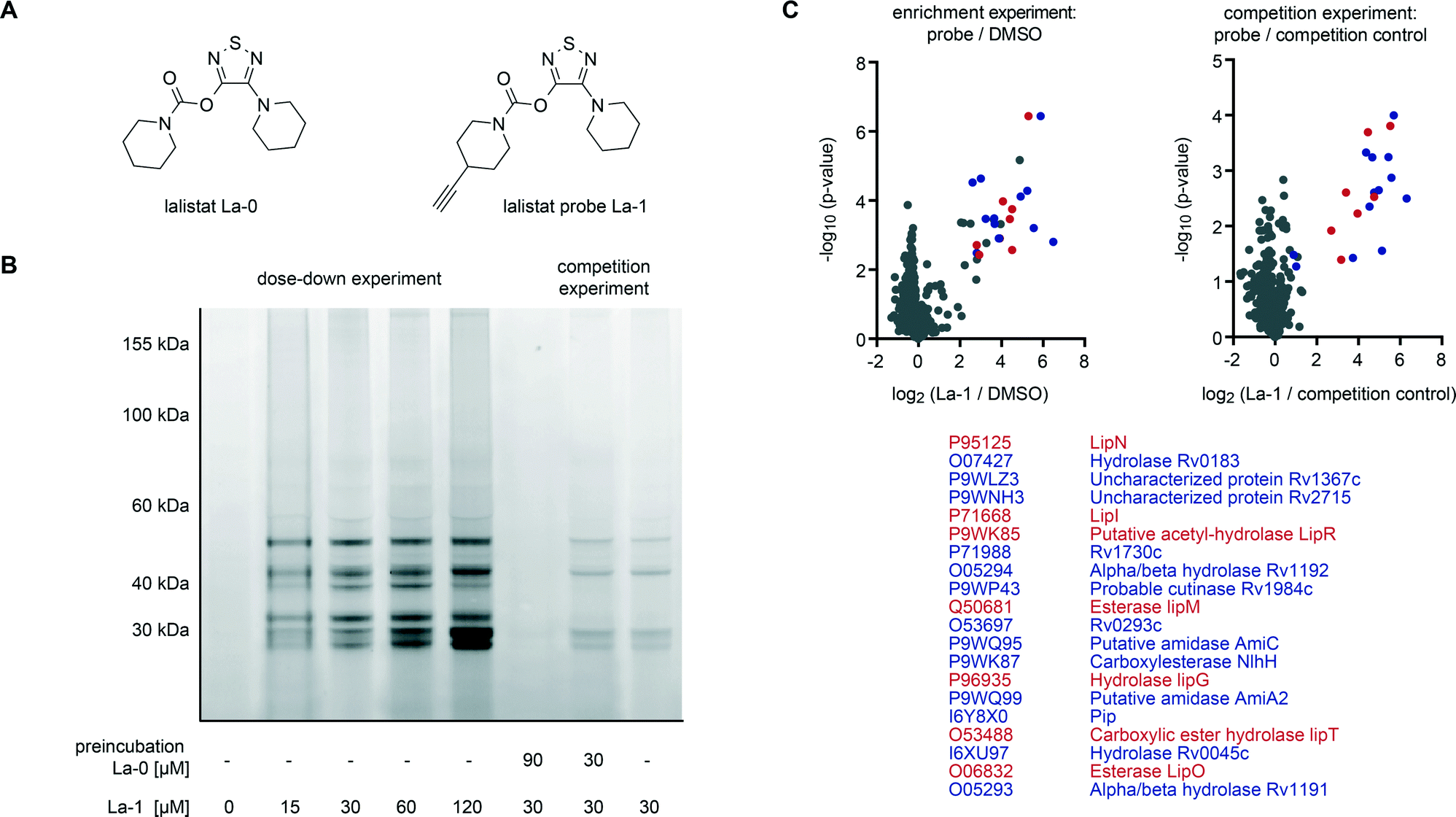 | ||
| Fig. 2 In vivo target identification of lalistat in M. tuberculosis H37Rv via chemical proteomics. (A) Structure of lalistat and the corresponding probe. (B) Concentration dependent labelling and competition experiment visualized via SDS-PAGE and fluorescent scanning. (C) Enrichment (6 biological replicates) and competition (4 biological replicates) target identification (ABPP) volcano plot representations and corresponding list of common hits (blue and red). Proteins of the Lip family are marked in red, all others in blue. | ||
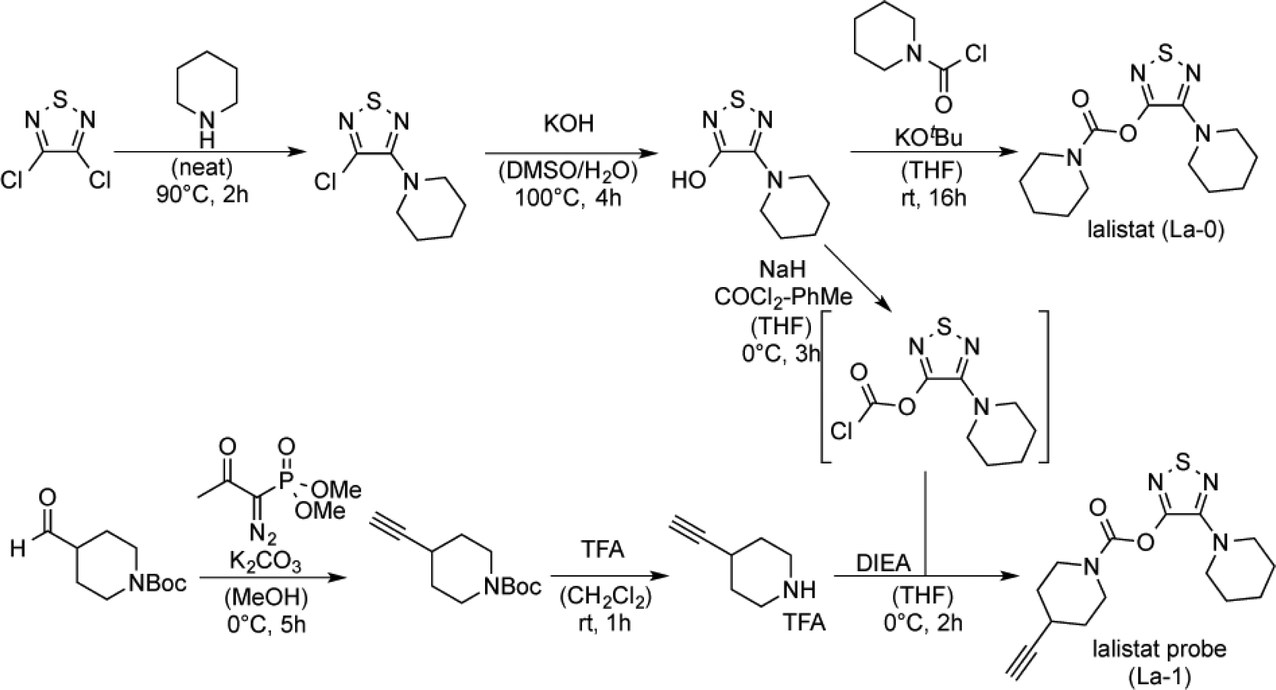 | ||
| Scheme 1 Synthesis of lalistat (La-0) and lalistat probe (La-1). DMSO: dimethylsulfoxide, THF: tetrahydrofuran, Boc: tert-butyloxycarbonyl-, TFA: trifluoroacetic acid, DIEA: N,N-diisopropylethylamine. | ||
Satisfyingly, La-1 retained activity and reduced M. tuberculosis growth comparable to the parent molecule La-0 (Fig. S1‡). Next, target identification experiments were conducted. M. tuberculosis cells were grown to log phase and incubated with La-1 or DMSO as a control. After cell lysis a rhodamine tag was attached to the alkyne moiety of La-1 via bioorthogonal click-reaction37–39 to visualize target proteins on SDS-PAGE gel by fluorescence scanning. Protein labelling at different probe concentrations are displayed in Fig. 2B. Interestingly, proteins around a molecular size of 30 kDa showed increased fluorescent signal response with raising probe concentration while others (∼50 kDa) only marginally changed intensity. A concentration between 30 μM and 60 μM was sufficient to achieve saturated labelling. We thus proceeded with gel-free proteomics and performed labeling studies with 30 μM La-1 in M. tuberculosis H37Rv to unravel the identities of target proteins. For these studies a biotin tag served as a handle to enrich labeled proteins on avidin beads (Fig. S2‡). After on-bead digestion primary amines were modified by heavy, medium and light isotopes using dimethyl labeling (DL).40 Labelling ratios of probe versus DMSO were normalized, z-score and log2(x) transformed. Target proteins with a high enrichment and statistical significance (see ESI‡ for details) are located in the upper right area of the enrichment volcano plot in Fig. 2C. To validate these enriched hits we performed competition experiments where cells were pre-incubated with La-0 (60 μM) and subsequently treated with 30 μM La-1 probe (Fig. 2C). Strikingly, spectra of enrichment and competition experiments include 7 Lip enzymes (LipN, -I, -R, -M, -G, -T and -O) that selectively react with lalistat.
A comparison of lalistat hits with results of previous orlistat proteome labeling studies in M. bovis (BCG)21 revealed Lip M, O, N, I and G as shared targets. Interestingly, while Lip R and T were selectively captured by orlistat-like probes suggesting a complementary target profile by both lipase inhibitors due to their different physico-chemical properties. It is thus intriguing to speculate that the shared preference for a common set of Lip enzymes may contribute to the growth inhibitory effects of both compounds.
LipR as one of the most strongly enriched hits by La-1, was exemplary confirmed as target by a heterologous expression and subsequent labeling with the probe. A strong fluorescent band indicated specific binding (Fig. 3). Beside members of the Lip family we identified six additional enzymes with lipolytic activity: Rv0183, a highly conserved monoalcylglycerol lipase,41,42 Rv1984 (ref. 43 and 44) a cutinase preferentially active against monoacylglycerols, as well as the uncharacterized proteins Rv2715, Rv1192, Rv1399c (characterized esterase)45 and Rv0045c46–48 which are suggested to participate in lipid hydrolysis in mycobacterial metabolism.
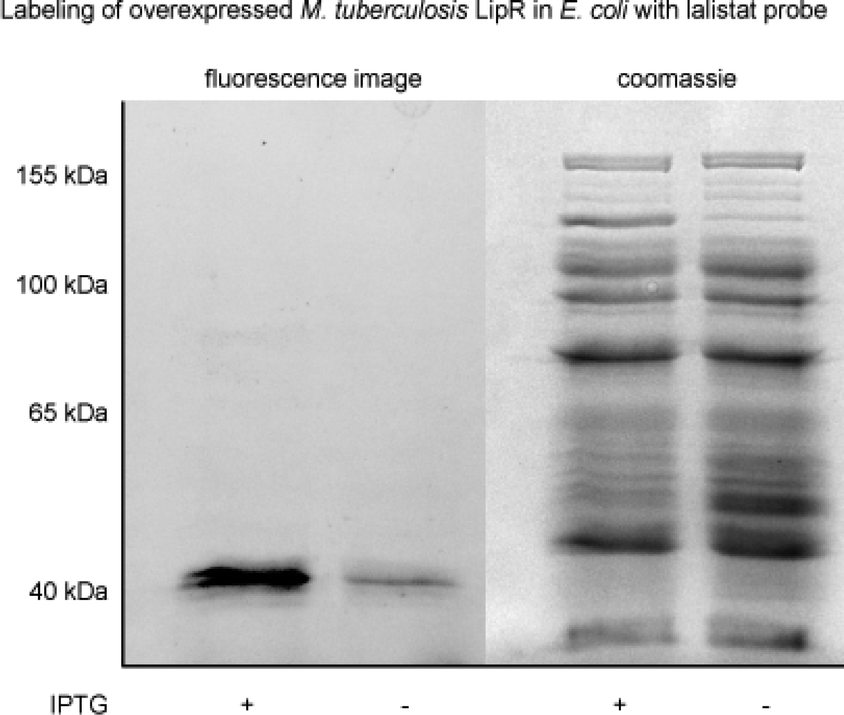 | ||
| Fig. 3 Validation of protein target LipR. LipR was overexpressed in E. coli and labelled with lalistat probe La-1. Rhodamine-azide was attached via click reaction after cell lysis. The experiment was conducted with and without induction by IPTG (indicated with “+” and “−”, respectively). | ||
We also detected few putative non-lipolytic enzymes bound by lalistat: Rv1367 and Rv1730, both possibly involved in cell wall biosynthesis, as well as amidases (Rv2888c and Rv2363), iminopeptidase (Rv0840c) and two proteins of unknown function (Rv0293c, Rv1191). To date it is not clear whether mycobacterial lipases hydrolysing short-chain substrates, like LipR, would participate in non-lipolytic metabolic processes because it remains unknown whether they have additional phospholipase, thioesterase or protease activity.49
Conclusions
Taken together, the specificity of lalistat for a suite of mycobacterial lipases is intriguing. Given the importance of these enzymes for M. tuberculosis viability during infection they may represent promising drug targets. Future studies need to further dissect and characterize the exact function and mechanism of these enzymes in order to design customized inhibitors suited to interfere with essential metabolic processes specifically in the bacteria. Here, orlistat and lalistat probes represent interesting chemical tools that allow simultaneous and selective detection of lipase activities in living mycobacteria. With regard to the paucity of potent anti-mycobacterial inhibitors future tuberculosis treatment approaches could largely benefit form adding these compounds into treatment regimens. They could also pave the way for the development of more specific drugs targeting mycobacterial lipases. Their use in combination with known anti-mycobacterial agents may offer an urgently needed opportunity to improve and shorten TB therapy.Acknowledgements
The work was funded by the Deutsche Forschungsgemeinschaft (DFG) SI1096/6-1 to SAS. JL was supported by the Studienstiftung des deutschen Volkes. NR was funded by a grant of the DFG Excellence Cluster EXC306 “Inflammation at Interfaces”. KK was supported by a doctoral fellowship of the Verband der Deutschen Chemischen Industrie e.V. (VCH). We gratefully acknowledge Lisa Niwinski and Carolin Golin (FZ Borstel), Mona Wolff and Katja Bäuml (TUM) for excellent technical support. We thank Eric Rubin for helpful advice and support.Notes and references
- WHO, Global tuberculosis report, 2015 Search PubMed.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- M. Gengenbacher and S. H. Kaufmann, FEMS Microbiol. Rev., 2012, 36, 514–532 CrossRef CAS PubMed.
- D. G. Russell, B. C. VanderVen, W. Lee, R. B. Abramovitch, M. J. Kim, S. Homolka, S. Niemann and K. H. Rohde, Cell Host Microbe, 2010, 8, 68–76 CAS.
- N. J. Garton, M. Gilleron, T. Brando, H. H. Dan, S. Giguere, G. Puzo, J. F. Prescott and I. C. Sutcliffe, J. Biol. Chem., 2002, 277, 31722–31733 CrossRef CAS PubMed.
- G. Singh, G. Singh, D. Jadeja and J. Kaur, Crit. Rev. Microbiol., 2010, 36, 259–269 CrossRef CAS PubMed.
- H. Christensen, N. J. Garton, R. W. Horobin, D. E. Minnikin and M. R. Barer, Mol. Microbiol., 1999, 31, 1561–1572 CrossRef CAS PubMed.
- WHO, Tuberculosis fact sheet, 2010, p. 104 Search PubMed.
- J. Daniel, H. Maamar, C. Deb, T. D. Sirakova and P. E. Kolattukudy, PLoS Pathog., 2011, 7, e1002093 CAS.
- C. Deb, C. M. Lee, V. S. Dubey, J. Daniel, B. Abomoelak, T. D. Sirakova, S. Pawar, L. Rogers and P. E. Kolattukudy, PLoS One, 2009, 4, e6077 Search PubMed.
- H. Bloch and W. Segal, J. Bacteriol., 1956, 72, 132–141 CAS.
- D. G. Russell, P. J. Cardona, M. J. Kim, S. Allain and F. Altare, Nat. Immunol., 2009, 10, 943–948 CrossRef CAS PubMed.
- P. Peyron, J. Vaubourgeix, Y. Poquet, F. Levillain, C. Botanch, F. Bardou, M. Daffe, J. F. Emile, B. Marchou, P. J. Cardona, C. de Chastellier and F. Altare, PLoS Pathog., 2008, 4, e1000204 Search PubMed.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS PubMed.
- L. Dedieu, C. Serveau-Avesque, L. Kremer and S. Canaan, Biochimie, 2013, 95, 66–73 CrossRef CAS PubMed.
- C. Dye, S. Scheele, P. Dolin, V. Pathania and M. C. Raviglione, JAMA, J. Am. Med. Assoc., 1999, 282, 677–686 CrossRef CAS PubMed.
- J. Hamilton, I. Jones, R. Srivastava and P. Galloway, Clin. Chim. Acta, 2012, 413, 1207–1210 CrossRef CAS PubMed.
- P. Y. Yang, K. Liu, M. H. Ngai, M. J. Lear, M. R. Wenk and S. Q. Yao, J. Am. Chem. Soc., 2010, 132, 656–666 CrossRef CAS PubMed.
- L. Kremer, C. de Chastellier, G. Dobson, K. J. Gibson, P. Bifani, S. Balor, J. P. Gorvel, C. Locht, D. E. Minnikin and G. S. Besra, Mol. Microbiol., 2005, 57, 1113–1126 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- M. S. Ravindran, S. P. Rao, X. Cheng, A. Shukla, A. Cazenave-Gassiot, S. Q. Yao and M. R. Wenk, Mol. Cell Proteomics, 2014, 13, 435–448 CAS.
- M. Fonovic and M. Bogyo, Expert Rev. Proteomics, 2008, 5, 721–730 CrossRef CAS PubMed.
- L.-N. Zhang, J. Vincelette, D. Chen, R. D. Gless, S.-K. Anandan, G. M. Rubanyi, H. K. Webb, D. E. MacIntyre and Y.-X. Wang, Eur. J. Pharmacol., 2011, 654, 68–74 CrossRef CAS PubMed.
- A. Iyer, D. P. Fairlie, J. B. Prins, B. D. Hammock and L. Brown, Nat. Rev. Endocrinol., 2010, 6, 71–82 CrossRef CAS PubMed.
- B. Borgstrom, Biochim. Biophys. Acta, 1988, 962, 308–316 CrossRef CAS.
- E. K. Weibel, P. Hadvary, E. Hochuli, E. Kupfer and H. Lengsfeld, J. Antibiot., 1987, 40, 1081–1085 CrossRef CAS PubMed.
- A. I. Rosenbaum, C. C. Cosner, C. J. Mariani, F. R. Maxfield, O. Wiest and P. Helquist, J. Med. Chem., 2010, 53, 5281–5289 CrossRef CAS PubMed.
- P. Hadvary, W. Sidler, W. Meister, W. Vetter and H. Wolfer, J. Biol. Chem., 1991, 266, 2021–2027 CAS.
- R. S. Reis, I. Neves Jr., S. L. Lourenco, L. S. Fonseca and M. C. Lourenco, J. Clin. Microbiol., 2004, 42, 2247–2248 CrossRef CAS PubMed.
- S. Sungkanuparph, R. Pracharktam, A. Thakkinstian, B. Buabut and W. Kiatatchasai, J. Med. Assoc. Thai, 2002, 85, 820–824 Search PubMed.
- J. C. Palomino, A. Martin, M. Camacho, H. Guerra, J. Swings and F. Portaels, Antimicrob. Agents Chemother., 2002, 46, 2720–2722 CrossRef CAS PubMed.
- R. J. Wallace Jr., D. R. Nash, L. C. Steele and V. Steingrube, J. Clin. Microbiol., 1986, 24, 976–981 CAS.
- K. R. Caleffi-Ferracioli, F. G. Maltempe, V. L. Siqueira and R. F. Cardoso, Tuberculosis, 2013, 93, 660–663 CrossRef CAS PubMed.
- D. F. Bruhn, M. S. Scherman, J. Liu, D. Scherbakov, B. Meibohm, E. C. Bottger, A. J. Lenaerts and R. E. Lee, Sci. Rep., 2015, 5, 13985 CrossRef CAS PubMed.
- A. Michelucci, T. Cordes, J. Ghelfi, A. Pailot, N. Reiling, O. Goldmann, T. Binz, A. Wegner, A. Tallam, A. Rausell, M. Buttini, C. L. Linster, E. Medina, R. Balling and K. Hiller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7820–7825 CrossRef CAS PubMed.
- N. Reiling, S. Homolka, K. Walter, J. Brandenburg, L. Niwinski, M. Ernst, C. Herzmann, C. Lange, R. Diel, S. Ehlers and S. Niemann, mBio, 2013, 4(4), e00250-13 CrossRef PubMed.
- R. Huisgen, Proc. Chem. Soc., 1961, 357–396 Search PubMed.
- V. V. Rostovtsev, J. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- J. L. Hsu, S. Y. Huang, N. H. Chow and S. H. Chen, Anal. Chem., 2003, 75, 6843–6852 CrossRef CAS PubMed.
- P. Saravanan, V. K. Dubey and S. Patra, Chem. Biol. Drug Des., 2012, 79, 1056–1062 CAS.
- K. Cotes, R. Dhouib, I. Douchet, H. Chahinian, A. de Caro, F. Carriere and S. Canaan, Biochem. J., 2007, 408, 417–427 CrossRef CAS PubMed.
- L. Dedieu, C. Serveau-Avesque and S. Canaan, PLoS One, 2013, 8, e66913 CAS.
- M. Schue, D. Maurin, R. Dhouib, J. C. Bakala N'Goma, V. Delorme, G. Lambeau, F. Carriere and S. Canaan, FASEB J., 2010, 24, 1893–1903 CrossRef CAS PubMed.
- S. Canaan, D. Maurin, H. Chahinian, B. Pouilly, C. Durousseau, F. Frassinetti, L. Scappuccini-Calvo, C. Cambillau and Y. Bourne, Eur. J. Biochem., 2004, 271, 3953–3961 CrossRef CAS PubMed.
- X. Zheng, J. Guo, L. Xu, H. Li, D. Zhang, K. Zhang, F. Sun, T. Wen, S. Liu and H. Pang, PLoS One, 2011, 6, e20506 CAS.
- C. P. Savas, A. Gehring, R. J. Johnson and G. Hoops, FASEB J., 2013, 27, 559.2 Search PubMed.
- J. Guo, X. Zheng, L. Xu, Z. Liu, K. Xu, S. Li, T. Wen, S. Liu and H. Pang, PLoS One, 2010, 5 Search PubMed.
- V. Delorme, S. V. Diomande, L. Dedieu, J. F. Cavalier, F. Carriere, L. Kremer, J. Leclaire, F. Fotiadu and S. Canaan, PLoS One, 2012, 7, e46493 CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00231e |
| § These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2016 |