
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the chemical space of hydrophobic pharmacophores: the role of hydrophobic substructures in the development of novel transcription modulators†
Shinya
Fujii
Institute of Molecular and Cellular Biosciences, The University of Tokyo, 1-1-1 Yayoi, Bunko-ku, Tokyo 113-0032, Japan. E-mail: fujiis@iam.u-tokyo.ac.jp
First published on 20th April 2016
Abstract
Interactions between biologically active compounds and their targets often involve hydrophobic interactions, and hydrophobicity also influences the pharmacokinetic profile. Nevertheless, comparatively less attention has been given to the utilization of hydrophobic substructures other than hydrocarbons in the design of biologically active compounds. Herein the author reviews the development of novel transcription modulators incorporating three types of substructures, i.e., silyl and germyl functionalities, hydrophobic boron clusters, and cyclopentadiene-based organometallic sandwich compounds, as hydrophobic components.
 Shinya Fujii | Shinya Fujii was born in Toyota City, Japan, in 1977. He received his PhD from The University of Tokyo. He served as a researcher in Kyowa Hakko Kogyo Co., Ltd. (2002–2004), a research associate at Tokushima-Bunri University (2004–2007) and an assistant professor at Tokyo Medical and Dental University (2007–2013), and he is currently a lecturer at The University of Tokyo. His research interests include medicinal chemistry and elemental chemistry focusing on the chemistry of hydrophobicity. |
Introduction
Biologically active compounds often show not only hydrogen bonding and other polar interactions with their targets, but also hydrophobic interactions. For example, hydrophobic interactions between protein residues and a single methyl group of a small molecule can stabilize complex formation as effectively as polar contacts.1 Thus, the hydrophobic moieties of biologically active molecules can be considered as pharmacophores. Nevertheless, in contrast to the wide variety of hydrophilic substructures, almost all hydrophobic substructures currently used in biologically active compounds are simple hydrocarbons or aromatic rings. Therefore, introduction of novel hydrophobic substructures would enormously expand the chemical space of hydrophobic pharmacophores, thereby increasing options for molecular design and structural development of novel agents. In addition, they would impact on the pharmacokinetic properties and toxicity.One approach to expanding the chemical space of hydrophobic pharmacophores would be the introduction of inorganic and organometallic functionalities. The strategy of introducing a range of novel substructures has mainly been applied in the development of compounds bearing hydrophilic components. However, there are also dozens of hydrophobic chemical species with sufficient stability in aqueous and physiological environments, and incorporation of these substructures into hydrophobic pharmacophores would be a potentially powerful methodology for developing novel biologically active compounds and drug candidates. In particular, hydrophobicity is an important feature of nuclear signal-regulating compounds, such as nuclear receptor ligands and other transcription factor modulators.
The author reviews herein the development of novel transcription modulators, focusing on the introduction of three classes of hydrophobic substructures: silyl and germyl functionalities, hydrophobic boron clusters, and cyclopentadiene-based organometallic sandwich compounds.
Alkylsilane and other group 14 atom-containing functionalities
Carbon–silicon exchange (sila-substitution) of hydrocarbons is a widely utilized strategy for the development of biologically active compounds bearing novel hydrophobic substructures.2–5 Regarding their application towards transcription modulators, silicon-containing retinoids, i.e. nuclear retinoic acid receptor (RARs) ligands, and rexinoids, i.e. retinoid X receptor (RXRs) ligands, have been intensively investigated. The key structural features of retinoids and rexinoids are a carboxyl group and a hydrophobic pharmacophore, and various retinobenzoic acids have been developed (Fig. 1).6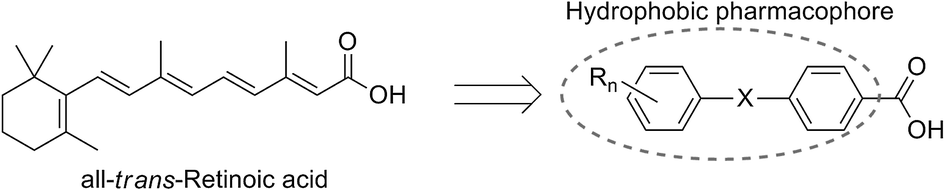 | ||
| Fig. 1 Design scheme of retinobenzoic acids. | ||
Carboxylic acid derivatives of benzanilide (Am compounds)7 and chalcone (Ch compounds)8 by Shudo and co-workers are potent RAR ligands. Hydrophobic substituents on the benzoyl moiety significantly affect the activity of the compounds, and sila-substitution of the hydrophobic substructure effectively enhances the activity. Replacement of the two tert-butyl groups of Am555 (1) with trimethylsilyl groups, giving Am555S (2, TAC101), i.e. di-sila-substitution of Am555, resulted in approximately ten-fold enhancement of the biological activity. Sila-substitution of chalcone derivative Ch55 (3) to afford silicon derivative Ch55S (4) also increased the activity. In addition, germa-substituted Ch55G (5) is also more potent than Ch55.9 Am555S had been developed for treatment of advanced hepatocellular carcinoma (Fig. 2).10
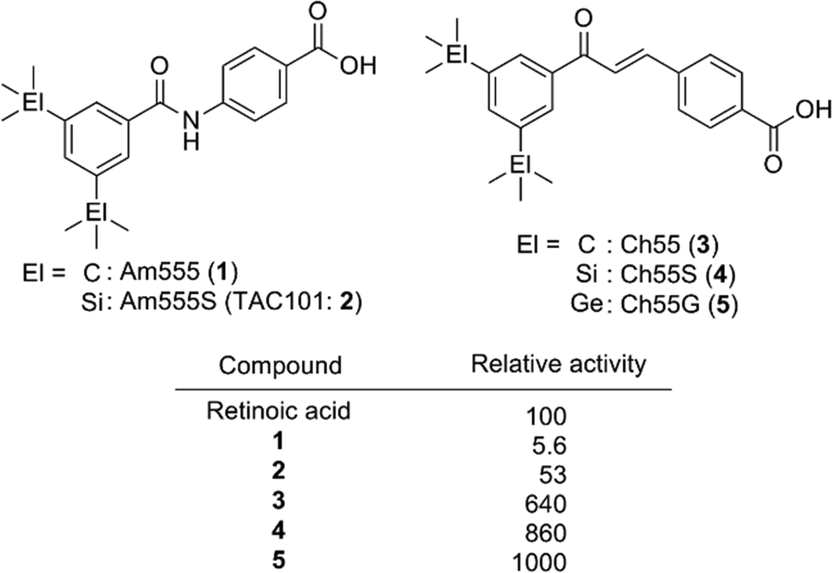 | ||
| Fig. 2 Structure and biological activity of representative bis(trialkylsilyl) and bis(trialkylgermyl) retinobenzoic acid derivatives by Shudo et al. | ||
etramethyltetrahydronaphthalene derivatives tamibarotene (Am80, 6)11,12 and bexarotene (12)13,14 are representative synthetic ligands for RAR and RXR, respectively, and have been approved for treatment of acute promyelocytic leukemia (APL) and cutaneous T cell lymphoma (CTCL), respectively. Tacke and co-workers have extensively investigated the di-sila-substituted derivatives of retinoids, rexinoids, and related compounds (Fig. 3). In the case of the RAR ligands, sila-substitution significantly affected the subtype selectivity. Silicon-containing RAR ligands 7 and 9 exhibited a potent activity toward RARα with almost the same potency as the parent compounds 6 and 8, respectively, whereas silicon derivatives 7 and 9 exhibited a more potent activity toward RARβ and RARγ.15 Di-sila-substitution of stilbene-based pan-RAR agonist TTNPB (disila-TTNPB, 11) resulted in a pan-RAR agonistic activity with potency similar to that of the parent TTNPB (10).16 As for the RXR ligands, disila-bexarotene (13) exhibited a similar activity to the parent bexarotene (12),17 and dioxolane-based disila-SR11237 (15) also exhibited similar potency to the parent SR11237 (14). On the other hand, di-sila-substituted 17 was ten-fold more potent than the carbon analog, indane derivative 16. Crystal structure analysis suggested that enlargement of the hydrophobic bicyclic structure by incorporation of silicon atoms enables additional hydrophobic interactions between the compound and the receptor (PDBID: 2ZXZ for 16-hRXRα and 2XY0 for 17-hRXRα).16,18
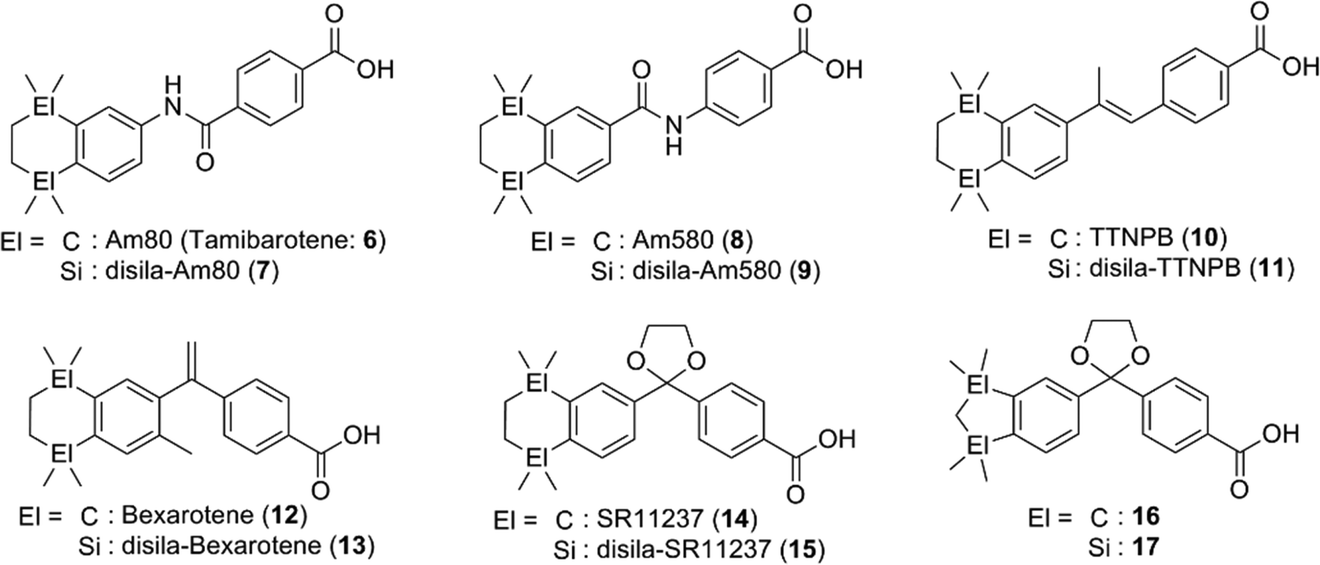 | ||
| Fig. 3 Representative disilacyclic derivatives developed by Tacke et al. as RAR or RXR ligands. | ||
Disilacyclic compounds were also developed as ligands for retinoic acid receptor-related orphan receptor (RORs). Benzanilide derivative 19 inhibits the constitutive activity of RORα and RORγ, indicating that it acts as an inverse agonist toward these RORs, whereas the activity of the corresponding carbon analog 18 was significantly less potent than that of the silicon analog. Enlargement of the hydrophobic structure and change of the electrostatic potential are possible reasons for the greater potency of the silicon analog (Fig. 4).19
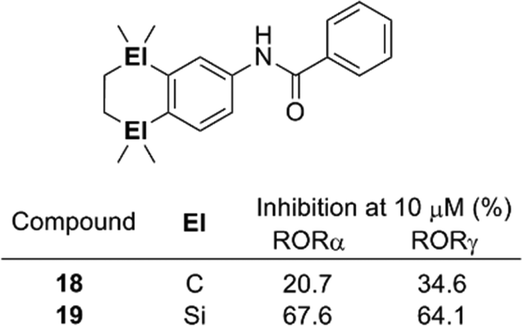 | ||
| Fig. 4 Structure and biological activity of benzanilide-based ROR ligands. | ||
In addition to the case of retinoids such as 7 and 9, switching of selectivity was also observed in the case of the diphenylsilane derivatives. Diphenylpentane derivative LG190155 (22) was developed as a derivative of vitamin D receptor (VDR) agonist LG190178 (20),20 and exhibited a VDR agonistic activity and moderate androgen receptor (AR) antagonistic activity.21 Sila-substitution of the central carbon atom of these compounds, giving sila-LG190178 (21) and sila-LG190155 (23), resulted in significant loss of the VDR agonistic activity and increase of the AR antagonistic activity (Fig. 5). This VDR/AR selectivity switching can be attributed to enlargement of the skeletal structure.22
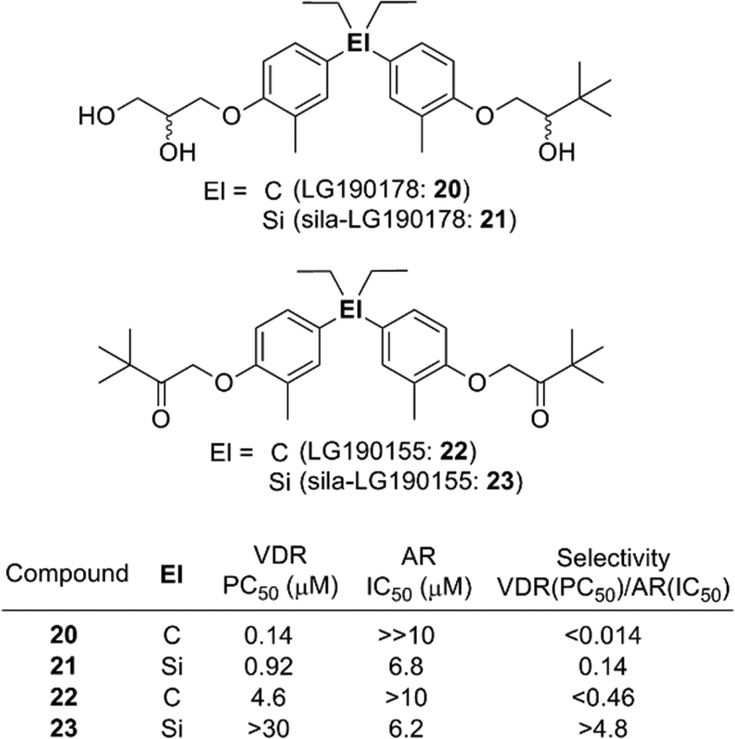 | ||
| Fig. 5 Selectivity switching by Si/C exchange. | ||
The characteristic properties of silicon- and germanium-containing derivatives such as Am555S (2) and compound 17 result at least partly from the change of inherent hydrophobicity compared with the corresponding carbon analogues. In order to understand the characteristics of silicon- and germanium-containing compounds, systematic determination of the hydrophobicity parameters of these heavier group 14 atom-containing substituents was performed. With 4-substituted phenol as a fundamental structure, hydrophobicity parameters (i.e. log Po/w values and Hansch–Fujita hydrophobic parameter π) of various alkylsilyl and alkylgermyl functionalities were determined.23 The silicon analogs exhibited larger log Po/w values than the corresponding carbon analogs, with differences in the log Po/w values of approximately 0.6. This difference in the log Po/w values between the silicon analogs and the corresponding carbon analogs was essentially independent of the alkyl substituents. On the other hand, the differences in the log Po/w values between the silicon analogues and the corresponding germanium analogues were small (Fig. 6). This suggests that the difference in the hydrophobicity can be mainly attributed to the difference in the covalent radius of the central atom (C, 77 pm; Si, 117 pm; Ge, 122 pm).
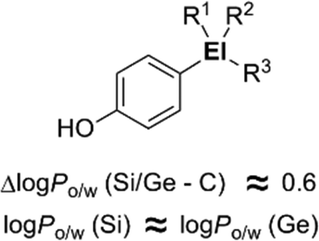 | ||
| Fig. 6 Increase in hydrophobicity upon sila- and germa-substitution. | ||
The 4-substituted phenol structure is a pharmacophore of estrogen receptor (ER) ligands, and therefore these trialkylsilyl- and trialkylgermylphenols should function as ER ligands. Evaluation of the estrogenic activity by means of an ERα-dependent MCF-7 cell proliferation assay revealed that the silyl and germyl derivatives exhibited a more potent activity than the corresponding carbon analogs. For example, triethylsilylphenol 25 and triethylgermylphenol 26 showed EC50 values of 3.4 and 2.1 nM (carbon analogue 24, EC50 = 23 nM) (Fig. 7).23 Thus, sila-substitution and germa-substitution are promising approaches to develop simple and potent bioactive compounds.
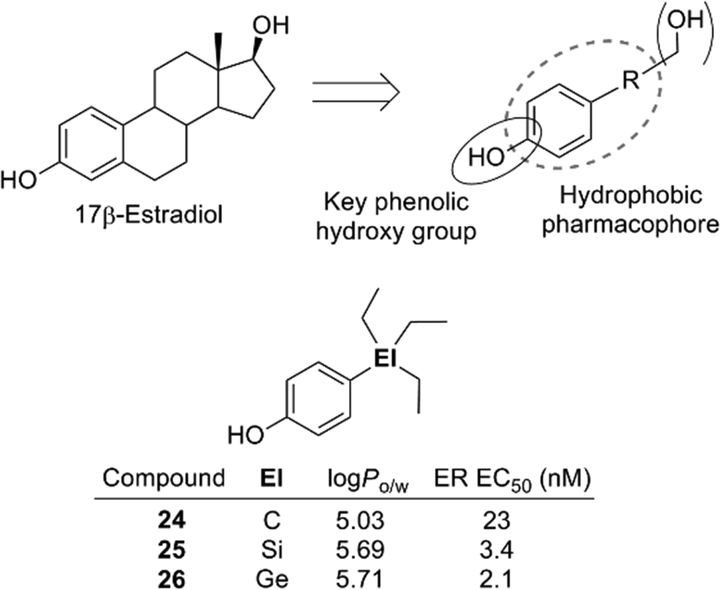 | ||
| Fig. 7 Pharmacophore of estrogen receptor ligands and structure of ER ligands. | ||
Recently, application of silyl groups, rather than simple sila-substitution (carbon–silicon exchange), has been proposed. The covalent radius of a silicon atom is larger than that of carbon, and therefore silyl groups could be geometrically equivalent to the diatomic substructure. Indeed, the diphenylsilane structure could function as an alternative to the cis-stilbene core structure in the case of tubulin polymerization inhibitors, indicating that silyl groups could be a bioisostere of a cis-olefinic double bond.24 There are many biologically active compounds bearing cis-olefin, including various nuclear receptor modulators. A representative example is oleoylethanolamide (OEA), an endogenous ethanolamide lipid of the C18:1 fatty acid. OEA exhibits various biological activities including a peroxisome proliferator-activated receptor alpha (PPARα) agonistic activity.25 Molecular orbital calculation indicated that the distance (d in Fig. 8) in the silane is similar to that of the cis-olefin, whereas the distance in the corresponding alkane is significantly shorter. Thus, replacement of the cis-olefin of fatty acid derivatives with a silyl functionality would be a reasonable conversion (Fig. 8). As a result of structural development, it was found that ethanolamide lipid 27a, containing a silyl moiety instead of a cis-olefinic double bond, exhibited a moderate PPARα agonistic activity as well as PPARδ agonistic activity (Fig. 9).26 Silicon-containing lipids could be an interesting category of biologically active compounds.
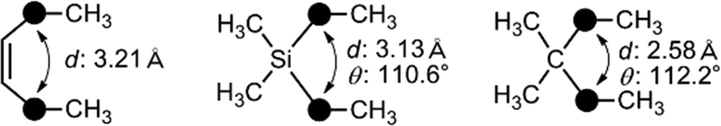 | ||
| Fig. 8 Similarity in the atomic distance in cis-olefin and silane. | ||
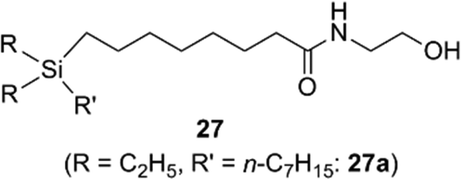 | ||
| Fig. 9 Silicon-containing ethanolamide lipids as PPAR ligands. | ||
In addition to cis-olefin, a cis-amide moiety could be replaced by a silyl functionality. The cis-amide-containing tricyclic heterocycle phenanthridin-6-one is a versatile scaffold for medicinal chemistry, and various bioactive phenanthridin-6-one derivatives, including nuclear liver X receptor (LXR) ligands and ROR ligands, have been reported.27 In the case of the ROR ligands, docking simulation suggested that the amide moiety of phenanthridin-6-one was surrounded by hydrophobic amino acids and did not contribute to hydrogen bonding to the receptor. Therefore, the cis-amide moiety of phenanthridin-6-one-based ROR ligands could be replaced by a hydrophobic silyl functionality. Structural development afforded dibenzosilole derivatives such as 28a that exhibited a significant ROR inverse agonist activity, nearly equal to that of the parent phenanthridin-6-one derivatives (Fig. 10).28 Silicon-containing heterocycles could be also versatile scaffolds for biologically active compounds.
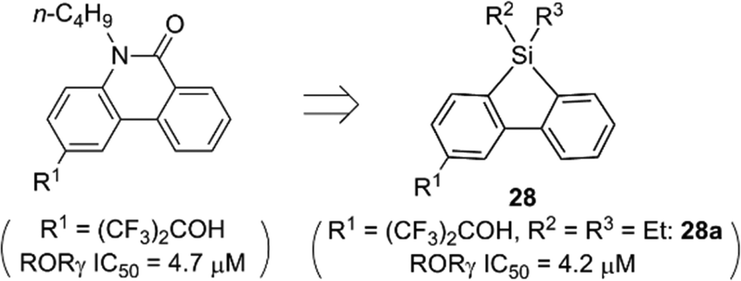 | ||
| Fig. 10 Design of dibenzosilole derivatives as ROR inverse agonists. | ||
Hydrophobic boron clusters
Higher boranes (boron hydrides) form clusters, and closo-borane compounds ([BnHn]2−) have closed structures such as octahedron and icosahedron. Since the carbon atom has one more valence electron than the boron atom, the C–H unit is isoelectronic and isolobal with [B–H]−. Thus, the two-carbon-containing analogs of [BnHn]2− are the neutral compounds C2Bn−2Hn, and these carbon-containing boron clusters are called carboranes or carbaboranes.29 The icosahedral twelve-vertex dicarba-closo-dodecaboranes (C2B10H12) have been extensively investigated, and are usually simply called carboranes. Carboranes have high chemical and thermal stability and exhibit high hydrophobicity, as well as having a bulky spherical structure.30,31Fig. 11 shows the structure of three isomers of carborane, namely ortho- (o-), meta- (m-) and para- (p-) carboranes (Fig. 11).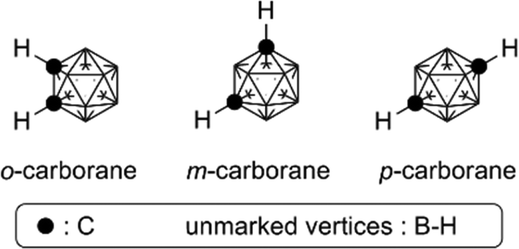 | ||
| Fig. 11 Structure of the three isomers of dicarba-closo-dodecaboranes (carboranes). | ||
In the field of medicinal chemistry, carboranes have been investigated as boron carriers for boron neutron capture therapy (BNCT) of cancer, based on the nuclear reaction of boron-10 (10B) with thermal neutrons to generate high linear energy transfer (LET) particles having destructive path lengths of only approximately one cell diameter (thermal neutrons themselves cause little damage because of their low energy). BNCT is considered a promising modality for treatment of brain cancer and prostate cancer.32,33
On the other hand, development of carborane-based biologically active compounds utilizing the characteristic features of carboranes, i.e. high hydrophobicity and spherical geometry, is at an early stage.34 A milestone in the field of carborane-based biologically active compounds was the development of the carboranylphenol derivatives as ERα super-agonists by Endo and co-workers. p-Carboranylphenol derivative 29 exhibited a potent ERα agonistic activity exceeding that of endogenous estrogen 17β-estradiol (E2). A simple carboranylphenol 30 also exhibited a significant ERα agonistic activity comparable to that of E2 (Fig. 12).35,36 These results suggested that the carborane cage can function as a hydrophobic core structure as effectively as hydrocarbons (such as a steroidal skeleton).
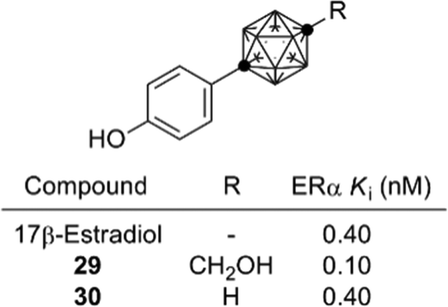 | ||
| Fig. 12 Structure and activity of carborane-based potent ER agonists. | ||
Based on the structural features of developed ER modulators tamoxifen and raloxifene, a series of carborane-based bisphenols and related derivatives were developed. Bis(4-hydroxyphenyl)-o-carborane 31, which exhibited an ERα antagonistic activity in vitro, is one of the most intensively investigated carborane derivatives. Interestingly, compound 31 significantly prevented bone loss in ovariectomized mice (OVX mice), suggesting that it acts as a selective estrogen receptor modulator (SERM) in vivo.37m-Carborane-based bisphenol is also a versatile scaffold for development of novel ER modulators. Simple bisphenol compound 32 exhibited a potent ERα agonistic activity. Modification of one of the phenolic hydroxyl groups of compound 32 resulted in significant changes of the activity. 3-Dimethylaminopropyl analog 34 exhibited a full agonistic activity, whereas 2-dimethylaminoethyl analog 33 exhibited a weak ERα partial agonistic activity. 2-Piperidinylethyl derivative 35 also exhibited an ERα partial agonistic activity. Surprisingly, compound 36, a B-phenyl o-carborane derivative having almost the same structure as 32, exhibited no significant activity.38,39 Thus, the difference in hydrophobicity between these two isomers significantly altered the biological activity. p-Carborane-based derivatives 37 exhibited an ERα agonistic activity (Fig. 13).40 Carboranes have been also used for the development of ERβ selective ligands such as 38–41(Fig. 14).41,42
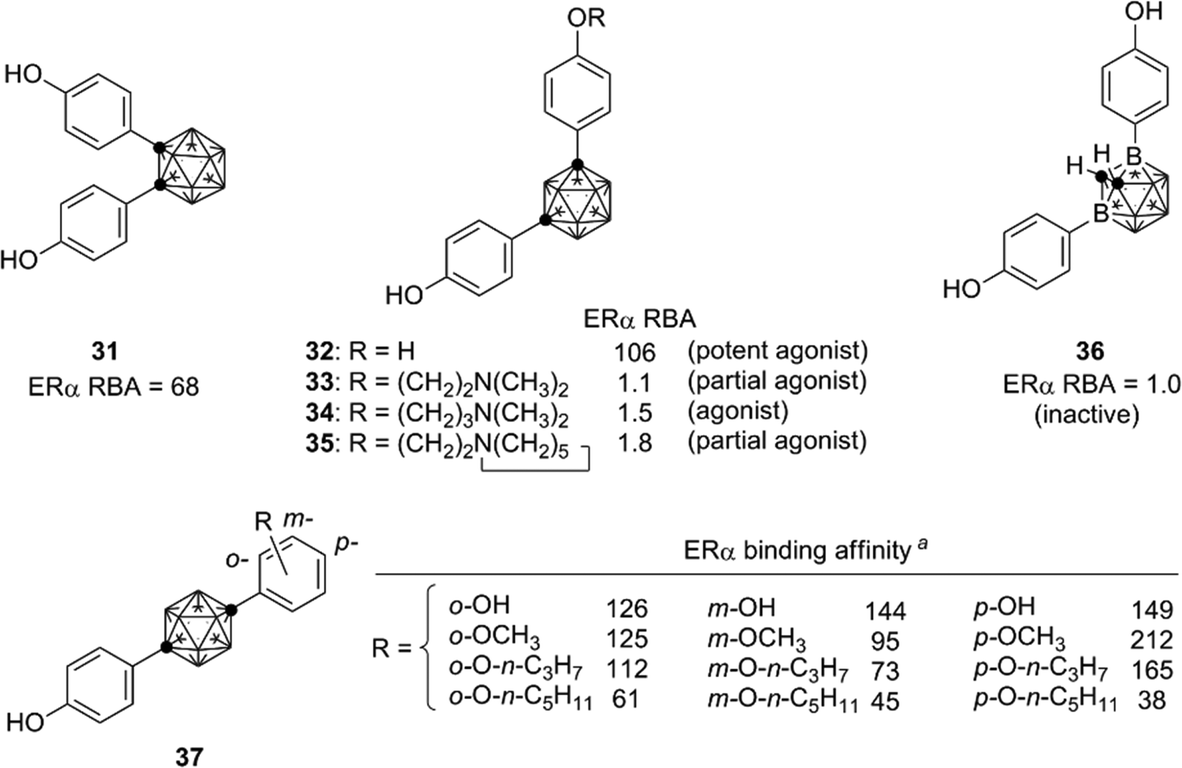 | ||
| Fig. 13 Representative ER ligands bearing a diphenylcarborane scaffold, developed by Endo et al. RBA: Relative binding affinity with respect to that of estradiol taken as 100. aERα binding affinity of the p-carborane derivatives is represented as percentage displacement of radiolabelled estradiol specific binding to hERα by each compound with a concentration of 4 nM (estradiol taken as 100). | ||
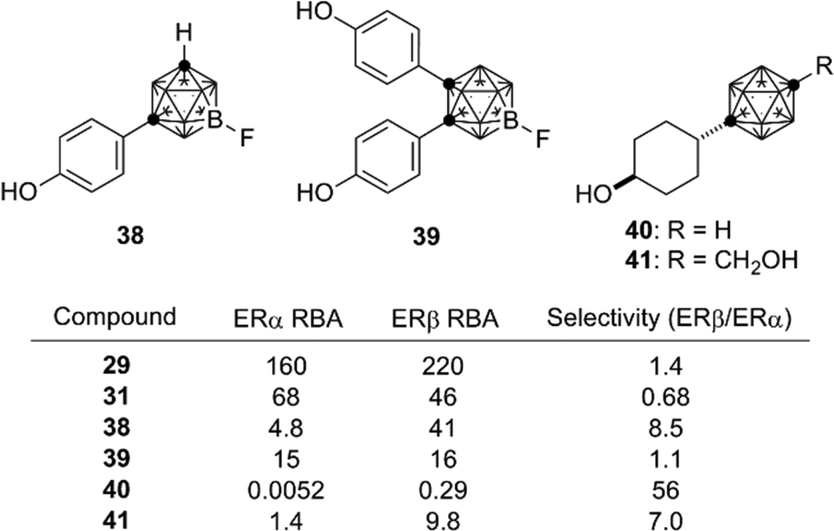 | ||
| Fig. 14 Structure and biological activity of some ERβ ligands. RBA: Relative binding affinity with respect to that of estradiol taken as 100. | ||
o-Carborane analogs of tamoxifen were also synthesized. Though isomerization of compound 42 to 43 was reported (Fig. 15), the carborane derivatives are less susceptible to degradation than the parent tamoxifen. Carborane analogs 42 and 43 showed a growth-inhibitory activity toward human breast cancer cell lines.43
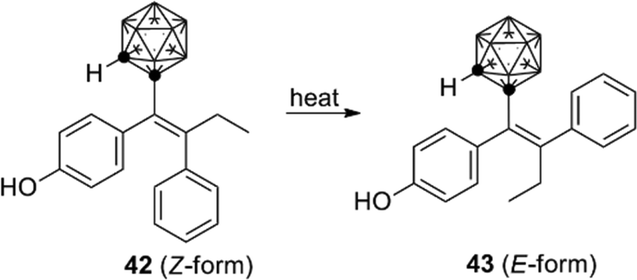 | ||
| Fig. 15 Structure and isomerization of the o-carborane analog of hydroxytamoxifen (Valliant et al.). | ||
Following the success in obtaining ER ligands, development of other nuclear steroid hormone receptor ligands was investigated. The androgen receptor (AR) is a nuclear receptor for androgens such as steroid hormones testosterone and dihydrotestosterone (DHT). Based on the findings during the development of carborane-containing ER ligands, cyclohexylcarborane derivatives such as 44 were designed and synthesized as AR ligand candidates. The cyclohexylcarborane derivatives exhibited a moderate AR antagonistic activity.44 On the other hand, phenylcarborane derivatives such as 45, which were designed as hybrids of 44 and developed AR antagonists such as flutamide and bicalutamide, exhibited a potent AR antagonistic activity.45 These results indicate that the phenylcarborane substructure is a promising scaffold for development of nuclear steroid hormone receptor ligands. Based on the structure of compound 45, a series of carborane-based AR antagonists have been developed. Ether, sulfide and sulfone derivatives such as 46 (ref. 46) and amide derivatives such as 47 (ref. 47) were developed as anti-androgens effective toward T887A-mutated AR. T877A mutation is one of the major cause of castration-resistant prostate cancer (CRPC),48,49 and therefore these carborane-based AR antagonists are possible lead compounds for development of drugs to treat CRPC (Fig. 16).
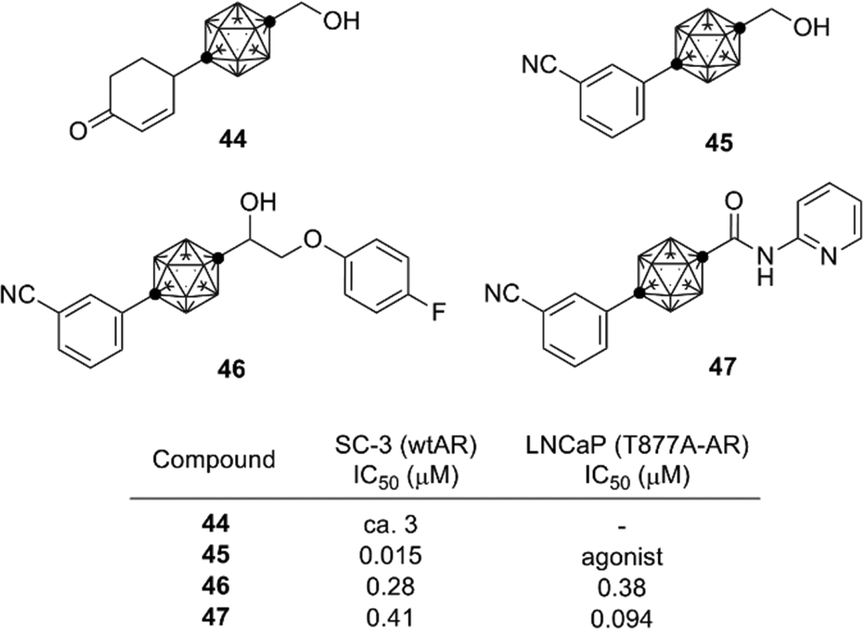 | ||
| Fig. 16 Carborane-based AR antagonists. | ||
During the development of AR antagonists, incorporation of carborane species other than dicarba-closo-dodecaboranes (C2B10H12) was also investigated. For example, ten-vertex p-carborane, specifically 1,10-dicarba-closo-decaborane (C2B8H10), is one of the smaller dicarba-closo-boranes, bearing a hydrophobic surface with ten vertices. Ten-vertex p-carborane has an overall ellipsoidal geometry and a smaller molecular volume in comparison to that of the highly symmetrical twelve-vertex carboranes. Ten-vertex p-carborane derivative 48 exhibited an AR partial agonistic activity, while the corresponding twelve-vertex carborane derivative 44 exhibited an AR full antagonistic activity.50 These results suggest that a slight difference in the spatial volume of the carborane cage can cause activity switching of the carborane-based AR ligands. Compound 49 exhibited potent binding affinity toward AR. In addition, compounds containing carba-closo-dodecaborate ([CB11H12]−), an anionic boron cluster with twelve vertices, have been developed. Compound 50 exhibits significant binding affinity toward the AR ligand-binding domain (LBD) (Fig. 17).51
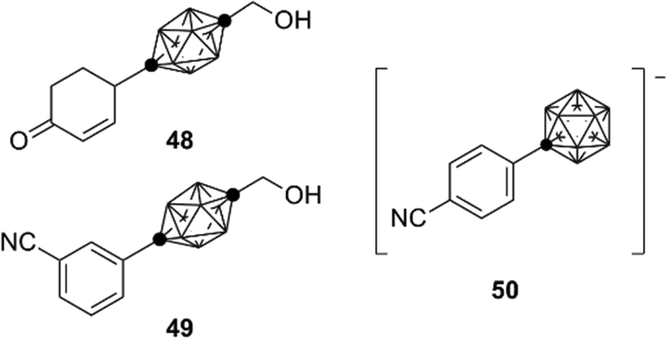 | ||
| Fig. 17 Ten-vertex p-carborane derivatives and carba-closo-dodecaborate derivative. | ||
AR-LBD shows high sequence identity with the nuclear progesterone receptor (PR), and thus the developed carborane-based AR ligands are also candidates as lead compounds for the development of PR ligands. Based on the structure of the developed AR antagonists, novel PR antagonists such as compounds 51 and 52, with selectivity for PR over AR and other steroid hormone receptors, were developed (Fig. 18).52,53
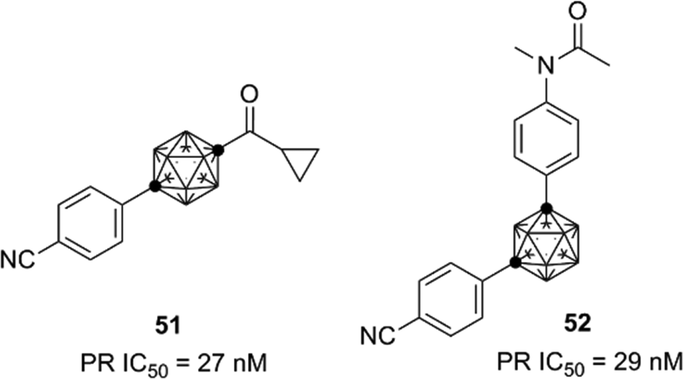 | ||
| Fig. 18 Carborane-based PR antagonists. | ||
Development of the steroid hormone receptor ligands suggested that the phenylcarborane scaffold is a promising alternative structure to the steroidal skeleton, and the carborane cage itself could be an alternative to the CD-ring of the steroidal framework. A secosteroidal bioactive compound, 1α,25-dihydroxyvitamin D3 (1α,25(OH)2D3), is the endogenous ligand of the vitamin D receptor (VDR). Almost all developed VDR ligands with high potency have the same secosteroidal skeleton, consisting of the A-ring bearing two hydroxyl groups, a conjugated olefin moiety, the CD-ring, and a side chain. However, the studies on the ER, AR and PR ligands suggested that the CD-ring of the secosteroidal structure could be replaced with a carborane cage. On the basis of these considerations, novel nonsecosteroidal VDR ligands such as compound 53 were developed. These VDR ligands exhibited a potent vitamin D activity, comparable to that of the natural hormone, despite their flexible structure (Fig. 19).
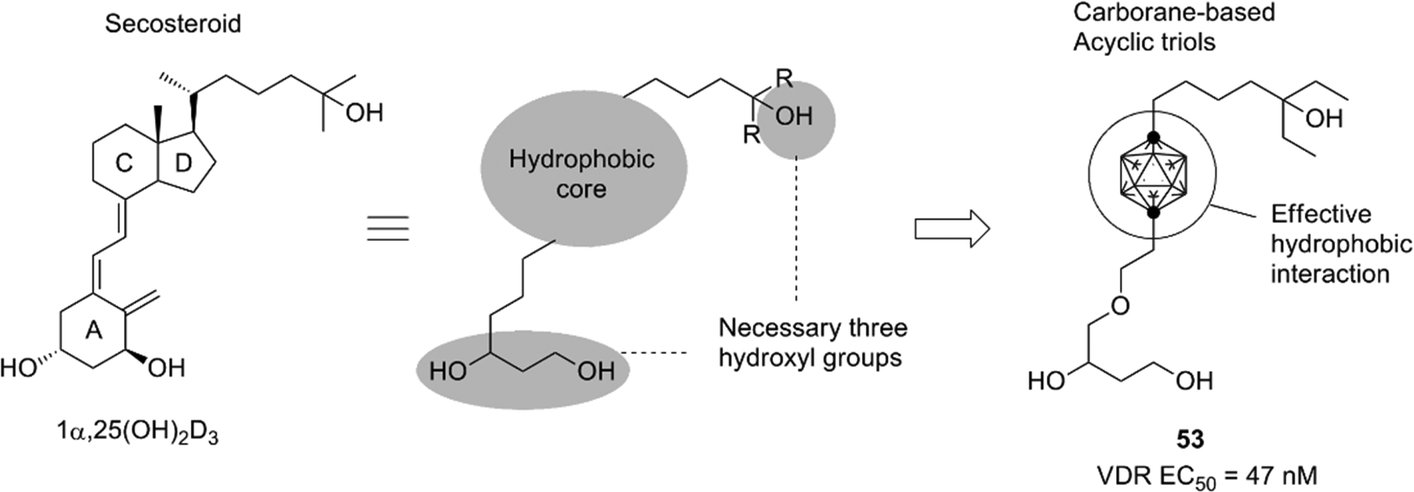 | ||
| Fig. 19 Design of carborane-based nonsecosteroidal VDR ligands. | ||
X-ray crystallographic analysis indicated that the hydrophobic surface of the carborane functions as a potent hydrophobic anchor, binding the ligand to the protein surface (PDBID: 3VJS for S-53 with rVDR and 3VJT for R-53 with rVDR). Thus, effective hydrophobic interactions appeared to counteract the entropic disadvantage involved in the interactions of a flexible molecule.54
Based on the crystallographic analysis, the structural modification was investigated, and highly potent compounds such as 54 and 55 were developed.55,56 Compound 54 is one of the most potent nonsecosteroidal VDR ligands currently available.56 Interestingly, bicyclo[2.2.2]octane derivatives such as 56 exhibited a markedly lower VDR agonistic activity than the corresponding carborane derivatives.57 These results indicate that hydrophobic interactions involving the carborane cage are essential for ligand potency (Fig. 20).
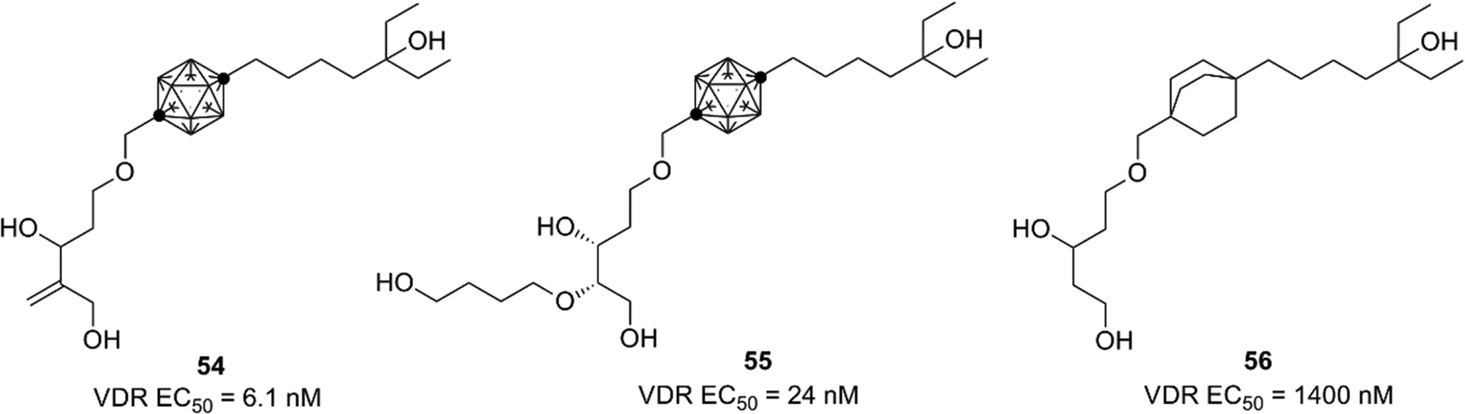 | ||
| Fig. 20 VDR ligands bearing p-carborane or bicyclo[2.2.2]octane as the hydrophobic core structure. (All compounds were evaluated as racemic mixtures.) | ||
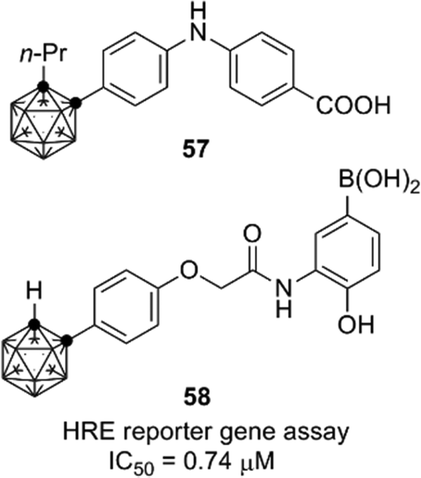
Retinoids and HIF-1 (hypoxia inducible factor 1) inhibitors containing an o-carborane cage as a hydrophobic substructure have also been developed. Diphenylamine-based retinoid 57 exhibited a cell differentiation-inducing activity toward human leukemia cell line HL-60 comparable to that of all-trans retinoic acid, and also exhibited a potent RAR agonistic activity with selectivity for RARα/β.58 RXR antagonists were developed by structural modification of the RAR agonists.59 As for HIF-1α, potent carborane-containing HIF-1α inhibitors, such as 58, were developed by Nakamura and co-workers.60 A chaperone protein HSP60 was identified as a primary target molecule of compound 58,61 and the o-carborane cage of 58 was essential for the inhibitory activity toward HSP60.62 These retinoids and HIF-1 inhibitors are expected to be versatile compounds for development of chemical probes and therapeutics for cancer (Fig. 21).
 | ||
| Fig. 21 Examples of carborane-containing retinoids and HIF-1α inhibitors. | ||
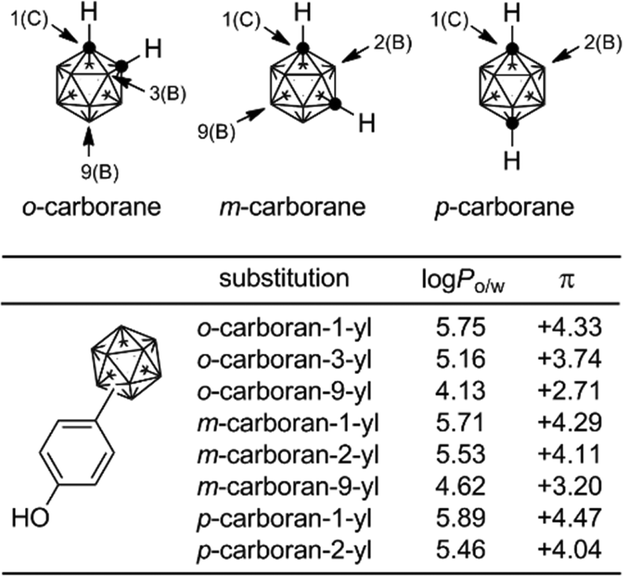
The hydrophobicity (log Po/w values) of carborane derivatives is significantly affected by the position of substitution at the carborane cage, as well as by the nature of the carborane isomer.63,64C-Substituted carboranyl groups are more hydrophobic than the corresponding B-substituted carboranyl groups. Among the B-substituted carboranyl groups, m-carboran-9-yl and o-carboran-9-yl groups are significantly less hydrophobic than the corresponding C-substituted carboranyl groups. It is suggested that the CH moiety on the carborane cage has a weak hydrophilic interaction with water. This characteristic, i.e. quite different hydrophobicity with the same geometry, is a unique feature of carborane derivatives (Fig. 22).
 | ||
| Fig. 22 Systematic determination of hydrophobicity of carboranylphenols. | ||
Organometallic and coordination compounds
Since the discovery of ferrocene, a stable hydrophobic coordination compound, various applications of this sandwich compound have been investigated. Applications of metallocenes in medicinal chemistry have also been widely studied.65,66 One of the most successful examples of ferrocene-containing compounds in medicinal chemistry is ferroquine, an antimalarial compound derived from chloroquine.67,68In addition, a few metallocene derivatives have been developed as nuclear receptor ligands.
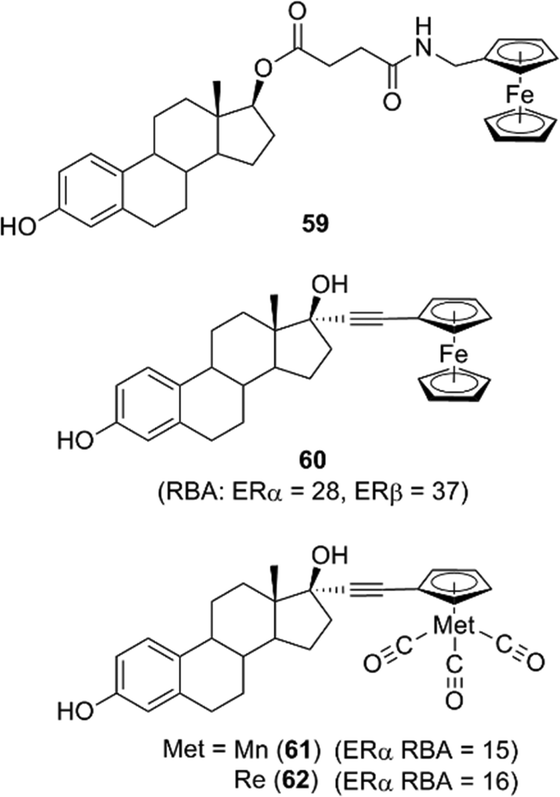
Among hormone–metallocene conjugates bearing metallocene as a substituent on an endogenous ligand of receptors, estradiol–metallocene conjugates including compound 59 have been most intensively investigated. Compounds bearing metallocene or other coordinated substituents at the 17α position, such as compounds 60–62 (Fig. 23), exhibited significant binding affinity toward ERα or ERβ.69–72 These compounds can be used as tracers in a metallo-immunoassay using metal-specific detection methods such as atomic absorption and IR spectroscopy. One example is the carbonyl metallo-immunoassay (CMIA).73 Radioactive coordination compounds, e.g. containing technetium, have also been developed as radiopharmaceuticals.74
 | ||
| Fig. 23 Examples of conjugates of estradiol and ferrocene or other organometallic derivatives. RBA: Relative binding affinity with respect to that of estradiol taken as 100. | ||
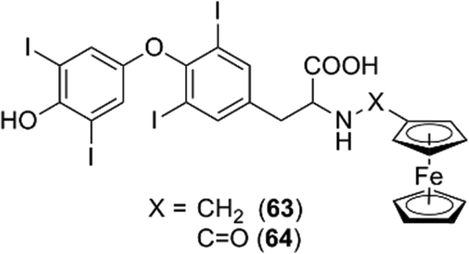
Ferrocene conjugates of thyroid hormones were developed for metallo-immunoassays. Ferrocene-linked triiodothyronine (T3) and thyroxin (T4) derivatives such as compounds 63 and 64 were synthesized (Fig. 24),75,76 and it was shown that the T4 derivatives can act as sensors of T4.
 | ||
| Fig. 24 Ferrocene thyroid hormone conjugates. | ||
Ferrocene and related coordination compounds exhibit remarkable hydrophobicity, and therefore can be used as hydrophobic substructures of biologically active compounds. For example, ferrocifen derivatives 65, which are ferrocene analogs of tamoxifen, have been widely investigated by Jaouen and co-workers. Ferrocifens contain a ferrocene moiety as a hydrophobic substructure instead of the benzene ring of tamoxifen, and were designed as candidate SERMs.77 Ferrocifens have dual modes of action, namely they show an anti-estrogenic activity toward estrogen-dependent cell line MCF-7 and an inhibitory activity toward estrogen-independent MDA-MB231 cell line.78,79 On the other hand, ruthenocene analogs 66 and osmosene analogs 67 of ferrocifens exhibited a low activity toward MDA-MB231.80,81 Ferrociphenol (68), a bisphenol derivative containing a ferrocene moiety as a hydrophobic substructure, exhibited moderate binding affinities toward ERα/β and a potent cell proliferation–inhibitory activity toward both MCF-7 and MDA-MB231 cell lines. On the other hand, a ferrocene analog of diethylstilbestrol 71, which is a regioisomer of ferrociphenol 68, scarcely affected the proliferation of these cell lines.82 Ruthenocene analog 69 and osmocene analog 70 of ferrociphenol also exhibited a quite low anti-proliferative activity toward these cell lines (Fig. 25).81
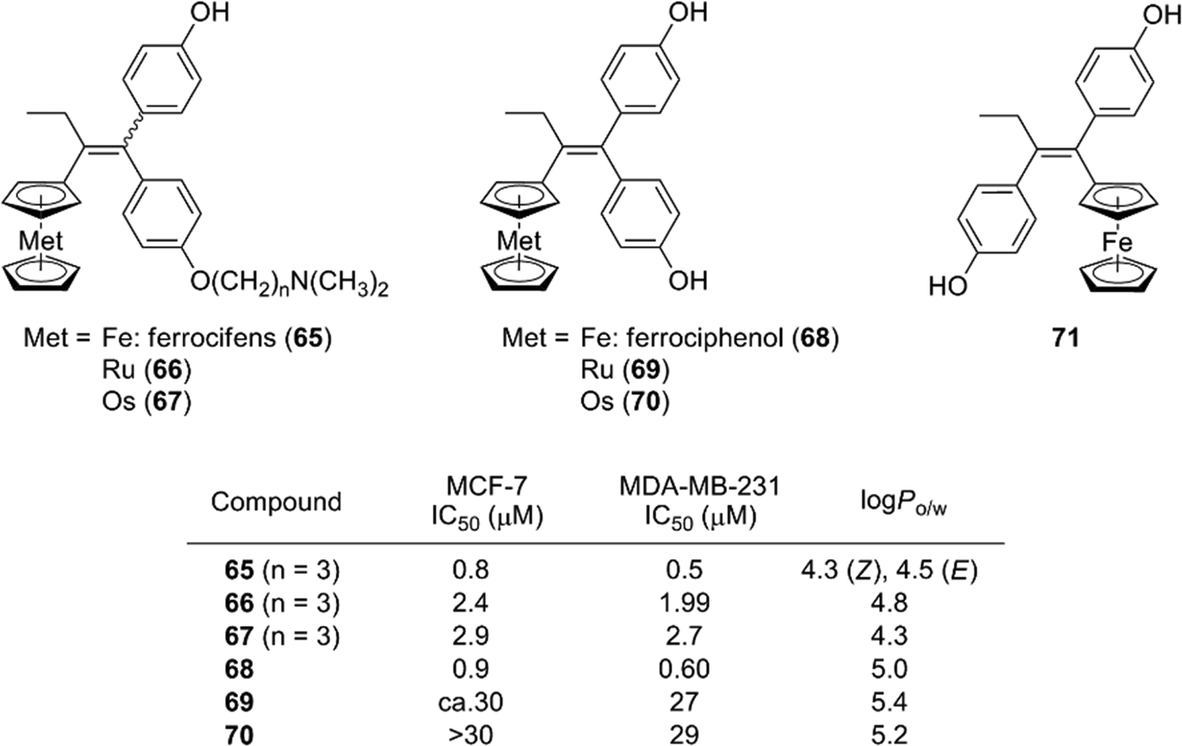 | ||
| Fig. 25 Structure and properties of ferrocifen, ferrociphenol and related compounds. | ||
Further structural development has been investigated, including the ansa series (72, 73),83 hydroxypropyl series and the corresponding quinone methide species (Fig. 26),84 and the mechanism of action of ferrocifens has been intensively studied. It was proposed that cytotoxic quinone methide 74 is formed by oxidation of 68 and that the redox properties of ferrocene contribute to the oxidation process. Changing the metal atom can significantly alter the redox properties. Experiments using quinone methide derivatives suggested that thioredoxin reductases are possible targets of the cytotoxicity of ferrocifens.85
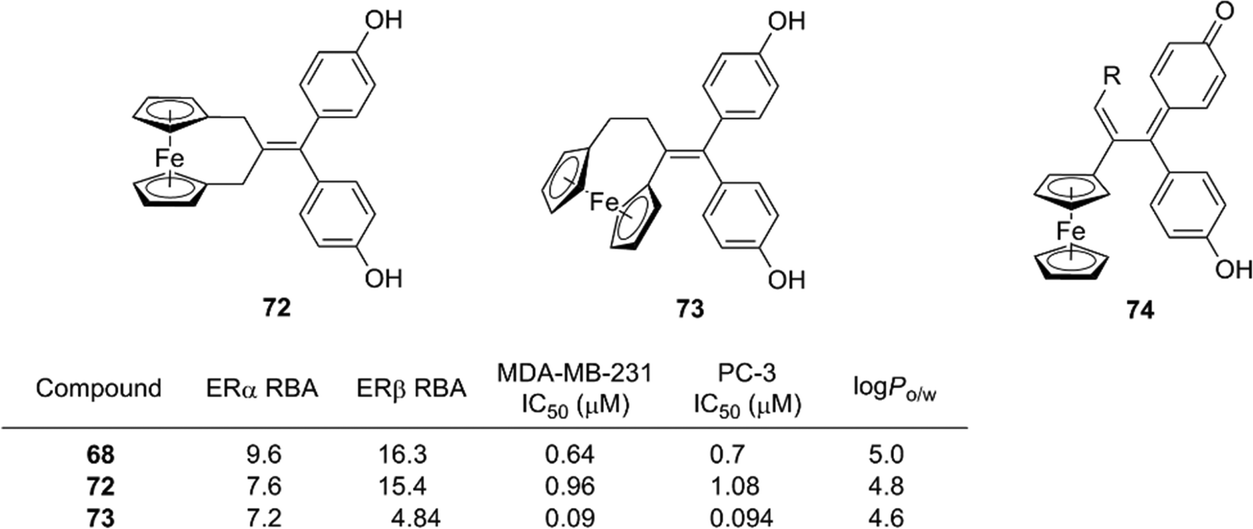 | ||
| Fig. 26 Structure and properties of ansa ferrociphenol derivatives and quinone methide species. RBA: Relative binding affinity with respect to that of estradiol taken as 100. | ||
In addition to antiestrogens, ferrocene-containing antiandrogens have been developed. Several ferrocene conjugates of nilutamide, such as 75 and 76, were synthesized based on the structure of nilutamide, a representative nonsteroidal AR antagonist.86 Among them, compound 76 exhibited a potent anti-androgenic activity toward the androgen-dependent LNCaP cell line, as well as a potent proliferation–inhibitory activity toward the androgen-independent PC-3 cell line. Compound 77 bearing an anisyl moiety instead of ferrocene also showed a significant activity toward both cell lines. Therefore, in contrast to ferrocifens, the cytotoxic effect of compound 76 is probably attributable to the aromatic or hydrophobic character of ferrocene, rather than to its redox characteristics (Fig. 27).
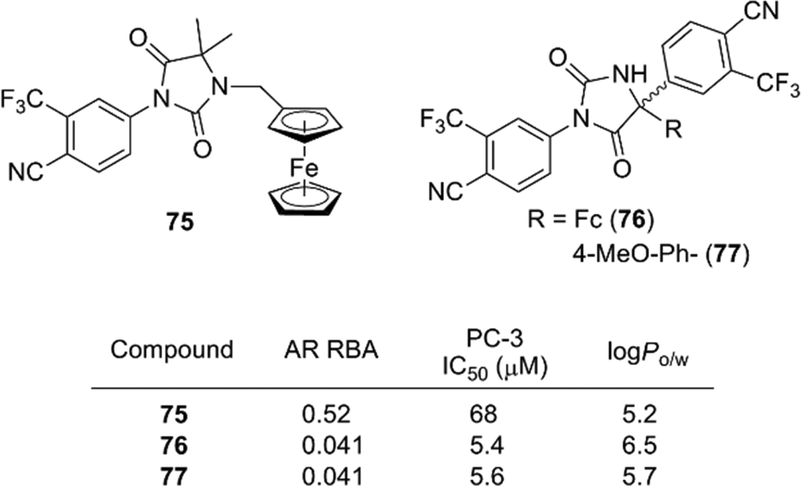 | ||
| Fig. 27 Ferrocene derivatives of AR antagonist nilutamide (Jaouen et al.). RBA: Relative binding affinity with respect to that of DHT taken as 100. | ||
Jaouen and co-workers determined the hydrophobicity parameter, log Po/w, of ferrocifens and nilutamide conjugates. Ferrociphenol 68 showed a log Po/w value of 5.0, which is larger by 0.6 than that of the corresponding phenyl analog.81 The difference in log Po/w between 76 (6.5) and 77 (5.7) is 0.8.86 Compounds with an ansa-ferrocene moiety have somewhat smaller log Po/w values (compound 72; 4.8, compound 73; 4.6) than the parent acyclic ferrociphenol 68 (5.0).83 The hydrophobicity of ruthenocene derivative 69 (5.4) and osmocene derivative 70 (5.2) is somewhat larger than that of ferrociphenol, and rhenium carbonyl derivatives of ferrocifen are more hydrophobic than the corresponding ferrocene analogs.74,82 Thus, ferrocene and other hydrophobic coordination species can serve as hydrophobic moieties with various functions, such as redox promotion and immunoassay labels.
Future perspective
Utilization of hydrophobic substructures other than simple hydrocarbons has the potential to greatly expand the chemical space of hydrophobic pharmacophores in medicinal chemistry. Herein, the author reviews the design of transcriptional modulators, focusing on three types of inorganic and organometallic hydrophobic structural motifs. Application of silicon or germanium functionalities is one approach to enhance the biological activity and/or target selectivity. Simple C/Si exchange, sila-substitution, has been applied not only to transcriptional modulators, but also to other targets, such as enzyme inhibitors and membrane receptor ligands, and is becoming a standard strategy. Sila-substitution has also been extended to mimicking diatomic substructures, and the introduction of hypervalent silicon functionalities is another possibility. The second approach is application of carboranes as unique spherical hydrophobic substructures. Compound libraries of carborane derivatives could be powerful tools for finding unique biological active compounds. So far, most carborane-incorporating biologically active compounds are dicarba-closo-dodecaborane derivatives, and investigation of other types of carboranes is a promising approach. In addition, there is interest in exploiting the B–N substructure as an isostere of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.87 For example, it was recently proposed that benzazaborinine can function as a bioisostere of naphthalene.88 In the third category of metallocenes, the redox properties of the metal atom play a major role. Metallocenes are not only merely hydrophobic substructures, but also functional chemical species, and may allow the development of polyfunctional bioactive compounds. One common issue to be addressed in applying all of the above hydrophobic fragments in drug discovery is their influence on ADMET profiles. Though some in vivo characterisation has been investigated in a few cases including compounds 2 (ref. 10) and 31,37 and in vitro metabolism of ferrocifens has been also investigated,89 there is a great need for systematic investigation of metabolic and toxicity profiles. Overall, novel hydrophobic substructures have great potential for enlarging the chemical space of hydrophobic pharmacophores in the field of medicinal chemistry.
C double bonds.87 For example, it was recently proposed that benzazaborinine can function as a bioisostere of naphthalene.88 In the third category of metallocenes, the redox properties of the metal atom play a major role. Metallocenes are not only merely hydrophobic substructures, but also functional chemical species, and may allow the development of polyfunctional bioactive compounds. One common issue to be addressed in applying all of the above hydrophobic fragments in drug discovery is their influence on ADMET profiles. Though some in vivo characterisation has been investigated in a few cases including compounds 2 (ref. 10) and 31,37 and in vitro metabolism of ferrocifens has been also investigated,89 there is a great need for systematic investigation of metabolic and toxicity profiles. Overall, novel hydrophobic substructures have great potential for enlarging the chemical space of hydrophobic pharmacophores in the field of medicinal chemistry.
Notes and references
- The Practice of Medicinal Chemistry, ed. C. G. Wermuth, Academic Press, Cambridge, 2nd edn, 2003 Search PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS PubMed.
- J. S. Mills and G. A. Showell, Expert Opin. Invest. Drugs, 2004, 13, 1149–1157 CrossRef CAS PubMed.
- S. Gately and R. West, Drug Dev. Res., 2007, 68, 156–163 CrossRef CAS.
- H. Kagechika and K. Shudo, J. Med. Chem., 2005, 48, 5875–5883 CrossRef CAS PubMed.
- H. Kagechika, E. Kawachi, Y. Hashimoto, T. Himi and K. Shudo, J. Med. Chem., 1988, 31, 2182–2192 CrossRef CAS PubMed.
- H. Kagechika, E. Kawachi, Y. Hashimoto and K. Shudo, J. Med. Chem., 1989, 32, 834–840 CrossRef CAS PubMed.
- T. Yamakawa, H. Kagechika, E. Kawachi, Y. Hashimoto and K. Shudo, J. Med. Chem., 1990, 33, 1430–1437 CrossRef CAS PubMed.
- K. B. Higginbotham, R. Lozano, T. Brown, Y. Z. Patt, T. Arima, J. L. Abbruzzese and M. B. Thomas, J. Cancer Res. Clin. Oncol., 2008, 134, 1325–1335 CrossRef CAS PubMed.
- K. Ohnishi, Int. J. Clin. Oncol., 2007, 12, 313–317 CrossRef CAS PubMed.
- A. Takeshita, K. Shinagawa, M. Adachi, T. Ono, T. Kiguchi and T. Naoe, Expert Opin. Orphan Drugs, 2014, 2, 961–969 CrossRef CAS.
- M. F. Boehm, L. Zhang, B. A. Badea, S. K. White, D. E. Mais, E. Berger, C. M. Suto, M. E. Goldman and R. A. Heyman, J. Med. Chem., 1994, 37, 2930–2941 CrossRef CAS PubMed.
- T. Tanaka and L. M. De Luca, Cancer Res., 2009, 69, 4945–4947 CrossRef CAS PubMed.
- R. Tacke, V. Müller, M. W. Büttner, W. P. Lippert, R. Bertermann, J. O. Daiß, H. Khanwalkar, A. Furst, C. Gaudon and H. Gronemeyer, ChemMedChem, 2009, 4, 1797–1802 CrossRef CAS PubMed.
- M. W. Büttner, C. Burschka, J. O. Daiss, D. Ivanova, N. Rochel, S. Kammerer, C. Peluso-Iltis, A. Bindler, C. Gaudon, P. Germain, D. Moras, H. Gronemeyer and R. Tacke, ChemBioChem, 2007, 8, 1688–1699 CrossRef PubMed.
- J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, I. Fleming, C. Gaudon, D. Ivanova, H. Gronemeyer and R. Tacke, Organometallics, 2005, 24, 3192–3199 CrossRef CAS.
- W. P. Lippert, C. Burschka, K. Götz, M. Kaupp, D. Ivanova, C. Gaudon, Y. Sato, P. Antony, N. Rochel, D. Moras, H. Gronemeyer and R. Tacke, ChemMedChem, 2009, 4, 1143–1152 CrossRef CAS PubMed.
- H. Toyama, M. Nakamura, M. Nakamura, Y. Matsumoto, M. Nakagomi and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 1948–1959 CrossRef CAS PubMed.
- M. F. Boehm, P. Fitzgerald, A. Zou, M. G. Elgort, E. D. Bischoff, L. Mere, D. F. Mais, R. P. Bissonnette, R. A. Heyman, A. M. Nadzan, M. Reichman and E. A. Allegretto, Chem. Biol., 1999, 6, 265–275 CrossRef CAS PubMed.
- S. Hosoda, A. Tanatani, K. Wakabayashi, Y. Nakano, H. Miyachi, K. Nagasawa and Y. Hashimoto, Bioorg. Med. Chem. Lett., 2005, 15, 4327–4331 CrossRef CAS PubMed.
- M. Nakamura, M. Makishima and Y. Hashimoto, Bioorg. Med. Chem., 2013, 21, 1643–1651 CrossRef CAS PubMed.
- S. Fujii, Y. Miyajima, H. Masuno and H. Kagechika, J. Med. Chem., 2013, 56, 160–166 CrossRef CAS PubMed.
- D. Kajita, M. Nakamura, Y. Matsumoto, M. Makishima and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 2244–2252 CrossRef CAS PubMed.
- G. Astarita, B. Di Giacomo, S. Gaetani1, F. Oveisi, T. R. Compton, S. Rivara, G. Tarzia, M. Mor and D. Piomelli, J. Pharmacol. Exp. Ther., 2006, 312, 563–570 CrossRef PubMed.
- D. Kajita, M. Nakamura, Y. Matsumoto, M. Ishikawa, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem. Lett., 2015, 25, 3350–3354 CrossRef CAS PubMed.
- Y. Nishiyama, M. Nakamura, T. Misawa, M. Nakagomi, M. Makishima, M. Ishikawa and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 2799–2808 CrossRef CAS PubMed.
- H. Toyama, M. Nakamura, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2015, 23, 2982–2988 CrossRef CAS PubMed.
- The borane, carborane, carbocation continuum, ed. J. Casanova, Wiley Interscience, New York, 1998 Search PubMed.
- V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS.
- A. H. Soloway, W. Tjarks and J. G. Barnum, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed.
- M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1993, 32, 950–984 CrossRef.
- G. L. Locher, Am. J. Roentgenol., Radium Ther. Nucl. Med., 1936, 36, 1–13 CAS.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed.
- Y. Endo, T. Iijima, Y. Yamakoshi, M. Yamaguchi, H. Fukasawa and K. Shudo, J. Med. Chem., 1999, 42, 1501–1504 CrossRef CAS PubMed.
- Y. Endo, T. Iijima, Y. Yamakoshi, H. Fukasawa, C. Miyaura, M. Inada, A. Kubo and A. Itai, Chem. Biol., 2001, 8, 341–355 CrossRef CAS PubMed.
- Y. Endo, T. Yoshimi and C. Miyaura, Pure Appl. Chem., 2003, 75, 1197–1205 CrossRef CAS.
- T. Ogawa, K. Ohta, T. Yoshimi, H. Yamazaki, T. Suzuki, S. Ohta and Y. Endo, Bioorg. Med. Chem. Lett., 2006, 16, 3943–3946 CrossRef CAS PubMed.
- K. Ohta, T. Ogawa, T. Suzuki, S. Ohta and Y. Endo, Bioorg. Med. Chem., 2009, 17, 7958–7963 CrossRef CAS PubMed.
- T. Ogawa, K. Ohta, T. Iijima, T. Suzuki, S. Ohta and Y. Endo, Bioorg. Med. Chem., 2009, 17, 1109–1117 CrossRef CAS PubMed.
- K. Ohta, T. Ogawa, A. Kaise and Y. Endo, Bioorg. Med. Chem. Lett., 2013, 23, 6555–6558 CrossRef CAS PubMed.
- K. Ohta, T. Ogawa, A. Oda, A. Kaise and Y. Endo, Bioorg. Med. Chem. Lett., 2015, 25, 4174–4178 CrossRef CAS PubMed.
- M. L. Beer, J. Lemon and J. F. Valliant, J. Med. Chem., 2010, 53, 8012–8020 CrossRef CAS PubMed.
- S. Fujii, Y. Hashimoto, T. Suzuki, S. Ohta and Y. Endo, Bioorg. Med. Chem. Lett., 2005, 15, 227–230 CrossRef CAS PubMed.
- S. Fujii, T. Goto, K. Ohta, Y. Hashimoto, T. Suzuki, S. Ohta and Y. Endo, J. Med. Chem., 2005, 48, 4654–4662 CrossRef CAS PubMed.
- T. Goto, K. Ohta, S. Fujii, S. Ohta and Y. Endo, J. Med. Chem., 2010, 53, 4917–4926 CrossRef CAS PubMed.
- S. Fujii, A. Yamada, K. Tomita, M. Nagano, T. Goto, K. Ohta, T. Harayama, Y. Endo and H. Kagechika, Med. Chem. Commun., 2011, 2, 877–880 RSC.
- H. I. Scher, G. Steineck and W. K. Kelly, Urology, 1995, 46, 142–148 CrossRef CAS PubMed.
- H. Miyamoto, M. M. Rahman and C. Chang, J. Cell. Biochem., 2003, 91, 3–12 CrossRef PubMed.
- S. Fujii, K. Ohta, T. Goto, A. Oda, H. Masuno, Y. Endo and H. Kagechika, Med. Chem. Commun., 2012, 3, 680–684 RSC.
- J. Kanazawa, R. Takita, A. Jankowiak, S. Fujii, H. Kagechika, D. Hashizume, K. Shudo, P. Kaszyński and M. Uchiyama, Angew. Chem., Int. Ed., 2013, 52, 8017–8021 CrossRef CAS PubMed.
- S. Fujii, A. Yamada, E. Nakano, Y. Takeuchi, S. Mori, H. Masuno and H. Kagechika, Eur. J. Med. Chem., 2014, 84, 264–277 CrossRef CAS PubMed.
- S. Fujii, E. Nakano, N. Yanagida, S. Mori, H. Masuno and H. Kagechika, Bioorg. Med. Chem., 2014, 22, 5329–5337 CrossRef CAS PubMed.
- S. Fujii, H. Masuno, Y. Taoda, A. Kano, A. Wongmayura, M. Nakabayashi, N. Ito, M. Shimizu, E. Kawachi, T. Hirano, Y. Endo, A. Tanatani and H. Kagechika, J. Am. Chem. Soc., 2011, 133, 20933–20941 CrossRef CAS PubMed.
- S. Fujii, A. Kano, H. Masuno, C. Songkram, E. Kawachi, T. Hirano, A. Tanatani and H. Kagechika, Bioorg. Med. Chem. Lett., 2014, 24, 4515–4519 CrossRef CAS PubMed.
- S. Fujii, R. Sekine, A. Kano, H. Masuno, C. Songkram, E. Kawachi, T. Hirano, A. Tanatani and H. Kagechika, Bioorg. Med. Chem., 2014, 22, 5891–5901 CrossRef CAS PubMed.
- A. Wongmayura, S. Fujii, S. Ito, A. Kano, Y. Taoda, E. Kawachi, H. Kagechika and A. Tanatani, Bioorg. Med. Chem. Lett., 2012, 22, 1756–1760 CrossRef CAS PubMed.
- Y. Endo, T. Iijima, K. Yaguchi, E. Kawachi, N. Inoue, H. Kagechika, A. Kubo and A. Itai, Bioorg. Med. Chem. Lett., 2001, 11, 1307–1311 CrossRef CAS PubMed.
- K. Ohta, T. Iijima, E. Kawachi, H. Kagechika and Y. Endo, Bioorg. Med. Chem. Lett., 2004, 14, 5913–5918 CrossRef CAS PubMed.
- K. Shimizu, M. Maruyama, Y. Yasui, H. Minegishi, H. S. Ban and H. Nakamura, Bioorg. Med. Chem. Lett., 2010, 20, 1453–1456 CrossRef CAS PubMed.
- H. S. Ban, K. Shimizu, H. Minegishi and H. Nakamura, J. Am. Chem. Soc., 2010, 132, 11870–11871 CrossRef CAS PubMed.
- H. Nakamura, Y. Yasui, M. Maruyama, H. Minegishi, H. S. Ban and S. Sato, Bioorg. Med. Chem. Lett., 2013, 23, 806–810 CrossRef CAS PubMed.
- K. Yamamoto and Y. Endo, Bioorg. Med. Chem. Lett., 2001, 11, 2389–2392 CrossRef CAS PubMed.
- Y. Endo, K. Yamamoto and H. Kagechika, Bioorg. Med. Chem. Lett., 2003, 13, 4089–4092 CrossRef CAS PubMed.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo and D. Dive, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- D. Dive and C. Biot, ChemMedChem, 2008, 3, 383–391 CrossRef CAS PubMed.
- M. Cais, S. Dani, Y. Eden, O. Gandolfi, M. Horn, E. E. Isaacs, Y. Josephy, Y. Saar, E. Slovin and L. Snarsky, Nature, 1977, 270, 534–535 CrossRef CAS PubMed.
- M. Cais, E. Slovin and L. Snarsky, J. Organomet. Chem., 1978, 160, 223–230 CrossRef CAS.
- D. Osella, C. Nervi, F. Galeotti, G. Cavigiolio, A. Vessières and G. Jaouen, Helv. Chim. Acta, 2001, 84, 3289–3298 CrossRef CAS.
- S. Top, H. El Hafa, A. Vessières, J. Quivy, J. Vaissermann, D. W. Hughes, M. J. McGlinchey, J.-P. Mornon, E. Thoreau and G. Jaouen, J. Am. Chem. Soc., 1995, 117, 8372–8380 CrossRef CAS.
- A. Vessières, J. Organomet. Chem., 2013, 734, 3–16 CrossRef.
- S. Top, A. Vessières, P. Pigeon, M.-N. Rager, M. Huché, E. Salomon, C. Cabestaing, J. Vaissermann and G. Jaouen, ChemBioChem, 2004, 5, 1104–1113 CrossRef CAS PubMed.
- G. A. Robinson, G. Martinazzo and G. C. Forrest, J. Immunoassay, 1986, 7, 1–15 CrossRef CAS PubMed.
- T. Yao and G. A. Rechnitz, Biosensors, 1988, 3, 307–312 CrossRef CAS.
- S. Top, J. Tang, A. Vessières, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- S. Top, A. Vessières, C. Cabestaing, I. Laios, G. Leclercq, C. Provot and G. Jaouen, J. Organomet. Chem., 2001, 637–639, 500–506 CrossRef CAS.
- S. Top, A. Vessières, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huché and G. Jaouen, Chem. – Eur. J., 2003, 9, 5223–5236 CrossRef CAS PubMed.
- P. Pigeon, S. Top, A. Vessières, M. Huché, E. A. Hillard, E. Salomon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS PubMed.
- H. Z. S. Lee, O. Buriez, F. Chau, E. Labbé, R. Ganguly, C. Amatore, G. Jaouen, A. Vessières, W. K. Leong and S. Top, Eur. J. Inorg. Chem., 2015, 4217–4226 CrossRef CAS.
- A. Vessières, S. Top, P. Pigeon, E. Hillard, L. Boubeker, D. Spera and G. Jaouen, J. Med. Chem., 2005, 48, 3937–3940 CrossRef PubMed.
- D. Plażuk, A. Vessières, E. A. Hillard, O. Buriez, E. Labbé, P. Pigeon, M.-A. Plamont, C. Amatore, J. Zakrzewski and G. Jaouen, J. Med. Chem., 2009, 52, 4964–4967 CrossRef PubMed.
- Y. Wang, P. Pigeon, S. Top, M. J. McGlinchey and G. Jaouen, Angew. Chem., Int. Ed., 2015, 54, 10230–10233 CrossRef CAS PubMed.
- A. Citta, A. Folda, A. Bindoli, P. Pigeon, S. Top, A. Vessières, M. Salmain, G. Jaouen and M. P. Rigobello, J. Med. Chem., 2014, 57, 8849–8859 CrossRef CAS PubMed.
- O. Payen, S. Top, A. Vessières, E. Brulé, M.-A. Plamont, M. J. McGlinchey, H. Müller-Bunz and G. Jaouen, J. Med. Chem., 2008, 51, 1791–1799 CrossRef CAS PubMed.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- F. J. R. Rombouts, F. Tovar, N. Austin, G. Tresadern and A. A. Trabanco, J. Med. Chem., 2015, 58, 9287–9295 CrossRef CAS PubMed.
- M.-A. Richard, D. Hamels, P. Pigeon, S. Top, P. M. Dansette, H. Z. S. Lee, A. Vessières, D. Mansuy and G. Jaouen, ChemMedChem, 2015, 10, 981–990 CrossRef CAS PubMed.
Footnote |
| † The author declares no competing interests. |
| This journal is © The Royal Society of Chemistry 2016 |