
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and application of ligand-based NMR screening assays for γ-butyrobetaine hydroxylase†‡
A.
Khan§
,
R. K.
Leśniak§
,
J.
Brem
,
A. M.
Rydzik
,
H.
Choi
,
I. K. H.
Leung¶
 ,
M. A.
McDonough
,
C. J.
Schofield
* and
T. D. W.
Claridge
*
,
M. A.
McDonough
,
C. J.
Schofield
* and
T. D. W.
Claridge
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk; tim.claridge@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 275658
First published on 17th February 2016
Abstract
γ-Butyrobetaine hydroxylase (BBOX) is a 2-oxoglutarate (2OG) dependent oxygenase that catalyses the stereoselective C-3 hydroxylation of γ-butyrobetaine (GBB) to give L-carnitine. L-Carnitine is involved in fatty acid metabolism in all animals and in some prokaryotes, and BBOX is a current drug target for the treatment of myocardial infarction. We describe the development and application of 1H NMR GBB/2OG reporter based assays employing paramagnetic relaxation enhancement to monitor inhibitor binding to the BBOX active site. In a single experiment, the method assesses inhibitors for competitive binding with 2OG or GBB, or both. The method was exemplified with a set of isoquinoline-based inhibitors; the results reveal structure–activity relationships that were not predicted from crystallographic studies, with some inhibitors competing 2OG only and some competing both 2OG and GBB. The method will also be applicable to work on the inhibition of other 2OG oxygenases.
Introduction
Ferrous ion (FeII) and 2-oxoglutarate (2OG) dependent oxygenases play multiple biologically important roles in humans and other animals, including in lipid metabolism.1,2 2OG oxygenases play a central role in the metabolism of the chlorophyll metabolite phytanic acid1 and in the biosynthesis of L-carnitine,1,2 which is required for fatty acid transport into mitochondria.3 Two steps in L-carnitine biosynthesis are catalysed by 2OG oxygenases, i.e. the C-3 hydroxylation of trimethyllysine (TML) and of γ-butyrobetaine (GBB), which are catalysed by trimethyllysine hydroxylase (TMLH) and γ-butyrobetaine hydroxylase (BBOX), respectively (Scheme 1).3 BBOX (and, maybe, TMLH) are inhibited by mildronate (THP or Met88),4 which is given to patients after myocardial infarction to suppress fatty acid metabolism.5 As shown by NMR and MS studies, mildronate is a competitive BBOX substrate, undergoing oxidation and subsequent degradation including via a Stevens type rearrangement;6 it may also inhibit uptake of dietary L-carnitine. There is, therefore, interest in the development of other types of BBOX inhibitors, for example compounds acting as reversibly binding inhibitors that are competitive with 2OG and/or GBB, but which are not substrates.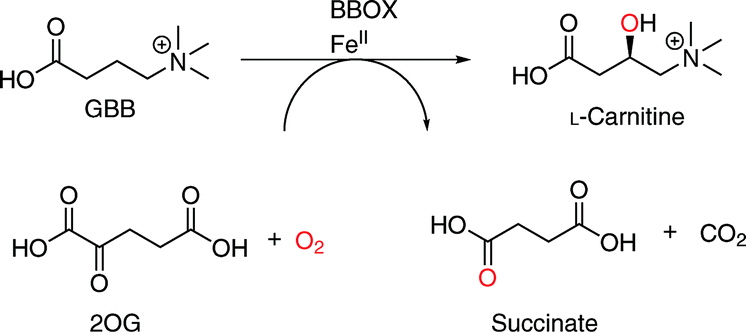 | ||
| Scheme 1 BBOX catalyses the stereoselective C-3 hydroxylation of GBB to give L-carnitine. The co-substrates are 2OG and oxygen and the co-products are succinate and CO2. | ||
We are interested in developing solution-based NMR methods to complement the extensive crystallographic studies on 2OG oxygenases, in order to help enable the development of clinically useful inhibitors.2 Work using NMR spectroscopy on the 2OG dependent hypoxia inducible factor (HIF) prolyl hydroxylase PHD2, has shown that crystallographically observed inhibitor binding modes do not always reflect those occurring in solutions7 and that inhibitors which might be expected to compete with substrates do not always do so.7–9
Direct ligand observation, monitoring the attenuation or broadening of ligand NMR resonances in the presence of a target protein, can be an efficient method for analysing the binding of ligands to a protein and may employ conventional 1H NMR10–12 and/or transverse relaxation-edited 1H NMR.13,14 Site specific ligand binding may also be assessed through ligand competition, i.e. by monitoring the sharpening and recovery of the NMR resonances of displaced ligands, often referred to as spy or reporter molecules.15
We describe the development of an NMR assay for recombinant bacterial BBOX from Pseudomonas sp. AK1 (psBBOX)16 that simultaneously uses both 2OG and GBB as reporter molecules. We validate the method using a set of isoquinoline-based derivatives, which are established inhibitors of 2OG oxygenases, including human BBOX (hBBOX).2,17 The results reveal that even within the same series of 2OG oxygenase inhibitors, unexpected structure–activity relationships can emerge. The results should help in efforts to develop new BBOX inhibitors and to promote the use of NMR spectroscopy in 2OG oxygenase medicinal chemistry. In developing the assay we employed psBBOX rather than hBBOX, since it can be readily prepared on a large scale and is thought to be structurally closely related to hBBOX.16
Several crystal structures have been reported for hBBOX,18–20 although none for psBBOX. hBBOX dimerises in an unusual manner using its N-terminal zinc binding domain to interact with the oxygenase domain of the other monomer in the dimer.19,20 The general active site architecture of BBOX is very similar to those of other 2OG oxygenases with the active site FeII (to which the oxalyl group of 2OG binds in a bidentate manner) being ligated by three protein residues.2 GBB binds via interactions involving both its trimethylammonium21 and carboxylate groups (Fig. 1). The trimethylammonium group is located in an aromatic cage formed by the side chains of Tyr-177, Tyr-194, Trp-181, Tyr-205 and Tyr-366, while the carboxylate group is positioned to interact with the side chains of Asn-292, Asn-191 and the backbone amide of Tyr-205 (Fig. 1).
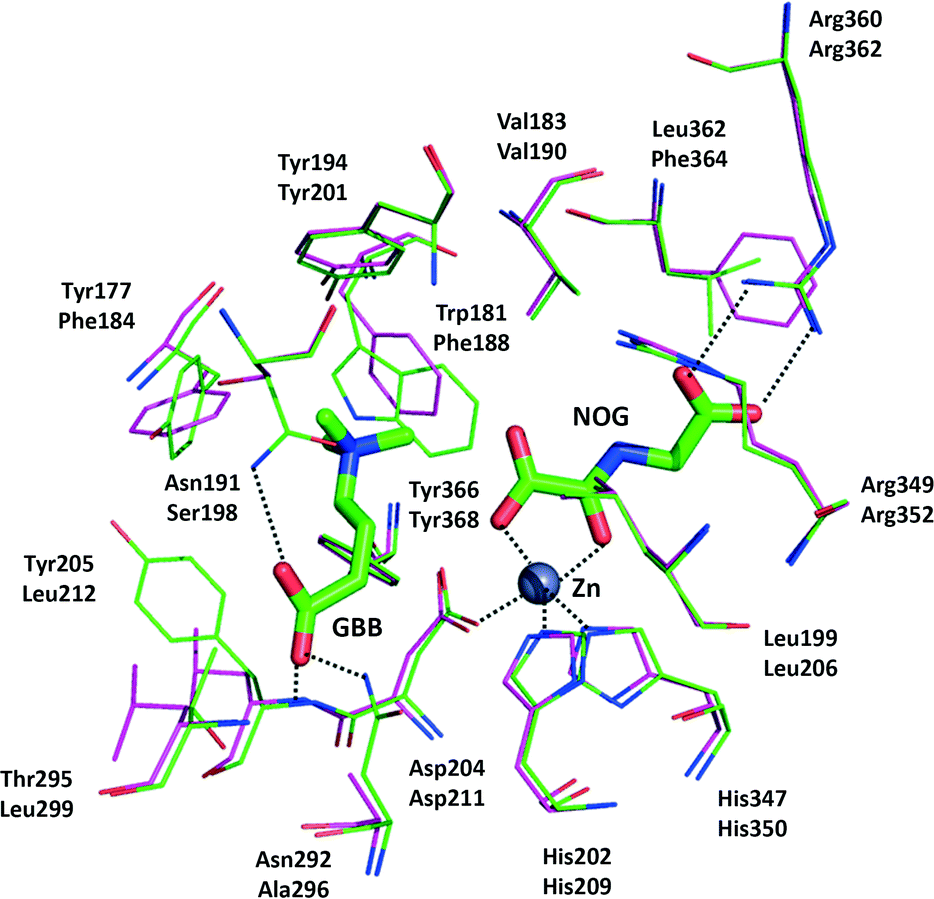 | ||
| Fig. 1 View from a hBBOX crystal structure (green sticks, upper residues numbers, PDB ID: 3O2G)20 and psBBOX model (pink sticks, lower residue numbers) showing binding of GBB and N-oxalylglycine (NOG), an unreactive 2OG analogue. ZnII replaces native FeII. | ||
Results and discussion
Initially, we optimised the assay conditions to enable detection of binding of both GBB and 2OG (as reporter molecules) to psBBOX in a single Carr-Purcell-Meiboom-Gill (CPMG)-edited 1H NMR ligand-observe experiment monitoring line shape changes in the presence of psBBOX13,14 (Fig. 2). The CPMG-edited 1H NMR experiments were used in order to improve NMR sensitivity to ligand binding by enhancing signal attenuation of broadened resonances. To prevent psBBOX-catalysed GBB hydroxylation and uncoupled 2OG decarboxylation into succinate, we replaced the FeII with the ‘non-catalytic’ metals ZnII or MnII. We observed that in the presence of psBBOX and ZnII, the GBB and 2OG signals were broadened and attenuated, demonstrating their fast-exchange binding interaction with psBBOX (Fig. 2a). However, despite adding high psBBOX concentrations, only relatively moderate broadening was observed. We envisaged that greater sensitivity to ligand binding might be achieved by employing paramagnetic relaxation enhancement (PRE).22,23 For this purpose either a paramagnetic metal co-factor such as CoII (ref. 24) or MnII (ref. 25, 26) or a protein covalently tagged with a spin label27 can be used. When a ligand binds to a protein containing a paramagnetic centre (within ∼15 Å), its nuclear spin relaxation rates are enhanced and its NMR signals are significantly broadened. Accordingly, when paramagnetic MnII, rather than diamagnetic ZnII, was added to the BBOX assay, significant increases in the line widths of both GBB and 2OG were observed in the presence of substantially lower amounts of psBBOX as compared to the ZnII assay (Fig. 2b). Control experiments (without psBBOX) revealed that at the concentrations employed, free MnII in solution did not affect the GBB line shape; it did, however, broaden the resonances of 2OG since this directly ligates MnII. Despite this broadening, the resonance intensities (1H NMR peak areas) remained similar to those prior to the addition of the metal (Fig. S1†). In a separate control experiment, when the GBB/2OG/MnII/psBBOX sample was heated (373 K, 5 min) to cause BBOX denaturation, recovery of the reporter signals was observed (Fig. S2†), implying that the enhanced broadening of the reporter signals upon the addition of psBBOX was due to binding to the protein. The binding affinities of GBB for both ZnII and MnII forms of BBOX in the presence of 2OG were similar with KD values of 5 ± 1 (Fig. S3†) and 4 ± 1 μM (Fig. S4†), respectively, as determined by titrating apo-psBBOX into a solution with constant GBB and metal concentrations. The MnII system was therefore chosen for further screening of inhibitors by the NMR dual-reporter displacement assay. In this, competition from a ligand for the GBB and/or 2OG binding sites could be established through the observed recovery of the reporter ligand resonances upon addition of the inhibitor to the GBB/2OG/MnII/psBBOX sample.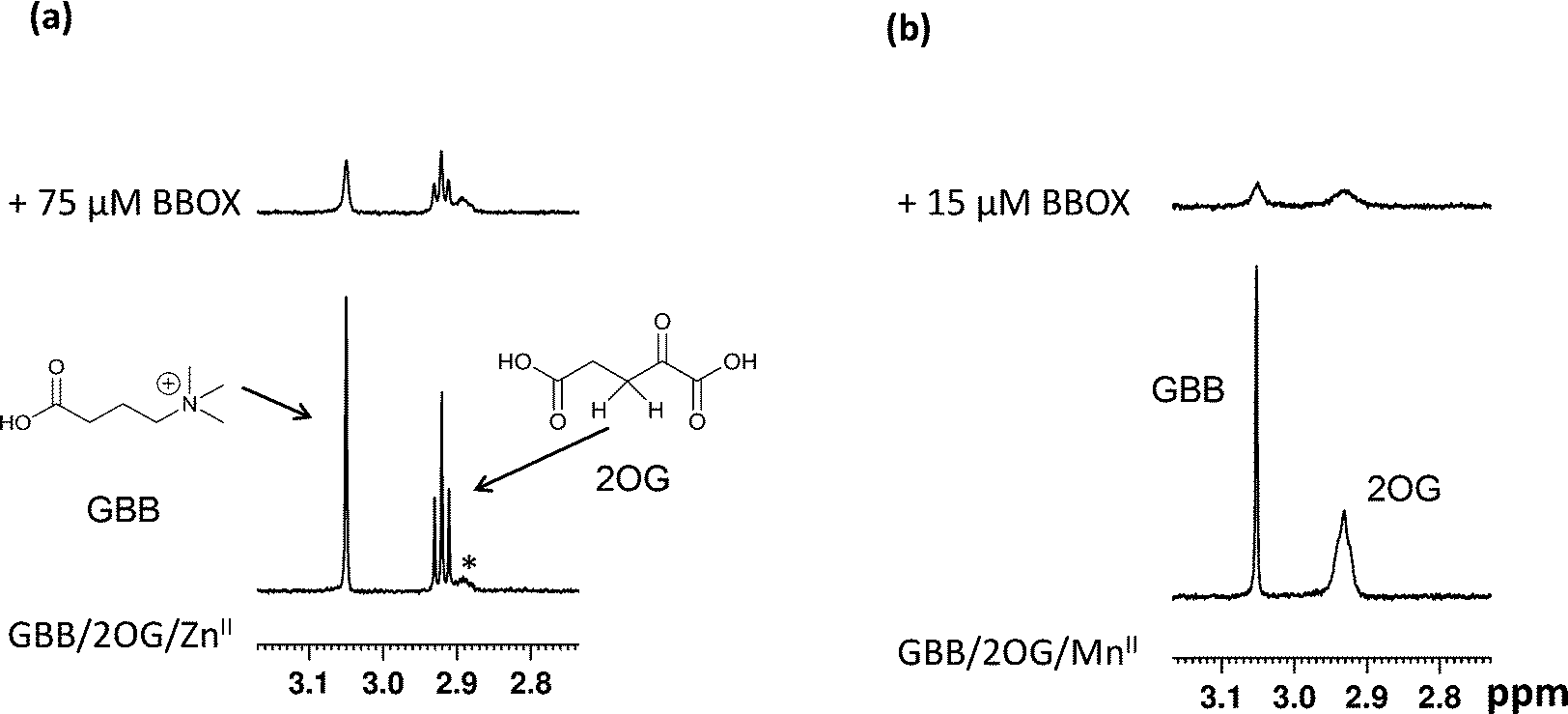 | ||
| Fig. 2 Monitoring the binding of GBB and 2OG with psBBOX as determined by NMR direct ligand-observation. The GBB (Me3 singlet at 3.05 ppm) and 2OG (CH2 triplet at 2.91 ppm) signals from CPMG edited 1H NMR experiments are shown. (a) GBB and 2OG binding to BBOX-ZnII (b) GBB and 2OG binding to BBOX-MnII. The assay mixture contains 25 μM GBB, 300 μM 2OG, 150 μM MII, 80 mM KCl, in 50 mM Tris-D11 buffer, pH 7.5, in D2O. The asterisk indicates an impurity. | ||
A set of 15 isoquinoline-based compounds17 were screened for their binding to psBBOX using simultaneously both GBB and 2OG as reporter molecules (Fig. 3). All these ligands shared the same isoquinoline core skeleton, but differed in their amino-acid derived side chains (Fig. 4). Exemplary results from the assay are presented in Fig. 3 for two of these isoquinoline-based ligands, one possessing the Gly (1) and the other the L-Trp (11) derived side chains. The spectra of GBB and 2OG with MnII were first recorded without and with psBBOX (Fig. 3a and b), then the isoquinoline-ligand was added to the latter sample. With 1, only the signals of 2OG were observed to recover (Fig. 3c), implying that 1 competes efficiently with 2OG, but not GBB. In contrast, with 11, we observed recovery of both 2OG and GBB signals (Fig. 3d), implying competition with both 2OG and GBB. The combined results of this assay for all the isoquinoline-ligands tested (at 25 μM) are summarised in Fig. 4.
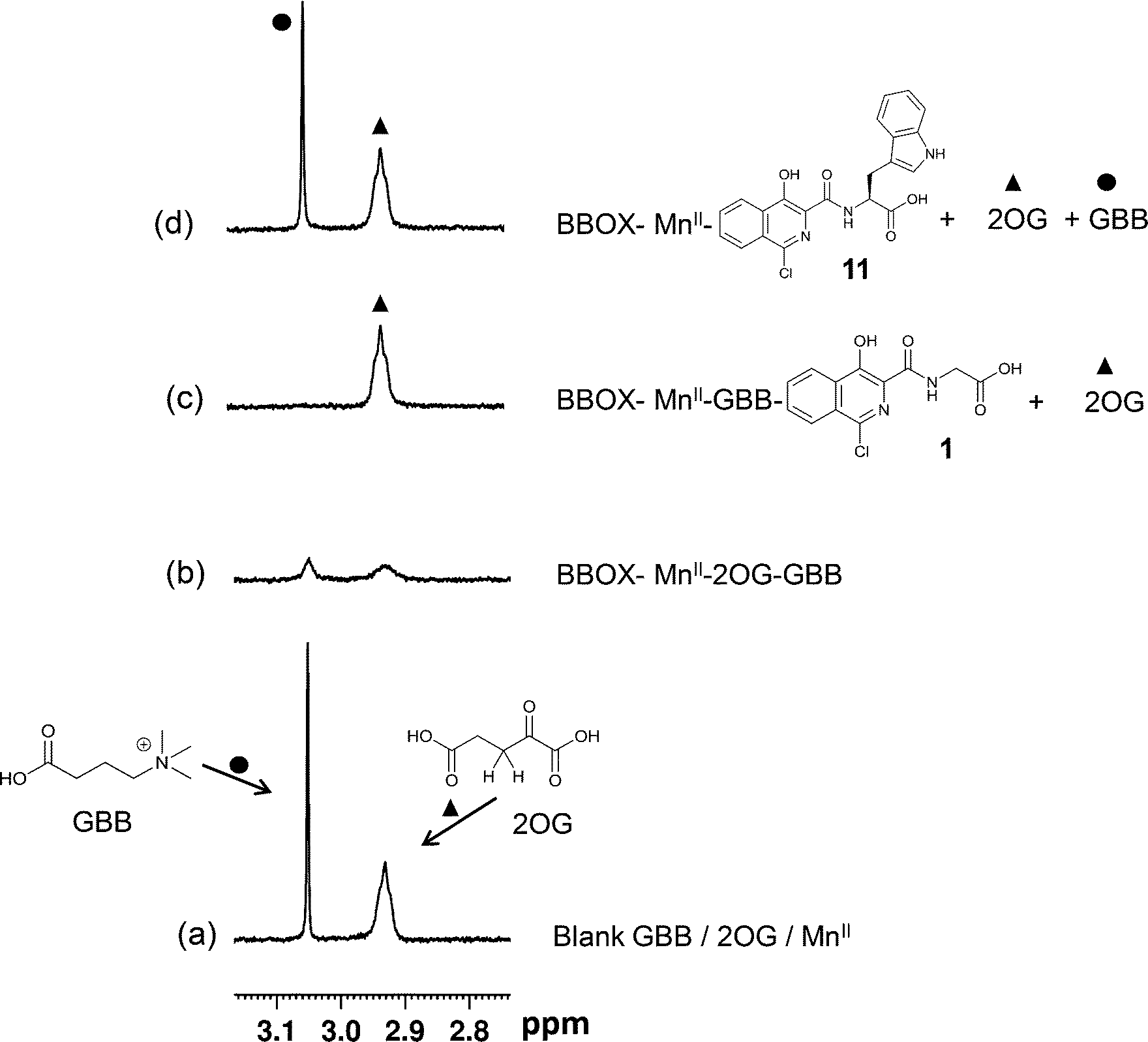 | ||
| Fig. 3 Studying the binding of isoquinoline ligands 1 and 11 with psBBOX using the NMR dual-reporter displacement assay. 2OG and GBB are used as reporter ligands in CPMG edited 1H NMR experiments. (a) Reporter ligands GBB and 2OG with MnII in solution in the absence of psBBOX. (b) After the addition of psBBOX. (c) Addition of 1 to sample of b. (d) Addition of 11 to sample of b. Symbols (●, ▲) indicate reporter signal recovery due to displacement. The final assay mixture contained 25 μM GBB, 300 μM 2OG, 150 μM MnII, 15 μM BBOX, 80 mM KCl and 25 μM isoquinoline-based ligand in 50 mM Tris-D11 buffer, pH 7.5, in D2O. | ||
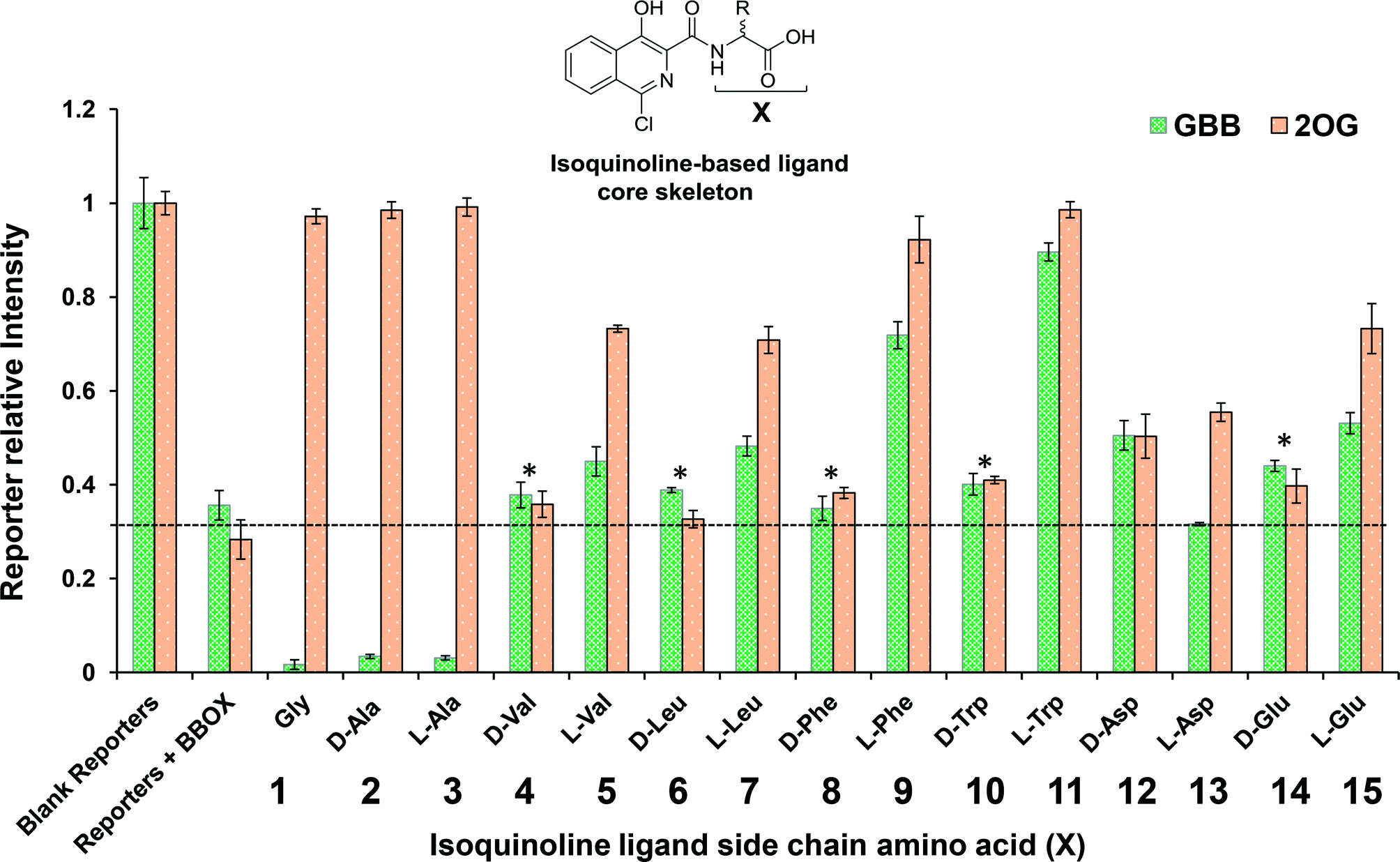 | ||
| Fig. 4 The chart compares the signal intensity of GBB (green) and 2OG (orange) in the absence of psBBOX (1st left set of columns; intensity set to unit intensity) with their intensity after the addition of psBBOX (2nd left set of columns) and with their intensity after the addition of isoquinoline-based inhibitors 1–15 (side-chain amino acid X varying). The threshold line represents the relative intensity of the reporters in the presence of psBBOX. After the addition of isoquinoline-ligand, the level above this line represents reporter displacement; the level below represents enhancement of reporter binding. Asterisks highlight isoquinoline-ligands whose addition results in a negligible difference on GBB/2OG under these conditions. The final assay mixture contained 25 μM GBB, 300 μM 2OG, 150 μM MnII, 15 μM BBOX, 80 mM KCl and 25 μM isoquinoline-based ligand in 50 mM Tris-D11 buffer, pH 7.5, in D2O. The error bars represent standard deviations from three separate measurements. | ||
The results imply that the isoquinoline-ligands containing the Gly (1) or Ala (both D, 2 and L, 3) derived side chains compete with 2OG, but not GBB. The full displacement of 2OG by these ligands suggests that they are likely strong binders of psBBOX. The results also imply that binding of these compounds enhanced GBB binding relative to that in the presence of 2OG. On increasing the size of the steric bulk of the side chain, i.e. for those derived from Val (4, 5) and Leu (6, 7), the ligands appeared to bind less favourably as demonstrated by lower levels, or even a lack, of 2OG displacement compared to 1, 2 and 3. The apparent displacement of both 2OG and GBB by 5 and 7, albeit at low levels, suggests that they attenuate GBB binding. Notably, binding was observed only for the L-forms (5, 7), but not for the D-forms (4, 6), of the Val and Leu derived compounds. This preference for binding by the L-enantiomer was preserved through most of the results where substantial reporter displacement was observed. These results encouraged us to analyse compounds with more hydrophobic or polar side chains (7–15). Ligands with the D- and L-Asp and Glu derived side chains (12–15) were mostly weak 2OG competitors relative to the inhibitors with hydrophobic side chains (5, 7, 9 and 11). 12 and 15 competed weakly with both 2OG and GBB, whereas, 13 competed with only 2OG, suggesting differences in binding modes, though care should be taken in interpreting results with weakly binding ligands. The results with the more bulky Phe (8, 9) and Trp (10, 11) derived side chain ligands were striking. The L-enantiomers 9 and 11 displaced both 2OG and GBB clearly contrasting with the ligands with smaller side chains (1–3).
The NMR binding results were then compared with those for the same compounds obtained using an intrinsic fluorescence-based binding assay.17,28 The results of fluorescence based assay (Table 1) demonstrate that ligands with Gly 1, D-Ala 2, L-Ala 3, L-Phe 9 and L-Trp 11 are the strongest binders, i.e. they agree with the 1H NMR assay results. They also support the proposal that in general the L-enantiomers bind more tightly than the D-enantiomers. The combined NMR and fluorescence assay results reveal that the level of 2OG displacement correlates with the binding affinity, notably 1, 2, 3, 9 and 11 all fully displace 2OG (within our limits of detection) and have approximately similar binding affinities (KD = 22, 20, 15, 26 and 29 μM, respectively). Similarly, ligands 5, 7 and 15, all compete relatively moderately with 2OG compared to ligands 1, 2, 3, 9 and 11, and have similar binding affinities (KD = 55, 55 and 64 μM respectively), which are weaker than 1, 2, 3, 9 and 11. Compounds 12 and 13, which appear to displace 2OG to a similar extent, also have similar affinities (KD = 101 and 91 μM respectively).
| Compound no. | Amino acid (X) | K D (μM) by fluorescence assay | IC50 (μM) by 1H NMR assay | IC50 (μM) by F− release assay |
|---|---|---|---|---|
| 1 | Gly | 22 ± 4 | 0.21 ± 0.01 | 1.2 ± 0.1 |
| 2 | D-Ala | 20 ± 4 | 0.20 ± 0.02 | 0.9 ± 0.03 |
| 3 | L-Ala | 15 ± 3 | 0.22 ± 0.01 | 0.7 ± 0.02 |
| 4 | D-Val | 144 ± 6 | 191 ± 6 | >1000 |
| 5 | L-Val | 55 ± 3 | 23 ± 2 | 130 ± 14 |
| 6 | D-Leu | 103 ± 4 | 94 ± 8 | >1000 |
| 7 | L-Leu | 55 ± 3 | 26 ± 2 | 125 ± 6 |
| 8 | D-Phe | 82 ± 3 | 38 ± 0.6 | 101 ± 6 |
| 9 | L-Phe | 26 ± 4 | 1 ± 0.03 | 13 ± 0.7 |
| 10 | D-Trp | 168 ± 37 | 18 ± 1 | >1000 |
| 11 | L-Trp | 29 ± 5 | 0.9 ± 1 | 26 ± 2 |
| 12 | D-Asp | 101 ± 1 | 38 ± 1 | 160 ± 61 |
| 13 | L-Asp | 91 ± 2 | 76 ± 2 | 245 ± 62 |
| 14 | D-Glu | 89 ± 6 | 86 ± 5 | >1000 |
| 15 | L-Glu | 64 ± 2 | 22 ± 2 | 147 ± 21 |
We then carried out catalytic turnover assays employing 1H NMR20 (Fig. 5) and fluoride ion release29 assays to study BBOX inhibition by these isoquinoline-based compounds (Table 1). For the NMR assay, reactions (60 s) were quenched by the addition of 1 M HCl (Fig. S5†); the concentrations of GBB and L-carnitine were measured by integration of their Me3 resonances (Fig. 5, S5†). The fluoride ion release assay29 employs BBOX catalysed hydroxylation of (3S)-3-fluoro-4-(trimethylammonio)butanoate (GBBF) to give an unstable product which fragments to give a fluoride ion that can subsequently deprotect a tert-butyldimethylsilyl-protected fluorescein to provide measurable fluorescence. Differences in the absolute IC50 values of 1H NMR and fluoride release assays, particularly for weak inhibitors, are likely due to the different experimental conditions used. The fluoride release assay was initially developed for use on hBBOX inhibition;29 however, when applied to psBBOX we found the assay required higher concentrations of psBBOX to observe detectable fluorescence when compared to the hBBOX protocol (1 μM vs. 0.2 μM, respectively). This concentration difference is notable when considering the amount of enzyme used for the NMR assay (0.14 μM). In addition, the NMR assay uses the natural substrate GBB, whereas the fluoride assay necessarily uses GBBF whose affinity for psBBOX is notably different (KM = 2.4 mM and 0.623 mM respectively).16 Nonetheless the results from both assays (Table 1) suggest that isoquinoline-based ligands 1, 2, 3, 9 and 11 are the strongest inhibitors in the series, consistent with their ability to compete with 2OG. Similarly, ligands 5, 7 and 15 are medium potency inhibitors, whereas, ligands 4, 6, 8, 10, 12, 13, and 14 are weak inhibitors (Fig. 4, Table 1).
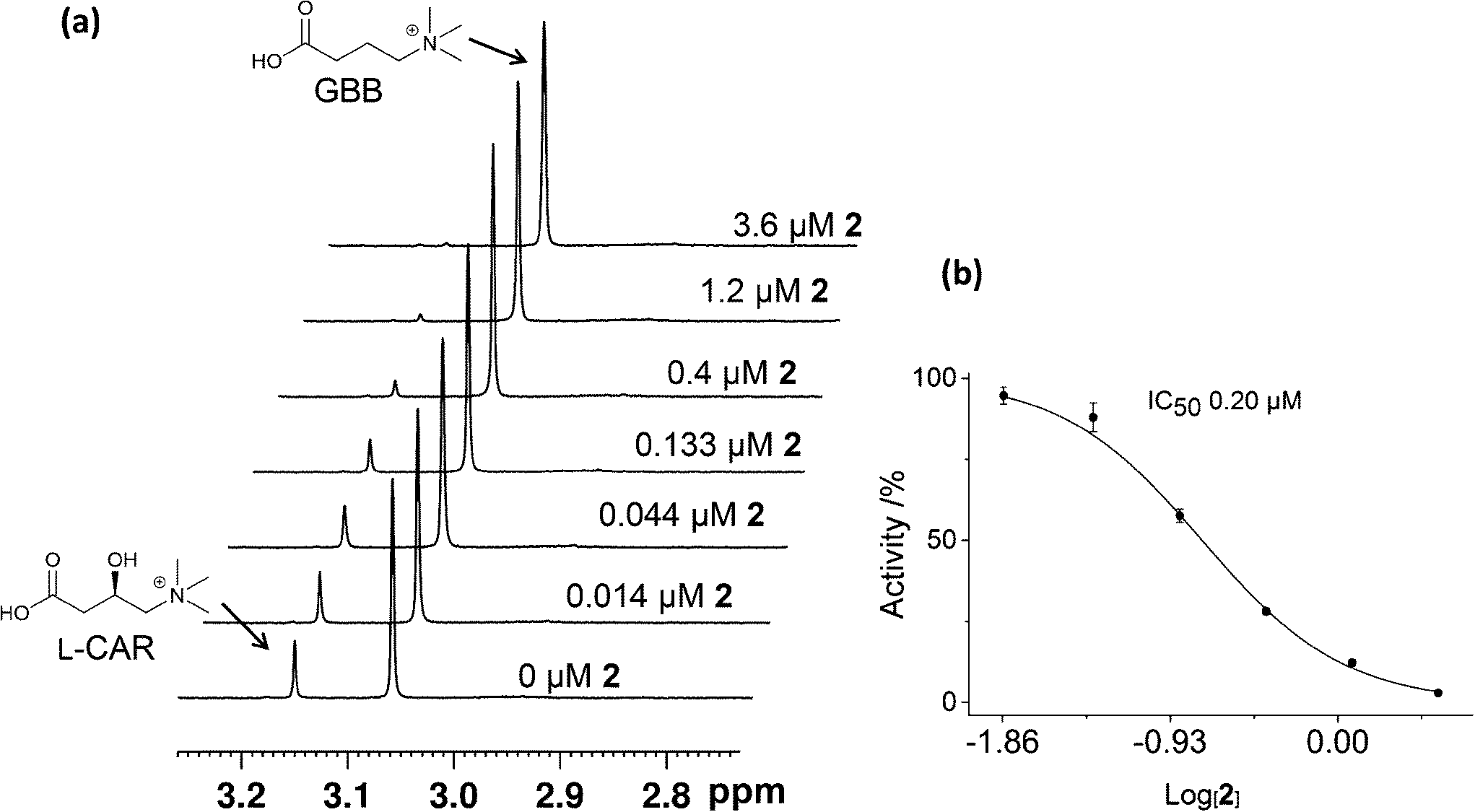 | ||
| Fig. 5 Measurements of the IC50 value of the isoquinoline-ligand 2 by 1H NMR. (a) Bottom to top; psBBOX catalysed GBB turnover into L-carnitine (L-CAR) in the presence of increasing amounts of 2. (b) Dose response curve of (a). Error bars represent standard deviations from three separate measurements. The assay mixture contained 200 μM GBB, 600 μM 2OG, 100 μM FeII, 500 μM L-ascorbate, 80 mM KCl and 0.140 μM BBOX in 50 mM Tris-D11, pH 7.5 in D2O. | ||
Although we have not yet been able to obtain a BBOX crystal structure with an isoquinoline-based inhibitor, manual docking and energy minimisation studies using a model of psBBOX based on a crystal structure of human BBOX (PDB ID: 3O2G)20 suggest a possible structural explanation for the observed trends (Fig. 6). Studies by Rydzik et al.17 on hBBOX using the fluoride ion release-based assay have shown that for hBBOX, as for psBBOX, the L-configured ligands 3 and 11 are better inhibitors than the corresponding D-forms (2 and 10) (IC50 6 μM vs. 73 μM, and 11 μM vs. 33 μM, respectively). The docking results suggest that 1 binds psBBOX possibly by chelating the active site metal via its pyridinium nitrogen and amide carbonyl oxygen, as observed for other 2OG oxygenases, i.e. PHD2 (PDB ID: 2G1M)30 and the fat mass and obesity protein FTO (PDB ID: 4 IE6),31 although more than one chelation mode has been observed for related compounds with PHD2.7 Removal of the isoquinoline inhibitor pyridinium nitrogen or the hydroxyl group leads to a significant loss in potency against hBBOX.17 In our proposed binding mode for 1, the phenyl ring of the isoquinoline projects towards the GBB binding pocket, causing movement of Phe184, but likely does not disrupt the pocket to the extent that GBB binding is ablated, i.e. it agrees with the NMR observations for 1–3 showing that 2OG but not GBB displacement occurs.
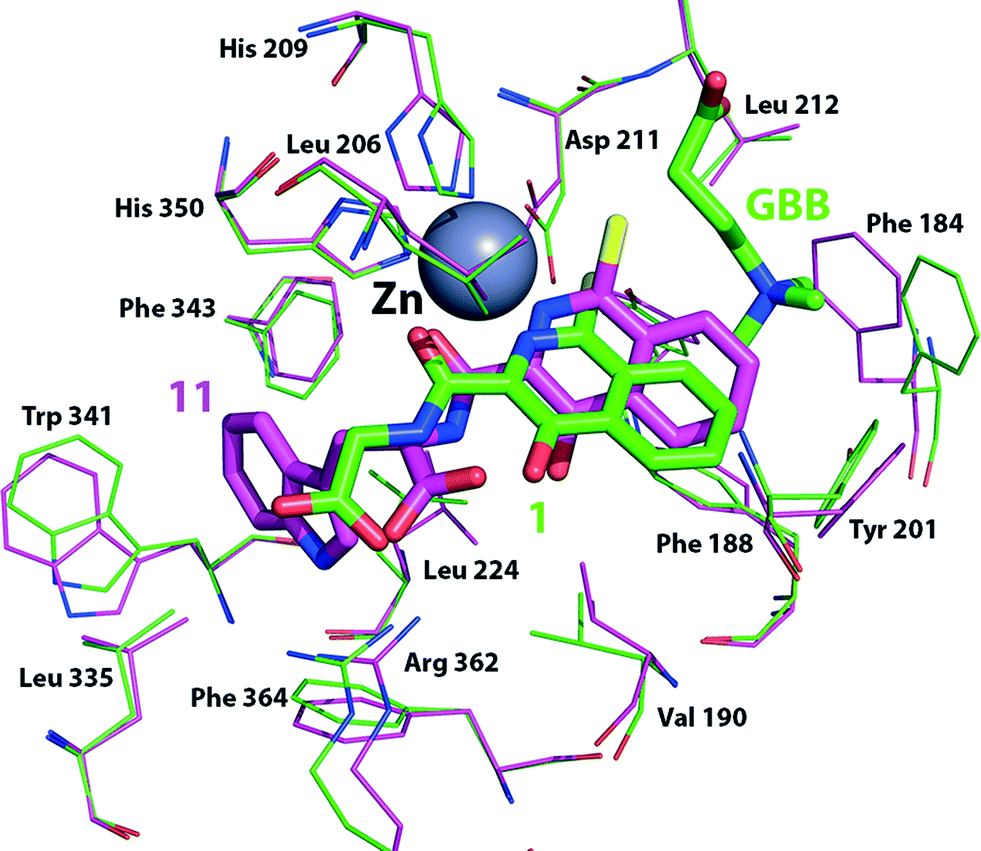 | ||
| Fig. 6 Docking simulation showing possible conformational changes as a result of binding inhibitors 1 (green sticks, Cl in yellow) or 11 (pink sticks, Cl in yellow) to a model of the psBBOX active site based on a hBBOX X-ray crystal structure (PDB ID: 3O 2G).20 | ||
The observation that some ligands with larger hydrophobic side chains (5, 7, 9, 11) compete with both 2OG and GBB, is interesting. Manual docking of 11 to the psBBOX model followed by energy minimisation suggests that the bicyclic ring system of these inhibitors is tilted with respect to that of the docked/minimised compound 1 while maintaining its metal coordination; this binding mode results in the chlorine and phenyl ring of the inhibitor protruding deeper into the GBB binding site. As a result the ‘aromatic cage’ residue Phe184 can move approximately 2.5 Å closer to the aromatic inhibitor thus changing the shape of the aromatic cage and also sterically blocking binding of the GBB trimethylamino group,21 consistent with the NMR results, i.e. this inhibitor competes with 2OG and GBB. Although, alternative binding/chelation modes cannot be ruled out, the modelling studies imply that the larger side chains bind in a largely hydrophobic pocket adjacent to the 2OG pocket (defined by the side chains of Val-190, Ala-200, Leu-206, Ser-236, Leu-224, Leu-335, Trp-341, Phe-343, Arg-352 and Phe-364); they also imply that binding in this pocket prefers the L-, rather than the D-, stereochemistry as observed (Table 1, Fig. 6) due to a potential clash between the carboxylate of 10 (D-stereochemistry) and Leu-206. It is notable that a loop (residues 190–205) involved in GBB binding also forms part of the hydrophobic pocket and biophysical studies have revealed the occurrence of conformational changes in BBOX catalysis.17 The binding of inhibitor 11 may thus elicit changes in the behaviour of this loop and so hinder GBB binding.
Conclusions
We have developed an efficient ligand-based 1H NMR competitive binding assay for determining inhibitor binding to psBBOX that is operationally simple and requires amounts of materials comparable to other biophysical techniques. The method readily reveals whether an inhibitor disrupts the binding of the co-substrate 2OG, the substrate GBB, or both. We envisage the assay will aid in the development of new BBOX inhibitors, including those selective for BBOX over other 2OG oxygenases. More generally, the results show how a readily applied NMR-based method can reveal mechanistic and structural insights not readily determined either by crystallography or by classical kinetic analyses.Experimental
Materials
Chemicals were purchased from Sigma Aldrich, Alfa Aesar and Cortecnet unless otherwise stated. Details of the synthesis of the isoquinoline derivatives have been previously reported.17,32Production and purification of psBBOX AK1
Recombinant psBBOX AK1 was produced and purified according to the protocol of Rydzik et al.16NMR Experiments
All NMR experiments were performed at a temperature of 298 K using a Bruker Avance III 700 MHz spectrometer equipped with a TCI inverse cryoprobe or a Bruker Avance III 600 MHz spectrometer equipped with a 5 mm BB-F/1H Prodigy N2 cryoprobe. The PROJECT – CPMG pulse sequence (90°x − [τ − 180°y − τ − 90°y − τ − 180°y − τ]n − acq)33 was used to attenuate broad resonances, using a total echo time of 32 ms. All spectra were processed with a Lorentzian line broadening of 0.3 Hz and were referenced to an internal standard (1,1,1 trifluoroacetone at 1.49 ppm). BBOX titration data for measuring the KD of GBB were fitted using OriginPro 9.0 (Origin lab, Northampton, MA, USA). Bruker MATCH (3 mm diameter) and 5 mm NMR tubes with a total sample volume of 160 μL and 500 μL, respectively, were used. The solutions were buffered in 50 mM Tris-D11·DCl, pH 7.5, in D2O. For each sample the pulse tip-angle calibration was carried out using the single-pulse nutation method.34 Each experiment was recorded with three separately prepared samples.NMR IC50 measurements
The GBB turnover to L-carnitine was initiated by the addition of psBBOX to the assay mixture in a 1.5 mL plastic Eppendorf tube at ambient temperature (294 K), and was quenched after 60 s by addition of 10 μl 1 M HCl. The assay mixture contained 200 μM GBB, 600 μM 2OG, 100 μM FeII (from Fe(NH4)2(SO4)2 salt, which was prepared as a 250 mM stock solution in 20 mM HCl which was further diluted to a 2.5 mM solution in MilliQ purified water), 500 μM L-ascorbate, and 80 mM KCl in 50 mM Tris-D11, pH 7.5 in D2O, leading to a final psBBOX concentration of 0.14 μM. The psBBOX activity (%) at a given inhibitor concentration was measured as ([CAR]P+I/[CAR]P) × 100, where P = protein, I = inhibitor. Response curves were fitted using OriginPro 9.0 (Origin lab, Northampton, MA, USA).Binding assay (KD) – intrinsic fluorescence quenching17,28
K D values were determined from the quenching of intrinsic tryptophan fluorescence measured at 294 K using a Pherastar FS plate reader (BMG labtech) with 96-well plates (Greiner, black, bottom: flat, clear). The following conditions were used: excitation 280 nm, emission 350 nm, 5 μM psBBOX, 50 μM MnII and varying concentrations of inhibitors to a final well volume of 50 μL in 50 mM Tris buffer pH 7.5, supplemented with 200 mM NaCl. Fluorescence readings were obtained in triplicate with errors represented as standard deviations. KD values were obtained from the raw data by plotting inhibitor concentration against ΔFobs/Fmax (ΔFobs is the decrease in observed fluorescence and Fmax is the observed fluorescence signal without any inhibitor present).Fluoride release assay (IC50)
IC50 measurements were obtained using a fluoride release assay, as previously applied to hBBOX.29 The fluorescence signal was detected using a Pherastar FS plate reader (BMG labtech) fitted with a FITC FP 485/30 (485 nm, bandwidth 30 nm) and FITC FP 520/40 emission (520 nm, bandwidth 40 nm) filters, using 384-well plates (black, clear, flat bottom, Grenier BioOne). GBBF and the TBS-protected fluorescein probe were synthesised according to the published protocol.29 The following conditions were used: 50 μM GBBF, 500 μM 2OG, 250 μM ascorbate, varying concentrations of inhibitors, 50 μM FeII (from Fe(NH4)2(SO4)2 salt, which was prepared as a 100 mM stock solution in 20 mM HCl which was further diluted to a 250 μM solution in MilliQ purified water before being added at the start of the measurements). Initiation of the assay was carried out by addition of psBBOX to each well containing all inhibitors and cofactors, to a concentration of 1 μM. Reactions were carried out in a final well volume of 10 μL in 50 mM Tris buffer pH 7.5 at 294 K, supplemented with 200 mM NaCl. After 10 minutes, the reaction was quenched by addition of 40 μL of TBS-protected fluorescein probe (final concentration 5 μM). The plates were then sealed and incubated for 1 hour at room temperature. Following this, 10 μL of HEPES buffer pH 7.0 was added and the fluorescence signal read up to 5 minutes after the addition of HEPES. Fluorescence data were recorded as quadruplicates and errors represented as standard deviations. Fluorescence signals were normalized by subtracting fluorescence values obtained from control wells which contained all reagents, but without enzyme or without inhibitor. IC50s were obtained from the raw data using the Prism four parameter logistic dose–response model (sigmoidal, variable slope).Acknowledgements
We thank the following for financial support: Abdul Wali Khan University, Mardan (AWKUM) Pakistan (A. K.), Biotechnology and Biological Sciences Research Council (BBSRC), the British Heart Foundation, Wellcome Trust and Cancer Research UK.References
- 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- N. R. Rose, M. A. McDonough, O. N. King, A. Kawamura and C. J. Schofield, Chem. Soc. Rev., 2011, 40, 4364–4397 RSC.
- F. M. Vaz and R. J. Wanders, Biochem. J., 2002, 361, 417–429 CrossRef CAS PubMed.
- B. Z. Simkhovich, Z. V. Shutenko, D. V. Meirena, K. B. Khagi, R. J. Mezapuke, T. N. Molodchina, I. J. Kalvins and E. Lukevics, Biochem. Pharmacol., 1988, 37, 195–202 CrossRef CAS PubMed.
- M. Dambrova, E. Liepinsh and I. Kalvinsh, Trends Cardiovasc. Med., 2002, 12, 275–279 CrossRef CAS PubMed.
- L. Henry, I. K. H. Leung, T. D. W. Claridge and C. J. Schofield, Bioorg. Med. Chem. Lett., 2012, 22, 4975–4978 CrossRef CAS PubMed.
- L. Poppe, C. M. Tegley, V. Li, J. Lewis, J. Zondlo, E. Yang, R. J. Kurzeja and R. Syed, J. Am. Chem. Soc., 2009, 131, 16654–16655 CrossRef CAS PubMed.
- M. C. Chan, O. Atasoylu, E. Hodson, A. Tumber, I. K. H. Leung, R. Chowdhury, V. Gomez-Perez, M. Demetriades, A. M. Rydzik, J. Holt-Martyn, Y. M. Tian, T. Bishop, T. D. W. Claridge, A. Kawamura, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, PLoS One, 2015, 10, e0132004 Search PubMed.
- I. K. H. Leung, M. Demetriades, A. P. Hardy, C. Lejeune, T. J. Smart, A. Szollossi, A. Kawamura, C. J. Schofield and T. D. W. Claridge, J. Med. Chem., 2013, 56, 547–555 CrossRef CAS PubMed.
- M. D. Shortridge, D. S. Hage, G. S. Harbison and R. Powers, J. Comb. Chem., 2008, 10, 948–958 CrossRef CAS PubMed.
- M. Sarrazin, J. C. Sari, M. Bourdeaux-Pontier and C. Briand, Mol. Pharmacol., 1979, 15, 71–77 CAS.
- J. J. Fischer and O. Jardetzky, J. Am. Chem. Soc., 1965, 87, 3237–3244 CrossRef CAS PubMed.
- C. Dalvit, A. D. Gossert, J. Coutant and M. Piotto, Magn. Reson. Chem., 2011, 49, 199–202 CrossRef CAS PubMed.
- P. J. Hajduk, E. T. Olejniczak and S. W. Fesik, J. Am. Chem. Soc., 1997, 119, 12257–12261 CrossRef CAS.
- W. Jahnke, P. Floersheim, C. Ostermeier, X. Zhang, R. Hemmig, K. Hurth and D. P. Uzunov, Angew. Chem., Int. Ed., 2002, 41, 3420–3423 CrossRef CAS.
- A. M. Rydzik, I. K. H. Leung, G. T. Kochan, N. D. Loik, L. Henry, M. A. McDonough, T. D. W. Claridge and C. J. Schofield, Org. Biomol. Chem., 2014, 12, 6354–6358 CAS.
- A. M. Rydzik, R. Chowdhury, G. T. Kochan, S. T. Williams, M. A. McDonough, A. Kawamura and C. J. Schofield, Chem. Sci., 2014, 5, 1765–1771 RSC.
- K. Tars, J. Leitans, A. Kazaks, D. Zelencova, E. Liepinsh, J. Kuka, M. Makrecka, D. Lola, V. Andrianovs, D. Gustina, S. Grinberga, E. Liepinsh, I. Kalvinsh, M. Dambrova, E. Loza and O. Pugovics, J. Med. Chem., 2014, 57, 2213–2236 CrossRef CAS PubMed.
- K. Tars, J. Rumnieks, A. Zeltins, A. Kazaks, S. Kotelovica, A. Leonciks, J. Sharipo, A. Viksna, J. Kuka and E. Liepinsh, Biochem. Biophys. Res. Commun., 2010, 398, 634–639 CrossRef CAS PubMed.
- I. K. H. Leung, T. J. Krojer, G. T. Kochan, L. Henry, F. von Delft, T. D. W. Claridge, U. Oppermann, M. A. McDonough and C. J. Schofield, Chem. Biol., 2010, 17, 1316–1324 CrossRef CAS PubMed.
- J. J. A. G. Kamps, A. Khan, H. Choi, R. K. Lesniak, J. Brem, A. M. Rydzik, M. A. McDonough, C. J. Schofield, T. D. W. Claridge and J. Mecinović, Chem. – Eur. J., 2016, 22, 1270–1276 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, ChemBioChem, 2005, 6, 1536–1549 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat and G. Parigi, Solution NMR of paramagnetic molecules: applications to metallobiomolecules and models, Elsevier, Amsterdam, 2001 Search PubMed.
- I. Bertini, M. Fragai, Y. M. Lee, C. Luchinat and B. Terni, Angew. Chem., Int. Ed., 2004, 43, 2254–2256 CrossRef CAS PubMed.
- I. K. H. Leung, E. Flashman, K. K. Yeoh, C. J. Schofield and T. D. W. Claridge, J. Med. Chem., 2010, 53, 867–875 CrossRef CAS PubMed.
- I. Bertini, M. Fragai, C. Luchinat and E. Talluri, Angew. Chem., Int. Ed., 2008, 47, 4533–4537 CrossRef CAS PubMed.
- W. Jahnke, S. Rudisser and M. Zurini, J. Am. Chem. Soc., 2001, 123, 3149–3150 CrossRef CAS PubMed.
- A. M. Rydzik, J. Brem, W. B. Struwe, G. T. Kochan, J. L. Benesch and C. J. Schofield, Bioorg. Med. Chem. Lett., 2014, 24, 4954–4957 CrossRef CAS PubMed.
- A. M. Rydzik, I. K. H. Leung, G. T. Kochan, A. Thalhammer, U. Oppermann, T. D. W. Claridge and C. J. Schofield, ChemBioChem, 2012, 13, 1559–1563 CrossRef CAS PubMed.
- M. A. McDonough, V. Li, E. Flashman, R. Chowdhury, C. Mohr, B. M. Lienard, J. Zondlo, N. J. Oldham, I. J. Clifton, J. Lewis, L. A. McNeill, R. J. Kurzeja, K. S. Hewitson, E. Yang, S. Jordan, R. S. Syed and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9814–9819 CrossRef CAS PubMed.
- W. Aik, M. Demetriades, M. K. Hamdan, E. A. Bagg, K. K. Yeoh, C. Lejeune, Z. Zhang, M. A. McDonough and C. J. Schofield, J. Med. Chem., 2013, 56, 3680–3688 CrossRef CAS PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- J. A. Aguilar, M. Nilsson, G. Bodenhausen and G. A. Morris, Chem. Commun., 2012, 48, 811–813 RSC.
- P. S. Wu and G. Otting, J. Magn. Reson., 2005, 176, 115–119 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00004e |
| ‡ The authors declare no competing interests. |
| § These authors contributed equally to this work. |
| ¶ Current address: School of Chemical Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. |
| This journal is © The Royal Society of Chemistry 2016 |