
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN,N′-Disubstituted thiourea and urea derivatives: design, synthesis, docking studies and biological evaluation against nitric oxide synthase†
Mariem
Chayah
a,
M. Encarnación
Camacho
*a,
M. Dora
Carrión
*a,
Miguel A.
Gallo
a,
Miguel
Romero
b and
Juan
Duarte
b
aDepartamento de Química Farmacéutica y Orgánica, Facultad de Farmacia, Universidad de Granada, Spain. E-mail: ecamacho@ugr.es; dcarrion@ugr.es; Tel: +34 958 243844 Tel: +34 958 240728
bDepartamento de Farmacología, Facultad de Farmacia, Universidad de Granada, Spain
First published on 4th January 2016
Abstract
The synthesis and biological evaluation of new types of N,N′-disubstituted thiourea and urea derivatives as inhibitors of both neuronal nitric oxide synthase (nNOS) and inducible nitric oxide synthase (iNOS) are described. These compounds have been designed by reduction of the carbonyl group in the thiourea and urea kynurenamine derivatives 3 previously synthesized by our research group. The synthetic route performed to this new family also allows us to obtain the molecules 3 with less synthetic steps and higher global yield. Regarding the biological results, in general, the new derivatives 4a–q inhibit the neuronal NOS isoform better than the inducible one. Furthermore, thioureas exhibit higher inhibition than ureas for both isoenzymes. Among all the tested compounds, 4g shows significant nNOS (80.6%) and iNOS (76.6%) inhibition values without inhibiting eNOS. This molecule could be an interesting starting point for the design of new inhibitors with application in neurological disorders where both isoenzymes are implicated such as Parkinson's disease.
Introduction
Nitric oxide synthase (NOS) is a family of isoenzymes that convert L-arginine to L-citrulline with nitric oxide (NO) release. There are three human NOS isoforms: endothelial NOS (eNOS) which regulates blood pressure and flow, inducible NOS (iNOS) which is involved in immune response, and neuronal NOS (nNOS) which is essential for neurotransmission.1 Nonetheless, several studies demonstrated the implication of NOS in many neurodegenerative,2–7 chronic inflammatory8,9 and cardiovascular10,11 diseases. Indeed, NO overproduction, by nNOS or iNOS, leads to cellular and tissue damage through nitrosative and oxidative stress.12–16 The underproduction of NO by eNOS affects the vascular tone and blood flow controls and causes hypertension. Therefore, the inhibition of nNOS and iNOS but not eNOS is a viable therapeutic strategy to treat and prevent disorders and pathologies as previously mentioned.A comparison of the NOS structures reveals a huge similarity especially in their substrate binding sites.17 This fact represents a real challenge in designing selective NOS inhibitors.
In previous efforts to find selective and potent i/nNOS inhibitors, our research group has synthesized and published several families of compounds. Fig. 1 shows some of them with analogous structures: the kynurenine 1,18 kynurenamine 2,19 and urea and thiourea kynurenamine 3 (ref. 20) derivatives. These last ones have mostly better inhibition results versus iNOS. Docking and molecular dynamic studies demonstrated the urea group implication especially in the selectivity process. In addition, both the aromatic ring and the amine group interact with the enzyme through π–cation interaction and hydrogen bonds, respectively. However, the carbonyl group does not show a clear role. Therefore, we decided to act at this point, changing this hydrogen bond acceptor residue to another donor such as hydroxyl.
 | ||
| Fig. 1 Some kynurenamine derivatives as NOS inhibitors synthesized by our research group. | ||
This way, herein we present a new series of compounds 4a–q where the carbonyl group, present in the three families of the above derivatives, has been replaced by a hydroxyl one, in order to increase the interaction with the enzyme and improve the inhibition. A new synthetic pathway was developed to obtain this series of molecules. Subsequent biological assays and docking studies were performed in this new family of compounds to evaluate the results against NOSs' isoforms.
In addition, compounds 2a–l previously published and evaluated as nNOS inhibitors19 were screened against iNOS, and the inhibition data are presented and discussed in this work. N,N′-Disubstituted thioureas and ureas 4a–q were designed from the kynurenamines with general skeleton 2, introducing two structural modifications: replacement of the terminal acyl group by a thiourea or urea one, isosteric to the final guanidine moiety of the NOS substrate (L-Arg) and substitution of the carbonyl group with a hydroxyl residue.
Results and discussion
Chemistry
The general synthetic pathway of all the final compounds 4a–q is represented in Scheme 1. 5-Methoxy-2-nitrobenzaldehyde 5b (synthesized from 5-hydroxy-2-nitrobenzaldehyde by reaction with MeI in the presence of K2CO3/THF),21 the commercially available 2-nitrobenzaldehyde 5a and 5-chloro-2-nitrobenzaldehyde 5c were transformed into the β-hydroxynitriles 6a–c by treatment with BuLi and acetonitrile in dry THF22 (70–90% yield). The nitrile group of these last intermediates was reduced selectively with borane in THF23 to give the 3-amino-1-(2-nitrophenyl)propan-1-ol derivatives 7a–c. Nucleophilic addition of either alkyl or phenyl isothiocyanate or isocyanate in situ, using the microwave (MW) technique, gave the intermediates 8a–q in good yields (70–85%). The application of the MW technique shortened the reaction time from 18 h (ref. 20 and 24) to 20 min, with respect to the previously described synthesis. Finally, reduction of the phenyl nitro group, with Pd/C in the flow hydrogenator, resulted in the final derivatives 4a–q (75–91% yield). | ||
| Scheme 1 Synthesis of N,N′-disubstituted thiourea and urea derivatives 4a–q. a) CH3CN, BuLi, THF, −78 °C, then RT; b) BH3-THF, 0 °C, then 4 h RT; c) XCNR2, CH2Cl2, 20 min (MW); d) 10% Pd/C, MeOH (flow hydrogenation), 60 °C, 60 bar; e) CrO3, CH3COCH3, H2SO4, 10 min; f) Fe/FeSO4, H2O, 95 °C, 3 h. | ||
On the other hand, this new synthetic route is useful to obtain urea and thiourea kynurenamines 3. Thereby, as an example, we carried out the oxidation of the hydroxyl group to a carbonyl one in the derivative 8o (R1 = Cl, R2 = Et, X = O), using the Jones reagent followed by the nitro reduction to an amino group20 (Scheme 1). This alternative synthesis improves the previously described one20 by shortening the synthetic route (from 8 to 5 steps) and doubling the global yield.
iNOS and nNOS inhibition
Compounds 4a–q were evaluated in vitro as inhibitors of iNOS and nNOS using recombinant isoenzymes. The assays were made at 1 mM concentration of each compound in order to identify the more active and selective derivatives. Besides, IC50 values were measured for the most interesting compounds.Table 1 illustrates the inhibition percentages versus iNOS and nNOS (the kynurenamine derivatives 3a–b, previously described,20 were introduced as references).
| Compound | R1 | R2 | X | % iNOS inhibitiona | % nNOS inhibitiona |
|---|---|---|---|---|---|
| a Values are the mean ± SEM of the percentage of iNOS and nNOS inhibition produced by 1 mM concentration of each compound. Each value is the mean of three experiments performed in triplicate using recombinant iNOS and nNOS enzymes. b 3a–b were used as references.20 | |||||
| 3a | OMe | Me | S | 78.20 ± 2.43 | 46.04 ± 2.85 |
| 3b | Cl | Et | O | 78.63 ± 1.34 | 9.86 ± 3.17 |
| 4a | H | Me | S | 20.74 ± 1.15 | 70.17 ± 0.77 |
| 4b | H | Et | S | 19.36 ± 3.33 | 44.84 ± 0.02 |
| 4c | H | Pr | S | 31.10 ± 0.55 | 51.74 ± 1.35 |
| 4d | H | Et | O | 18.34 ± 0.43 | 7.81 ± 3.06 |
| 4e | H | Pr | O | 19.04 ± 1.45 | 7.00 ± 0.32 |
| 4f | H | Ph | O | 27.86 ± 1.15 | 17.69 ± 1.32 |
| 4g | OMe | Me | S | 76.55 ± 0.33 | 80.55 ± 2.29 |
| 4h | OMe | Pr | S | 25.39 ± 1.71 | 6.02 ± 1.61 |
| 4i | OMe | Et | O | 26.64 ± 2.05 | 43.02 ± 0.48 |
| 4j | OMe | Pr | O | 14.66 ± 0.19 | 15.81 ± 1.03 |
| 4k | OMe | Ph | O | 12.33 ± 1.30 | 12.10 ± 1.08 |
| 4l | Cl | Me | S | 22.13 ± 0.28 | 70.53 ± 4.60 |
| 4m | Cl | Et | S | 60.42 ± 2.31 | 70.61 ± 3.54 |
| 4n | Cl | Pr | S | 54.98 ± 3.21 | 32.40 ± 2.24 |
| 4o | Cl | Et | O | 33.85 ± 1.86 | 15.22 ± 2.33 |
| 4p | Cl | Pr | O | 19.61 ± 3.04 | 23.81 ± 1.34 |
| 4q | Cl | Ph | O | 3.11 ± 2.36 | 38.62 ± 1.63 |
In general, compounds 4a–q show better values of inhibition versus nNOS than iNOS, since five compounds exhibit good nNOS percentage inhibition (4g, 80.6%; 4m, 70.6%; 4l, 70.5%; 4a, 70.2% and 4c, 51.7%) and only three compounds show good inhibitory activity versus iNOS (4g, 76.6%; 4m, 60.4% and 4n, 55.0%). Regarding the R1 radical, no clear structure–activity relationship can be concluded. Independently of the substituent nature ((R1 = H), electron-donating (R1 = OCH3) or electron withdrawing (R1 = Cl)), different inhibition levels can be observed. It seems that the inhibitory activity depends more on X and R2. Thus, derivatives with a thiourea residue (X = S) show better inhibition data for both isoenzymes than those bearing urea (X = O). In addition, compounds with a methyl substituent as R2 produce higher inhibition for iNOS such as 4g and for nNOS such as 4a, 4g and 4l. It is noteworthy that compound 4m shows good inhibition having R2 = Et. Finally, 4g stands out as the best inhibitor of both isoforms.
Furthermore, Table 2 shows the iNOS inhibition values of kynurenamines 2a–l previously synthesized (not published before), as well as their nNOS values already described19 in order to compare them with the new N,N′-disubstituted thioureas and ureas 4a–q.
| Compound | R1 | R2 | % iNOS inhibitiona | % nNOS inhibitionb |
|---|---|---|---|---|
| a Values are the mean ± SEM of the percentage of iNOS and nNOS inhibition produced by 1 mM concentration of each compound. Each value is the mean of three experiments performed in triplicate using recombinant iNOS and nNOS enzymes. b See ref. 19. | ||||
| 2a | OMe | Me | 30.74 ± 2.92 | 65.36 ± 5.6 |
| 2b | OMe | Et | 0.24 ± 1.95 | 50.87 ± 1.9 |
| 2c | OMe | Pr | 7.35 ± 4.48 | 42.81 ± 1.9 |
| 2d | OMe | Bu | 14.11 ± 0.89 | 39.68 ± 1.17 |
| 2e | OMe | c-C3H5 | 2.66 ± 1.4 | 40.44 ± 1.84 |
| 2f | OMe | c-C4H7 | 0.01 ± 1.52 | 33.75 ± 1.39 |
| 2g | OMe | c-C5H9 | 0.61 ± 2.49 | 45.04 ± 1.97 |
| 2h | OMe | c-C6H11 | 9.24 ± 2.1 | 48.24 ± 2.42 |
| 2i | OMe | Ph | 10.86 ± 3.73 | 46.47 ± 2.36 |
| 2j | Cl | Me | 19.70 ± 0.57 | 17.70 ± 1.13 |
| 2k | Cl | c-C3H5 | 5.37 ± 2.59 | 9.66 ± 4.17 |
| 2l | Cl | Ph | 26.60 ± 4.52 | 7.20 ± 1.29 |
The best nNOS inhibitors of derivatives 2a–l were 2a (R1 = OMe, R2 = Me, 65.4%) and 2b (R1 = OMe and R2 = Et, 50.9%); however, no compound with relevant iNOS inhibition value was found, so they can be considered as selective inhibitors of nNOS versus iNOS. Kynurenamine 2a, the N1-methyl-5-methoxykynurenamine, is the main brain metabolite of melatonin hormone in mammalians, that mediates some of the actions of this indolamine. Both of them inhibited nNOS activity in vitro in a dose-related manner.25
If we compare the derivatives 4a–q with the kynurenamines 2a–l, we can see that both series inhibit better nNOS than iNOS (compounds 4a–q having greater potency), unlike the corresponding molecules 3 showing better iNOS inhibition values. Besides, the best inhibitors of the first two families carry a methoxy group as R1 and a methyl group as R2 (urea 4g and kynurenamine 2a, respectively), while the urea-kynurenamine 3b which has a chlorine group as R1 and an ethyl group as R2 is the best inhibitor of this family and the most selective one. Finally, 4g has the highest percentage of inhibition of all the tested derivatives with similar structures.
Table 3 includes the IC50 data of the most interesting derivatives 4a–q. Compound 4g stands out as one with the best values of inhibition, 130 and 180 μM, versus nNOS and iNOS, respectively, confirming the potency of this molecule.
eNOS inhibition
We investigated the eNOS inhibitory activity of compound 4g, using HUVECs incubated with 100 μmol L−1 of 4g or DMSO and measuring the NO production stimulated by the known eNOS activator A23187. This agent increased, in a time dependent manner, NO production. No significant differences (p > 0.05) were observed in A23187-stimulated NO production in the presence of 4g, showing that this compound did not inhibit eNOS (Fig. 2a). Moreover, the endothelium-dependent relaxation to acetylcholine was not affected by 4g (Fig. 2b), confirming the absence of eNOS inhibition of this compound. However, L-NAME suppressed this NO-dependent relaxant response.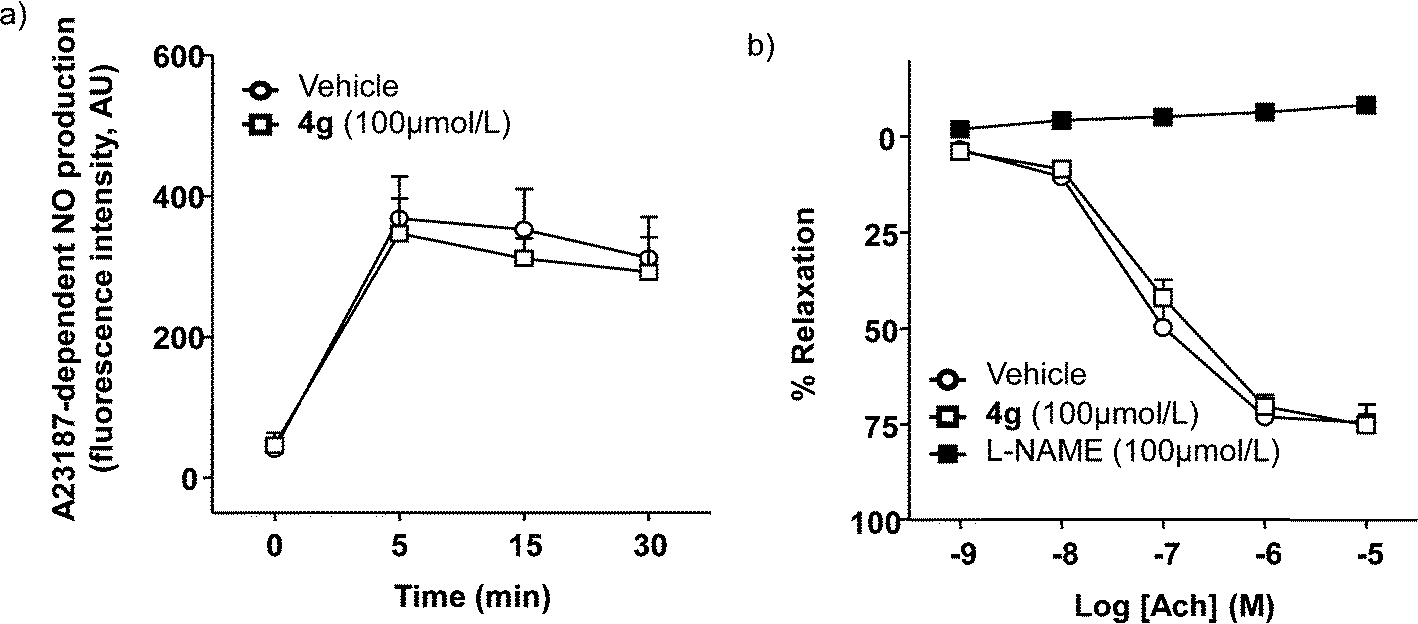 | ||
| Fig. 2 Effects of 4g on eNOS activity. a) A23187-dependent NO production in HUVECs incubated with DMSO or 4g (100 μmol L−1) for 30 min (n = 12). b) Acetylcholine-evoked relaxation in aortic rings with endothelium contracted with 1 μM noradrenaline in the presence of DMSO, 4g (100 μmol L−1), or L-NAME (100 μmol L−1) for 30 min (n = 5). The data are expressed as the mean ± SEM of n experiments. | ||
Cell viability
We investigated the cell viability activity of compound 4g, using HUVECs. We found that this compound has little toxicity, since only at concentrations >1 mM is cell viability weakly (approximately 20%) reduced (Fig. 3).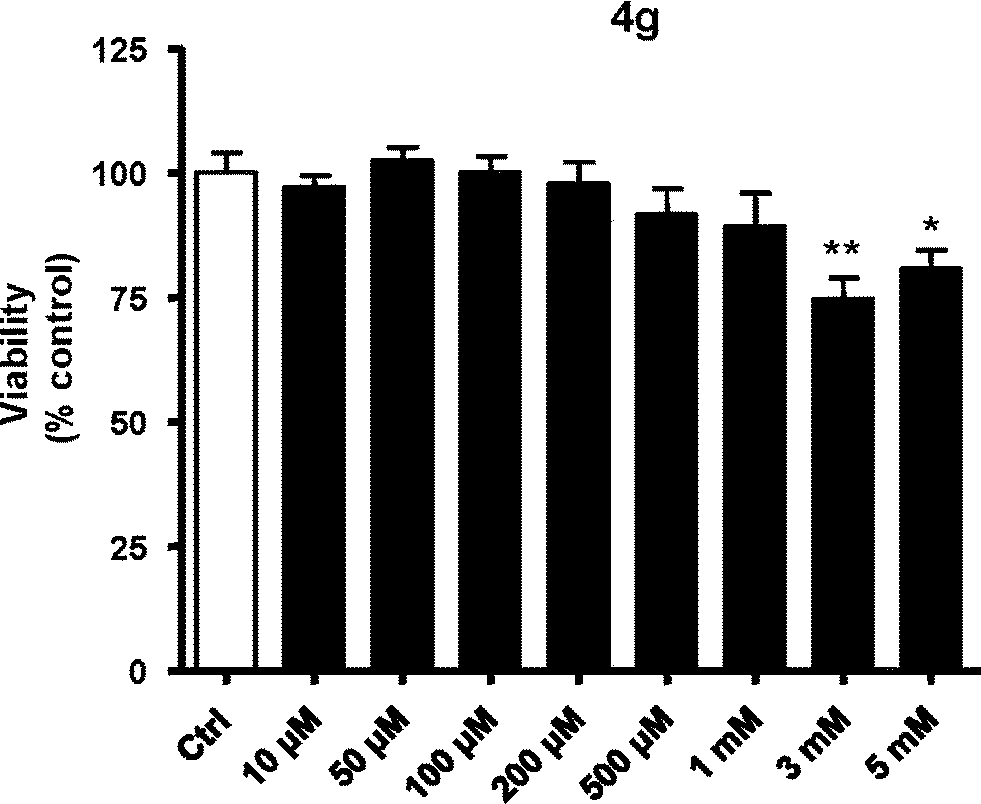 | ||
| Fig. 3 Study of cell viability of 4g using MTT assay. *p < 0.05 vs. control (ctrl); **p < 0.01 vs. ctrl. | ||
Docking studies
Docking studies were performed to propose and understand the binding mode of N,N′-disubstituted thiourea and urea derivatives 4a–q inside nNOS and iNOS.Fig. 4 illustrates the main poses obtained for these compounds in the nNOS (PDB id: 1QW6)26 binding site. All the ligands interact with Glu592 and a carboxylate moiety of the heme group through the hydroxyl group and both thiourea/urea nitrogens, respectively. Compound 4a (Fig. 4a) shows additional interactions of the amino group, forming two more hydrogen bonds, one with Trp587 and the other with Glu592. Moreover 4a has a good orientation in the binding site: the phenyl ring points to the lipophilic region (Phe584 and Pro565) being situated below the heme group establishing a π–cation interaction, the aliphatic chain is directed to Val575 establishing VdW interactions and the thiourea moiety is oriented to a polar pocket formed by three arginines (Arg596, Arg603 and Arg481). In addition, more VdW interactions are established with Gln478.
 | ||
| Fig. 4 Detailed view of the main poses obtained for compounds 4a (a) and 4j (b) in the nNOS binding site. The dotted lines indicate hydrogen bond interactions between the ligand and the residues of the enzyme. | ||
On the other hand, compound 4j (Fig. 4b) shows no additional hydrogen bonds in the binding site. It presents a similar orientation to 4a, but it is longer, consequently it adopts a more constricted conformation which results more unstable and reduces the π–cation interaction, due to the inclination of the aromatic ring with respect to the heme group. This fact can explain the loss of activity of some compounds.
Fig. 5 shows the most common poses obtained for hydroxypropyl derivatives in the iNOS binding site. Compound 4g (Fig. 5a) forms three hydrogen bonds, one with Glu371 through the hydroxyl group and two with a heme propionate moiety through both thiourea nitrogens. It is noteworthy that, although 4a has more favourable H-bond contribution inside nNOS, 4g seems to have higher VdW stabilising contribution inside iNOS. This contribution is performed through the aliphatic chain which interacts with the nearby residues Val346, Pro344 and Gln257, and especially through the thiourea moiety which is oriented to Gln257 and to the arginine pocket.
 | ||
| Fig. 5 Detailed view of the main poses obtained for compounds 4g (a) and 4l (b) in the iNOS binding site. The dotted lines indicate hydrogen bond interactions between the ligand and the residues of the enzyme. | ||
Other compounds such as 4l (Fig. 5b) demonstrate weaker interaction with the iNOS (PDB id: 3NW2)27 binding site. In this case, two hydrogen bonds are established, one between the amino group and a propionate of the heme group and the other between a thiourea nitrogen and Glu371. Moreover, the conformation adopted by 4l moves away the phenyl ring and the thiourea moiety reducing the π–cation interaction with the heme group and the VdW interactions which decrease the stability and efficiency of the binding process.
Conclusions
In summary, we have designed and successfully synthesized a series of novel N,N′-disubstituted thiourea and urea derivatives. The synthetic pathway of these new compounds can be used to prepare kynurenamine-thioureas and ureas more easily, with less synthetic steps and higher global yield than the previously described route. Their biological evaluation was performed versus nNOS and iNOS isoforms for all compounds and versus eNOS isoform for 4g. In general, compounds 4a–q inhibit the neuronal NOS isoform better than the inducible one, in which the thioureas exhibit higher inhibition than the ureas. The derivatives 4a and 4g are the most active compounds with IC50 values of 350 and 130 μM, respectively, versus nNOS, and 180 μM versus iNOS for the second compound. In addition, 4g does not inhibit eNOS which is necessary to avoid hypertension. Moreover, the cell viability test demonstrated the absence of cell toxicity in compound 4g at the IC50 value. Both in silico and in vitro studies reveal promising properties for these compounds which could be a reference to design new nNOS and iNOS inhibitors without cytotoxicity and adverse vascular effects, useful in neurodegenerative disorders such as Parkinson's disease, where both isoforms are involved.Experimental section
Chemistry
Melting points were determined using a capillary melting point apparatus and were uncorrected. Analytical thin layer chromatography was performed using Merck Kieselgel 60F254 aluminum sheets, and the spots were developed with UV light (λ = 254 nm). Flash chromatography was carried out using silica gel 60, 230–240 mesh (Merck), and the eluents used are reported within parentheses. 1H NMR and 13C NMR spectra were recorded on a Varian Inova Unity 300 spectrometer operating at 300.20 for 1H and 75.49 MHz for 13C, on a Varian direct drive 400 spectrometer operating at 400.57 MHz for 1H and 100.73 MHz for 13C, on a Varian Inova Unity 500 spectrometer operating at 499.79 for 1H and 125.68 MHz for 13C and on a Varian direct drive 600 spectrometer operating at 600.25 MHz for 1H and 150.95 MHz for 13C in the deuterated solvents indicated at ambient temperature. Chemical shifts are reported in ppm (δ) and are referenced to the residual solvent peak. IR spectra were recorded on a Perkin Elmer 782 spectrometer. High-resolution mass spectrometry (HRMS) was carried out using a Waters LCT Premier Mass Spectrometer. Elemental analyses were within ±0.4% of the theoretical values. Small scale microwave-assisted synthesis was carried out using an Initiator 2.0 single-mode microwave instrument producing controlled irradiation at 2.450 GHz (Biotage AB, Uppsala). Reaction time refers to holding time at 90 °C, not to total irradiation time. The temperature was measured using an IR sensor outside the reaction vessel. Anhydrous CH2Cl2 was used as solvent.General synthetic method of 3-hydroxy-3-(2-nitro-5-substituted phenyl)propanenitrile, 6a–c
BuLi (1.6 M/hexane, 20.3 mL) was added to dry THF (37.5 mL) cooled to −78 °C under argon. Then, a solution of acetonitrile (1.7 mL) in dry THF (4.9 mL) was added dropwise. The mixture was stirred at −78 °C for 1 h. Afterward, a solution of the corresponding benzaldehyde 5a–c (16.16 mmol) in dry THF (4.9 mL) was added dropwise. The mixture was stirred again at −78 °C for 30 min and then warmed to RT. The reaction was agitated for 15 min at RT and was quenched with cold water (25 mL), diluted with diethyl ether (30 mL) and washed with 2% aqueous HCl (15 mL). The aqueous layer was extracted with diethyl ether (3 × 15 mL). Finally, the organic phase was washed with NaHCO3 saturated solution (10 mL) and brine (15 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The crude mixture was purified by flash chromatography (EtOAc/hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).
:2).
General synthetic method of 3-amino-1-(2-nitro-5-substituted phenyl)propan-1-ol, 7a–c
3-Hydroxy-3-(2-nitro-5-substituted phenyl)propanenitrile derivatives 6a–c (1.7 mmol) were treated dropwise with a solution of BH3 (12.3 mL) in THF at 0 °C under argon. The mixture was stirred for 4 h at RT. Afterward, the reaction mixture was cooled to 0 °C and an ice-cold solution of 6 N HCl (7.5 mL) was added carefully. The THF was evaporated, and the aqueous phase was basified with 4 N NaOH to pH = 10 and extracted with EtOAc (3 × 15 mL). The organic phase was washed with brine, dried over Na2SO4 and concentrated. The crude was used for the next step without purification.General synthetic method of N-alkyl or phenyl-N′-[3-hydroxy-3-(2-nitro-5-substituted phenyl)propyl]-thioureas and ureas, 8a–q
Thioisocyanate or isocyanate (0.9 mmol) was added, under argon, to a solution of 3-amino-1-(2-nitro-5-substituted phenyl)propan-1-ol derivatives 7a–c (0.6 mmol) in dry CH2Cl2. The reaction mixture was irradiated under microwave conditions at 90 °C for 20 min. The crude mixture was purified by flash chromatography (EtOAc/hexane, 1:1).
General synthetic method of N-alkyl or phenyl-N′-[3-(2-amino-5-substituted phenyl)-3-hydroxypropyl]-thioureas and ureas, 4a–q
A solution of each of the nitro precursors 8a–q (0.5 mmol) in MeOH (10 mL) was passed through the flow hydrogenator under the following conditions: 60 °C, 60 bar, 0.2 mL min−1 flow rate and 10% Pd/C as a catalyst. After evaporation of the solvent, the crude mixture was purified by recrystallization (diethyl ether) or by flash chromatography (EtOAc or EtOAc/hexane, 1:1).
General synthetic method of N-[3-(2-amino-5-chlorophenyl)-3-oxopropyl]-N′-ethylurea 3b from N-[3-(5-chloro-2-nitrophenyl)-3-hydroxypropyl]-N′-ethylurea 8o
The Jones reagent (0.418 mL), freshly prepared (a mixture of 2.67 g of chromic anhydride and 2.3 mL of H2SO4 dissolved in 10 mL of water), was added to a solution of 8o (0.742 mmol) in acetone (3 mL). After 10 min stirring, the reaction was quenched with ice-water (25 mL) and saturated NaHSO4 solution (5 mL). The resulting mixture was extracted with ethyl acetate, filtered, dried (Na2SO4) and concentrated under vacuum. The crude mixture was purified by flash chromatography (EtOAc/hexane, 3:1). The next step was the nitro group reduction to amino one using Fe/FeSO4 in water as previously described.20
Biological procedures
In vitro nNOS and iNOS activity determination
L-Arginine, L-citrulline, N-(2-hydroxymethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), DL-dithiothreitol (DTT), hypoxantine-9-β-D-ribofuranosid (inosine), ethylene glycol-bis-(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), bovine serum albumin (BSA), Dowex-50 W (50 × 8-200), FAD, NADPH and 5,6,7,8-tetrahydro-L-biopterin dihydrochloride (H4-biopterin), tris-(hydroxymethyl)-aminomethane (Tris-HCl) and calcium chloride were obtained from Sigma-Aldrich Química (Spain). L-[3H]-arginine (47.4 Ci mmol−1) was obtained from Perkin Elmer (Spain). Calmodulin from bovine brain and recombinant iNOS and nNOS were obtained from Enzo Life Sciences (Spain).The i/nNOS activity was measured by the Bredt and Snyder method,29 monitoring the conversion of L-[3H]-arginine to L-[3H]-citrulline. The final incubation volume was 100 μL and consisted of 10 μL of an aliquot of recombinant i/nNOS added to a buffer with a final concentration of 25 mM Tris-HCl, 1 mM DTT, 4 μM H4-biopterin, 10 μM FAD, 0.5 mM inosine, 0.5 mg mL−1 BSA, 0.1 mM CaCl2, 10 μM L-arginine, 10 μg mL−1 calmodulin (only for nNOS) and 50 nM L-[3H]-arginine, at pH 7.6. The reaction was started by the addition of 10 μL of 7.5 mM NADPH and 10 μL of each of the derivatives 4a–q in ethanol (10%) to give a final concentration of 1 mM. The tubes were vortexed and incubated at 37 °C for 30 min. Control incubations were performed by the omission of NADPH. The reaction was halted by the addition of 400 μL of cold 0.1 M HEPES, 10 mM EGTA, and 0.175 mg mL−1L-citrulline, pH 5.5. The reaction mixture was decanted into a 2 mL column packet with Dowex-50 W ion-exchange resin (Na+ form) and eluted with 1.2 mL of water. L-[3H]-citrulline was quantified by liquid scintillation spectroscopy. The retention of L-[3H]-arginine in this process was greater than 98%. Specific enzyme activity was determined by subtracting the control value, which usually amounted to less than 1% of the radioactivity added. The nNOS activity was expressed as picomoles of L-[3H]-citrulline produced (per mg of protein per min).
eNOS inhibition
Quantification of NO in human umbilical vein endothelial cells (HUVECs)
The investigation has been conducted according to the principles expressed in the Declaration of Helsinki and has been approved by the Ethics Committee for the Human Investigation, at the University of Granada, Granada, Spain (Approval no. 201302400001491). Subjects have been properly instructed and have indicated that they consent to participate by signing the appropriate informed consent paperwork. Endothelial cells were isolated from human umbilical cord veins using a previously reported method with several modifications.30 The cells were cultured (Medium 199 + 20% fetal bovine serum + penicillin/streptomycin 2 mmol L−1 + amphotericin B 2 mmol L−1 + glutamine 2 mmol L−1 + HEPES 10 mmol L−1 + endothelial cell growth supplement 30 μg mL−1 + heparin 100 mg mL−1) under 5% CO2 at 37 °C. HUVECs were then used to measure NO production by diaminofluorescein-2 (DAF-2) fluorescence, as described previously.30 Briefly, cells were incubated for 30 min in the presence of 4g at a concentration of 100 μmol L−1. After this period, cells were washed with PBS and then were pre-incubated with L-arginine (100 μmol L−1 in PBS, 5 min, 37 °C). Subsequently, DAF-2 (0.1 μmol L−1) was incubated for 2 min, and then the calcium ionophore calimycin (A23187, 1 μmol L−1) was added for 30 min and cells were incubated in the dark at 37 °C. Then the fluorescence (arbitrary units) was measured using a spectrofluorimeter (Fluorostart, BMG Labtechnologies, Offenburg, Germany). The auto-fluorescence was subtracted from each value. In some experiments, NG-nitro-L-arginine methyl ester (L-NAME, 100 μmol L−1) was added 15 min before the addition of L-arginine. The difference between fluorescence signal without and with L-NAME was considered NO production.Tissue preparation and measurement of tension
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and with the principles outlined in the Declaration of Helsinki and approved by the Ethics Committee for the welfare of experimental animals, at the University of Granada, Granada, Spain (Approval no. 459-bis-CEEA-2012). Male Wistar rats (250–300 g), obtained from Harlam Laboratories SA (Barcelona, Spain), were euthanized by a quick blow on the head followed by exsanguination. The descending thoracic aortic rings were dissected, and the rings were then mounted in organ chambers filled with Krebs solution (composition in mmol L−1: NaCl, 118; KCl, 4.75; NaHCO3, 25; MgSO4, 1.2; CaCl2, 2; KH2PO4, 1.2; and glucose, 11) and were stretched to 2 g of resting tension by means of two L-shaped stainless-steel wires inserted into the lumen and attached to the chamber and to an isometric force-displacement transducer (UF-1, Cibertec, Madrid, Spain), and the data were recorded using a recording and analysis system (MacLab AD Instruments), as described previously.30 After equilibration, aortic rings with a functional endothelium were incubated with DMSO, 4g (100 μmol L−1), or L-NAME (100 μmol L−1) for 30 min and contracted with phenylephrine (1 μmol L−1). Once a plateau contraction was reached, a concentration–response curve was constructed by cumulative addition of acetylcholine. The results are expressed as percentage of phenylephrine-evoked contraction. The data are expressed as the mean ± SEM, and n reflects the number of aortic rings from different rats.Cell viability tests
HUVECs in suspension were seeded at 1 × 104 cells per well in a 96-well microtiter plate, and these cells were incubated at 37 °C for up to 24 hours. The cell supernatants were removed and replaced by fresh medium before MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) analysis. Then the cells were exposed to serial dilutions of 4g (10 μM–5 mM) for 30 minutes. At the chosen time, 20 μL of 5 mg mL−1 MTT in PBS was added to the cells and further incubated at 37 °C. After washing, 100 μL of DMSO were added into each well, and the absorbance at 570 nm was measured using a multi-well plate reader (Model 680XR, BIO-RAD) with subtraction of the blank value at 630 nm and was compared with control, untreated cells.Statistical analysis
The data are expressed as the mean ± SEM. Statistically significant differences between groups were calculated by Student's t test for unpaired observations or for multiple comparisons by an ANOVA followed by a Newman Keuls test. p < 0.05 was considered statistically significant.Docking studies
The suite of programs Maestro (Schrödinger, LCC31) was used for the docking studies. The Cartesian coordinates for the two proteins iNOS and nNOS were obtained from the available X-ray structures with PDB id. 3NW2 and 1QW6, respectively (Protein Data Bank). These receptors were prepared using the Protein Preparation Wizard module32 implemented in Maestro. The charge and coordination sphere of the Fe(II) ion within the heme prosthetic group were manually redefined before replacing and minimizing all hydrogens in the receptor. LigPrep33 program was used to generate the 3D structures of a set of conformers of 4a–q. Geometries of this set were optimized using the MacroModel module. A rigid docking protocol was followed using Glide program in Standard Precision (SP) mode and taking into account the best 5 solutions. Figures were built using PyMOL (v1.3, Schrödinger, LLC).34Acknowledgements
We are very grateful to Dr. Pedro A. Sánchez-Murcia for his help. This work was partially supported by the Instituto de Salud Carlos III through grant FI11/00432 and by Ministerio de Economía y Competitividad, Instituto de Salud Carlos III (RIC RD12/0042/0011).References
- R. G. Knowles and S. Moncada, Biochem. J., 1994, 298, 249–258 CrossRef CAS PubMed.
- V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butterfield and A. M. Giuffrida Stella, Neuroscience, 2007, 8, 766–775 CAS.
- D. Cho, T. Nakamura, J. Fang, P. Cieplak, A. Godzik, Z. Gu and S. A. Lipton, Science, 2009, 324, 102–105 CrossRef CAS PubMed.
- M. A. Smith, M. Vasak, M. Knipp, R. J. Castellani and G. Perry, Free Radical Biol. Med., 1998, 25, 898–902 CrossRef CAS PubMed.
- G. T. Liberatore, V. Jackson-Lewis, S. Vukosavic, A. S. Mandir, M. Vila, W. G. McAuliffe, V. L. Dawson, T. M. Dawson and S. Przedborski, Nat. Med., 1999, 5, 1403–1409 CrossRef CAS PubMed.
- P. J. Norris, H. J. Waldvogel, R. L. Faull, D. R. Love and P. C. Emson, Neuroscience, 1996, 72, 1037–1047 CrossRef CAS PubMed.
- N. K. Wong and M. J. Strong, Eur. J. Cell Biol., 1998, 77, 338–343 CrossRef CAS PubMed.
- C. O. Bingham III, J. Rheumatol., Suppl., 2002, 65, 3–9 Search PubMed.
- K. D. Kröncke, K. Fehsel and V. Kolb-Bachofen, Clin. Exp. Immunol., 1998, 113, 147–156 CrossRef.
- S. Taddei, A. Virdis, L. Ghiadoni, I. Sudano and A. Salvetti, J. Cardiovasc. Pharmacol., 2001, 38(Suppl.2), S11–S14 CrossRef CAS PubMed.
- Napolia, F. de Nigrisa, S. Williams-Ignarrod, O. Pignalosaa, V. Sicaa and L. J. Ignarroe, Nitric Oxide, 2006, 15, 265–279 CrossRef PubMed.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- F. Torreilles, S. Salman-Tabcheh, M. Guerin and J. Torreilles, Brain Res. Rev., 1999, 30, 153–163 CrossRef CAS PubMed.
- D. G. Hirst and T. Robson, Front. Biosci., 2007, 12, 3406–3418 CrossRef CAS.
- T. Uehara, T. Nakamura, D. Yao, Z. Q. Shi, Z. Gu, Y. Ma, E. Masliah, Y. Nomura and S. A. Lipton, Nature, 2006, 441, 513–517 CrossRef CAS PubMed.
- L. A. Ridnour, D. D. Thomas, D. Mancardi, M. G. Espey, K. M. Miranda, N. Paolocci, M. Feelisch, J. Fukuto and D. A. Wink, Biol. Chem., 2004, 385, 1–10 CrossRef CAS PubMed.
- T. O. Fischmann, A. Hruza, X. D. Niu, J. D. Fossetta, C. A. Lunn, E. Dolphin, A. J. Prongay, P. Reichert, D. J. Lundell, S. K. Narula and P. C. Weber, Nat. Struct. Biol., 1999, 6, 233–242 CrossRef CAS PubMed.
- E. Camacho, J. Leon, A. Carrión, A. Entrena, G. Escames, H. Khaldy, D. Acuña-Castroviejo, M. A. Gallo and A. Espinosa, J. Med. Chem., 2002, 45, 263–274 CrossRef CAS PubMed.
- A. Entrena, M. E. Camacho, D. Carrión, L. C. López-Cara, G. Velasco, J. León, G. Escames, D. Acuña-Castroviejo, V. Tapias, M. A. Gallo, A. Vivó and A. Espinosa, J. Med. Chem., 2005, 48, 8174–8181 CrossRef CAS PubMed.
- M. Chayah, M. D. Carrión, M. A. Gallo, R. Jiménez, J. Duarte and M. E. Camacho, ChemMedChem, 2015, 10, 874–882 CrossRef CAS PubMed.
- A. Fürstner, D. N. Jumbam and G. Seidel, Chem. Ber., 1994, 127, 1125–1130 CrossRef.
- D. A. Claremon, L. Zhuang, K. Leftheris, C. M. Tice, Z. Xu, Y. Ye, S. B. Singh, S. Cacatian, W. Zhao and F. Himmelsbach, Pat. WO 2009017664A1, 2009 Search PubMed.
- R. B. Silverman, G. R. Lawron, H. R. Ranaivo, L. K. Chico, J. Seo and D. M. Watterson, Bioorg. Med. Chem., 2009, 17, 7593–7605 CrossRef CAS PubMed.
- J. S. Fortin, J. Lacroix, M. Desjardins, A. Patenaude, E. Petitclerc and R. C. Gaudreault, Bioorg. Med. Chem., 2007, 15, 4456–4469 CrossRef CAS PubMed.
- J. León, G. Escames, M. I. Rodríguez, L. C. López, V. Tapias, A. Entrena, E. Camacho, M. D. Carrión, M. A. Gallo, A. Espinosa, D.-X. Tan, R. J. Reiter and D. Acuña-Castroviejo, J. Neurochem., 2006, 98, 2023–2033 CrossRef PubMed.
- R. Fedorov, E. Hartmann, D. K. Ghosh and I. Schlichting, J. Biol. Chem., 2003, 278, 45818–45825 CrossRef CAS PubMed.
- U. Grädler, T. Fuchß, W.-R. Ulrich, R. Boer, A. Strub, C. Hesslinger, C. Anézo, K. Diederichs and A. Zaliani, Bioorg. Med. Chem. Lett., 2011, 21, 4228–4232 CrossRef PubMed.
- S. S. Labadie and J. K. Stille, Tetrahedron, 1984, 40, 2329–2336 CrossRef CAS.
- D. S. Bredt and S. H. Snyder, 682–685., Proc. Natl. Acad. Sci. U. S. A., 1990, 87 Search PubMed.
- R. Jiménez, M. Sánchez, M. J. Zarzuelo, M. Romero, A. M. Quintela, R. López-Sepúlveda, P. Galindo, M. Gómez-Guzmán, J. M. Haro, A. Zarzuelo, F. Pérez-Vizcaíno and J. Duarte, J. Pharmacol. Exp. Ther., 2010, 332, 554–561 CrossRef PubMed.
- Schrödinger Suite 2012 Update 2 Search PubMed.
- Schrödinger Suite 2012 Protein Preparation Wizard, Epik version 2.3, Schrödinger, LLC, New York, NY, 2012 Search PubMed; Impact version 5.8, Schrödinger, LLC, New York, NY, 2012 Search PubMed; Prime version 3.1, Schrödinger, LLC, New York, NY, 2012 Search PubMed.
- Lig Prep, version 2.5, Schrödinger, LLC, New York, NY, 2012 Search PubMed.
- PyMOL Molecular Graphics System, Version 1.4, Schrödinger. LLC Search PubMed.
Footnote |
| † The authors declare no competing interests. |
| This journal is © The Royal Society of Chemistry 2016 |