
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A 18F-labeled dibenzocyclooctyne (DBCO) derivative for copper-free click labeling of biomolecules†‡
K.
Kettenbach
 a and
T. L.
Ross
*ab
a and
T. L.
Ross
*ab
aInstitute of Nuclear Chemistry, Johannes Gutenberg-University Mainz, 55128 Mainz, Germany
bRadiopharmaceutical Chemistry, Department of Nuclear Medicine, Hannover Medical School, 30625 Hannover, Germany. E-mail: ross.tobias@mh-hannover.de
First published on 12th January 2016
Abstract
The new prosthetic group 18F-TEG-DBCO (dibenzocyclooctyne) can be prepared within a total reaction time of 60 min including purification with an overall yield (n.d.c.) of 34 ± 5%. Copper-free click cycloadditions with an azido-cRGD, a folate-azide and two α-MSH analogue azido-peptides resulted in very high RCYs and fast reaction kinetics.
For non-invasive in vivo imaging of processes and pharmacokinetics of radiolabeled biomolecules, the positron emission tomography (PET) is one of the most powerful methods.1 For PET applications, fluorine-18 has ideal nuclear characteristics and is the most commonly applied radionuclide in PET. The relatively long half-life of 110 minutes enables multi-step radiosyntheses, and the rather low β+-energy ensures a very high spatial resolution in tomography.2 The challenge for nuclear chemists consists in finding appropriate 18F-labeling strategies, especially for sensitive biomolecules. Most of them are sensitive to the commonly used harsh conditions in direct 18F-labeling reactions such as high temperatures and strong basic conditions.3,4 As a result, the development of indirect labeling strategies via18F-prosthetic groups, which can subsequently be attached to biomolecules under mild reaction conditions, is needed.5–7 Besides, the radiolabeling reaction should allow bioorthogonal 18F-labeling to treat the multitude of functional groups in bioactive compounds with respect. The most prominent example of such reactions, which fulfills all the mentioned criteria, is the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) first published by Sharpless et al. in 2001.8 This variant of the Huisgen 1,3-dipolar cycloaddition of terminal alkynes and azides enables 18F-labeling with high specificity and excellent yields under mild conditions.9,10 In the past decade, a widespread spectrum of PET tracers has been synthesized using the CuAAC method for 18F-labeling of bioactive compounds.11 One of the latest developments is based on an amino acid, which is thought to minimize the influence on the pharmacokinetic properties of the intended radiotracer. As an amino acid derived 18F-prosthetic group, it is particularly suitable for peptides and proteins.12 However, with all the advantages of the copper(I)-catalyzed cycloaddition, there is one major disadvantage. The need for cytotoxic copper species as a catalyst in the click reaction causes an extensive work-up guaranteeing the complete removal of copper for in vivo applications. This fact led to the necessity of alternative fast and copper-free click reaction strategies. By using strained alkynes instead of terminal alkynes, copper is no longer needed to catalyze the click reaction. These so-called strain-promoted click reactions were first reported by Baskin et al.13 and can be carried out between cyclooctyne derivatives and azides or tetrazines as 3 + 2 cycloaddition.11 The use of azadibenzocyclooctynes for copper-free click reactions was first reported by Kuzmin et al. in 2010.14 Recently, Arumugam et al. have published the development of a 18F-labeled azadibenzocyclooctyne for 18F-labeling of peptides via a strain-promoted click reaction without the use of a copper species, showing the high potential of this concept for 18F-labeling of biomolecules.15 Our aim was to develop a new 18F-prosthetic group based on (aza)dibenzocyclooctyne (DBCO) for radiolabeling of biomolecules such as peptides and microproteins. For reduced lipophilicity, we introduced a triethylene glycol spacer to the (aza)dibenzocyclooctyne. Two different leaving groups, different bases, base concentrations and precursor amounts during radiolabeling were evaluated for optimized 18F-labeling. Consequently, two DBCO-based precursors and the non-radioactive reference compound were synthesized, and the 18F-labeling reaction was optimized. Finally, we performed a proof-of-principle click reaction with the new 18F-labeled prosthetic group and an azido-functionalized cyclic Arg-Gly-Asp (cRGD) peptide as a model system. This peptide is used as the gold-standard vector in targeting the αVβ3 integrin.16,17 Furthermore, we carried out further copper-free click reactions using a folate-azide for targeting the folate receptor and two α-MSH analogue azido-functionalized peptides with high specificities to the melanocortin receptor 1 (MC1R).
The syntheses of reference compound 11 and the 18F-labeling precursors 9 and 10 are depicted in Scheme 1. The synthesis started from commercially available triethylene glycol 2. In the first step, 2 was reacted with tert-butylacrylate 1 to create a carboxylic acid function enabling the desired amide coupling.18 Compound 3 was then reacted with either p-toluenesulfonyl chloride19 or methanesulfonyl chloride20 to transfer the hydroxyl function into suitable leaving groups for the nucleophilic radiofluorination reaction. Subsequently, protected intermediates 4 and 6 were deprotected by trifluoroacetic acid in dichloromethane at room temperature to yield 5 and 7.21 Both linker groups were coupled via an amide bond to the dibenzocyclooctyne (DBCO)-amine 8, using HBTU N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uranium-hexafluorophosphate (HBTU) as coupling reagent and N,N-diisopropylethylamine (DIPEA) as base. The coupling was performed at room temperature for 12 h to yield the desired precursors for the 18F-fluorination reaction in overall yields of 28% (for precursor 9) and 56% (for precursor 10) over four steps. Due to the quite high costs for DBCO-amine 8, we aimed to insert this component in the last synthesis step. In relation to the amounts of 8, the yields were good to very high, leading to 56% and 87%, respectively. The reference compound was synthesized through 19F-fluorination of 9 using tetrabutylammonium fluoride (TBAF) at 120 °C for 2 h to yield 11 in excellent yields of 82%.
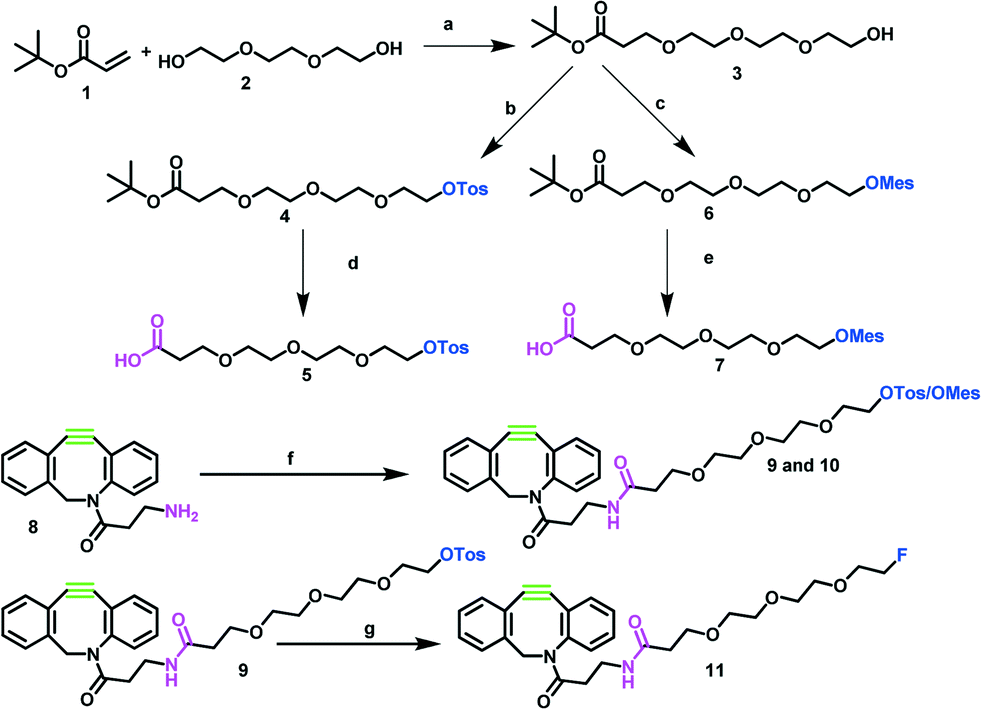 | ||
| Scheme 1 Synthesis of alkyne-functionalized reference compound 11 and labeling precursors 9 and 10. Regents and conditions: a) sodium, THF, 24 h, rt; b) TEA, p-toluenesulfonyl chloride, DCM, 1 h, 0 °C, rt; c) TEA, methanesulfonyl chloride, 1 h, 0 °C, rt; d) TFA, DCM, 4 h, rt; e) TFA, DCM, 4 h, rt; f) TEG-carboxylic acid, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uranium-hexafluorophosphate (HBTU), N,N-diisopropylethylamine (DIPEA), DMF, 24 h, rt; g) tetrabutylammonium fluoride, THF, 2 h, 80 °C. | ||
The radiofluorination of precursor 9 is depicted in Scheme 2. The radiolabeling of precursors 9 and 10 was optimized using different parameters such as various bases, base concentrations, reaction times and different amounts of precursors. Initially, the use of two different bases, tetrabutylammonium hydroxide (TBA-OH) and tetraethylammonium bicarbonate (Et4NHCO3), in acetonitrile was screened. The use of TBA-OH caused decomposition of the precursors, and a RCY of only 30% was achievable. The use of precursor 9 (7.5 mg, 12 μmol) in acetonitrile and tetraethylammonium bicarbonate gave the highest RCY of ≥90% within 10 minutes. For further evaluation of precursors 9 and 10, tetraethylammonium bicarbonate was used as base. With a base amount below 17 μmol, no 18F-labeling was observed, while increasing the base amount to higher than 17 μmol resulted in reduced yields. Besides, the amount of precursor played an important role. Reaction kinetics were monitored for 2.5, 5.0 and 7.5 mg (4, 8 and 12 μmol) of precursor 9. By increasing the amount of precursor (12 μmol), RCYs of ≥90% after 10 min were observed. Between the two leaving groups we did not observe significant differences in RCYs.
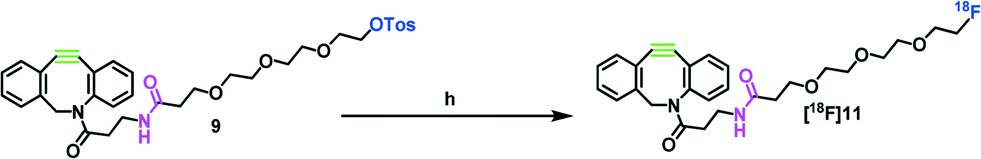 | ||
| Scheme 2 Synthesis of 18F-prosthetic group [18F]11. Reagents and conditions: h) n.c.a. 18F−, Et4NHCO3, MeCN, 100 °C, 10 min, RCY 91%. | ||
Isolation of the final 18F-labeled prosthetic group was performed by fixation of the product fraction obtained from semi-preparative HPLC on a C18 reversed phase cartridge, followed by elution of the 18F-prosthetic group from the resin with acetonitrile (1 mL). An exemplary radio-HPLC chromatogram of the crude mixture after radiolabeling of [18F]11 is shown in the ESI.‡ The solvent was removed under reduced pressure, and the 18F-prosthetic group was resolved in the desired solvent to perform the subsequent click reaction. The new 18F-prosthetic group was synthesized and isolated within only 60 min in an excellent overall yield (n.d.c.) of 34 ± 5%, ready for copper-free click reactions with azido-functionalized biomolecules. For the lipophilicity of the 18F-prosthetic group, a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D value of 1.20 ± 0.07 was calculated using the octanol–water distribution coefficient.
D value of 1.20 ± 0.07 was calculated using the octanol–water distribution coefficient.
To test the viability of [18F]11, it was used in the copper-free cycloaddition with azido-functionalized cRGD peptide 12 (1 mg, 1.1 μmol), as shown in Scheme 3, as a model system.
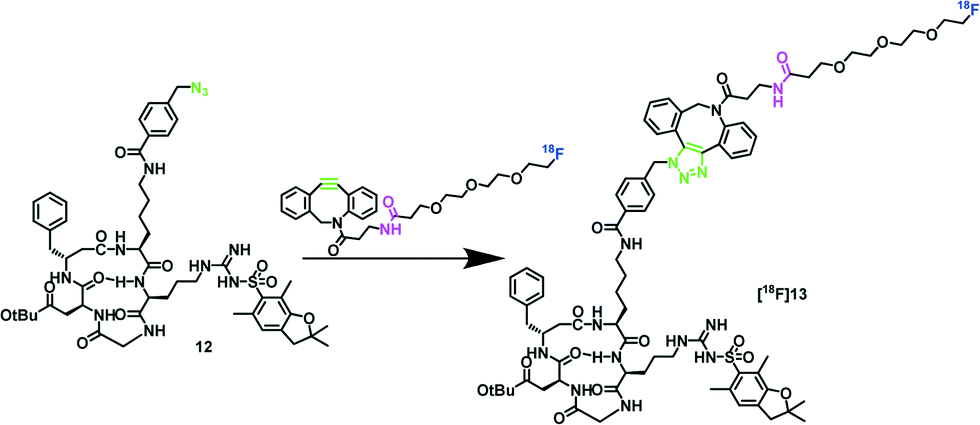 | ||
| Scheme 3 SPAAC of protected azido-functionalized cRGD 18 and the new prosthetic group [18F]11. Click reaction conditions: PBS buffer/acetonitrile (1:1), 25 °C or 40 °C, 5 min, RCY 93%. | ||
The copper free 18F-click reaction gave the desired peptide [18F]13 in excellent RCY of 93% within 5 min, which shows the particularly high potential of the new prosthetic group for 18F-labeling of sensitive biomolecules under very mild conditions (25 °C, phosphate-buffered saline (PBS, pH 7.4), 5 min). An exemplary radio-HPLC chromatogram of [18F]13 in comparison to the 18F-prosthetic group [18F]11 is shown in the ESI.‡
Furthermore, an azido-functionalized folate derivative as a well-known tumor targeting vector was 18F-labeled in a copper free click reaction using the new 18F-prostehtic group. Remarkably, quantitative 18F-click labeling was observed after a few minutes at room temperature and a good (low) logD value of 0.6 ± 0.07 was determined for the final 18F-folate. The 18F-labeled folate can be separated from unreacted folate-azide by HPLC and C18 SPE. The stability of the 18F-folate was analyzed in human serum at 37 °C. After 1.0 h and 1.5 h, ≥95% intact 18F-folate was observed. To our best knowledge, this is the first report on a new 18F-folate labeled via a copper-free click approach.
Two different azido-functionalized α-MSH analogue peptides, N3-TEG-Gly-Gly-Nleu-Gly-His-DPhe-Arg-Trp-NH2 and N3-TEG-Gly-Gly-Nleu-[Cys-His-DPhe-Arg-Trp-Gly-Cys]-NH2, with high specificities to the MC1R (melanocortin receptor 1) were prepared by using solid phase peptide synthesis (SPPS).22 Both peptides were radiolabeled with the 18F-prostehtic group in a copper-free click cycloaddition. For the linear peptide, RCY of up to 79% after 20 min was observed for 0.4 μmol peptide at 40 °C. For the cyclic α-MSH analogue, the click reaction proceeded with excellent RCY of 92% even with a lower amount of only 0.2 μmol peptide at 40 °C.
Summarized conditions, RCY and kinetics for the four biomolecule-azides, which were tested in copper-free click reactions with the new 18F-prosthetic group, are shown in the ESI.‡ The RCYs are also displayed as a bar chart in Scheme 4.
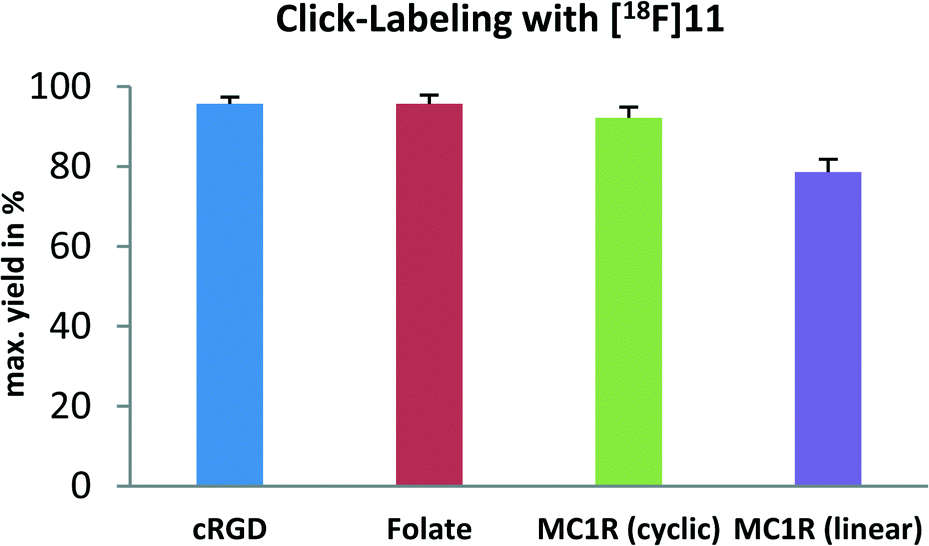 | ||
| Scheme 4 Radiolabeling of various azides with [18F]11. RCYs after 20 min are displayed in a bar chart. Errors are given as standard deviation representing n = 3. | ||
Especially for the radiolabeling of sensitive biomolecules, the use of 18F-prosthetic groups is of particular interest, where the use of harsh conditions for direct 18F-labeling reactions is excluded. Due to the toxicity of copper, the attachment of the 18F-prosthetic groups via copper(I)-catalyzed cycloaddition is no longer the first choice. The use of strained alkynes for copper-free cycloaddition enables selective radiolabeling of azido-functionalized biomolecules under very mild conditions.
The herein reported synthesis strategy of a 18F-labeled DBCO-based prosthetic group enables copper-free 18F-click labeling of various biomolecule-azides under very mild conditions and with outstanding efficacy. The organic syntheses provided the two precursors in good to high yields over four steps. The organic syntheses are robust and very reliable, and in reference to DBCO-amine, the strategy and yields were optimized for economic reasons. High to excellent results were obtained for the 18F-labeling of the two different precursors, which are available and ready to use for subsequent 18F-click reactions within only 60 min and in high yields of 34% (n.d.c.).
The new 18F-prosthetic group performs outstandingly in copper-free click reactions with different biomolecule-azides, which are known as excellent tumor targeting vectors of common interest.16,17,23–25 All click reactions proceeded with excellent to even quantitative yields under very mild conditions (water or PBS, RT or 40 °C) with very fast reaction kinetics. The tested biomolecule-azides (RGD, linear MSH-peptides and folic acid) were not achievable in such good yields with conventional copper(I)-catalyzed click cycloaddition.11
In cases of non-quantitative 18F-click labeling, the 18F-labeled products were easily separated by radio-HPLC from the unreacted 18F-prostehtic group. For the 18F-labeled folate derivative, a low logD value of 0.6 ± 0.07 was determined. High stability was observed in human serum at 37 °C over a period of 1.5 h. Further in vitro and in vivo evaluation of the new 18F-folate using human KB cells and PET imaging is ongoing. Similarly, investigations and evaluation using the other new 18F-tracers derived from copper-free 18F-click labeling in in vivo PET imaging are planned.
Notes and references
- N. Johnsson and K. Johnsson, ACS Chem. Biol., 2007, 2(1), 31 CrossRef CAS PubMed.
- J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2013, 56(3–4), 225 CrossRef CAS PubMed.
- H. H. Coenen, R. H. Elsinga, R. Iwata, M. R. Kilbourn, M. R. A. Pillai, G. R. Rajan, H. N. Wagner and J. J. Zaknun, Nucl. Med. Biol., 2010, 37(7), 727 CrossRef CAS PubMed.
- L. Lang and W. C. Eckelman, Appl. Radiat. Isot., 1994, 45(12), 1155 CrossRef CAS PubMed.
- S. Okarvi, Eur. J. Nucl. Med., 2001, 28(7), 929 CrossRef CAS.
- T. Poethko, M. Schottelius, G. Thumshirn, U. Hersel, M. Herz, G. Henriksen, H. Kessler, M. Schwaiger and H. Wester, J. Nucl. Med., 2004, 45, 892 CAS.
- T. Priem, C. Bouteiller, D. Camporese, X. Brune, J. Hardouin, A. Romieu and P.-Y. Renard, Org. Biomol. Chem., 2013, 11(3), 469 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2(10), 565 CrossRef.
- J. Marik and J. L. Sutcliffe, Tetrahedron Lett., 2006, 47(37), 6681–6684 CrossRef CAS.
- K. Kettenbach, H. Schieferstein and T. L. Ross, BioMed Res. Int., 2014, 1 CrossRef PubMed.
- H. Schieferstein and T. L. Ross, Eur. J. Org. Chem., 2014, 3546 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(43), 16793–16797 CrossRef CAS PubMed.
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS PubMed.
- S. Arumugam, J. Chin, R. Schirmacher, V. V. Popik and A. P. Kostikov, Bioorg. Med. Chem. Lett., 2011, 21(23), 6987 CrossRef CAS PubMed.
- A. Almutairi, R. Rossin, M. Shokeen, A. Hagooly, A. Ananth, B. Capoccia, S. Guillaudeu, D. Abendschein, C. J. Anderson, M. J. Welch and J. M. J. Fre, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(3), 685 CrossRef CAS PubMed.
- Z.-B. Li, Z. Wu, K. Chen, F. T. Chin and X. Chen, Bioconjugate Chem., 2007, 18(6), 1987 CrossRef CAS PubMed.
- O. Seitz and H. Kunz, J. Org. Chem., 1997, 62, 813 CrossRef CAS.
- S. A. Campos, J. D. Harling, A. H. Miah and I. E. D. Smith, Pat., WO 2014108452A1, 2014 Search PubMed.
- S. Keil, C. Claus, W. Dippold and H. Kunz, Angew. Chem., Int. Ed., 2001, 40(2), 366 CrossRef CAS.
- Y. Hirata, S. Hosoe, M. Maemoto, M. Sugawara, A. Yanagisawa and J. Ouchi, Pat., WO 2013129435A1, 2013 Search PubMed.
- M. Amblard, J. Fehrentz, J. Mertinez and G. Subra, Mol. Biotechnol., 2006, 33, 239 CrossRef CAS.
- C. P. Leamon and P. S. Low, Drug Discovery Today, 2001, 6446(2), 44 CrossRef.
- W. Siegrist, F. Solca, S. Stutz, L. Giuffre, S. Carrel, J. Girard and A. Eberle, Cancer Res., 1989, 49, 6352 CAS.
- F. Salazar-Onfray, M. López, A. Lundqvist, A. Aguirre, A. Escobar, A. Serrano, C. Korenblit, M. Petersson, V. Chhajlani, O. Larsson and R. Kiessling, Br. J. Cancer, 2002, 87(4), 414 CrossRef CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00508f |
| This journal is © The Royal Society of Chemistry 2016 |