
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of transporter ectodomains in drug recognition and binding: phlorizin and the sodium–glucose cotransporter†
M.
Raja‡
a,
T.
Puntheeranurak‡
b,
H. J.
Gruber
c,
P.
Hinterdorfer
c and
R. K. H.
Kinne
*a
aMax Planck Institute of Molecular Physiology, Dortmund, Germany. E-mail: rolf.kinne@mpi-dortmund.mpg.de
bDepartment of Biology, Center of Nanoscience, Faculty of Science, Mahidol University, Ratchathevi, Bangkok, Thailand
cInstitute for Biophysics, Christian Doppler Laboratory of Nanoscopic Methods in Biophysics, Johannes Kepler University of Linz and Center for Advanced Bioanalysis GmbH (CBL), Linz, Austria
First published on 15th April 2016
Abstract
This article reviews the role of segments of SLCs located outside the plasma membrane bilayer (ectodomains) using the inhibition of SGLTs (SLC5 family) by the aromatic glucoside phlorizin as a model system. Phlorizin has been the lead substance for the development of SGLT2 (SLC5A2) inhibitors that have been introduced recently for the treatment of type 2 diabetes. Using mainly biophysical methods, it is shown that three ectodomains form well-defined substructures, such as short helices, that are arranged in a vestibule and undergo significant conformational changes during binding of phlorizin. From these data, tentative structures of inhibitor/ectodomain complexes are derived by molecular modeling. The ectodomains provide an additional binding site for aglucones, which cooperates with the binding site for the sugar moiety of phlorizin in the sugar translocation pathway, buried inside the membrane. They play a significant role in determining the specificity, selectivity, and affinity of sugar transport inhibitors and might even explain the difference in sensitivity of various members of the SGLT family, located in different tissues and organs of the human body. Similar binding modes are suggested for several other SLCs, in particular the monoamine transporter (SLC6 family), that belong to the sodium/neurotransmitter cotransporter family.
1. Introduction
Although a membrane transporters comprise about 25% of the human genome and despite their pivotal importance in health and disease, only a few of them are targets for drugs. Notable (and very successful) exceptions are diuretics which inhibit either the sodium–chloride cotransporter (SCC; SLC12A3)1 or the sodium–potassium–chloride cotransporter (NKCC; SLC12A1–2).2 They were developed without even knowing their molecular targets, and their (sometimes surprising) action was discovered during animal screening experiments. Other examples are antidepressants that act on the serotonin transporter and inhibit reuptake of the neurotransmitter from synapses. In this instance, the pharmacologically active molecule was known, and by careful and systematic chemical modifications, drugs were developed. Again details on the structure and the translocation mechanism were unknown at that time. A third group of drugs, which have been recently introduced into the market, are compounds like dapagliflozin acting on one of the renal sodium–glucose cotransporters, SGLT2 (SLC5A2).3,4 Here, the transporter had been cloned before, and tentative substrate binding and transport models had been developed by homology from bacterial transport systems.5–7Detailed information at the molecular level is now available for almost all transporters. It is derived mostly from X-ray analysis of transporter molecules crystallized in outward-facing, occluded or inward-facing configurations combined with studies in which putative substrate binding sites have been mutated. Due to the complex structures of membrane proteins, these studies describe mostly those parts which are embedded in the phospholipid bilayer (endodomains) and can be captured in the “frozen” state of a crystal. However, the parts of transporters outside the membrane (ectodomains) can rarely be resolved because of their high flexibility and inherent mobility. Recently, molecular modelling studies on drug binding and specificity8–10 as well as single molecule recognition and force spectroscopy studies by atomic force microcopy (AFM)11–14 have revealed the importance of vestibules formed by extramembranous loops on the cell surface in target recognition, in defining drug specificity and in governing binding processes. The overall idea of exploiting ectodomain–ligand interactions to achieve target specificity and enhanced binding affinity is a novel potential general strategy for inhibitor/drug development.
The current review summarizes the information available on the role of extracellular ectodomains in the interaction of inhibitors with SLCs. It focuses mainly on the inhibitory action of phlorizin, a glucoside and the leading substance for developing SGLT2 inhibitors. The data obtained in single molecule analysis by AFM on intact cells and in studies of isolated ectodomains in solution are used to model the SGLT–inhibitor interactions. Investigations on other SLCs, in particular SERT (SLC6A4), are briefly mentioned in which homology modeling, induced fit docking calculation, and molecular dynamics simulation combined with site directed mutagenesis also suggest potential interactions of inhibitors with ectodomains. These studies complement, enrich and deepen our knowledge on drug–target interactions and may provide new tools for the development of drugs targeting specifically biologically important membrane transporters.
2. The SGLTs: function, substrates, and inhibitors
Sodium–glucose cotransporters (SGLT) belong to the SLC5A gene family which employs the chemical sodium gradient and electrical potential across the plasma membrane to drive glucose and other nutrients into the cells.5 Major physiological roles have been assigned to SGLT1 (SLC5A1) and SGLT2 (SLC5A2). SGLT1 is the main transporter for the absorption of D-glucose and D-galactose (for chemical formulae, see Fig. 1) in the small intestine.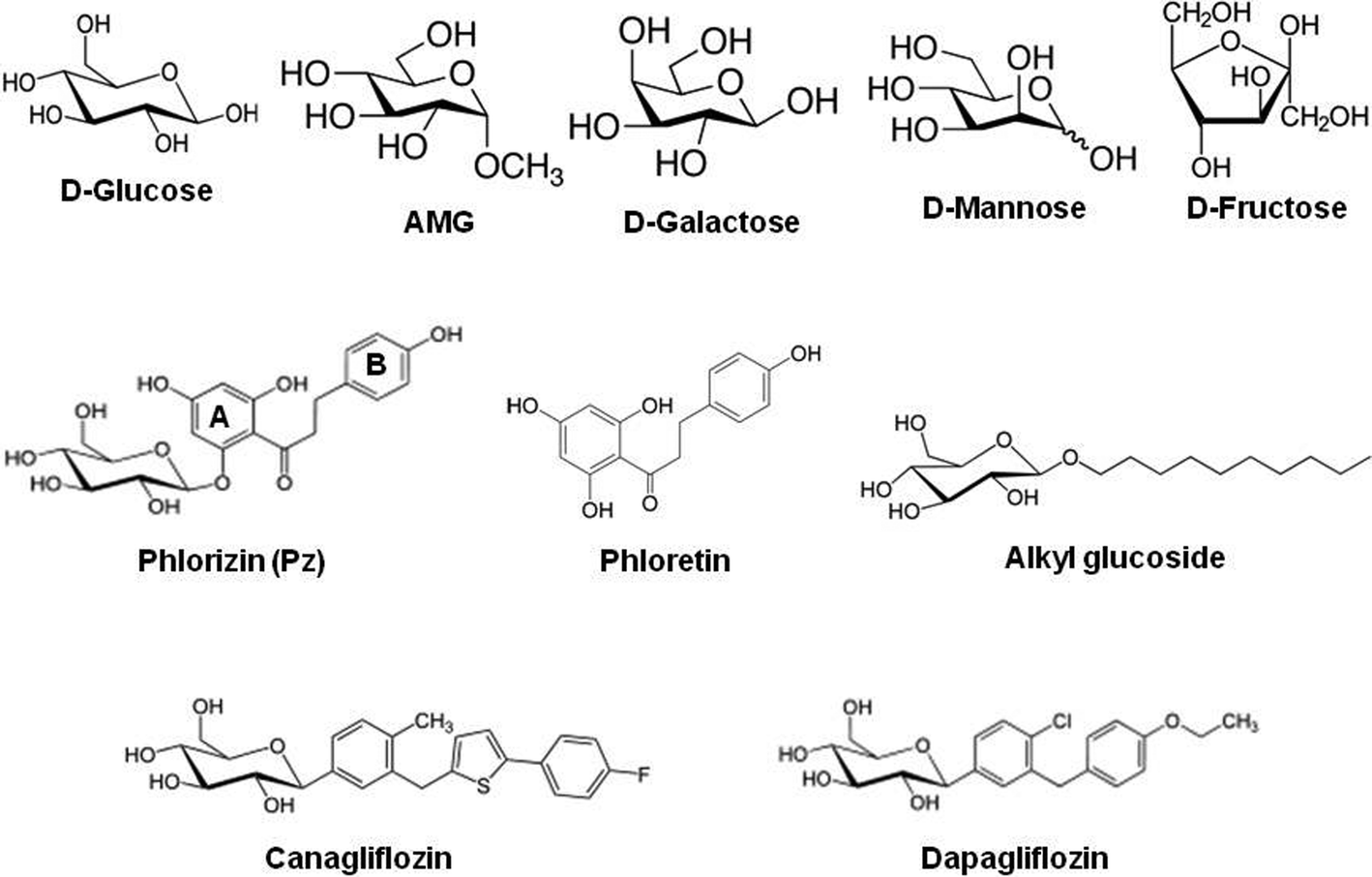 | ||
| Fig. 1 Chemical structures of monosaccharides and sodium–D-glucose cotransporter inhibitors. D-Glucose, alpha-methyl-D-glucose (AMG) and D-galactose are transported by SGLT; D-mannose and D-fructose are shown as a comparison for non-transported sugars. | ||
SGLT2 is responsible for bulk reabsorption of D-glucose from the primary urine in early parts of the renal proximal tubule, and the residual sugar is almost completely removed by SGLT1 in the late proximal tubule.15 Isoforms SGLT3 to SGLT5 have been found in a variety of other tissues (for a complete list, see ref. 5). In functional studies, SGLT1 and SGLT2 have been found to be inhibited by aromatic glucosides. The most extensively studied glucoside is phlorizin in which D-glucose is linked to the aglucone phloretin via a beta-glycosidic bond (see Fig. 1 for chemical structures). In addition, alkyl-glucosides inhibit the transporter (see Fig. 1 for chemical structures).16
SGLT2 has recently been identified as a target for the treatment of diabetes;3 based on phlorizin as the leading substance, drugs such as canagliflozin and dapagliflozin (see Fig. 1 for chemical structures) have been developed and introduced into the market.4 Another exciting discovery was that some pancreatic and prostatic cancer cells express SGLT2 and their metabolism and growth depend on the uptake of D-glucose via this transport pathway, suggesting the possibility for an effective treatment.17 Kinetic studies on intact cells or plasma membranes have shown that phlorizin and other SGLT2 inhibitors act from the extracellular side.18,19 The interaction of phlorizin with the SGLT transporter involves the sugar-binding site of the transporter and aglucone-binding sites.20 The affinity of SGLT1 for phlorizin is about 1000-fold higher than that for glucose.21
A bacterial homologue of SGLT has been crystallized, and its structure has been determined. The model proposed for the translocation of sugars, integrating kinetic and structural data, contains the following steps.7 First, Na+ binds to the ligand-free outside-facing state of the transporter to open the outward-facing gate, permitting outside sugar to bind and be subsequently trapped in the substrate binding site of the transporter in an occluded state, where egress to the outside solution is blocked. This is followed by a conformational change from the outward-occluded state to an inward-occluded state. On opening the inward gate, the Na+ ions and sugar are released into the cell interior. The transport cycle is completed by the change in conformation from the inward-facing ligand-free state to the outward-facing ligand-free state.
Structural and biophysical data predict that formation of the phlorizin–SGLT complex creates an additional, highly condensed conformational state which makes translocation of sugars impossible.20,22 The bacterial sodium glucose cotransporter is not inhibited by phlorizin, and until now, no phlorizin–SGLT complex has been crystallized; however, several biochemical, genetic and biophysical studies have been performed. The following sections will summarize the information currently available on phlorizin binding sites, in particular on the ectodomains of the SGLT transporter facing the outside of glucose transporting cells.
3. Topology of SGLT ectodomains
Determining the topology of a transporter in the membrane usually starts from sequence data and homology screening. This preliminary model is then tested by introducing reporter sites into the molecule such as glycosylation sites, new epitopes for antibodies or reactive groups which can be modified selectively. All the latter methods involve a modification of the transporter amino acid sequence, and thus, there is a danger that the arrangement of the transmembrane helices (endodomains) and/or extramembranous loops (ectodomains) will be changed. Therefore, during recent years, methods have been developed to analyze the spatial geometry and properties of transporters in the native, unaltered state and in their natural membrane in living cells. One of these methods is atomic force microscopy using cantilevers carrying specific antibodies, substrates or drugs.14 In the following section, this methodology will be briefly described, and the results obtained with SGLT expressing cells will be reviewed.12,23–253.1. Biophysical analysis in intact cells: biosensors and AFM
Atomic force microscopy (AFM) is one of the powerful approaches for resolving nanostructures in both biological and physical sciences. It has been used on versatile surfaces including cells and membrane proteins.26,27 The advantages of AFM are that it can achieve high resolution imaging, real-time monitoring and fluid imaging, even under environmentally controlled conditions mimicking closely the physiological situations. The potential of AFM to measure ultra-low (a few pN) forces at high lateral resolution allows the investigation of single-molecule recognition processes such as binding of ligands to surface macromolecules. Advanced biochemical modifications of the cantilever tips make it possible to study for example membrane protein interactions with drugs, substrates and antibodies, which provide high resolution information on the topology of the protein and molecular dynamics of the binding processes. The principle of single-molecule-recognition AFM is illustrated in Fig. 2. | ||
| Fig. 2 Schematic representation of an atomic force microscopy force–distance cycle. The tip is moved toward the surface of the cells grown on a flat support (dotted line, 1–2) and subsequently retracted (solid line) at a constant lateral position. If during the approach the ligand attached to the cantilever interacted with a specific binding site on the surface of the cell, during retraction a force signal of a distinct shape (3) (representing a deflection of the cantilever) is observed. The force increases until dissociation occurs (4) at an unbinding force fu. For further details, see ref. 25. | ||
In a “force–distance cycle”, the cantilever tip tethered with the ligand is first approached towards the surface whereupon a single receptor–ligand complex is formed based on the specific ligand–receptor recognition. Subsequently, the cantilever is retracted from the surface. An increasing bending force is exerted on the cantilever due to the ligand–receptor connection until the interaction bonds break at a critical force (the so-called unbinding force, fu, calculated by using Hooke's law: F = kΔx, where k is the spring constant of the cantilever, and Δx is the cantilever deflection). From such experiments, the specifics of the ligand–receptor interaction such as affinity, binding, unbinding rate constants, and structure of the binding pocket can be delineated (see below).
Various methods to firmly attach ligands at a low surface density to cantilever tips are depicted in Fig. 3.
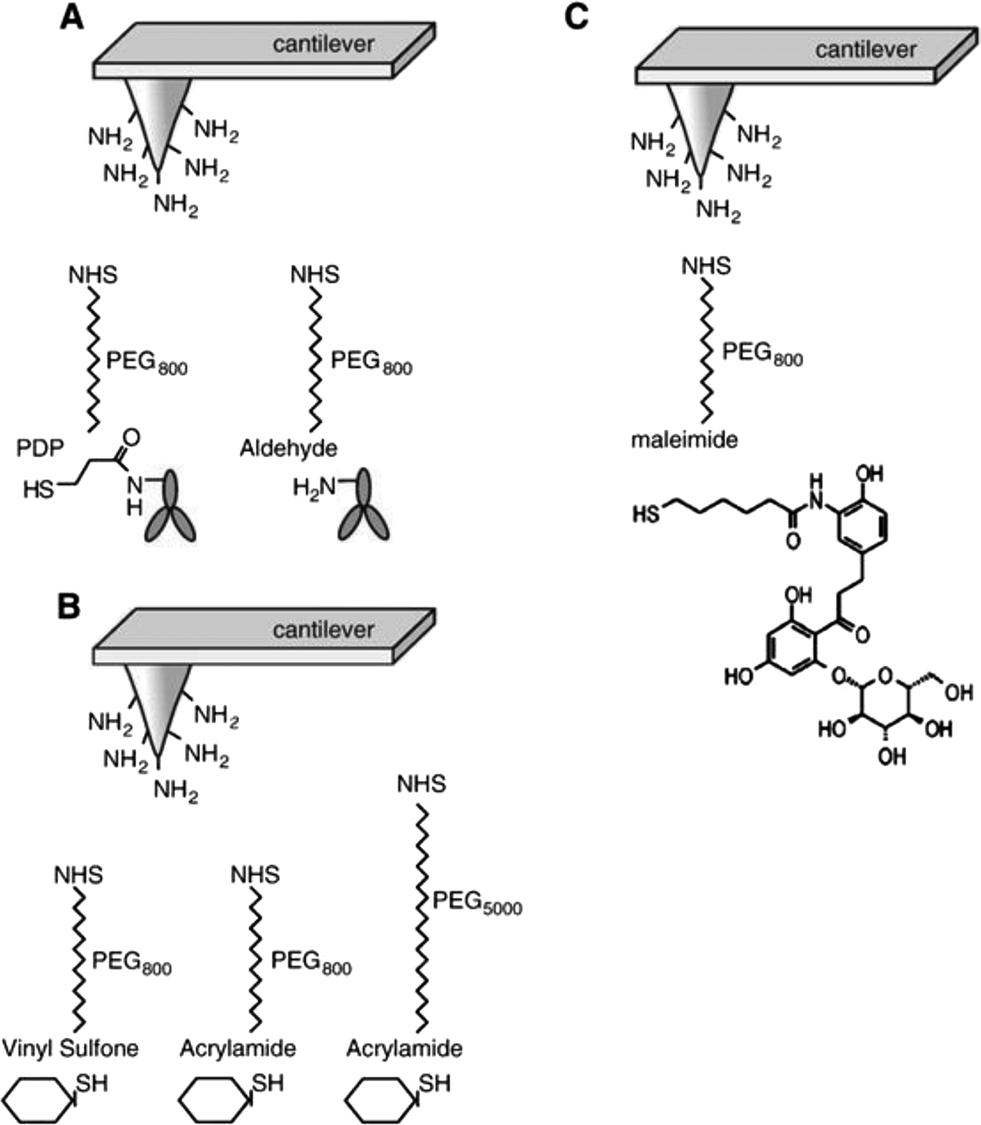 | ||
| Fig. 3 Linkage of ligands to AFM cantilever tips. (A) Specific antibodies were covalently coupled to AFM tips via heterobifunctional polyethylene glycol (PEG) linkers, PDP–PEG800–NHS (left) and aldehyde–PEG800–NHS (right). (B) Sugars were covalently linked to AFM tips via vinyl sulfone–PEG800–NHS (large end group) or acrylamide–PEG800 and PEG5000–NHS (small end groups). (C) Maleimide–PEG800–NHS was used to couple phlorizin to AFM tips. In all instances, the NHS ends of the PEG linkers were covalently bound to amines on the functionalized cantilever tips. Abbreviations: PDP = 2-pyridyldithiopropionyl residue, NHS = N-hydroxy-succinimidyl residue.25 | ||
In order to provide the flexibility of ligands required to interact with complementary macromolecules, a long poly(ethylene glycol) (PEG) chain is inserted between the tip and the probe molecule. PEG is a water soluble, non-toxic, and chemically and physically inert polymer. The PEG linker can vary in length. We commonly use a PEG linker of an extended length of 6 nm; however, longer cross linkers are also available to probe deeper into proteins.24 To construct AFM-tip sensors, the cross linker contains two different functional groups at its ends, i.e. one end couples to the tip of the cantilever and the other to the ligand. Various heterobifunctional cross linkers have been developed. In our laboratory, the cross linker widely used for antibody coupling has an N-hydroxy-succinimidyl (NHS) residue on one end, which reacts to amines on the ethanolamine coated cantilever tip and a 2-pyridyldithiopropionyl (PDP) residue on the other end, which can covalently bind to thiols of proteins (Fig. 3A). This sulfur chemistry is highly advantageous, since it is very reactive and renders site-directed coupling possible. If no free thiols for coupling are available on the native ligands, they can be generated by modification with SATP [N-succinimidyl-3-(S-acetylthio)propionate]. Another linker widely used for protein coupling in our laboratory is aldehyde–PEG800–NHS which interacts with free amino groups of peptide chains28 (Fig. 3A).
Glucose tethered cantilevers were created by using thioglucose, vinyl sulfone–PEG800–NHS, acrylamide–PEG800–NHS or acrylamide–PEG5000–NHS (Fig. 3B). In this AFM tip sensor, the OH groups known to be essential for translocation of D-glucose (i.e. at C2, C3, and C4) were not modified. Maleimide–PEG800–NHS was employed to couple a modified phlorizin to cantilever tips (Fig. 3C). In the following sections, we will describe how these specific AFM cantilevers can be used as biosensors to explore the topology and dynamics of ectodomains, such as conformational changes and substrate–transporter and inhibitor–transporter interactions of the SGLT1 protein in living cells at the single molecule level.
3.2. Interaction of antibody-primed AFM cantilevers with SGLT in intact cells
The structure of SGLT1 has been proposed to contain 14 transmembrane α-helices and 13 ectodomains or loops connecting the transmembrane segments. However, the structural orientation of ectodomains of SGLT1, especially of the large C-terminal loop, connecting TMH (transmembrane helices) 13 and 14, is still under debate.20,29–32 Here, the topology, arrangement, and function of the three large ectodomains of SGLT1 were investigated by using specific antibody-tethered AFM tips under near-physiological conditions.23,25 Specific antibodies (named PAN2-2, QIS30, and PAN3-2) against the three ectodomains, i.e. subdomain I (loop between TMH 6 and 7), subdomain II (loop between TMH 8 and 9), and subdomain III (loop between TMH 13 and 14), respectively, were separately linked to AFM tips via a flexible PEG cross linker as described above (Fig. 3A). Single-molecule force spectroscopy experiments were performed on both non-SGLT1-expressing cells and SGLT1-expressing cells, with the assistance of a CCD camera for localization of the AFM cantilever. At a fixed lateral position above the living cells, force–distance cycles were carried out with a sweep amplitude of 1000 nm at 1 Hz sweep rate. The number of typical specific recognition events from all three antibody-tagged AFM tips is depicted as binding probabilities in Fig. 4.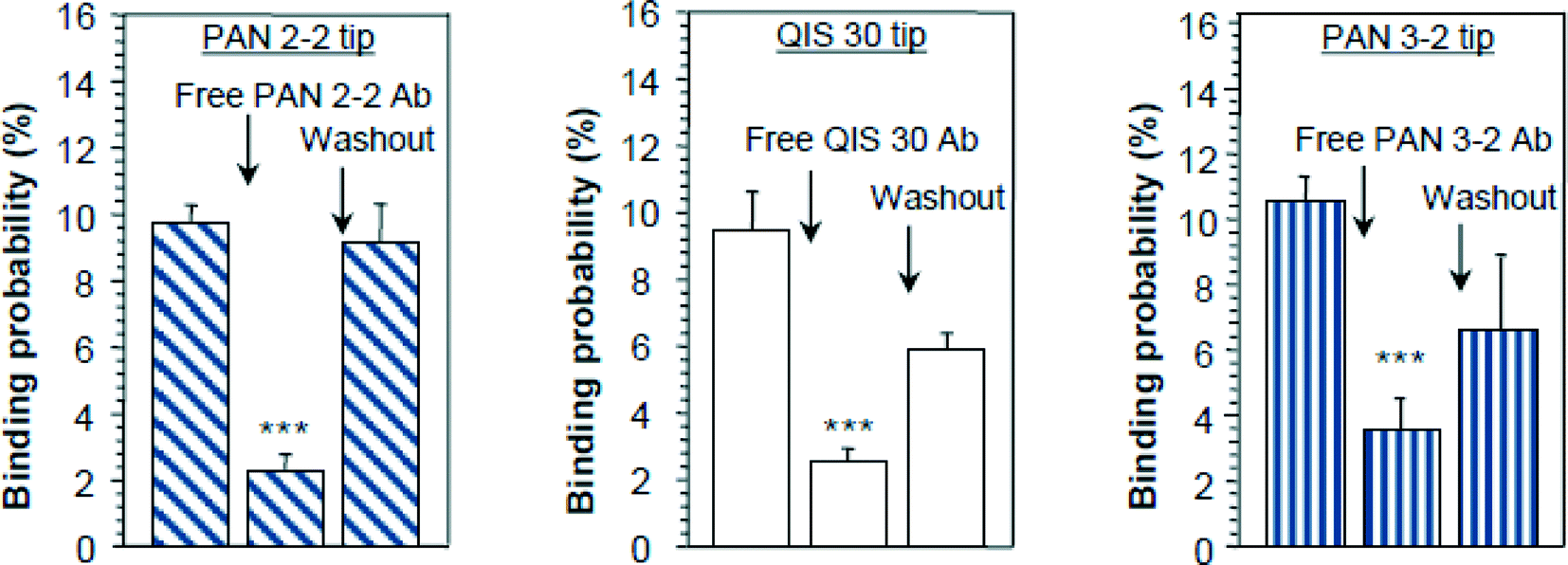 | ||
| Fig. 4 Recognition of ectodomains of SGLT1 on the surface of intact cells. AFM cantilever tips were tagged with epitope-specific antibodies against subdomain I = PAN 2-2 = loop 7, subdomain II = QUIS 30 = loop 9, and subdomain III = PAN 3-2 = late loop 13 of rbSGLT1 (rabbit isoform of SGLT1). Quantitative comparison of binding probabilities to cells expressing rbSGLT1 in the absence or presence of free specific antibodies in the medium. Triple asterix indicate statistical significance, for further details see ref. 25. | ||
Specificity of recognition was confirmed in “blocking experiments”, either by saturating the surface of cells with free antibodies against SGLT or by blocking antibodies attached to the AFM tip with free antigens. Thereby, recognition events disappeared (Fig. 4) and the force–distance cycles were similar to the ones obtained from non-expressing cells (data not shown). To achieve statistical significance, up to 500–1000 force–distance cycles were performed for each location on the surface of cells, and up to four locations (different cells) were investigated for each condition. Thus, it could be demonstrated, at the molecular level, that these three ectodomains of SGLT1 can be specifically recognized on the surface of living cells by epitope-specific antibodies. This provides strong evidence that they are located on the outer membrane surface of the transporter. Interestingly, our results also show that at least the late part of subdomain III is accessible from the extracellular space, suggesting a model for the membrane topology of the transporter shown in Fig. 5, which contains a membrane-crossing loop between TMH 13 and 14.
 | ||
| Fig. 5 Membrane topology of SGLT1. Three ectodomains (subdomains I to III) are exposed to the extracellular medium; the dotted lines represent the epitopes of the specific antibodies. They are connected by S–S bonds (not shown) and form a vestibule in the access pathway of D-glucose to the translocation site of SGLT1.23 | ||
Furthermore, transport studies with cysteine mutants suggested the presence of a disulfide bridge between Cys255 in subdomain I and Cys608 in subdomain III. This bridge brings subdomain II in close vicinity to the other subdomains creating a vestibule for initial sugar binding to the transporter. This initial binding reaction studied with AFM-thioglucose exhibits a different stereo-specificity than that found in transport studies, suggesting several consecutive selectivity filters of the transporter (for details see below).23
4. Function and dynamics of SGLT ectodomains
4.1. Transport inhibition studies in intact cells
In order to define potential interaction sites of SGLT1 with phlorizin, mutagenesis studies were performed in a hydrophobic region of subdomain III (aa 604–610), located extracellularly, close to the C-terminus. COS 7 cells were transiently transfected with SGLT1 mutants, and kinetic parameters of alpha-methyl-D-glucopyranoside (AMG) (see Fig. 1 for chemical structures) uptake into the cells were investigated. Replacement of the respective amino acids with the charged amino acid lysine lowered the sensitivity for phlorizin at least by a factor of 5 compared to that for the wild-type. Most striking changes were observed for Y604K (16-fold decrease) and C608K (18-fold decrease). On the contrary, replacement of these amino acids with nonpolar amino acids (glycine, alanine or phenylalanine) instead of lysine yielded slightly higher affinities for phlorizin (see Fig. 6).33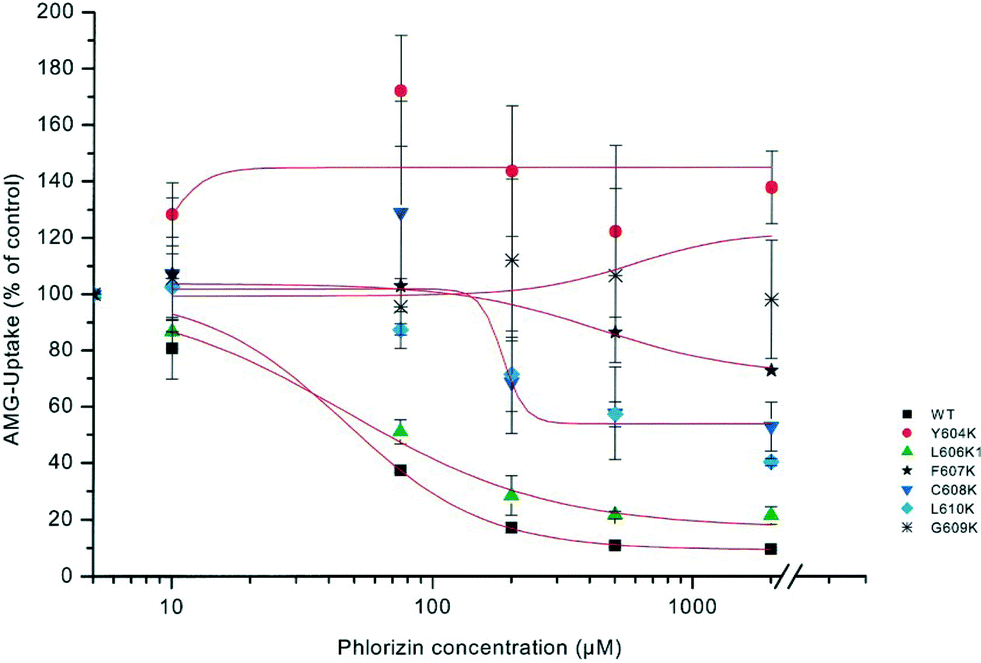 | ||
| Fig. 6 Dose–response curves of phlorizin inhibition of sodium-dependent AMG uptake into cells mutated in loop 13. COS 7 cells were transiently transfected with plasmids of rbSGLT1 (WT) or various mutants of SGLT1, in which lysine (K) had been introduced at different sites of subdomain III. AMG uptake into cells was measured in the presence of 5 mmol l−1 alpha-methyl-D-glucopyranoside with or without 120 mM NaCl. Uptake in the presence of various phlorizin concentrations was compared to the uptake in the absence of phlorizin set as 100%.33 | ||
In all mutants, the apparent affinity of sugar uptake for AMG was not statistically different from that of the wild type. These studies suggest that the region of subdomain III between amino acids 604 and 610 is involved in the interaction between SGLT1 and phlorizin, probably by providing a hydrophobic pocket for one of the aromatic rings of the aglucone moiety of the glycoside.33
4.2. Tryptophan fluorescence scanning of isolated hSGLT1 reconstituted into proteoliposomes
In order to gain understanding of the interaction of phlorizin with hSGLT1 (human isoform of SGLT1), single Trp residues were introduced into a functional hSGLT1 mutant devoid of Trps at positions that previously had been postulated to be involved in sugar recognition/translocation and/or phlorizin binding. The mutant proteins were expressed in Pichia pastoris, purified, and reconstituted into liposomes.34 In transport experiments, the putative sugar binding site mutants (W457hSGLT1 and W460hSGLT1) showed a drastic decrease in affinity toward AMG and also a strong decrease in the inhibitory effect of phlorizin. In Trp fluorescence studies, the position of the emission maxima of the mutants, their sensitivity to N-bromosuccinimide oxidation, and their interaction with water soluble quenchers demonstrate that in the presence of sodium Trp457 and Trp460 are in contact with the extravesicular environment (open outward-facing gate). In both mutants, Trp fluorescence was quenched significantly, but differently, by various glucose analogues. They also show significant protection by D-glucose and phlorizin against acrylamide, KI, or TCE quenching. In contrast, mutants W602hSGLT1 and W609hSGLT1 located in the putative aglucone binding site of subdomain III exhibit normal sugar and phlorizin affinity and show fluorescence properties which indicate that they are located in a very hydrophilic environment. Phlorizin and its aglucone phloretin, but not D-glucose, protect both mutants against collisional quenchers. Depth calculations using the parallax method suggest the locations of Trp457 and Trp460 at an average distance of 10.8 Å and 7.4 Å from the center of the lipid bilayer, while Trp602 and Trp609 are located outside the membrane. These results suggest that in the native carrier residues Gln at position 457 and Thr at position 460 reside in a hydrophilic access pathway extending 5–7 Å into the membrane to which sugars as well as the sugar moiety of inhibitory glucosides can bind. Residues Phe602 and Phe609 contribute by their hydrophobic aromatic residues toward binding of the aglucone part of phlorizin (see Fig. 7).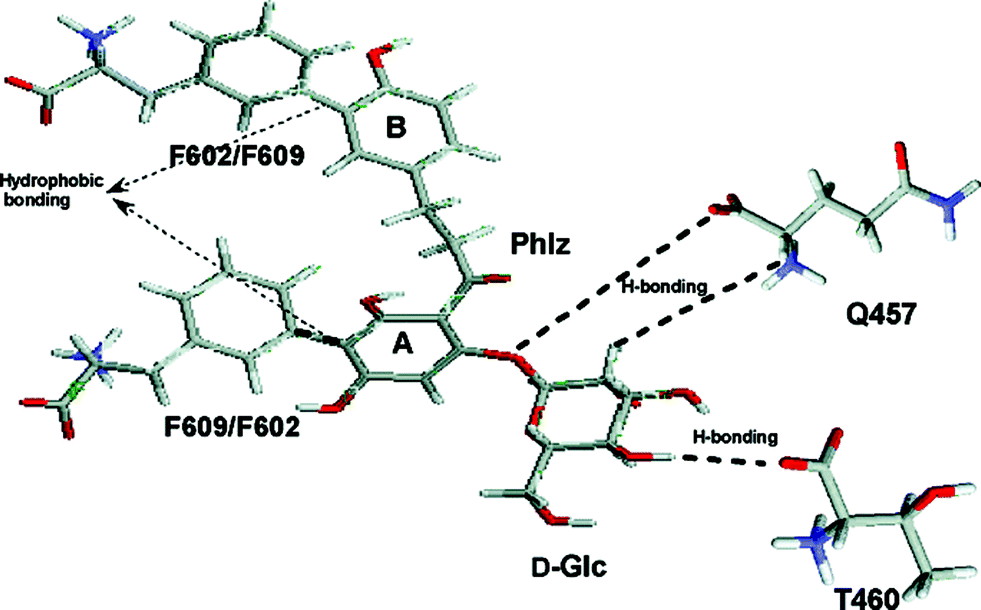 | ||
| Fig. 7 D-Glucose recognition and phlorizin-binding sites in isolated hSGLT1 (human isoform of SGLT1) incorporated in proteoliposomes. The hypothetical scheme of major interaction sites between phlorizin and hSGLT1. The sugar moiety of phlorizin interacts with residues Gln457 and Thr460 present in transmembrane helix IX probably by the same hydrogen bond interactions as D-glucose does; the aromatic rings A and B of the aglucone interact with Phe609/Phe602 in subdomain III.34 | ||
Thereby, in the phlorizin–carrier complex, a close vicinity between these two subdomains of the transporter is established, creating a phlorizin binding pocket with the previously estimated dimensions of 10 × 17 × 7 Å.20,35,36
4.3. Conformational changes in the presence of sugars and phlorizin in intact cells
The results illustrated in Fig. 8 show that specific unbinding events of 1-thio-glucose to the SGLT1 transporter can be detected only in the presence of sodium. By constructing an empirical probability density function (pdf) of the unbinding force, the maximum of the distribution was found to be about 40–50 pN (Fig. 8B) which was smaller than that obtained with the antibodies. This may due to a lower affinity of the sugar to SGLT1. Moreover, binding events were only detected in CHO cells overexpressing rbSGLT1.
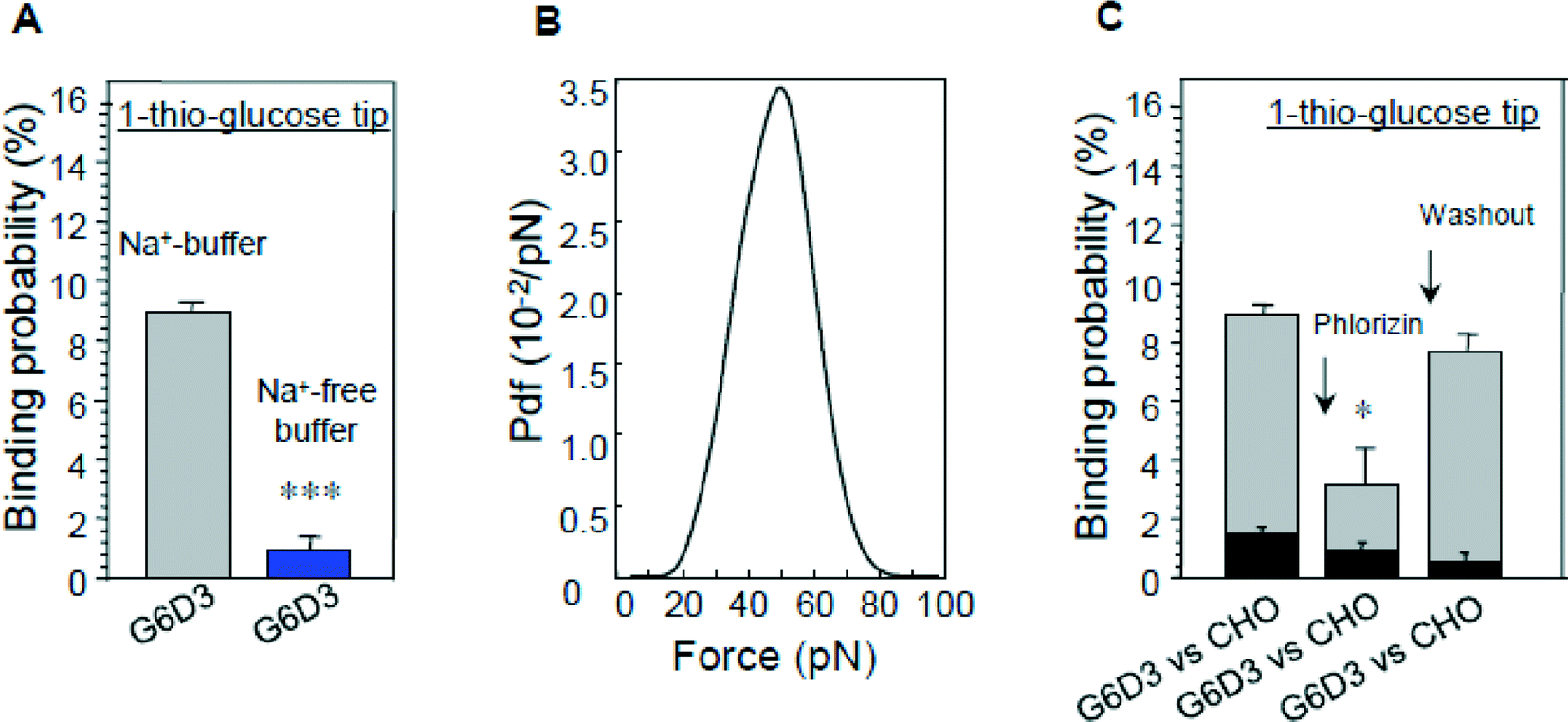 | ||
| Fig. 8 Interaction of glucose-primed AFM cantilevers with cells expressing rbSGLT1 (G6D3). (A) The sodium dependence of binding, (B) the most probable unbinding force fu and (C) the inhibition of binding by phlorizin; binding of cells not expressing rbSGLT1 (CHO) is shown as a comparison.25 | ||
The specificity of interaction could also be confirmed by blocking the sugar binding site with phlorizin (Fig. 8C). These data establish that the binding events recorded occur at the surface of SGLT1 molecules. The vinyl sulfone–PEG800–NHS cross-linker was purposely selected for this investigation since the bulky vinyl sulfone group present at the end of the linker most probably prevents a translocation, and therefore, only initial D-glucose binding events to SGLT1 should be observed. It is proposed that substrate transport by SGLT1 proceeds in several steps along the translocation pathway involving conformational alterations of the carrier.31,37,41,42 Our results demonstrate directly that the sugar binding site is accessible only when sodium is bound to the carrier, which opens the outward-facing gate of the transporter.7
When the effect of various sugars on the binding probability of the 1-thio-D-glucose tip to the transporter was investigated, a stereo-selectivity differing from that obtained in transport studies was found.23 The results suggest an initial binding site, which represents a first selectivity filter and selects sugars with regard to D- or L-conformation and the presence or positioning of OH groups at C1, C3, C4, and C6 but not at C2. The rejection of 2-deoxy-D-glucose, which is not transported, has to occur later during a subsequent binding/transport step. A similar discrepancy between initial binding and translocation has been observed previously in the rat kidney proximal tubule. 2-Deoxy-D-glucose was shown to inhibit binding of phlorizin to isolated brush border membrane vesicles but not transepithelial transport in microperfusion studies.43,44 Thus, the stereospecificity of transport is determined by at least two different selectivity filters, one located at the surface of the transporter, probably in or formed by the vestibule, and the other close to Gln457 and Thr406 in the translocation pathway.
Furthermore, an antibody against subdomain II partly inhibits the interaction of glucose with the transporter. Conversely, glucose inhibits the interaction of an antibody with subdomain II (see Fig. 9).
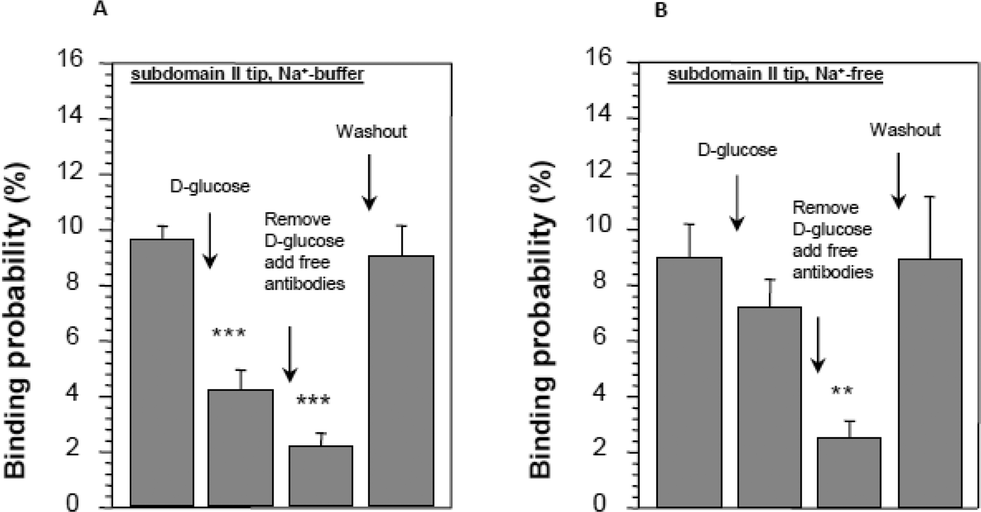 | ||
| Fig. 9 Conformational changes in subdomain II in the presence of D-glucose in SGLT1-expressing cells. Distance–force cycles were performed with the cantilever primed with an antibody against subdomain II. (A) The reduction in binding of the antibody in the presence of D-glucose and when the binding sites are saturated with free antibody in solution. In the absence of sodium, no significant effect of D-glucose was observed (B). Asterisks indicate statistical significance, for further details see ref. 23. | ||
These findings also support the idea that the three subdomains form a vestibule containing the initial outside-facing interaction sites of the transporter and undergo significant conformational changes during the binding process.
 | ||
| Fig. 10 Interaction of a phlorizin-primed AFM cantilever with right-side-out oriented brush border membrane vesicles isolated from the rat renal cortex. The upper panel shows reduced binding in the presence of free phlorizin (Pz), and the lower panel shows inhibition of binding by high concentrations of D-glucose.46 | ||
Experiments were also performed using an AFM cantilever tethered with an antibody directed against the epitope aa 603–630 of subdomain III. In the presence of phlorizin, the probability of antibody binding was drastically reduced, confirming a strong change in conformation of this ectodomain induced by phlorizin binding. Specific binding of an AFM cantilever primed with phlorizin to SGLT was also detected in SGLT 1 expressing cells.12 In addition, the changes in conformation of subdomain III after phlorizin binding were confirmed in intact cells.12
4.4. Kinetic properties of glucose and phlorizin binding to rbSGLT1 in intact cells
In a study combining experiments with glucose- and phlorizin-primed cantilevers, single molecule force spectroscopy was employed to investigate the dynamics of substrate and inhibitor binding at the single molecule level. Again cells stably expressing rbSGLT1 were probed by using atomic force microscopy tips carrying either thioglucose or amino-phlorizin. Experiments were performed at 10 and 37 °C to address different conformational states of SGLT1. Unbinding forces between ligands and SGLT1 were recorded at different loading rates by changing the retraction velocity, yielding the binding probability, width of the energy barrier of the binding pocket and kinetic off rate constant of the binding reaction (see experimental details in ref. 12). As shown in Table 1, with increasing temperature, the width of energy barrier and average lifetime increased for the interaction of SGLT1 with thioglucose but decreased for amino-phlorizin binding.12 The former indicates again that in the membrane-bound SGLT1 the pathway to sugar translocation involves several steps with different temperature sensitivities.The decrease in the average lifetime for phlorizin suggests that the aglucone binding sites for transport inhibitors have specific, temperature-sensitive conformations. These conformations will be investigated in the next sections.
5. Biophysical analysis of isolated ectodomains in vitro
5.1. Structural elements in ectodomains
All the experiments reported above indicate specific binding sites for the interaction of subdomain III with phlorizin, which would suggest that defined substructures exist in this domain. In order to investigate this question, we expressed subdomain III in E. coli and performed circular dichroism studies on the isolated peptide in solution. The CD spectrum showed characteristic minima at 222 and 208 nm from which the secondary structure was estimated to consist of 37% helical residues and 63% random coils. Calculations of secondary structure elements based on the amino acid sequence suggested the presence of small helical segments surrounded and linked by randomly coiled amino acid chains35 (see Fig. 11 below).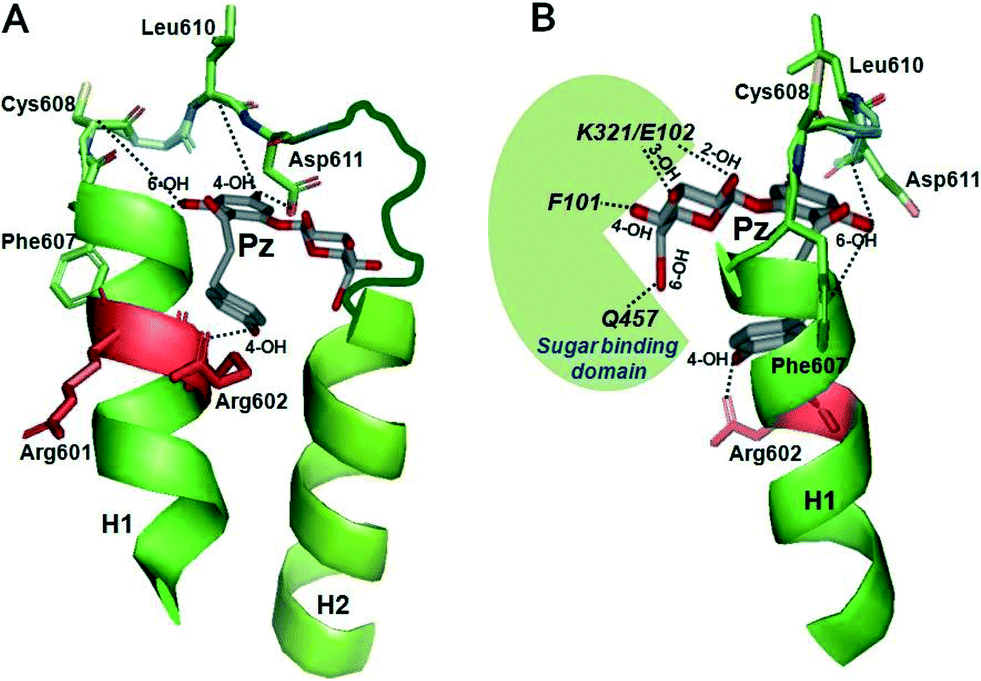 | ||
| Fig. 11 Modeling of interactions of subdomain III with phlorizin. The regions between TMH 12 and 13 of the vSGLT transporter structure (PDB ID: http://www.rcsb.org/pdb/explore.do?structureId=3DH4) were selected as templates for helices H1 and H2 (in green) as described in detail in ref. 48. Phlorizin (Pz) is shown in gray. (A, front view) The 4-OH of ring B of phlorizin is shown to interact with Arg-602 in H1, the 6-OH of ring B with residues 606–609 and the 4-OH of ring A with/around Asp-611. (B, side view) The glucose moiety of phlorizin is highlighted which is pointing towards the sugar binding domain depicted in green. Interactions between the OH groups of glucoside and several amino acids in the sugar binding domain of SGLT1 are also depicted. Modeling was performed using a PyMol computer modeling program (http://www.pymol.org/).48 | ||
5.2. Tryptophan scanning experiments with subdomain III in solution
6. Modeling the interaction between transport inhibitors and ectodomains of SGLT
6.1. Modeling the binding of phlorizin to subdomain III
Recently, modeling of the interaction of subdomain III with phlorizin has been performed by taking the bacterial SGLT (vSGLT) transporter structure as a template (Fig. 11). According to this modeling, 4- and 6-OH groups of aromatic ring A of phlorizin interact with the phlorizin binding domain (PBD) which is apparently located in an unstructured loop buried in between two helices H1 and H2, whereas ring B interacts with H1 at Arg-602. The modeling also predicted that in its most favorable configuration the glucoside moiety of phlorizin might point away from the aglucone binding pocket in order to interact with sugar binding domains. Hence, a condensed state of loop 13 followed by major conformational changes can be achieved via interaction of 2-OH and/or 3-OH of glucoside of phlorizin with K321 and E102 of SGLT1 (K294 and E88 in vSGLT) and 4-OH with F101 (Y87 in vSGLT) and 6-OH with Q457-SGLT1 (Q428-vSGLT).6.2. Modeling the binding of hexyl-glucoside to subdomain III
We have also constructed a model of a hexyl-glucoside interaction with subdomain III (Fig. 12) (M. Raja and R. K. Kinne, unpublished). The hydrophobic residues (sticks) in red and orange have been highlighted to depict the interaction of C2 with the peptide Gly-Phe-Phe-Arg (amino acid residues 598–601) in helix 1 and C6 with L628/L630 in helix 2. The two big red arrows indicate the flexible movement of the alkyl side chain with regard to the hydrophobic residues. In addition, the sugar binding domain is highlighted in green. | ||
| Fig. 12 Modeling of the interactions between hexyl-glucoside and subdomain III. The data were obtained in studies with tryptophan mutants of isolated subdomain III in solution.47 The modeling of the interactions of hexyl-glucoside with regions between TMH 12 and 13 of the vSGLT transporter structure (PDB ID: http://www.rcsb.org/pdb/explore.do?structureId=3DH4) was performed using a PyMol program as described in Fig. 11. The interaction of C2 of the alkyl chain of HG with the region around residues 598–600 of helix 1 and C6/C8 of the alkyl chain with the region between residues 624 and 630 in helix 2 is depicted. Similar to the phlorizin–subdomain III complex, the glucose moiety is free to interact with the sugar binding domain depicted in green. The red arrows indicate flexibility of the alkyl side chain for proper interaction in the binding. | ||
When both models (phlorizin and hexyl-glucoside models) are compared, the glucoside moiety of phlorizin is slightly above the plane of that of the hexyl-glucoside, agreeing well with the idea that aglucone binding domains are flexible to move with regard to the membrane plane.
If phlorizin or alkyl-glucosides interact with the accessible extramembranous part of loop 13 (subdomain III), they first interact via their aglucone moiety while pushing the aglucone–loop 13 complex into the membrane to interact with the sugar translocation pathway. These changes in conformation of subdomain III are in agreement with the inhibitory effect of phlorizin on the interaction of a cantilever primed with an antibody against subdomain III (see above). Thus, after phlorizin binding to the transporter, the epitope (aa 606–630) is no longer accessible to the antibody PAN3-2. This change could be due to an obstruction of the antibody binding sites by phlorizin itself or due to a conformational change in the transporter. Support for the former assumption is rather weak because the epitope is much larger than the region between amino acids 602 and 611, which, as detailed above, acts as a phlorizin-binding domain.35,46,47,49 Since it has been shown that endodomains of the C-terminal part (region between transmembrane helices 10–13) are responsible for sugar translocation, we rather assume that phlorizin induces a movement of subdomain III which brings it close to or even into the plasma membrane plane, thereby making the interaction with sugars and thus their translocation impossible.
7. Interaction of phlorizin with different members of the SGLT family
The studies on recognition/interaction of phlorizin with SGLT1-loop 13 have also provided valuable insights into how phlorizin and its high-affinity analogs could interact with closely related SGLT members, particularly SGLT2, 3 or 4. Taking advantage of molecular modeling of SGLT1-loop 13 with phlorizin, primary sequence analyses and secondary structure predictions, we found quite similar phlorizin binding domains in the C-termini of human SGLT2–4 (Fig. 13). | ||
| Fig. 13 Schematic representation of phlorizin interaction with subdomain III of various members of the SGLT family. Qualitatively similar folding patterns of phlorizin binding domains are predicted for SGLT1–4, except for vSGLT, which lacks secondary structure contents and does not exhibit a proper phlorizin binding pocket. The ranking of phlorizin interaction is depicted based on previously described phlorizin inhibition studies and our prediction analyses. The gradient filled arrow (in gray) represents the magnitude of phlorizin interaction/inhibition in various SGLTs.48 | ||
Furthermore, we found a strong correlation between the length of the C-terminus and phlorizin inhibition. The weakest phlorizin inhibition in vSGLT is probably caused by the absence of a long C-terminus as well as a proper phlorizin binding domain. However, SGLT1–4 carry an extended C-terminus/loop 13 as well as secondary structures in phlorizin binding domains, thereby exhibiting higher sensitivity. These predictions strongly agree with the previously reported phlorizin inhibition kinetics with the following ranking: SGLT2 > SGLT1 > SGLT4 > SGLT3 ≫ vSGLT. Interestingly, the sugar binding residues were also found to be quite conserved among these SGLT members, thereby indicating a common mechanism by which other SGLTs are inhibited by phlorizin or its high affinity analogs.48
Structurally similar analogs of phlorizin, like dapagliflozin and canagliflozin, exhibit better absorption in the gastrointestinal tract than phlorizin. Considering that these analogs share the same binding pocket as phlorizin,50 our predictions are very useful in understanding the mechanism of SGLT inhibition. Based on primary sequence analyses and computer modeling prediction, the increase in secondary structural contents (helices H1, H2 and H3) could promote high specificity and strong affinities of phlorizin and its analogs for SGLT2 as compared to SGLT1.48 These assumptions fit quite nicely with the recently illustrated homology modeling and molecular dynamics simulation studies predicting higher stability of SGLT2 binding complexes than SGLT1, and an increase in hydrophobicity might also promote hydrogen bonding within the binding pocket upon inhibitor binding.27 Furthermore, for SGLT2 an additional binding of the B ring of phlorizin to H3 is expected which could explain the high affinity of dapagliflozin, which has an extended CH2–CH3 chain at C6 of the B ring.
8. Potential role of ectodomains in the interaction of other SLCs with inhibitors
In reviewing the literature also, hints for a potential role of ectodomains in inhibitor–transporter interaction can be found. They usually point to a transient binding during the approach of the inhibitors into the substrate translocation channel before they bind stably to the substrate binding site. Those incidences are summarized in Table 2.| SLC | Literature name | Function | Drug/inhibitor | Potential ectodomain | Remarks | Ref. |
|---|---|---|---|---|---|---|
| SLC2 | GLUT1 (SLC2A1) | Glucose transport | Phloretin | Extracellular vestibule (infundibulum) | There is an additional intracellular binding site at the exit of the transport channel | 5, 51 |
| Forskolin | ||||||
| SLC6 | SERT (SLC6A4) | Na/Cl/monoamine cotransport | Tricyclic anti-depressants (TCA) | Extracellular vestibule | Transient, low affinity, noncompetitive interaction | 26, 52, 53 |
| DAT (SLC6A3) | Serotonin | High affinity binding in the substrate binding site | ||||
| NET (SLC6A2) | Dopamine | |||||
| Noradrenalin | ||||||
| SLC12 | NKCC1 (SLC12A1) | Na/K/2Cl cotransport | Loop diuretics: furosemide, bumetanide | Extracellular part of a transmembrane helix | Bumetanide: transient interaction at the extracellular entry point of TMH 3, high affinity binding at the inner end of TMH 3 at a chloride binding site. Furosemide: less affected by mutations suggesting additional binding sites | 2, 54 |
| SLC5 | SGLT (SLC5A1, SLC5A2) | Na/glucose cotransport | Phlorizin | Extracellular vestibule | The sugar moiety of the beta-glucoside phlorizin binds in addition to the sugar translocation site | This review |
| Phloretin |
Thus, in the sodium-independent glucose transporter, GLUT1, phloretin is found in homology building, molecular modeling and docking studies both in the extracellular vestibule and at the intracellular exit of the transport channel. The same holds for forskolin, whereas cytochalasin B acts only at an intracellular site.51 In the monoamine transporter, there is some evidence that a weak transient interaction of tricyclic antidepressants in the extracellular vestibule is followed by strong binding to the substrate binding site which is of competitive nature.26,52,53 For the sodium–potassium–chloride cotransporter, a transient interaction at the entry point of a transmembrane segment that is part of the ion translocation pathway might precede the competitive binding to a chloride binding site deep in the protein. Furosemide appears to have additional interactions with the transporter. It should be pointed out that in most of the studies mentioned above modeling and simulations are the basis of the conclusions and thus require further investigations such as crystallization of the complexes; direct experimental evidence for ectodomain–inhibitor binding has only been provided for SGLT1.
9. Synopsis and perspectives
There are several major points this review wants to make. One is that it is evident that phlorizin and other SGLT2 inhibitors can be added to the class of multivalent drugs, in which “substrate analogues” are linked by a mostly flexible chain to larger organic molecules. As is the case for phlorizin and its analogues, in the monoamine transporter, the substrate binding/translocation site is the major binding site for the inhibitor and there are additional sites in the vicinity.26,52,53 Similar suggestions have been made for example for the interaction of loop diuretics (furosemide, bumetanide and piretanide) with the sodium–potassium–chloride cotransporter2,54 and for the binding of thiazide diuretics to the electroneutral sodium–chloride cotransporter1 (SLC12A3). In all instances, there is an indication that the transport inhibitors block the cavity which leads to the translocation site within the membranes formed by endodomains. The extent to which ectodomains are actually involved remains to be determined.Thus, we would propose a common mechanism (including SGLT1) for inhibition. Recognition events take place at the ectodomain level, and then, the inhibitors travel through vestibules via multiple binding sites and finally bind strongly in the cavity of the transporter. In this regard, the crystal structure of SLC6 in the presence of an inhibitor55 nicely complements our functional/modelling studies and agrees well with the idea that inhibitors of the SLC family are recognized in the ectodomain regions before the second high affinity binding event takes place in the transmembrane region, e.g. phlorizin binding to loop 13 (ectodomain level) and its glucoside binding within the sugar binding pocket (transmembrane region), by repositioning of various parts of the transporter and the inhibitor during inhibition.
The studies presented above clearly indicate that ectodomains of the transporter molecule can have quite stable substructures which can provide selective binding sites for additional chemical entities of the inhibitors, not interacting with the substrate binding pockets. They, therefore, have to be included in modelling transport inhibitor complexes, docking experiments or virtual screening techniques.56
Functional roles of ectodomains have been described for a variety of transporters57–59 which point to additional potential target–inhibitor interaction sites. Another reason to take ectodomains close to the substrate binding pocket into account is that, in contrast to the high homology/similarity in the amino acid sequence of transport translocation pathways, ectodomains differ considerably amongst members of a transporter family, as could be demonstrated for the SGLT family in this review. This opens the possibility that different members of the transporter family, which are usually expressed in different organs, might be targeted by modification of the ectodomain binding entities of the drug. In more general terms, selectivity probably can be improved by targeting secondary binding pockets with extended ligands.
References
- D. B. Mount, E. Delpire, G. Gamba, A. E. Hall, E. Poch, R. S. Hoover and S. Hebert, J. Exp. Biol., 1998, 201, 2091–2102 CAS.
- S. Somasekharan, J. Tanis and B. Forbush, J. Biol. Chem., 2012, 287, 17308–17317 CrossRef CAS PubMed.
- R. K. Kinne and F. Castaneda, Handb. Exp. Pharmacol., 2011, 203, 105–126 CAS.
- L. A. Gallo, E. M. Wright and V. Vallon, Diabetes Vasc. Dis. Res., 2015, 12, 78–89 CrossRef CAS PubMed.
- M. Raja, T. Puntheeranurak, P. Hinterdorfer and R. K. Kinne, Curr. Top. Membr., 2012, 70, 29–76 CAS.
- E. M. Wright, Mol. Aspects Med., 2013, 34, 183–196 CrossRef CAS PubMed.
- M. Sala-Rabanal, B. A. Hirayama, D. D. Loo, V. Chaptal, J. Abramson and E. M. Wright, Am. J. Physiol., Cell Physiol., 2012, 302, C1293–C1305 CrossRef CAS PubMed.
- A. C. Pan, D. W. Borhani, R. O. Dror and D. E. Shaw, Drug Discovery Today, 2013, 18, 667–673 CrossRef CAS PubMed.
- Y. Shan, E. T. Kim, M. P. Eastwood, R. O. Dror, M. A. Seeliger and D. E. Shaw, J. Am. Chem. Soc., 2011, 133, 9181–9183 CrossRef CAS PubMed.
- R. O. Dror, A. C. Pan, D. H. Arlow, D. W. Borhani, P. Maragakis, Y. Shan, H. Xu and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13118–13123 CrossRef CAS PubMed.
- L. Wildling, C. Rankl, T. Haselgrübler, H. J. Gruber, M. Holy, A. H. Newman, M. F. Zou, R. Zhu, M. Freissmuth, H. H. Sitte and P. Hinterdorfer, J. Biol. Chem., 2012, 287, 105–113 CrossRef CAS PubMed.
- I. Neundlinger, T. Puntheeranurak, L. Wildling, C. Rankl, L. X. Wang, H. J. Gruber, R. K. Kinne and P. Hinterdorfer, J. Biol. Chem., 2014, 289, 21673–21683 CrossRef PubMed.
- C. Lamprecht, P. Hinterdorfer and A. Ebner, Expert Opin. Drug Delivery, 2014, 11, 1237–1253 CrossRef CAS PubMed.
- T. Puntheeranurak, I. Neundlinger, R. K. Kinne and P. H., Nat. Protoc., 2011, 6, 1443–1452 CrossRef CAS PubMed.
- T. Rieg, T. Masuda, M. Gerasimova, E. Mayoux, K. Platt, D. R. Powell, S. C. Thomson, H. Koepsell and V. Vallon, Am. J. Physiol. Renal Physiol., 2014, 306, F188–F193 CrossRef CAS PubMed.
- H. Kipp, J. T. Lin and R. K. Kinne, Biochim. Biophys. Acta, 1996, 1282, 125–130 CAS.
- C. Scafoglio, B. A. Hirayama, V. Kepe, J. Liu, C. Ghezzi, N. Satyamurthy, N. A. Moatamed, J. Huang, H. Koepsell, J. R. Barrio and E. M. Wright, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4111–E4119 CrossRef CAS PubMed.
- C. Ghezzi, B. A. Hirayama, E. Gorraitz, D. D. Loo, Y. Liang and E. M. Wright, Physiol. Rev., 2014, 2 Search PubMed.
- M. A. Firnges, J. T. Lin and R. K. Kinne, J. Membr. Biol., 2001, 179, 143–153 CrossRef CAS PubMed.
- A. Kumar, N. K. Tyagi, E. Arevalo, K. W. Miller and R. K. Kinne, Biochim. Biophys. Acta, 2007, 1774, 968–974 CrossRef CAS PubMed.
- E. M. Wright, D. D. Loo and B. A. Hirayama, Physiol. Rev., 2011, 91, 733–794 CrossRef CAS PubMed.
- N. K. Tyagi, W. Siess, P. Goyal, A. Kumar, D. Pandey and R. K. Kinne, Biochemistry, 2005, 44, 15514–15524 CrossRef CAS PubMed.
- T. Puntheeranurak, M. Kasch, X. Xia, P. Hinterdorfer and R. K. Kinne, J. Biol. Chem., 2007, 282, 25222–25230 CrossRef CAS PubMed.
- T. Puntheeranurak, R. K. Kinne, H. J. Gruber and P. Hinterdorfer, J. Korean Phys. Soc., 2008, 52, 1336–1340 CrossRef CAS.
- T. Puntheeranurak, L. Wildling, H. J. Gruber, R. K. Kinne and P. Hinterdorfer, J. Cell Sci., 2006, 119, 2960–2967 CrossRef CAS PubMed.
- J. Grouleff, L. K. Ladefoged, H. Koldso and B. Schiott, Front. Pharmacol., 2015, 6, 235 Search PubMed.
- J. Xu, H. Yuan, T. Ran, Y. Zhang, H. Liu, S. Lu, X. Xiong, A. Xu, Y. Jiang, T. Lu and Y. Chen, J. Mol. Recognit., 2015, 28, 467–479 CrossRef CAS PubMed.
- A. Ebner, L. Wildling, A. S. Kamruzzahan, C. Rankl, J. Wruss, C. D. Hahn, M. Holzl, R. Zhu, F. Kienberger, D. Blaas, P. Hinterdorfer and H. J. Gruber, Bioconjugate Chem., 2007, 18, 1176–1184 CrossRef CAS PubMed.
- E. Turk, C. J. Kerner, M. P. Lostao and E. M. Wright, J. Biol. Chem., 1996, 271, 1925–1934 CrossRef CAS PubMed.
- E. Turk and E. M. Wright, J. Membr. Biol., 1997, 159, 1–20 CrossRef CAS PubMed.
- D. G. Gagnon, A. Holt, F. Bourgeois, B. Wallendorff, M. J. Coady and J. Y. Lapointe, Biochim. Biophys. Acta, 2005, 1712, 173–184 CrossRef CAS PubMed.
- J. Lin, J. Kormanec, D. Homerova and R. K. Kinne, J. Membr. Biol., 1999, 170, 243–252 CrossRef CAS PubMed.
- R. Novakova, D. Homerova, R. K. Kinne, E. Kinne-Saffran and J. T. Lin, J. Membr. Biol., 2001, 184, 55–60 CrossRef CAS PubMed.
- N. K. Tyagi, A. Kumar, P. Goyal, D. Pandey, W. Siess and R. K. Kinne, Biochemistry, 2007, 46, 13616–13628 CrossRef CAS PubMed.
- M. M. Raja, N. K. Tyagi and R. K. Kinne, J. Biol. Chem., 2003, 278, 49154–49163 CrossRef CAS PubMed.
- S. Wielert-Badt, J. T. Lin, M. Lorenz, S. Fritz and R. K. Kinne, J. Med. Chem., 2000, 43, 1692–1698 CrossRef CAS PubMed.
- D. D. Loo, B. A. Hirayama, E. M. Gallardo, J. T. Lam, E. Turk and E. M. Wright, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7789–7794 CrossRef CAS.
- D. D. Loo, B. A. Hirayama, M. H. Karakossian, A. K. Meinild and E. M. Wright, J. Gen. Physiol., 2006, 128, 701–720 CrossRef CAS PubMed.
- X. Z. Chen, M. J. Coady and J. Y. Lapointe, Biophys. J., 1996, 71, 2544–2552 CrossRef CAS PubMed.
- A. Hazama, D. D. Loo and E. M. Wright, J. Membr. Biol., 1997, 155, 175–186 CrossRef CAS PubMed.
- B. A. Hirayama, D. D. Loo and E. M. Wright, J. Biol. Chem., 1997, 272, 2110–2115 CrossRef CAS PubMed.
- A. K. Meinild, B. A. Hirayama, E. M. Wright and D. D. Loo, Biochemistry, 2002, 41, 1250–1258 CrossRef CAS PubMed.
- R. K. Kinne, Curr. Top. Membr. Transp., 1976, 8, 209–267 CAS.
- H. Glossmann and D. M. Neville, Jr., J. Biol. Chem., 1972, 247, 7779–7789 CAS.
- J. T. Lin, K. D. Hahn and R. Kinne, Biochim. Biophys. Acta, 1982, 693, 379–388 CrossRef CAS.
- S. Wielert-Badt, P. Hinterdorfer, H. J. Gruber, J. T. Lin, D. Badt, B. Wimmer, H. Schindler and R. K. Kinne, Biophys. J., 2002, 82, 2767–2774 CrossRef CAS PubMed.
- M. M. Raja, H. Kipp and R. K. Kinne, Biochemistry, 2004, 43, 10944–10951 CrossRef CAS PubMed.
- M. Raja and R. K. Kinne, Biochimie, 2015, 115, 187–193 CrossRef CAS PubMed.
- X. Xia, J. T. Lin and R. K. Kinne, Biochemistry, 2003, 42, 6115–6120 CrossRef CAS PubMed.
- C. S. Hummel, C. Lu, J. Liu, C. Ghezzi, B. A. Hirayama, D. D. Loo, V. Kepe, J. R. Barrio and E. M. Wright, Am. J. Physiol., Cell Physiol., 2012, 302, C373–C382 CrossRef CAS PubMed.
- A. Salas-Burgos, P. Iserovich, F. Zuniga, J. C. Vera and J. Fischbarg, Biophys. J., 2004, 87, 2990–2999 CrossRef CAS PubMed.
- S. Sinning, M. Musgaard, M. Jensen, K. Severinsen, L. Celik, H. Koldso, T. Meyer, M. Bols, H. H. Jensen, B. Schiott and O. Wiborg, J. Biol. Chem., 2010, 285, 8363–8374 CrossRef CAS PubMed.
- H. Koldso, J. Grouleff and B. Schiott, Front. Pharmacol., 2015, 6, 208 Search PubMed.
- P. Isenring, S. C. Jacoby, J. Chang and B. Forbush, J. Gen. Physiol., 1998, 112, 549–558 CrossRef CAS PubMed.
- A. Penmatsa, K. H. Wang and E. Gouaux, Nat. Struct. Mol. Biol., 2015, 22, 506–508 CAS.
- O. V. Mortensen and S. Kortagere, Front. Pharmacol., 2015, 6, 223 Search PubMed.
- M. Grunewald and B. I. Kanner, J. Biol. Chem., 2000, 275, 9684–9689 CrossRef CAS PubMed.
- O. Kelly, C. Lin, M. Ramkumar, N. C. Saxena, T. R. Kleyman and D. C. Eaton, Am. J. Physiol. Renal Physiol., 2003, 285, F1279–F1290 CrossRef CAS PubMed.
- H. Murer, Kidney Int., 2002, 62, 375–382 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |