
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open PHACTS computational protocols for in silico target validation of cellular phenotypic screens: knowing the knowns†‡
D.
Digles§
*a,
B.
Zdrazil
 a,
J.-M.
Neefs
b,
H.
Van Vlijmen
b,
C.
Herhaus
c,
A.
Caracoti
d,
J.
Brea
e,
B.
Roibás
e,
M. I.
Loza
e,
N.
Queralt-Rosinach
f,
L. I.
Furlong
f,
A.
Gaulton
g,
L.
Bartek
h,
S.
Senger
h,
C.
Chichester
ij,
O.
Engkvist
k,
C. T.
Evelo
l,
N. I.
Franklin
m,
D.
Marren
n,
G. F.
Ecker
a and
E.
Jacoby§
*b
a,
J.-M.
Neefs
b,
H.
Van Vlijmen
b,
C.
Herhaus
c,
A.
Caracoti
d,
J.
Brea
e,
B.
Roibás
e,
M. I.
Loza
e,
N.
Queralt-Rosinach
f,
L. I.
Furlong
f,
A.
Gaulton
g,
L.
Bartek
h,
S.
Senger
h,
C.
Chichester
ij,
O.
Engkvist
k,
C. T.
Evelo
l,
N. I.
Franklin
m,
D.
Marren
n,
G. F.
Ecker
a and
E.
Jacoby§
*b
aDepartment of Pharmaceutical Chemistry, University of Vienna, Pharmacoinformatics Research Group, Althanstraße 14, 1090 Wien, Austria. E-mail: daniela.digles@univie.ac.at
bJanssen Research & Development, Turnhoutseweg 30, B-2340 Beerse, Belgium. E-mail: ejacoby@its.jnj.com
cMerck KGaA, Merck Serono R&D, Computational Chemistry, Frankfurter Straße 250, 64293 Darmstadt, Germany
dBIOVIA, a Dassault Systèmes brand, 334 Cambridge Science Park, Cambridge CB4 0WN, UK
eGrupo BioFarma-USEF, Departamento de Farmacología, Facultad de Farmacia, Campus Universitario Sur s/n, 15782 Santiago de Compostela, Spain
fResearch Programme on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM), Department of Experimental and Health Sciences, Universitat Pompeu Fabra, C/Dr Aiguader 88, E-08003 Barcelona, Spain
gEuropean Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK
hGlaxoSmithKline, Medicines Research Centre, Stevenage SG1 2NY, UK
iSwiss Institute of Bioinformatics, CALIPHO Group, CMU Rue Michel-Servet 1, 1211 Geneva 4, Switzerland
jNestlé Institute of Health Sciences SA, EPFL Innovation Park, Bâtiment H, 1015 Lausanne, Switzerland
kChemistry Innovation Centre, Discovery Sciences, AstraZeneca R&D Gothenburg, SE-431 83 Mölndal, Sweden
lDepartment of Bioinformatics – BiGCaT, P.O. Box 616, UNS50 Box19, NL-6200MD Maastricht, The Netherlands
mOpen Innovation Drug Discovery, Discovery Chemistry Eli Lilly and Company, Lilly Corporate Center, DC 1920, Indianapolis, IN 46285, USA
nEli Lilly and Company Ltd., Lilly Research Centre, Erl Wood Manor, Sunninghill Road, Windlesham, Surrey GU20 6PH, England, UK
First published on 11th May 2016
Abstract
Phenotypic screening is in a renaissance phase and is expected by many academic and industry leaders to accelerate the discovery of new drugs for new biology. Given that phenotypic screening is per definition target agnostic, the emphasis of in silico and in vitro follow-up work is on the exploration of possible molecular mechanisms and efficacy targets underlying the biological processes interrogated by the phenotypic screening experiments. Herein, we present six exemplar computational protocols for the interpretation of cellular phenotypic screens based on the integration of compound, target, pathway, and disease data established by the IMI Open PHACTS project. The protocols annotate phenotypic hit lists and allow follow-up experiments and mechanistic conclusions. The annotations included are from ChEMBL, ChEBI, GO, WikiPathways and DisGeNET. Also provided are protocols which select from the IUPHAR/BPS Guide to PHARMACOLOGY interaction file selective compounds to probe potential targets and a correlation robot which systematically aims to identify an overlap of active compounds in both the phenotypic as well as any kinase assay. The protocols are applied to a phenotypic pre-lamin A/C splicing assay selected from the ChEMBL database to illustrate the process. The computational protocols make use of the Open PHACTS API and data and are built within the Pipeline Pilot and KNIME workflow tools.
Introduction
Even though the discussion is still ongoing whether or not phenotypic screening was historically more productive for the discovery of first in class drugs than target-directed screening, and whether it continues to do so, it is clear that phenotypic screening opens new avenues to investigate new cellular biology.1–4 In the 1970s, phenotypic screening on physiological whole animal or organ testing with a limited number of compounds was very popular and successful. Drug hunters like Paul Janssen or James Black tested pharmacologically rich compounds systematically on a broad panel of such phenotypic assays across a spectrum of therapeutic areas and discovered breakthrough medicines like antipsychotics, beta-blockers, or anti-ulceratives.5 Also, phenotypic screening analysis of approved drugs can generate new insights. Recently, Lee et al. screened a large collection of approved drugs in phenotypic assays including models for osteoporosis, diabetes and cancer, identifying novel activities for several known compounds.6With the fantastic progress in molecular and cellular biology, cell-based phenotypic screening in primary or engineered cell-lines constitutes a promising avenue. New biology like for instance alternative splicing or translational read through becomes experimentally accessible using MTS/HTS approaches. The experiments deliver potentially potent and specific compounds for which it can be interesting to elucidate and validate the molecular mechanism. Next to experimental target validation including chemogenomics pull-down and knock-in/out experiments, the in silico assessment of the hit lists constitutes a key step.7–11 This analysis requires a high level of data integration in order for it to be complete and seamless. Such integration was recently achieved by the IMI Open PHACTS project12 resulting in the Open PHACTS Discovery Platform (http://www.openphacts.org).13 The Open PHACTS project uses semantic web technology for drug discovery by integrating relevant concept spaces of compound–target–pathway and disease (see Fig. 1 for concepts/URIs used in this work). This enables, as we will show herein, insightful interpretation of the phenotypic screening results to sustain target validation based on hitherto established drug discovery knowledge.
 | ||
| Fig. 1 Outline of data showing the links between the different key identifiers (URI, uniform resource identifiers) assessed for annotation of compounds and targets in the provided computational protocols. The data provenance is shown in brackets. | ||
Here we present six protocols, which could be useful to annotate the results of a phenotypic screening experiment. Protocol 1 retrieves known classifications for compounds of interest. Protocols 1 to 4 retrieve targets, which these compounds have recorded bioactivity values for, and subsequently retrieves additional data for these targets (ChEMBL classification, GO terms, pathways, and diseases, respectively). These protocols are depicted in Fig. 2 and the implementation of protocol 1 in Pipeline Pilot is shown in Fig. S1.‡ Protocol 5 retrieves all kinases and reported bioactivity values available in the ChEMBL database and returns an overlap with the compounds from the phenotypic screening (Fig. 3 and Fig. S2‡). A possibility to join the data retrieved from the Open PHACTS Discovery Platform with external data is shown in protocol 6 (Fig. 4 and Fig. S3‡).
 | ||
| Fig. 2 Schematic overview of protocols 1 to 4. | ||
 | ||
| Fig. 3 Schematic overview of protocol 5. | ||
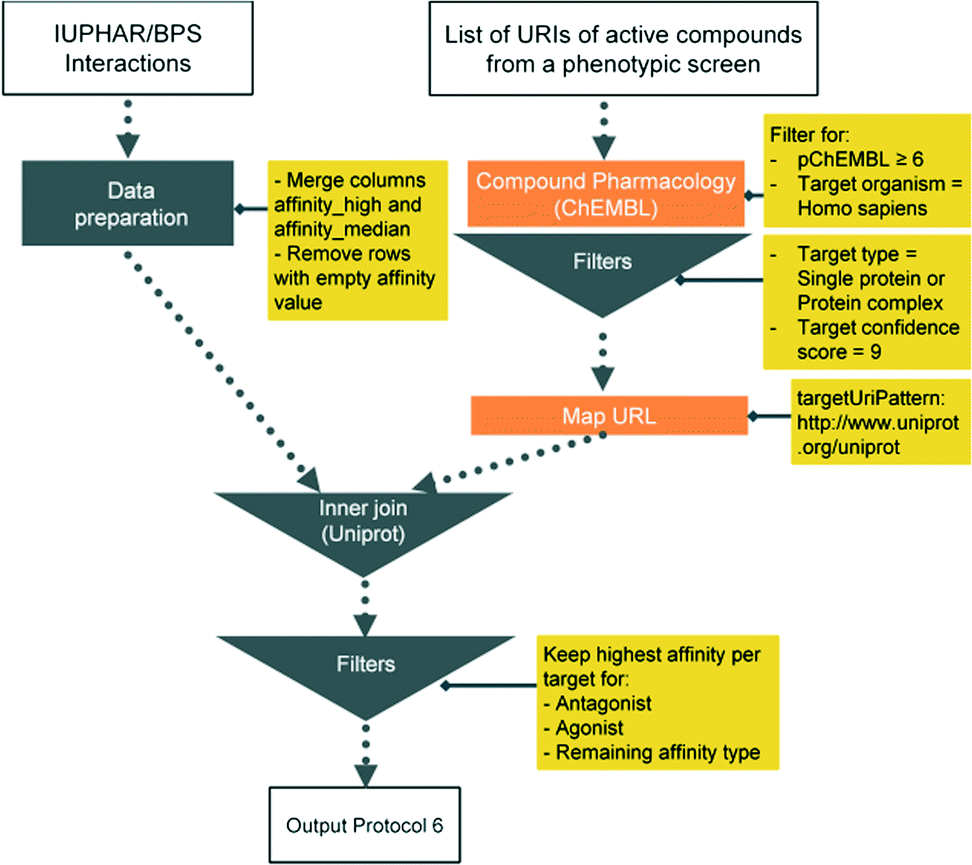 | ||
| Fig. 4 Schematic overview of protocol 6. | ||
Experimental
Software
Workflows were generated first with Pipeline Pilot from BIOVIA,14 and were then adapted for KNIME.15 Pipeline Pilot protocols were created in version 9.2 using version 2.0 of the Open PHACTS component collection, which was downloaded from the BIOVIA ScienceCloud Exchange.KNIME version 2.12.1 with installed JSON (KNIME Labs Extensions) and REST nodes (KNIME Community Contributions provided by Cenix BioScience) was used to create the workflows. Open PHACTS KNIME nodes (org.openphacts.utils.json_1.1.0) were retrieved from the github repository (https://github.com/openphacts/OPS-Knime).
Execution of API calls
An overview of API calls used in this study is provided in Table 1. Documentation of the Open PHACTS API13 is available at http://dev.openphacts.org/.| Protocol | Used API calls | Input | Output | Aggregation |
|---|---|---|---|---|
| 1-2 ChEMBL, GO | Compound Pharmacology: List, | List of Compound URIs | Cache 1: CompoundID and URICompound, canonical smiles | Join cache 3 and 2 based on target name; Join cache 1 on URICompound |
| Target Classifications | Cache 2: URI compound, URI assay, pChembl, TargetName, URITarget | Merge and group on classification | ||
| Cache 3: Target Name, URIClassification, Classification | ||||
| 1 Chebi | Compound Classifications | List of Compound URIs | Cache 1: CompoundID and URICompound, canonical smiles | Join cache 2 and 1 based on URICompound. |
| Cache 2: URI compound, URI Chebi, ChebiDescription | Merge and group on ChebiDescription | |||
| 3 Pathways | Compound Pharmacology: List, | List of Compound URI | Cache 1: CompoundID and URICompound, canonical smiles | Join cache 3 and 2 based on target name; Join cache 1 on URICompound |
| Pathways for Target: List | Cache 2: URI compound, URI assay, pChembl, TargetName, URITarget | Merge and group on PathwayName | ||
| Cache 3 : Target Name, URIPathway, PathwayID, PathwayName | ||||
| 4 Disease | Compound Pharmacology: List, | List of Compound URIs | Cache 1 : CompoundID and URICompound, canonical smiles | Join cache 3 and 2 based on target name; Join cache 1 on URICompound |
| Diseases for Target: List | Cache 2 : URI compound, URI assay, pChembl, TargetName, URITarget | Merge and group on DiseaseName | ||
| Cache 3 : Target Name, URIDisease, DiseaseName | To limit the runtime of the protocol merging is done directly on the data stream of Cache 3. | |||
| 5 Correlation Robot | Target Class Member: List, | Use CHEMBL_PC_6 kinase family key to launch query | Cache 1 : List of 455 human kinases for which ChEMBL holds data | Join Cache 2 based on INCHIKEY to each assay from Cache 1 data stream |
| Target Pharmacology: List | Cache 2 : Lamin A/C splicing assay data | |||
| 6 GtoPdb Box | Compound Pharmacology: List, | List of Compound URIs. Cache 1 : Read GtoPdb interaction file | Cache 2 Keep URITarget and URIUniprot and extract UniprotID from URIUniprot | Join cache 1 GtoPdb interaction file to cache 2 based UniprotID. Merge by ligand name |
| Map URL |
Data sources
Calls to the Open PHACTS Discovery Platform were made with the API version 2.0 (https://dev.openphacts.org/docs/2.0). Data collected in this study, which was accessed via the Open PHACTS Discovery Platform, includes ChEMBL16,17 version 20, ChEBI18 release 125, Gene Ontology (GO)19,20 annotations (accessed Feb. 2015), WikiPathways21 v20151118, and DisGeNET22,23 version 2.1.0.The pre-lamin A/C splicing assay data was selected from ChEMBL via the Open PHACTS Discovery Platform with the ChEMBL1293235 target ID (target pharmacology: list call) and subsequent filtering for the CHEMBL1614310 assay ID.
Protocol 6 includes interaction data from the IUPHAR/BPS Guide to PHARMACOLOGY (GtoPdb)24 downloaded from http://guidetopharmacology.org/DATA/interactions.csv (accessed Oct. 2015).
Results and discussion
Protocol design
In the Open PHACTS Discovery Platform, this information is accessible using again the ‘Target Classification’ API call, by inputting the protein target URIs in order to retrieve the GO classification trees. This information is highly complementary to the target annotation obtained in protocol 1. The information on the cellular component can be used to localize the site of action of the compounds provided that the underlying assay data is at the cellular level and not a cell-free biochemical format. The biological process and molecular mechanism information is essential for the assessment of the activities. Again, simple aggregation statistics at the level of individual assay activities, individual targets, or directly compounds allow an assessment of the relevance of the information. Observing, for instance, multiple highly potent compounds hitting different targets pointing to the same molecular process builds confidence of the relevance of this process for the observed phenotype. Given the richness of the GO terminology, its interpretation requires broad knowledge of general molecular and cellular biology and disease biology in order to assess the relevance.
Extending the biological knowledge of a hit list is possible using the WikiPathways annotation. Like in protocol 1 and 2, the workflow first retrieves the (poly)pharmacology data for the given compounds, resulting in a list of targets. The use of the ‘Pathways for Targets List’ API call, which yields to the associated WikiPathways URIs and Names for an inputted protein target URI list, gives a pathway-based summarizing view of the bioactivities.
With the integration achieved in the Open PHACTS Discovery Platform, it is directly feasible to retrieve all kinases from the ChEMBL classification tree (‘Target Class Member: List’ call) as well as the connected pharmacological data (‘Target Pharmacology: List’ call). Alternatively, the ‘Target Class Pharmacology: List’ call can be used directly. We opt here to apply the most simple correlation type by analysing the number of hits in common in the assays to be compared. The assay correlation robot might obviously be applied to other target families or to the entire pharmacological space. This analysis is complementary to the analysis provided in protocol 1.
This application demonstrates the flexibility by combining the Open PHACTS API and data workflow tools to integrate additional data sources. The protocol was designed in a manner to distinguish between agonist, antagonist, and other interaction types and to keep the most potent compound for each category for each target.
The Pipeline Pilot implementation makes full usage of the cache functionality which allows to store data using the API into data caches and then to join and aggregate it within a separate data stream. The design of the data pipelining protocols is made in a manner that at each step selected output data is cached and only the required input URIs are forward propagated to the next API calling node (see Fig. S1–S3‡). The outputs are then joined and grouped in a sequential manner to produce the desired information. This principle illustrates a key advantage for data mining. In this approach data is obtained collectively and the user sorts out and selects the desired information afterwards. Protocols 1 to 4 were combined into a single workflow in KNIME to reduce calculation time for redundant steps.
The resulting data for this application example from the Pipeline Pilot protocols are provided in the ESI.‡ Investigation of the target classifications (protocol 1) show that 47 kinase activities are observed based on 8 compounds on 27 targets. Interesting are the CGMC kinases DYRK1A and GSK3B and the MAP Kinases p38 α and β, c-Jun2 and 3 and ERK2. DYRK1A inhibitors are reported in the literature to modulate alternative pre-mRNA splicing of model gene transcripts in cells with submicromolar potencies.31 For the family A GPCR, 56 activities are observed based on 14 compounds on 19 targets. Most prominent are the monoamine receptor activities. 27 epigenetic regulator activities are observed based on 17 compounds on five targets.
Regarding GO component, 186 terms are found; 13 compounds are linked to the spliceosomal complex via the heterogeneous nuclear ribonucleoprotein A1 and the survival motor neuron protein. For GO process, 1287 terms are found. Multiple compounds are linked to various DNA related processes via the Bloom syndrome protein, while 13 compounds are linked to spliceosomal complex assembly. For GO function, 340 terms are found. The kinase assay correlation robot supports the hint to kinases and points to the MAP kinase ERK2 assay CHEMBL1613808 which has eight compounds in common. The underlying pathway is the MAPK signalling pathway, which is found in protocol 3 for 12 targets.
In general, 306 pathways are identified, with ‘GPCR downstream signaling’ and ‘GPCR ligand binding’ showing the highest count of identified targets (19), and ‘FAS pathway and Stress induction of HSP regulation’ and ‘Integrated Pancreatic Cancer Pathway’ showing the highest count of active molecules (37).
The DisGeNET annotation (protocol 4) provides links to 3631 diseases and side-effects; 89 of them have more than 20 potential efficacy targets links. Various neoplasms and cancers are prominent given the link via kinases. Spinal muscular atrophy is linked by 13 hits via the survival motor protein link. It will require further disease biology expertise to recognize relevant links to the observed phenotype.
ChEBI terms (protocol 1) associated with at least five compounds include five metabolites and nine antineoplastic agents among which fluorouracil, camptothecin and rotenone are listed. Rotenone is discussed in the literature to modulate splicing of several genes, e.g. alternative splicing of the X-linked NDUFB11 gene of the respiratory chain complex I.32
The analysis from protocol 6 suggests testing of 79 compounds in the phenotypic screening assay. Very prominent are monoamine receptor ligands and kinase inhibitors.
After having competed the in silico annotation, an obvious questions aims towards the modus operandi for follow-up experiments and drug discovery. A first obvious experiment is to test the tool compounds retrieved from the GtoPdb database in the phenotypic assay to verify if they produce the desired phenotype. Obviously, in vitro target validation through, for instance, CRISPR/cas9 experiments would complete the experimental target validation. In a similar perspective, ChEMBL biological annotations point to targets which enable such testing. The usage of the results from the correlation robot opens the possibility to substitute the phenotypic assay for the mechanistic target based assay for as the primary screening or optimization assay. This might allow for instance for higher throughput in screening. Given the low number of common hits found between the MAP kinase ERK2 assay and the pre-lamin phenotypic assay, we would recommend to test further ERK2 reference compounds in the phenotypic assay before taking a decision. A key difficulty relies in making sense of the GO, WikiPathways and DisGeNET annotations. Given that each putative target pulls potentially a multitude of these annotation categories, a clear navigation strategy is missing. One possible way forward could be the analysis of similarities between the annotations. Further work and domain expertise is needed to achieve this. Practically, the hint that 13 compounds are linked to the biology of the spliceosome complex increases the attractiveness of these compounds for follow-up chemistry lead optimization. Conversely, the link to activity on the Bloom syndrome protein flags a different set of compounds as potentially problematic, given the link to genomic instability of this protein.
Conclusions
Knowing the knowns about a phenotypic screening hit list is a first essential step in the analysis of every phenotypic screening project and contributes to the validation of the potential efficacy targets associated to the hits.33 Collected knowledge might help to decide whether the research team should optimize hit compounds based on a phenotypic readout or pursue with mechanistic target based assays. The interpretation of the provided information requires broad knowledge of general molecular and cellular biology, as well as disease biology in order to assess their relevance. Importantly, this data-driven collected knowledge also guides the assessment of potential off-biology, including pharmacological and toxicological side-effects early in the projects. Herein presented computational protocols focus on cellular phenotypic screening data where the link to molecular mechanisms is possible.A limitation of the here presented analysis could be a bias in the available data in the public domain databases used. In Open PHACTS we mainly use one data source for each of the data types (e.g. ChEMBL for bioactivities, DisGeNET for Diseases, WikiPathways for pathways). While most of these sources combine data from several places, this could lead to a bias in the data. The illustrated analyses will benefit from the inclusion of data beyond the Open PHACTS Discovery Platform, as for instance commercial data sources (like GOStar from GVKbio34) or patent extracted data (like SureChEMBL35). Additionally, API calls to other available data sources (e.g. the Entrez Utilities API Eutils) could be integrated into the protocols, to increase the coverage of the returned data. The corporate internal SAR data stores with massive amounts of fulldeck screening data will not only enable to include proprietary compounds into the analysis, but also to have access to more complete SAR data matrices. This will be of benefit especially for correlation analyses. The inclusion of negative screening results becomes equally possible with the corporate in house data. Negative data is of particular value given that the identified target proteins for phenotypic negatives cannot be dominant phenotypic targets. This is a very important point. In a typical in silico deconvolution effort, many active compounds will point to promiscuous targets, e.g. biogenic amine GPCRs,36 or point to generic pathways, e.g. ‘GPCR downstream signaling’. However, when putting these activities into context and showing that a similar or greater percentage of phenotypically inactive compounds are also hitting these targets, they can be removed from the list of potentially interesting efficacy targets. At this stage a rigorous statistical analysis will enable to distinguish a real signal from noise and help to interpret the results. It will thus be relevant to store the inactives of a phenotypic screen to enable such analysis.
Further restrictions on the suggested targets might appear from the inclusion of gene expression data. This is already feasible with the Open PHACTS Discovery Platform. The inclusion of protein complex information offers an equally interesting extension possibility of the pathway analysis protocol 3. A relevant question is: are multiple members of a given functional complex hit by different compounds? This aspect can be in part addressed by the GO cellular component annotation. Also, a combination of pathway and disease information could be worthwhile, to investigate the overlap of both.
Extending beyond the known knowns is possible by applying predictive chemogenomics SAR models which are currently being developed in academia and industry.37–40 Especially noteworthy are the predictive inference capabilities intrinsic to semantic approaches which allow integrating similarities among the data. Similarities between compounds, as well as similarities between proteins at the sequence level or even binding site level, can directly be coded in RDF. An extended version of the protocols, integrating experimental and predicted data would obviously top rank targets for which there is experimental evidence, and then, highlight the additional conclusions drawn from the predictions. The added value of the predictions is to potentially extend to the discovery of novel targets, not belonging to the known knowns.
Acknowledgements
We thank all colleagues participating in the Open PHACTS Scientific Task Force and Forum for discussions and insights leading to this consolidated view. We also want to thank the referees for their valuable feedback and suggestions for data analysis. The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115191, resources of which are composed of financial contributions from the EU's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies' in-kind contribution. L. I. F. received support from ISCIII-FEDER (PI13/00082, CP10/00524) and the EU H2020 Programme 2014-2020 under grant agreements no. 634143 (MedBioinformatics) and no. 676559 (Elixir-Excelerate). The Research Programme on Biomedical Informatics (GRIB) is a node of the Spanish National Institute of Bioinformatics (INB).References
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- D. C. Swinney, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS PubMed.
- J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed.
- B. T. Priest and G. Erdemli, Front. Pharmacol., 2014, 5, 264, DOI:10.3389/fphar.2014.00264.
- W. Sneader, Drug Discovery: A History, Wiley, 2005 Search PubMed.
- J. A. Lee, P. Shinn, S. Jaken, S. Oliver, F. S. Willard, St. Heidler, R. B. Peery, J. Oler, S. Chu, N. Southall, T. S. Dexheimer, J. Smallwood, R. Huang, R. Guha, A. Jadhav, K. Cox, C. P. Austin, A. Simeonov, G. S. Sittampalam, S. Husain, N. Franklin, D. J. Wild, J. J. Yang, J. J. Sutherland and C. J. Thomas, PLoS One, 2015, 10, e0130796 Search PubMed.
- S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744–2792 CrossRef CAS PubMed.
- A. Ursu and H. Waldmann, Bioorg. Med. Chem. Lett., 2015, 25, 3079–3086 CrossRef CAS PubMed.
- D. W. Young, A. Bender, J. Hoyt, E. McWhinnie, G. W. Chirn, C. Y. Tao, J. A. Tallarico, M. Labow, J. L. Jenkins, T. J. Mitchison and Y. Feng, Nat. Chem. Biol., 2008, 4, 59–68 CrossRef CAS PubMed.
- T. Cheng, Q. Li, Y. Wang and S. H. Bryant, J. Chem. Inf. Model., 2011, 51, 2440–2448 CrossRef CAS PubMed.
- M. Schirle and J. L. Jenkins, Drug Discovery Today, 2016, 21, 82–89 CrossRef CAS PubMed.
- A. J. Williams, L. Harland, P. Groth, S. Pettifer, C. Chichester, E. L. Willighagen, C. T. Evelo, N. Blomberg, G. Ecker, C. Goble and B. Mons, Drug Discovery Today, 2012, 17, 1188–1198 CrossRef PubMed.
- A. J. G. Gray, P. Groth, A. Loizou, S. Askjaer, C. Brenninkmeijer, K. Burger, C. Chichester, C. T. Evelo, C. Goble, L. Harland, S. Pettifer, M. Thompson, A. Waagmeester and A. J. Williams, Semant. Web, 2014, 5, 101–113 CrossRef.
- Pipeline Pilot 9.2 (BIOVIA – A Dassault Systèmes brand – 5005 Wateridge Vista Drive, San Diego, CA 92121 USA) Search PubMed.
- KNIME Analytics Platform (version 2.12.2), KNIME GmbH, Konstanz, Germany Search PubMed.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Kruger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, Nucleic Acids Res., 2014, 42, 1083–1090 CrossRef PubMed.
- S. Jupp, J. Malone, J. Bolleman, M. Brandizi, M. Davies, L. Garcia, A. Gaulton, S. Gehant, C. Laibe, N. Redaschi, S. M. Wimalaratne, M. Martin, N. Le Novere, H. Parkinson, E. Birney and A. M. Jenkinson, Bioinformatics, 2014, 30, 1338–1339 CrossRef CAS PubMed.
- J. Hastings, P. de Matos, A. Dekker, M. Ennis, B. Harsha, N. Kale, V. Muthukrishnan, G. Owen, S. Turner, M. Williams and C. Steinbeck, Nucleic Acids Res., 2013, 41, D456–D463 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- The Gene Ontology Consortium, Nucleic Acids Res., 2015, 43, D1049–D1056 CrossRef PubMed.
- M. Kutmon, A. Riutta, N. Nunes, K. Hanspers, E. L. Willighagen, A. Bohler, J. Mélius, A. Waagmeester, S. R. Sinha, R. Miller, S. L. Coort, E. Cirillo, B. Smeets, C. T. Evelo and A. R. Pico, Nucleic Acids Res., 2016, 44, D488–D494 CrossRef PubMed.
- J. Piñero, N. Queralt-Rosinach, À. Bravo, J. Deu-Pons, A. Bauer-Mehren, M. Baron, F. Sanz and L. I. Furlong, Database, 2015, bav028 CrossRef PubMed.
- N. Queralt-Rosinach, J. Piñero, À. Bravo, F. Sanz and L. I. Furlong, Bioinformatics, 2016 DOI:10.1093/bioinformatics/btw214.
- C. Southan, J. L. Sharman, H. E. Benson, E. Faccenda, A. J. Pawson, S. P. H. Alexander, O. P. Buneman, A. P. Davenport, J. C. McGrath, J. A. Peters, M. Spedding, W. A. Catterall, D. Fabbro, J. A. Davies and NC-IUPHAR, Nucleic Acids Res., 2016, 44, D1054–D1068 CrossRef PubMed.
- E. Jacoby, G. Tresadern, S. Bembenek, B. Wroblowski, C. Buyck, J.-M. Neefs, D. Rassokhin, A. Poncelet, J. Hunt and H. van Vlijmen, Drug Discovery Today, 2015, 20, 652–658 CrossRef CAS PubMed.
- J. M. Elkins, V. Fedele, M. Szklarz, K. R. Abdul Azeez, E. Salah, J. Mikolajczyk, S. Romanov, N. Sepetov, X. P. Huang, B. L. Roth, A. Al Haj Zen, D. Fourches, E. Muratov, A. Tropsha, J. Morris, B. A. Teicher, M. Kunkel, E. Polley, K. E. Lackey, F. L. Atkinson, J. P. Overington, P. Bamborough, S. Müller, D. J. Price, T. M. Willson, D. H. Drewry, S. Knapp and W. J. Zuercher, Nat. Biotechnol., 2016, 34, 95–103 CrossRef CAS PubMed.
- J. D. Moore, Drug Discovery Today, 2015, 20, 450–457 CrossRef CAS PubMed.
- C. A. Goble, J. Bhagat, S. Aleksejevs, D. Cruickshank, D. Michaelides, D. Newman, M. Borkum, S. Bechhofer, M. Roos, P. Li and D. De Roure, Nucleic Acids Res., 2010, 38, W677–W682 CrossRef CAS PubMed.
- National Center for Biotechnology Information, PubChem BioAssay Database; AID=1487, Source=NCGC, http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1487.
- D. Auld, M. Shen and C. Thomas, A High-throughput Screen for Pre-mRNA Splicing Modulators, Probe Reports from the NIH Molecular Libraries Program [Internet], Bethesda (MD): National Center for Biotechnology Information (US), 2010 - 2009 May 18 [updated 2010 Sep 2] Search PubMed.
- C. Schmitt, P. Miralinaghi, M. Mariano, R. W. Hartmann and M. Engel, ACS Med. Chem. Lett., 2014, 5, 963–967 CrossRef CAS PubMed.
- D. Panelli, F. P. Lorusso, F. Papa, P. Panelli, A. Stella, M. Caputi, A. M. Sardanelli and S. Papa, Biochim. Biophys. Acta, 2013, 29, 211–228 CrossRef PubMed.
- A. M. Wassermann, E. Lounkine, J. W. Davies, M. Glick and L. M. Camargo, Drug Discovery Today, 2014, 20, 422–434 CrossRef PubMed.
- GOSTAR (GVK BIO Online Structure Activity Relationship Database), GVK Biosciences Private Limited, S-1, Phase-1, T.I.E. Hyderabad, A.P, India, 2010 Search PubMed.
- G. Papadatos, M. Davies, N. Dedman, J. Chambers, A. Gaulton, J. Siddle, R. Koks, S. A. Irvine, J. Pettersson, N. Goncharoff, A. Hersey and J. P. Overington, Nucleic Acids Res., 2016, 44, D1220–D1228 CrossRef PubMed.
- J.-U. Peters, J. Hert, C. Bissantz, A. Hillebrecht, G. Gerebtzoff, S. Bendels, F. Tillier, J. Migeon, H. Fischer, W. Guba and M. Kansy, Drug Discovery Today, 2012, 17, 325–335 CrossRef PubMed.
- A. M. Afzal, H. Y. Mussa, R. E. Turner, A. Bender and R. C. Glen, J. Cheminf., 2015, 7, 24 Search PubMed.
- A. M. Wassermann, E. Lounkine, L. Urban, S. Whitebread, S. Chen, K. Hughes, H. Guo, E. Kutlina, A. Fekete, M. Klumpp and M. Glick, ACS Chem. Biol., 2014, 9, 1622–1631 CrossRef CAS PubMed.
- A. Koutsoukas, B. Simms, J. Kirchmair, P. J. Bond, A. V. Whitmore, S. Zimmer, M. P. Young, J. L. Jenkins, M. Glick, R. C. Glen and A. Bender, J. Proteomics, 2011, 74, 2554–2574 CrossRef CAS PubMed.
- S. Paricharak, I. Cortés-Ciriano, A. P. IJzerman, T. E. Malliavin and A. Bender, J. Cheminf., 2015, 7, 15 Search PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Pipeline Pilot protocols, xls file with the output of the Pipeline Pilot protocols, KNIME workflows, and supplementary figures showing the Pipeline Pilot protocols. See DOI: 10.1039/c6md00065g |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |