
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
eCF309: a potent, selective and cell-permeable mTOR inhibitor†‡
Craig
Fraser
,
Neil O.
Carragher
and
Asier
Unciti-Broceta
*
Edinburgh Cancer Research UK Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Crewe Road South, Edinburgh EH4 2XR, UK. E-mail: Asier.Unciti-Broceta@igmm.ed.ac.uk
First published on 22nd December 2015
Abstract
Kinase inhibitors capable of blocking the phosphorylation of protein substrates with high selectivity are essential to probe and elucidate the etiological role of such molecules and their signalling pathways. By addressing these biochemical questions in disease relevant cell-based and in vivo models, strong pharmacological evidence can be generated towards validating or disproving a target hypothesis. Pharmacological studies can also provide fundamental information to identify appropriate biomarkers and rational drug combination strategies and thereby facilitate clinical translation. However, due to the high number of kinases encoded by the human genome (>500) and their highly conserved catalytic domains, the development of such an elite class of inhibitors—a.k.a. high-quality chemical probes—represents a major challenge. Through a ligand-based inhibitor design, focused library synthesis and phenotypic screening to prioritize compounds with potent cell activity, we recently identified a cell cycle inhibitor with micromolar potency that inhibits mTOR kinase activity. Following a rapid lead optimization campaign, we report the development of eCF309, an mTOR inhibitor displaying low nanomolar potency both in vitro and in cells and an excellent selectivity profile (S-score (35%) = 0.01 at 10 μM).
Introduction
The mechanistic or mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that operates as the catalytic subunit of two essential protein complexes called mTORC1 and mTORC2.1 These complexes play a central role in several signal transduction cascades, acting as sensors that integrate multiple extracellular and intracellular signals to coordinate cell metabolism, proliferation, survival and migration.1 Increased mTOR signalling is found in many types of cancers, metabolic disorders and neurodegenerative diseases.2,3 In cancer, such augmented phosphorylative activity contributes to cancer pathogenesis and chemoresistance mechanisms, and is typically induced via genetic dysregulation of different upstream modulators of mTORC1 and/or mTORC2 such as EGFR, PI3K, PTEN, AKT, RAS or RAF.4–7 Interestingly, mTOR itself is rarely mutated in cancer.8,9 Oncogenic activation of mTORC1/2 is identified by studying the phosphorylation status of downstream molecules such as 4E-BP1,4 AKT or P70-S6K and its substrate S6,10 which can assist in the selection of patients that are likely to respond to mTOR-targeted therapy.11 Clinical evidence of increased mTOR activity is found in approximately half of all human malignancies,12 which make mTOR inhibition a very attractive strategy to treat a wide range of cancers.To date three drugs, rapamycin (or sirolimus), temsirolimus and everolimus, have been clinically approved for medical use as inhibitors of mTOR signalling.12,13 These compounds belong to a family of chemically related macrolides known as rapalogs that inhibit the kinase activity of the mTORC1 complex through an allosteric interaction – involving the FKBP12 protein – that do not occur in the mTORC2 complex,14 thus being active only in the former. However, the mTORC2 complex is an important driver of cancer cell proliferation and survival.12,13 Inhibitors that target the catalytic domain of mTOR are then expected to induce superior anticancer activity by the concurrent inhibition of both complexes.12
Via an iterative process consisting of ligand-based design and phenotypic screening of focused chemical libraries,15 we have recently discovered a novel pyrazolopyrimidine (5) with high antiproliferative activity in cells (Scheme 1). Compound 5 displays submicromolar inhibition of mTOR (IC50 = 328 nM, in biochemical assays) and selectivity over other family of kinases. Herein we report the optimization of lead 5 into the potent, highly selective mTOR inhibitor eCF309.
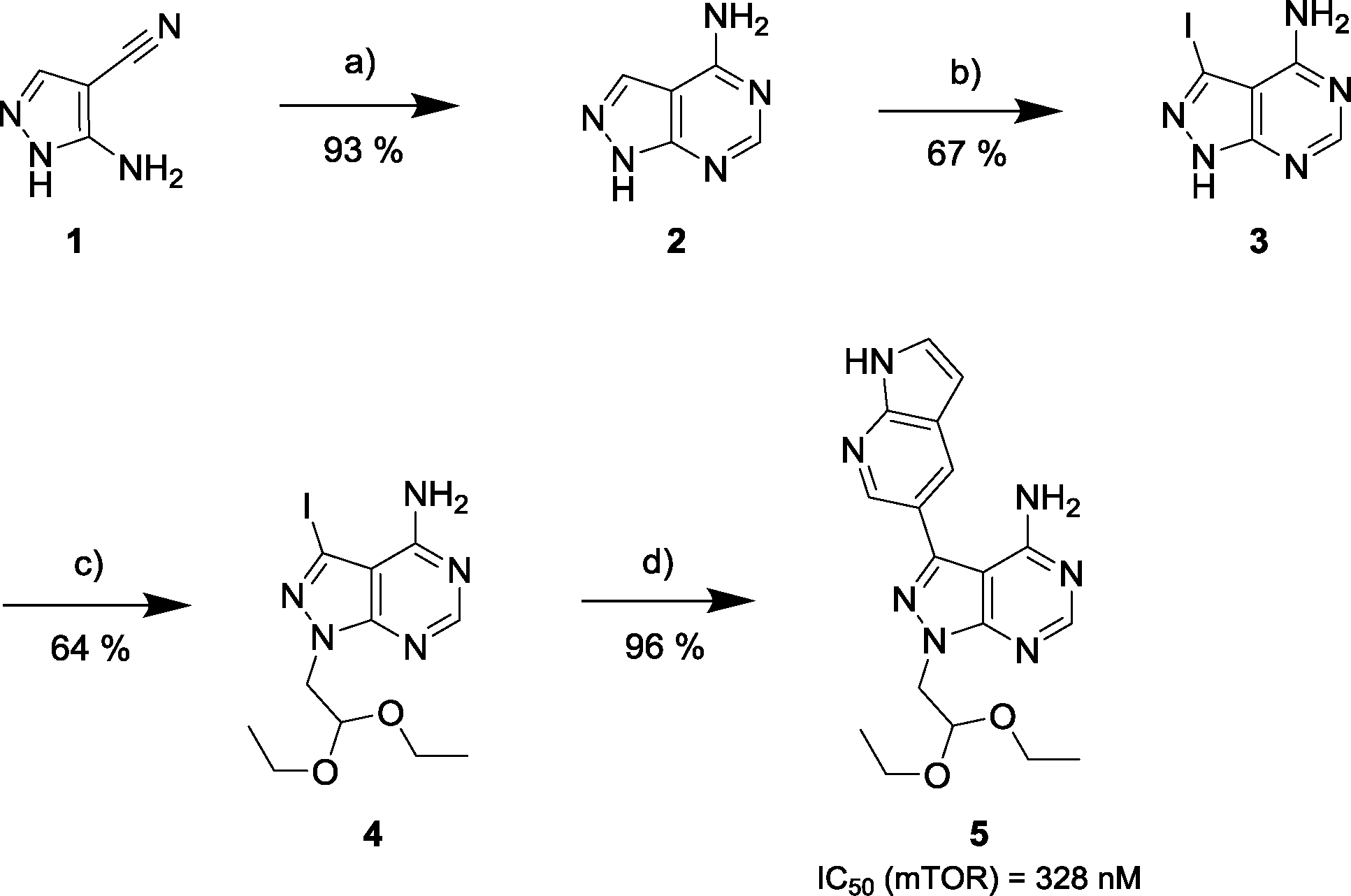 | ||
Scheme 1 Synthesis of mTOR inhibitor 5. a) Formamide, 180 °C, μw; b) NIS, 180 °C, DMF, μw; c) NaH, bromoacetaldehyde diethyl acetal, 150 °C, μw; d) 1H-pyrrolo[2,3-b]pyridine-5-boronic acid pinacol ester, K2CO3, Pd(OAc)2, PPh3, dioxane/water (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 120 °C, μw. :1), 120 °C, μw. | ||
Results and discussion
Design, synthesis and screening of novel derivatives of lead compound 5
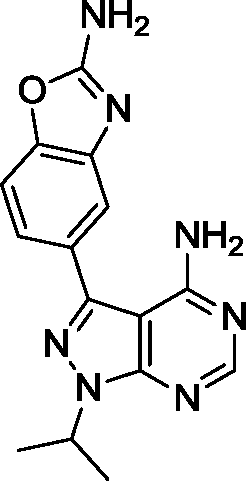
The 4-aminopyrazolo[3,4-d]pyrimidine scaffold has been widely explored for the development of bioactive small molecules and, particularly, for the design of ATP-competitive kinase inhibitors due to its resemblance to the structure of adenine.16 Medicinal chemistry campaigns on that scaffold have been mainly directed at the C3 position of the heterocycle, which have led to the discovery of inhibitors of different kinases, mostly tyrosine kinases e.g. SRC, ABL, RET, PDGFRs, IGF1R, VEGFRs or KIT,16–18 but also non-tyrosine kinases such as PI3Ks and mTOR.7,18Aiming to improve the potency of lead compound 5 against mTOR, we prepared a series of derivatives displaying different acetal groups at the N1 position and by introducing the C3 substituent of INK128 (now renamed as sapanisertib; see structure in Table 2), a 4-aminopyrazolo[3,4-d]pyrimidine analogue that is currently in clinical trials and reported to have excellent potency against mTOR (IC50 = 1 nM, in vitro).7 Following the synthesis route outlined in Scheme 2, new derivatives 10–14 and lead compound 5 were prepared. Briefly, the iodo-functionalized intermediates 4, 6 and 7 were synthesised by alkylation of common intermediate 3 with the corresponding alkyl bromide (Scheme 2). Compounds 10–14 were prepared by Suzuki–Miyaura cross-coupling with the corresponding arylboronic acid/boronate in moderate to excellent yields.
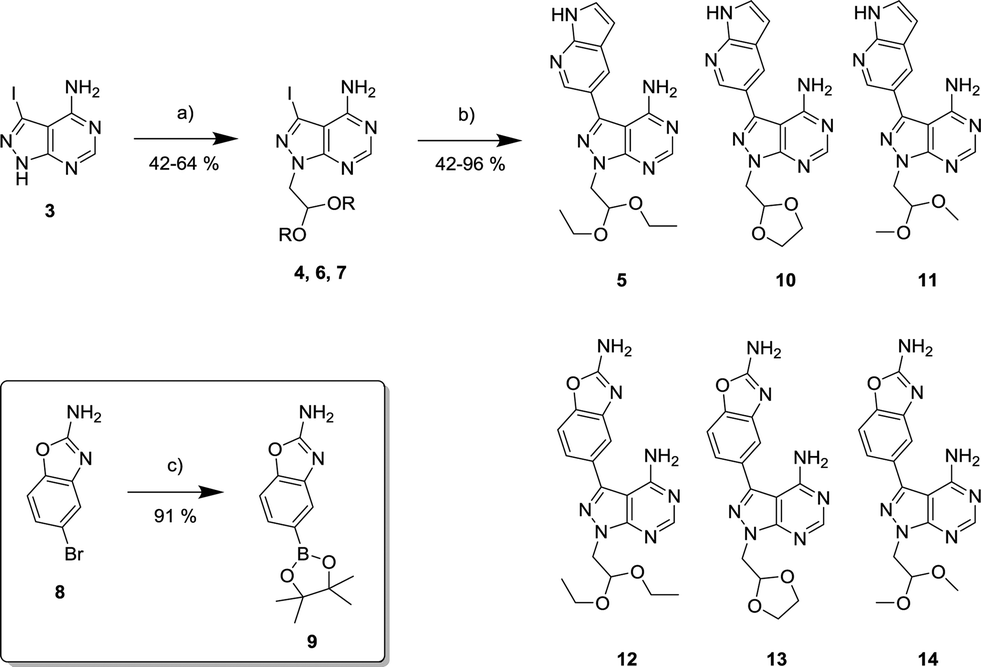 | ||
| Scheme 2 Synthesis of novel derivatives 10–14. a) Bromoacetaldehyde diethyl acetal, or 2-bromomethyl-1,3-dioxolane, or bromoacetaldehyde dimethyl acetal, NaH, DMF, 150 °C, μw. b) 1H-pyrrolo[2,3-b]pyridine-5-boronic acid pinacol ester or 2-amino-1,3-benzoxazole-5-boronic acid (9), K2CO3, Pd(OAc)2, PPh3, dioxane/water (10:1), 120 °C, μw. c) Bis(pinacolato)diboron, K2CO3, Pd(OAc)2, PPh3, dioxane/water (10:1), 120 °C, μw. | ||
Based on the potent antiproliferative activity displayed by lead compound 5 against HER2-positive MCF7 cells,15 the novel analogues were also tested against this breast cancer cell line. Besides compound 5, rapamycin, everolimus, INK128 and AZD2014 were tested as positive controls; all of which are potent mTOR inhibitors either clinically-approved or in clinical development.7,12,19 As previously discussed, rapamycin and everolimus are inhibitors of the mTORC1 complex, while both INK128 and AZD2014 are ATP-competitive mTOR inhibitors.
As shown in Fig. 1a, both compounds 10 and 11 showed slightly superior antiproliferative properties than lead 5. Remarkably, compounds 12–14 – all of which possess the 2-amino-1,3-benzoxazole moiety found at the C3 position of INK128 – led to an activity increment of over two orders of magnitude. 12, 13 and 14 exhibited EC50 values upon MCF7 breast cancer cell viability of 8.4, 7.6 and 6.7 nM, respectively, and superior potency than AZD2014, a drug candidate currently under phase I and II clinical development across several cancer indications.12 It is important to note that, although rapamycin and everolimus exhibited very high antiproliferative properties (EC50 < 1 nM), their activity plateaus at approximately 30% viability and thus did not result in the complete elimination of MCF7 cell viability, even at the highest dose (Fig. 1b). This is a well-established feature of mTORC1 inhibitors attributed to compensatory feedback mechanisms due to the lack of inhibition of the mTORC2 complex.12
 | ||
| Fig. 1 (a) EC50 values calculated after treating MCF7 cells with compounds 5, 12–14, rapamycin, everolimus, INK128 and AZD2014 for 5 d (dose range: 0.3–1000 nM). Cell viability was determined using the PrestoBlue® reagent. Error bars: ±SD from n = 3. (b) Dose response curves of MCF7 cell viability under treatment with compound 12, rapamycin and everolimus. Error bars: ±SD from n = 3. | ||
Kinase profiling of inhibitors 12–14
To determine the selectivity profile of the novel compounds, the most potent derivatives (12–14) were screened against a selected panel of recombinant kinases known to be inhibited by 4-aminopyrazolo[3,4-d]pyrimidine derivatives. Changes on kinase activity relative to DMSO were determined by measuring 33P incorporation on its substrate (poly[Glu,Tyr]4:1) by Reaction Biology Corp., USA. Compounds were tested at 10 different doses with 3-fold serial dilution starting from 10 μM and calculated IC50 values are shown in Table 1. Notably, the novel compounds displayed high inhibition of mTOR, several fold superior to that of the lead compound 5, and good-to-excellent selectivity over other kinases, including PI3Ks, a subfamily of kinases known to be targeted by most mTOR kinase inhibitors.12 While derivative 12 (a.k.a. eCF309) exhibited slightly lower antiproliferative potency against MCF7 cells than 13 and 14, it induced the strongest inhibition of mTOR kinase activity (IC50 = 15 nM) and displayed higher selectivity over PI3Ks. These results indicate that even small modifications at the acetal group of the N1 position of the ring can result in significant variations on the binding properties of the inhibitor.
| Kinase\hit |
5
|
12
|
13
|
14
|
|---|---|---|---|---|
| ABL | 1210 | >105 | 8630 | 9920 |
| BLK | >105 | >105 | >105 | >105 |
| FYN | 1230 | >105 | >105 | >105 |
| KIT | >105 | >105 | >105 | >105 |
| mTOR | 328 | 15 | 59 | 25 |
| PDGFRα | >105 | >105 | >105 | >105 |
| RET | 598 | >105 | >105 | >105 |
| SRC | 2450 | >105 | >105 | >105 |
| YES | 566 | >105 | >105 | >105 |
| PI3Kα | ND | 981 | 115 | 300 |
| PI3Kβ | ND | >105 | 3850 | 5600 |
| PI3Kγ | ND | 1340 | 302 | 99 |
| PI3Kδ | ND | 1840 | 271 | 505 |
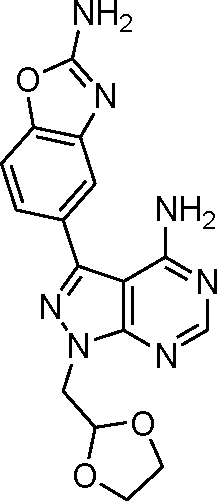

Encouraged by the high selectivity displayed by 12 against several kinases that are typically inhibited by 4-aminopyrazolo[3,4-d]pyrimidine derivatives, we extended the characterization of its kinome profile by performing a single dose inhibition study against 375 wild type and mutant kinases (Reaction Biology Corp., USA). Compound 12 was tested at 10 μM in duplicate and levels of enzymatic inhibition compared to DMSO (= 0% inhibition, see listed values in the ESI‡). Averaged results were plotted with a 65% cut-off value in a representation of the human kinome phylogenetic tree using TREEspot™ from DiscoveRx (Fig. 2). Kinome profiling identified only 5 hits, with an S-score = 0.01. mTOR was the only protein inhibited at very high levels, with an inhibition of its activity superior to 99%. In agreement with previous assays, two lipid kinases PI3Kγ and PI3Kα (E545K) were found among the hits with 85% and 65% inhibition, respectively. The screening only identified two new hits: DNA-PK, with 90%, and DDR1, with 77% inhibition. DNA-PK is a serine/threonine protein kinase involved in the repair of double-strand DNA and its inhibition has been proposed to be beneficial to cancer therapy.20 Similarly, DDR1 (discoidin domain receptor family member 1) is overregulated in several cancers and has also been nominated as an oncology target.21 Although several kinase inhibitors have shown cross-reactivity with mTOR, PI3K and DNA-PK,12 the inhibition of DDR1 is an unusual feature. Overall, the study shows that 12 (eCF309) is a potent mTOR inhibitor with remarkably low off-target activities. According to the literature, the selectivity profile of 12 is as good as or even better than that of any other selective mTOR inhibitor reported to date.7,12,18,22,23
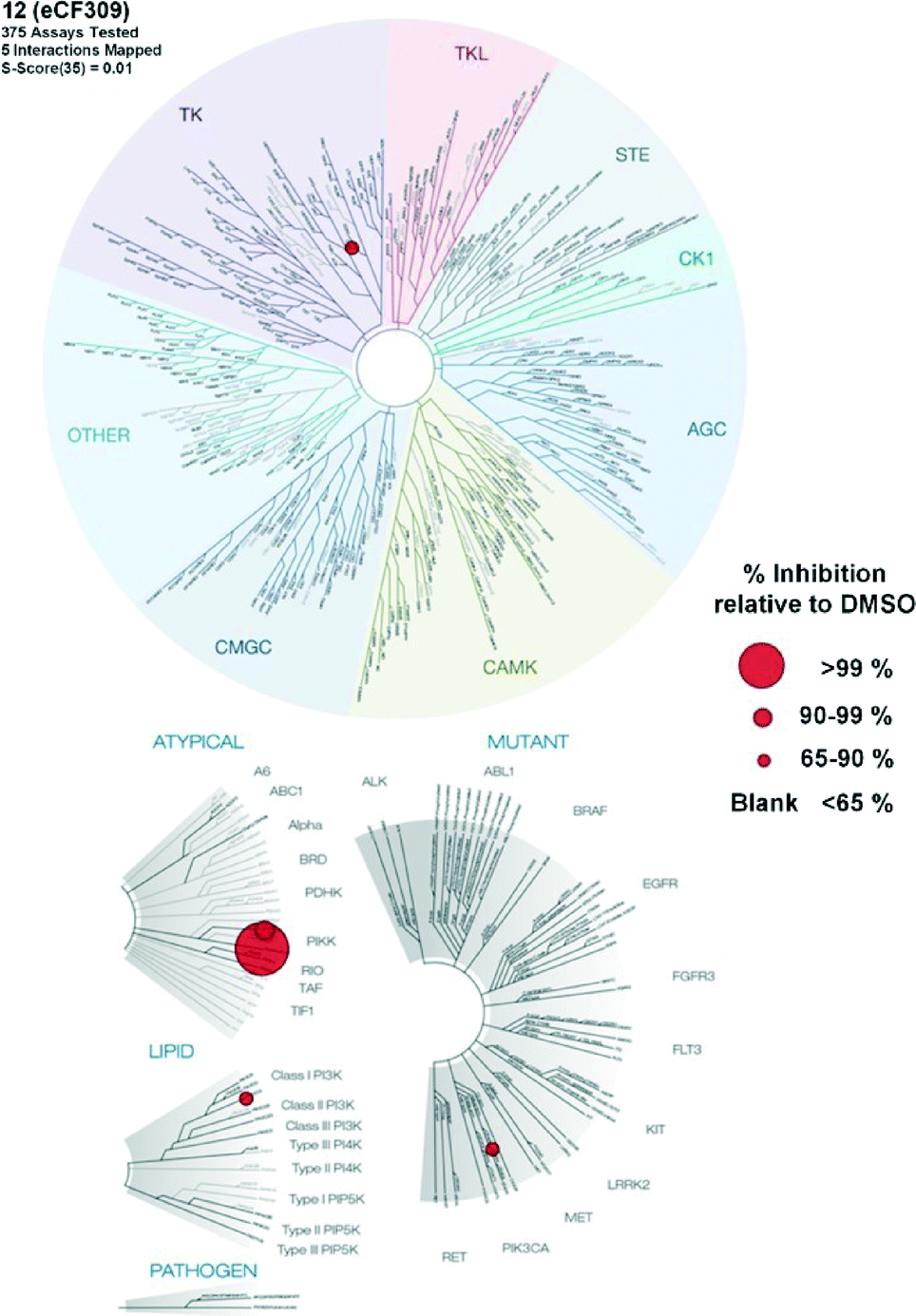 | ||
| Fig. 2 Map of the human kinome inhibited by compound 12 at 10 μM. Red circles denote inhibition of activity above 65%. The 5 interactions mapped are DDR1, DNA-PK, mTOR, PI3Kα (E545K) and PI3Kγ. S-score (35%) = 0.01. | ||
Western blot analysis
To verify that compound 12 inhibits both mTORC1 and mTORC2 signalling in cells by inhibition of the mTOR catalytic domain, we assessed the activation state (= phosphorylation) of downstream targets of both complexes by Western blot. Phosphorylation levels of the p70 ribosomal S6 kinase 1 (P70S6K) and its substrate S6 were analysed as substrates of mTORC1, and phospho-AKTY473 as an mTORC2 substrate. MCF7 cells were seeded in a 6-well plate and grown until 80% confluence. The cells were serum starved (0.1% FBS) for 24 h before incubation with the inhibitor (compound 12 and INK128, at varying concentrations) or DMSO (0.1% v/v) for 30 min, followed by 1 h of serum stimulation (10% FBS). The cells were then lysed and analysed by protein immunoblot. As shown in Fig. 3, compound 12 mediated a dose-dependent reduction of the pP70S6K, pS6 and pAKT levels in MCF7 cells, achieving almost complete inhibition of the phosphorylation of these targets at 30 nM. The potency of compound 12 was about an order of magnitude lower than that of INK128. These results are in good correlation with the IC50 values calculated in the biochemical assays and the phenotypic activities exhibited by both compounds.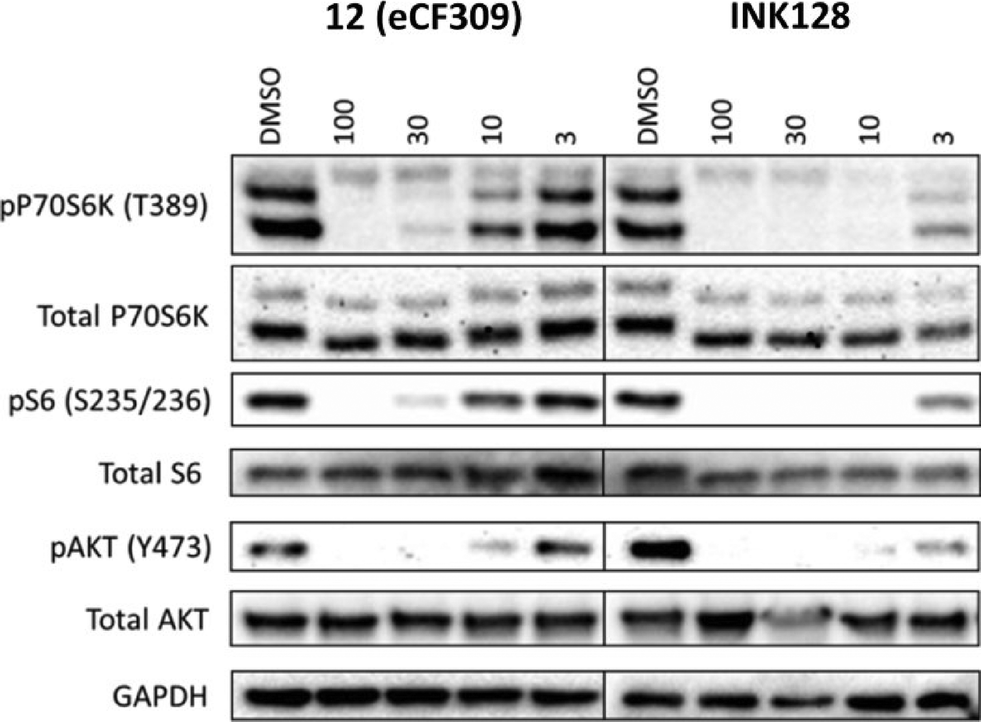 | ||
| Fig. 3 Western blot of MCF7 cell lysates after treatment with compound with 12 or INK128 at a dose range (3–100 nM). DMSO = negative control. | ||
Synthesis and biological screening of derivatives 17–19
Due to its potency and selectivity against mTOR, compound 12 was the most interesting derivative of the series. Nonetheless, the high activity displayed by compound 13 against both PI3Ks and mTOR kinases prompted us to explore further structure activity relationships. Maintaining the constrained conformation provided by the 1,3-dioxolane at the N1 position of compound 13, we studied the influence of removing one or both oxygen atoms from the saturated 5-member ring. Derivatives 17 and 18 were prepared as described in Scheme 3. These novel compounds contained either a methylcyclopentyl or 2-methyltetrahydrofuran group, respectively, at the N1 position of the pyrazolopyrimidine ring. Additionally, the aldehyde-containing compound 19 was synthesized by acetal deprotection of compound 12 in acidic conditions. Derivative 19 was prepared to evaluate whether this potential metabolite of inhibitor 12 could still mediate bioactivity in cells.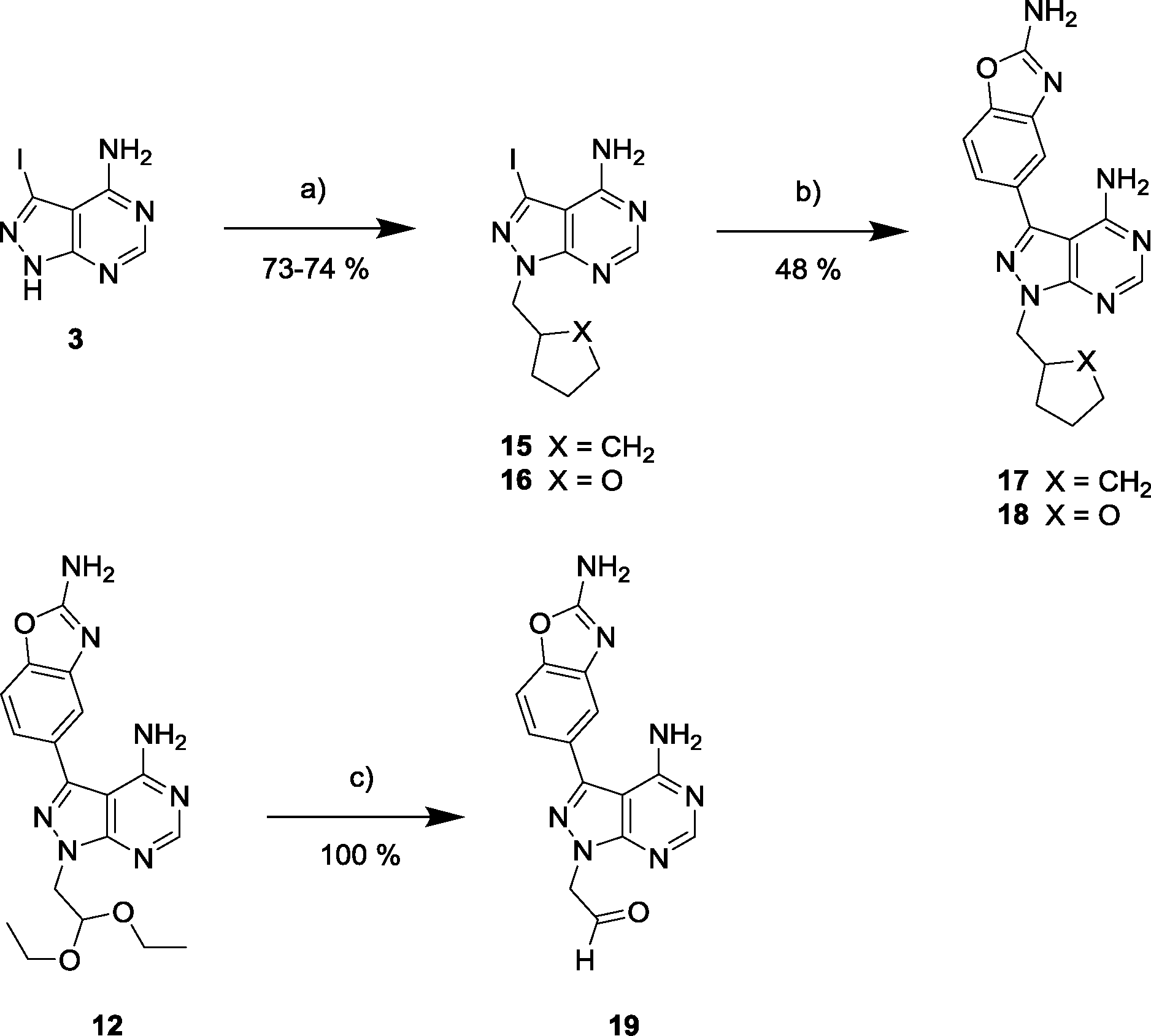 | ||
| Scheme 3 a) (Iodomethyl)cyclopentane or 2-(bromomethyl) tetrahydrofuran, NaH, DMF, 150 °C, μw; b) 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2-amine, K2CO3, Pd(OAc)2, PPh3, 1,4-dioxane/water (10:1), 120 °C, μw; c) TFA/water (1:1), 100 °C, μw. | ||
The antiproliferative properties of the new derivatives were screened in a panel of 3 cancer cell lines: breast cancer MCF7 cells (as before), MDA-MB-231 (triple negative breast cancer) and PC3 (prostate cancer). Cell viability was measured using the PrestoBlue® reagent and analysed by spectrofluorometry. EC50 values were calculated for compounds 12, 17–19 and INK128 using a 10-point half-log dose response study (0.3–1000 nM). As shown in Fig. 4, compounds 12, 17 and 18 exhibited highly potent antiproliferative activity against all three cell types. Notably, compound 17 was found to be the most potent of all the compounds tested, with low nanomolar EC50 values and superior activity than INK128.
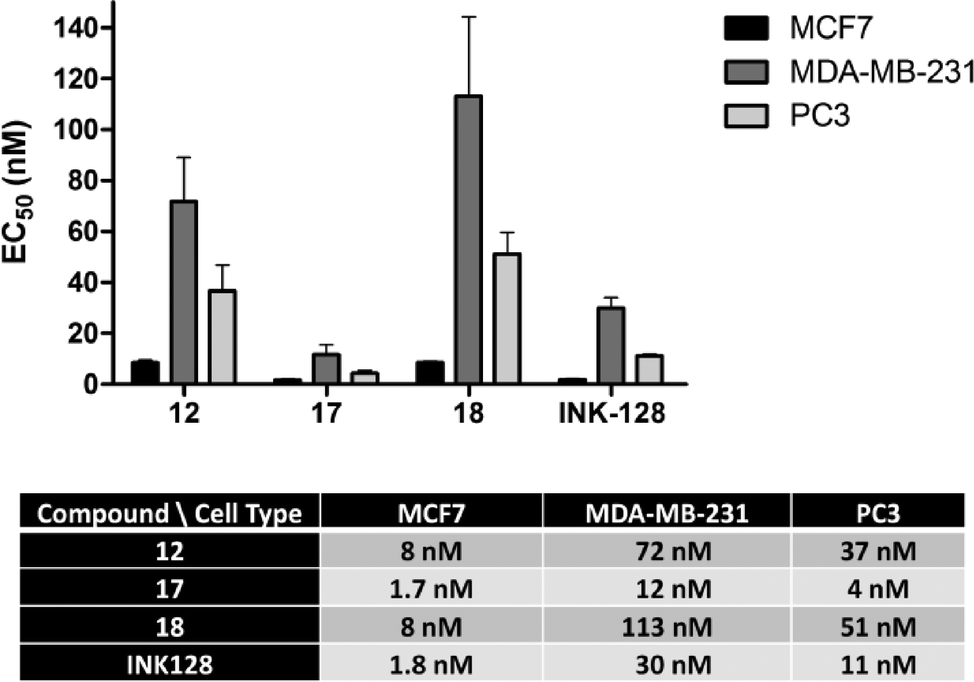 | ||
| Fig. 4 EC50 values calculated after treating MCF7, MDA-MB-231 and PC3 cells with compounds 12, 17, 18 and INK128 (positive control) for 5 d (dose range: 0.3–1000 nM). Cell viability was determined using the PrestoBlue® reagent. Error bars: ±SD from n = 3. | ||
The same trend in cell type sensitivity to treatment was observed for the four derivatives (MCF7 > PC3 > MDA-MB-231), thus suggesting that compounds 12, 17, 18 and INK128 target the same signalling pathway (PI3K-AKT-mTOR). These results also provides information about the level of dependence that each of these cell lines have on that pathway for driving cell growth. In contrast, aldehyde derivative 19 showed no activity at the concentrations tested (ESI‡), indicating that the acetal group exclusive to compound 12 is necessary for the observed phenotypic effect and thus stable in the cellular environment.
Phenotypic assessment of the antiproliferative properties of derivatives 12, 17 and 18
In cancers, upregulated mTOR signalling contributes to tumour cell proliferation through induction of cell cycle progression.1–3 Indeed, previous studies in MCF7 cells by our group have shown that the antiproliferative activity of lead compound 5 was mediated by inhibition of the cell cycle.15 Therefore, it was important to assess whether the effect exerted by the novel compounds 12, 17 and 18 was mediated by cell cycle arrest. MCF7, MDA-MB-231 and PC3 cells were treated with the 3 compounds and INK128 for 24 h using DMSO (0.1%) as negative control. Afterwards, cells were fixed with 4% paraformaldehyde and stained for DNA (Hoechst 33342), phospho-histone H3 (anti-pHH3 rabbit antibody with AlexaFluor®-594 goat anti-rabbit IgG) and cyclin B1 (anti-cyclin B1 antibody with AlexaFluor®-488 donkey anti-mouse IgG). Cells were imaged using ImageXpress High Content System (Molecular Devices, USA) and phenotypic response quantified automatically using MetaXpress image analysis software. Cells were subsequently classified according to their DNA content into G0/G1, S or G2 cell cycle phases. Mitotic cells were classified according to their cyclin B1 or phospho-histone H3 nuclear expression levels. As shown in Fig. 5a, all the compounds, in all the cell types tested, caused G0/G1 cell cycle arrest, as identified by the increase of the black population in the charts. A similar trend to that seen in the viability assays was found both in terms of compound potencies and cell selectivity, with MCF7 being the most responsive cell type and compound 17 being the most potent inhibitor. Notably, the cell cycle inhibition activity of all the compounds were in good correlation with the EC50 values calculated from the cell viability assays. Analysis of the increment of the G0/G1 cell population (Fig. 5b) showed that compounds 12 and 18 led to similar phenotypic effect in all the cell lines. The G0/G1 cell cycle arrest mediated by INK128 is in agreement with the published literature.7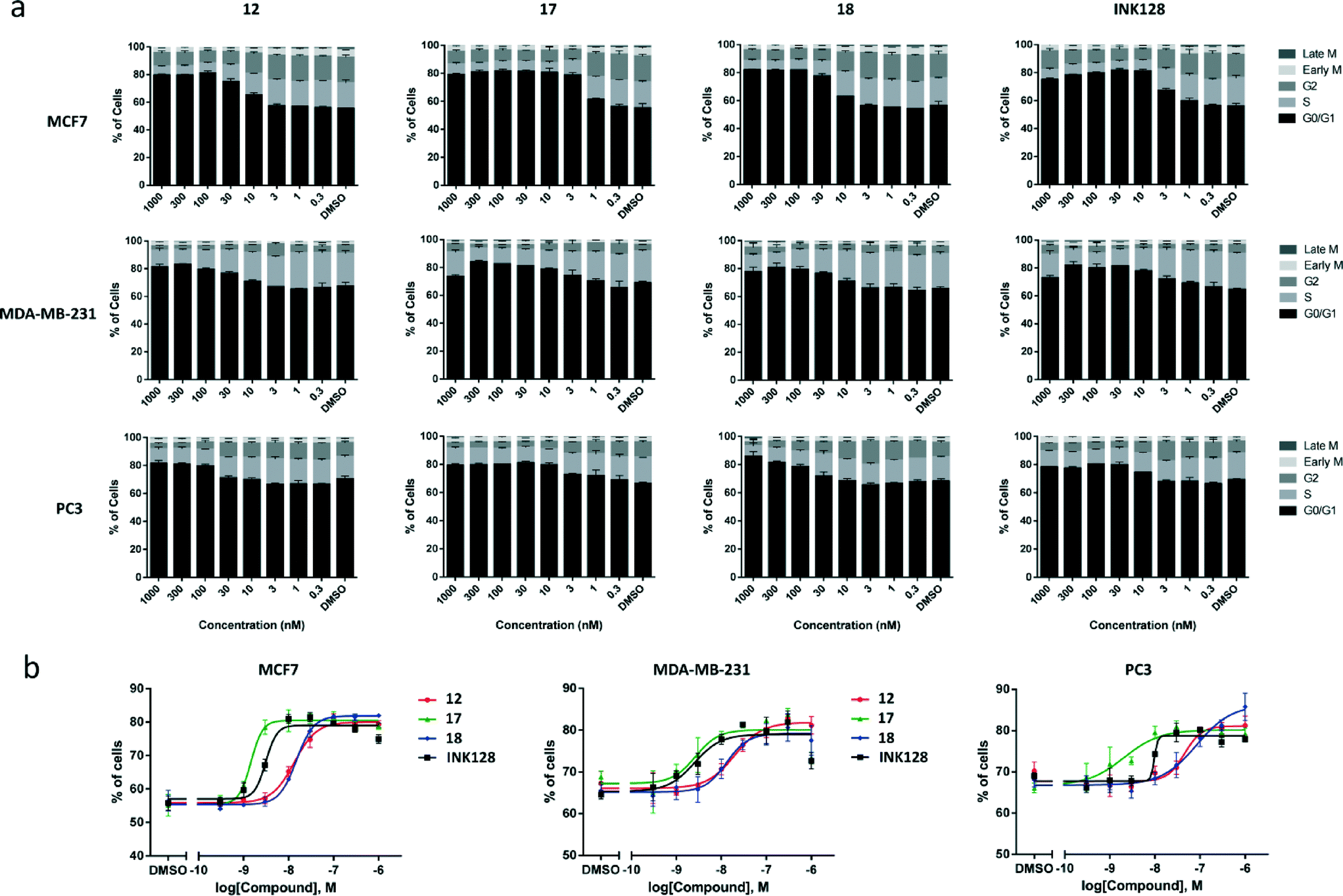 | ||
| Fig. 5 (a) Relative distribution of MCF7, MDA-MB-231 and PC3 cell populations by cell cycle phase under treatment with compounds 12, 17, 18 and INK128 (dose range 0.3–1000 nM). G0/G1 phase is in black and other stages in shades of grey. Error bars: ±SD from n = 3. (b) Dose response curves for the accumulation of cell population in the G0/G1 stage after treatment with 12, 17, 18 and INK128. Error bars: ±SD from n = 3. | ||
Kinase profiling of inhibitors 17 and 18
To assess the targets likely involved in the bioactivity of 17 and 18, their kinase inhibition profile was studied on a selected panel of recombinant kinases (Reaction Biology Corp., USA). In accordance with the kinome profiling of compound 12, kinase inhibition was evaluated for DDR1, DNA-PK, PI3Ks and mTOR. Table 2 shows the IC50 values found for compounds 12, 17 and 18, in comparison with the values reported for INK-128.7 From the novel derivatives, 12 (eCF309) was found to be the most selective mTOR inhibitor. After mTOR (with IC50 = 15 nM), the most inhibited kinase was DNA-PK with an IC50 of 320 nM, thus displaying >20-fold difference in activity. A gap greater than 65-fold was observed for the rest of kinases, remarkable properties that make compound 12 (eCF309) one of the mTOR inhibitors with higher selectivity ever reported.12| Kinase\hit |
12
|
17
|
18
|
INK128
|
|---|---|---|---|---|
| ND = Not determined | ||||
| DDR1 | 2110 | 4.7 | 137 | ND |
| DNA-PK | 320 | 3 | 48 | ND |
| mTOR | 15 | 1.8 | 5 | 17 |
| PI3Kα | 981 | 3.5 | 44 | 2197 |
| PI3Kβ | >105 | 89 | 1120 | 52907 |
| PI3Kγ | 1340 | 5 | 77 | 2307 |
| PI3Kδ | 1840 | 7.3 | 149 | 2217 |
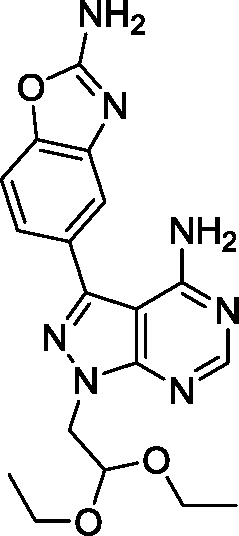
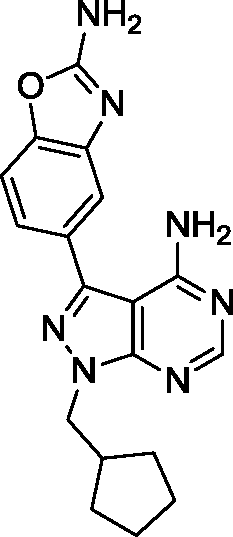
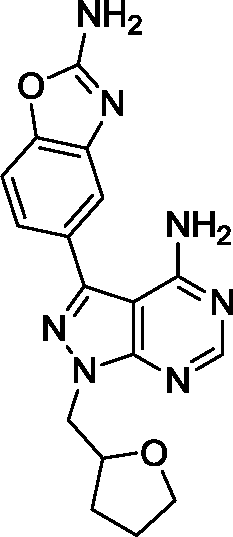
Surprisingly, compound 18 showed 3-fold higher potency against mTOR than derivative 12 and exhibited cross-reactivity for the rest of the kinases tested. The similar patterns shown by compound 12 and 18 in the cell viability and cell cycle inhibition assays may be therefore related to a superior capacity of compound 12 to cross cell membranes. In agreement with the cell assays, derivative 17 (a.k.a. eCF324) showed a substantial increase in potency against mTOR relative to 12, with an IC50 value of 1.8 nM, similar to that reported for compound INK-128.7 Interestingly, compound 17 was also highly potent (IC50 = 3–7 nM) against all the kinases tested except PI3Kβ. While this dual PI3K/mTOR inhibitory capability is not a novel feature,12 it is of relevance because it has been associated with increased clinical efficacy in cancers driven by the PI3K-AKT-mTOR pathway.18,23
Analysis of the kinase inhibition profile of 12, 17 and 18 indicates that the presence of oxygen atoms in the alkyl group at the position N1 of the ring results in increased mTOR selectivity. This, however, contrasts with the high selectivity reported for INK128 over PI3Ks,7,24 a compound that displays a small oxygen-free isopropyl group at N1. Intrigued by this, we tested INK128 for internal ranking purposes. Since the re-evaluation of the properties of INK128 is out of the scope of this work, these results are not shown. However, we feel the responsibility to implore scientists not to ignore the off-target effects mediated by small molecule inhibitors (e.g. INK128 over other related kinases such as DNA-PK and PI3Kα) to ensure the rigor of their biological conclusions upon target biology. This is particularly true for inhibitors targeting highly conserved protein families.
Conclusions
The development of potent, selective and cell-permeable small molecules that mediate dose-dependent inhibition of a target protein in its natural environment is essential to elucidate the role of such prospective targets in normal and pathological settings. In clinical treatment, highly-selective inhibitors are also important for the development of safer drug combination therapies.25 Although many chemical probes are currently available, customized chemical design of target selectivity combined with careful characterization of their biochemical and phenotypic properties is uncommon, even if that is essential to confirm the accuracy of any biological conclusions.26 This is particularly true in cancer, where multiple etiological factors can be typically involved in carcinogenesis.27In the search to optimize the cell potency and selectivity against mTOR of a lead compound identified in previous studies,15 we prepared and tested a series of novel pyrazolopyrimidines following a straightforward 5-step synthesis route. Phenotypic screening facilitated the identification of several compounds exhibiting low nanomolar antiproliferative activity against breast and prostate cancer cells via cell cycle arrest. Among the novel compounds, the most potent derivative was 17 (eCF324), which displayed single-digit nM potency against cancer cell proliferation via G0/1 phase cell cycle arrest and high in vitro inhibition of several kinases including mTOR, PI3Ks, DNA-PK and DDR1. Remarkably, 17 (eCF324) demonstrated superior potency in cells than INK128, an mTOR kinase inhibitor currently in clinical development.24
From these series, the most interesting inhibitor for biological studies was compound 12 (eCF309). Kinase profiling and Western blotting demonstrated that eCF309 is a potent inhibitor of mTOR signalling (IC50 = 10–15 nM, both in vitro and in cells) with very high selectivity (S-score (35%) = 0.01 at 10 μM). Its remarkable potency in cells together with its relatively simple synthesis, make compound eCF309 a highly valuable probe for chemical biology and biomedicine. Notably, its aldehyde derivative 19 exhibited no activity, proving that the acetal group of compound eCF309 is necessary for the compound's bioactivity and stable in the cytoplasm. Future studies will serve to evaluate whether this series of mTOR inhibitors are also suitable for cancer treatment.
Acknowledgements
We are grateful to the Wellcome Trust ISSF for financial support. C. F. is grateful to MRC for a DTP scholarship. N. O. C. and A. U. B. thanks RCUK and IGMM, respectively, for an Academic Fellowship.References
- M. Laplante and D. M. Sabatini, J. Cell Sci., 2009, 122, 3589–3594 CrossRef CAS PubMed
.
- M. Shimobayashi and M. N. Hall, Nat. Rev. Mol. Cell Biol., 2009, 10, 307–318 CrossRef PubMed
- M. Laplante and D. M. Sabatini, Cell, 2012, 149, 274–293 CrossRef CAS PubMed
- J. D. Richter and N. Sonenberg, Nature, 2005, 433, 477–480 CrossRef CAS PubMed
- N. Chapuis, J. Tamburini, A. S. Green, L. Willems, V. Bardet, S. Park, C. Lacombe, P. Mayeux and D. Bouscary, Leukemia, 2010, 24, 1686–1699 CrossRef CAS PubMed
- Y. J. Zhang, Y. J. Bao, Q. Dai, W. Y. Yang, P. Cheng, L. M. Zhu, B. J. Wang and F. H. Jiang, Ann. Surg. Oncol., 2011, 18, 580–588 CrossRef PubMed
- A. C. Hsieh, Y. Liu, M. P. Edlind, N. T. Ingolia, M. R. Janes, A. Sher, E. Y. Shi, C. R. Stumpf, C. Christensen, M. J. Bonham, S. Wang, P. Ren, M. Martin, K. Jessen, M. E. Feldman, J. S. Weissman, K. M. Shokat, C. Rommel and D. Ruggero, Nature, 2012, 485, 55–61 CrossRef CAS PubMed
- T. Sato, A. Nakashima, L. Guo, K. Coffman and F. Tamano, Oncogene, 2010, 29, 2746–2752 CrossRef CAS PubMed
- E. Dazert and M. N. Hall, Curr. Opin. Cell Biol., 2011, 23, 744–755 CrossRef CAS PubMed
- S. Menon and B. D. Manning, Oncogene, 2008, 27, S43–S51 CrossRef CAS PubMed
- T. O'Reilly and P. M. McSheehy, Transl. Oncol., 2010, 3, 65–79 CrossRef
- Y. Zhang, Y. Duan and S. Zheng, Drug Discovery Today, 2011, 16, 325–331 CrossRef CAS PubMed
- L. M. Ballou and R. Z. Lin, J. Chem. Biol., 2008, 1, 27–36 CrossRef PubMed
- L. A. Banaszynski, C. W. Liu and T. J. Wandless, J. Am. Chem. Soc., 2005, 127, 4715–4721 CrossRef CAS PubMed
-
C. Fraser, PhD Thesis, University of Edinburgh, 2015 Search PubMed
- M. Chauhan and R. Kumar, Bioorg. Med. Chem., 2013, 21, 5657–5668 CrossRef CAS PubMed
- P. Dinér, J. P. Alao, J. Söderlund, P. Sunnerhagen and M. Grøtli, J. Med. Chem., 2012, 55, 4872–4876 CrossRef PubMed
- B. Apsel, J. A. Blair, B. Gonzalez, T. M. Nazif, M. E. Feldman, B. Aizenstein, R. Hoffman, R. L. Williams, K. M. Shokat and Z. A. Knight, Nat. Chem. Biol., 2008, 4, 691–699 CrossRef CAS PubMed
- K. G. Pike, K. Malagu, M. G. Hummersone, K. A. Menear, H. M. E. Duggan, S. Gomez, N. M. B. Martin, S. L. Ruston and M. Pass, Bioorg. Med. Chem. Lett., 2013, 23, 1212–1216 CrossRef CAS PubMed
- F. Hsu, S. Zhang and B. P. C. Chen, Transl. Cancer Res., 2012, 1, 22–34 Search PubMed
- L. Miao, S. Zhu, Y. Wang, J. Li, J. Ding, J. Dai, H. Cai, D. Zhang and Y. Song, Med. Oncol., 2013, 30, 626 CrossRef PubMed
- A. A. Estrada, D. G. Shore, E. Blackwood, Y. H. Chen, G. Deshmukh, X. Ding, A. G. Dipasquale, J. A. Epler, L. S. Friedman, M. F. Koehler, L. Liu, S. Malek, J. Nonomiya, D. F. Ortwine, Z. Pei, S. Sideris, F. St-Jean, L. Trinh, T. Truong and J. P. Lyssikatos, J. Med. Chem., 2013, 56, 3090–3101 CrossRef CAS PubMed
- F. Chiarini, C. Evangelisti, J. A. McCubrey and A. M. Martelli, Trends Pharmacol. Sci., 2015, 36, 124–135 CrossRef CAS PubMed
- M. R. Janes, C. Vu, S. Mallya, M. P. Shieh, J. J. Limon, L. S. Li, K. A. Jessen, M. B. Martin, P. Ren, M. B. Lilly, L. S. Sender, Y. Liu, C. Rommel and D. A. Fruman, Leukemia, 2013, 27, 586–594 CrossRef CAS PubMed
- N. Carragher, A. Unciti-Broceta and D. Cameron, Future Med. Chem., 2012, 4, 87 CrossRef CAS PubMed
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed
- S. H. Myers, V. G. Brunton and A. Unciti-Broceta, J. Med. Chem., 2016 DOI:10.1021/acs.jmedchem.5b01273
Footnotes |
| † The authors declare no competing interest. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization of compounds 2–19, biological methods and supplementary Fig. S1 and Table S1. See DOI: 10.1039/c5md00493d |
| This journal is © The Royal Society of Chemistry 2016 |