
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethods of protein surface PEGylation under structure preservation for the emulsion-based formation of stable nanoparticles†‡
Lydia
Radi
a,
Matthias
Fach
a,
Mirko
Montigny
b,
Elena
Berger-Nicoletti
c,
Wolfgang
Tremel
b and
Peter R.
Wich
*a
aInstitut für Pharmazie und Biochemie, Johannes Gutenberg-Universität Mainz, 55128 Mainz, Germany. E-mail: wich@uni-mainz.de
bInstitut für Anorganische Chemie und Analytische Chemie, Johannes Gutenberg-Universität Mainz, 55128 Mainz, Germany
cInstitut für Organische Chemie, Johannes Gutenberg-Universität Mainz, 55128 Mainz, Germany
First published on 15th August 2016
Abstract
Proteins show remarkable versatility as multifunctional materials for therapeutic applications. They can be easily modified with the toolkit of bioorganic chemistry and are particularly attractive because of their degradability and biocompatibility. Herein, we evaluate different methods for the attachment of multiple PEG chains on the surface of the enzyme lysozyme. For this, we activated standard 2 kDa mPEG chains with four different electrophilic groups and tested their ability to react with different amino acids on the surface of our model protein. The aim was to find an effective and at the same time mild modification method that preserves the native structure and activity of the enzyme. The amphiphilic properties of PEG induce a solubility switch of the protein material which allows the formation of nanoparticles using a nano-emulsion technique in the size range of 100–130 nm. We found that, even though all produced materials are soluble in organic solvents, the amount of introduced PEG chains and the enzyme activity significantly vary depending on the chosen PEGylation method.
Introduction
Proteins are structurally well-defined biomacromolecules that have attracted increasing interest as source materials for biotechnological and therapeutic applications. These natural polymers have diverse roles in the body, like catalysis of biochemical reactions, transport of molecules and stimuli responses.1 Proteins possess unique chemical, physical and biological properties and can be easily modified with the toolkit of bioorganic chemistry. When used as materials for biotechnological applications, they have several advantages over synthetic polymers, including aspects of biocompatibility and degradability, as well as low antigenicity and low toxicity.1,2The chemical modification of proteins with other natural and synthetic macromolecules is well studied and has emerged as a valuable tool for the development of tailor-made materials and advanced therapeutics.3–6 Applications range from enhanced pharmacokinetic properties to new materials for tissue engineering and dynamic drug delivery systems. Protein–polymer biohybrids can help to increase the protein stability, alter the immune response, change the reactivity of enzymes or increase the blood circulation half-life, to name only a few examples.7
Most chemical bioconjugation methods to attach polymers to a protein rely on the reaction with nucleophilic amino acids, in particular cysteine, lysine, arginine, serine, threonine and tyrosine.8,9 Among them, lysine is the most abundant amino acid in protein sequences,10 and its side chain, the ε-amino group, is a common target for conjugation.11 The extent of modification depends on the size and structure of the native protein, as well as on the reactive group on the polymer.
Many linear and branched polymers have been used in the past for protein−polymer conjugates with improved in vitro and in vivo properties. Examples are N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers, poly(vinylpyrrolidones) (PVP) and poly(2-oxazolines), as well as various polysaccharides and polypeptides.12,13 Among all these materials, polyethylene glycol (PEG) is still the gold standard for stealth polymers and represents the only clinically approved protein conjugate.10,13,14 Its unique ability to be soluble in both aqueous and organic solvents makes it particularly interesting for conjugation to biological macromolecules under mild physiological conditions. Even though it is not biodegradable, PEG is generally regarded as safe by the FDA. Currently, ten PEGylated proteins are already used for therapeutic applications.3
PEGylated enzymes have also gained increasing attention as bioconversion tools in organic synthesis.15–17 Whereas native proteins will precipitate and denature in most organic solvents, PEGylation can increase their stability and solubility significantly.18 It is known that a high degree of PEGylation increases the hydrophobicity of proteins,19,20 making them soluble, for example, in organic solvents such as benzene, toluene, and chlorinated hydrocarbons. In addition, enzymatic activity is in many instances not just preserved, but can even be increased by several folds. This can be due to better solubility of substrates, less competitive interactions in binding sites, and increased structural dynamics that benefit the active sites.18,21 However, depending on the extent of surface PEGylation of the protein and the resulting shielding effects, the accessibility of bigger substrates might be impaired, resulting in a reduced activity.
To couple linear PEGs to biomacromolecules, it is necessary to activate one end by introducing a reactive functional group.8 The predominant reaction partners of proteins are nucleophilic amino acids and the N-terminal end of the peptide chain. Therefore, electrophilic and amine-selective functions on PEGs are preferred if a high surface modification is needed. These include various activated carbonates, like succinimidyl-, benzotriazole-, p-nitrophenyl- and chlorophenyl carbonates, aromatic chlorotriazines and carbonylimidazoles, as well as alkylating groups like aldehyde- or epoxy-modified PEGs.8,22 Linear PEGs are commercially available in a wide range of sizes. Low molecular weights (≤10 kDa) represent the best weight ratio for the modification of most proteins. Compared to higher weights, they are also more rapidly cleared in vivo in urine.8
The interest in protein-based materials, in particular as materials for drug delivery systems, has been considerable over the past few decades.2,23,24 In addition to the mentioned biocompatibility, especially, the well-defined peptide backbone provides attractive possibilities for bioconjugation and drug interaction. With or without polymer modification, various methods of particle preparation are possible, for example emulsification,25 desolvation,26 thermal gelation27 or spray drying.28 However, most methods require either dissolution or denaturation of the native proteins to obtain hydrophobic materials, or permanent chemical crosslinks to form stable particles are necessary.7
Alternatively, as previously shown by our group with lysozyme29 and modified polysaccharides,30 it is possible to prepare nanoparticles using mild nano-emulsion techniques31–33 with hydrophobic modified biopolymers. The use of ultrasonic methods for the preparation of oil-in-water emulsions is favorable for applications with biopolymers since they can be gentle enough to warrant their integrity.31 In our case, we applied a high surface PEGylation that made it possible to transfer proteins into organic solvents. By applying a single emulsion oil-in-water (O/W) solvent evaporation method, payloads can then be physically entrapped inside the particles between the biopolymer matrixes without the need for stabilizing crosslinking agents (Fig. 1).
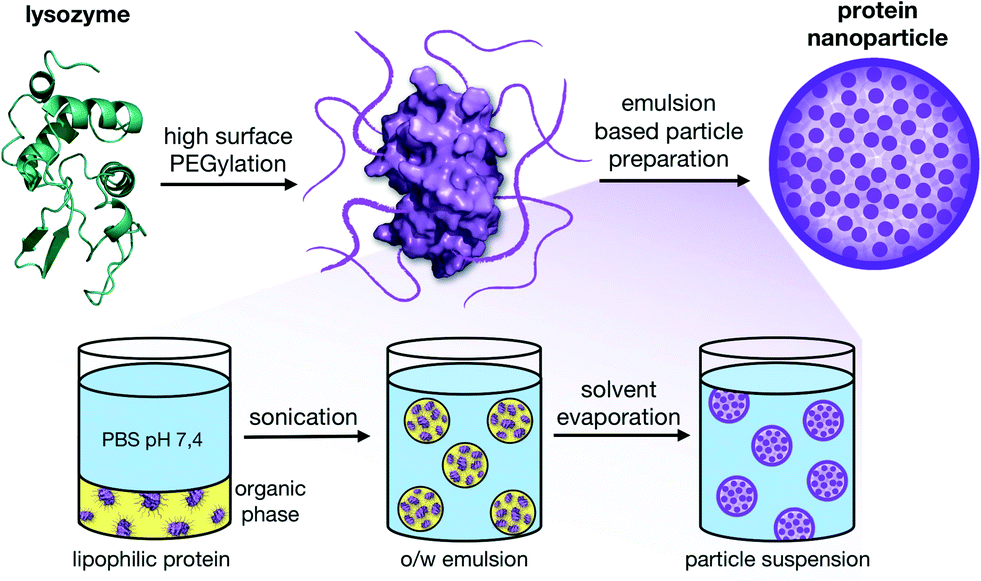 | ||
| Fig. 1 High surface PEGylation of native lysozyme results in single proteins that are soluble in organic solvents without denaturation. This lipophilic switch allows the application of a single emulsion solvent evaporation technique for the formation of stable nanoparticles without crosslinking. | ||
In our previous work, we reported the PEGylation of lysozyme using cyanuric chloride-modified PEG for the formation of nanoparticles and delivery of doxorubicin.29 Based on these results, we evaluated in the present study different methods for electrophilic PEG activation with the aim of finding a highly reactive and at the same time mild method for protein modification. We particularly focused on the preservation of the native structure and the enzymatic activity of lysozyme.
Results and discussion
mPEG activation
In order to activate methoxypolyethylene glycol (mPEG) for the reaction with nucleophilic groups on the protein surface, we functionalized mPEG with different electrophile linkers. The polymer activation was performed similar to previously published reactions with short and long ethylene glycol molecules.34–37For the formation of a reactive ester, we introduced tetrafluorophenol (TFP) to α-methoxy-ω-carboxy PEG (mPEG, 2000 g mol−1). We chose the tetrafluorophenyl ester (TFP-mPEG) over the corresponding NHS ester, since it is known to have better stability towards hydrolysis in aqueous solution, exhibiting a 10-fold increased half-life under slightly basic buffered conditions.38,39 The compound was isolated in 61% yield (see the ESI‡).
Using epichlorohydrin, the hydroxyl-functionalized mPEG can be activated, creating a glycidyl end group (epoxy-mPEG). The epoxy group is highly reactive towards primary amines on the surface of proteins. However, reactions with hydroxyl, imidazole and thiol groups may also take place.22 The yellow solid was isolated in good yield (92%). Despite the high reactivity, the compound is stable for 48 h in aqueous solution, allowing long reaction times for a high protein PEGylation.
Using p-nitrophenyl chloroformate, a reactive carbonate derivate of mPEG can be synthesized (carbonate-mPEG). The disadvantages of this compound are the relatively low stability towards hydrolysis and the need for an extensive purification procedure to remove p-nitrophenol after the reaction with the protein.
All three mentioned activation methods result in mono-functionalized mPEG according to size-exclusion chromatography (see the ESI‡).
As a fourth method, we evaluated the activation with cyanuric chloride (TsT). This linker strategy is well studied for polymer and PEG activation. The resulting materials show only minimal toxicity and have proven to be successful in various in vitro and in vivo therapeutic applications.40–44 As previously shown, mono-mPEG functionalized TsT has a high reactivity towards nucleophilic groups on protein surfaces.17,29,45 In order to increase the selectivity towards primary amines, we studied in the present work 2,4-mPEG modified triazines.8 The remaining single chlorine function should reduce the reactivity and allow a mild modification of lysine residues. According to elementary analysis and size-exclusion chromatography, it was possible to functionalize 88% of cyanuric chloride with two mPEG chains, leaving the rest mono-functionalized. As previously shown by our group, the cyanuric chloride-modified mPEG is a non-toxic material.29
Protein modification
The activated mPEGs were reacted with lysozyme (LYZ, 14.3 kDa), which we selected as our model enzyme system, to facilitate a solubility switch of the protein material. In order to achieve a high enough modification of the protein surface, the reactivity of the linear single stranded mPEGs had to be high enough to react with multiple accessible nucleophilic groups. This modification should lead to solubility of lysozyme in organic solvents with preservation of its native structure (Fig. 2).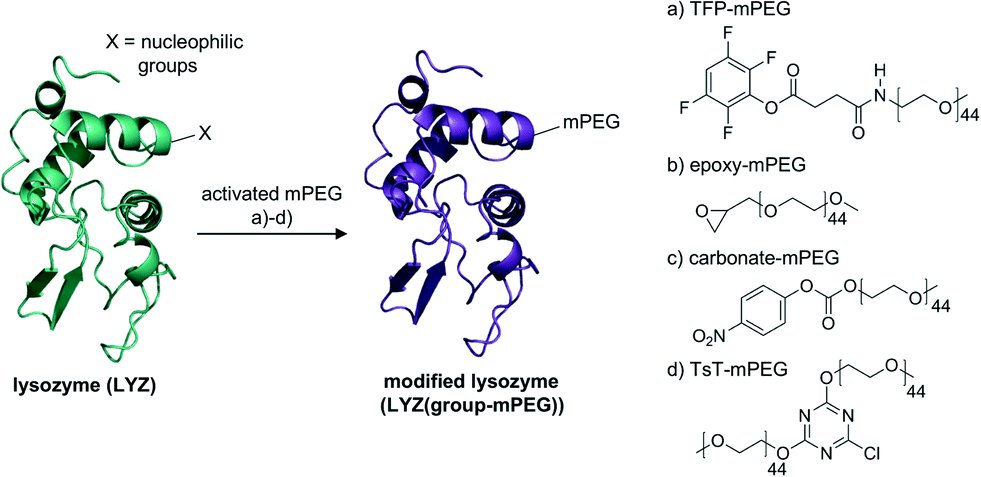 | ||
| Fig. 2 Protein modification with four different activated mPEGs (a–d). The nucleophilic groups on the protein surface can react with the PEGs without causing damage to the native protein structure. | ||
Most previously reported approaches focus on the site-specific (often a single PEG chain) modification of proteins to improve their pharmacokinetic properties.3 This study compares the effects of different high-amount surface PEGylations. We focus on the detailed analysis of the resulting lipophilic protein material, in particular the structural integrity and the remaining catalytic activity.
All modifications were performed in buffered aqueous solutions with a 3- to 20-fold excess of activated mPEG, followed by repeated dialysis to remove unreacted PEG. The activated mPEGs (a–d) led to lysozyme modifications with different molecular weights, indicating that the reactivity of each mPEG towards the protein surface varies. The use of TFP-mPEG (a) results in a lysozyme modification with a molecular weight of around 20–24 kDa (determined by SDS (Fig. 3) and MALDI-ToF MS (see the ESI‡)), introducing around four 2 kDa mPEG chains per protein (Table 1). We performed a fluorescamine assay to determine the remaining primary amines on the surface of the protein after the modification. In comparison with the native enzyme that bears seven free amines on the surface, we detected for the modification with TFP-mPEG only one free primary amine. The difference in the degree of modification determined via SDS and MALDI-ToF MS in comparison with this fluorescence-based assay underlines the limits of the fluorescamine assay for the analysis of highly PEGylated proteins. The attached polyethylene glycol chains shield off still available amines on the protein surface, resulting in false/higher modification efficiencies.
 | ||
| Fig. 3 SDS-PAGE (15%) of non-PEGylated (lanes 0 and 5) and PEGylated hen egg white lysozymes (lanes 1–4). PEGylation was performed with TFP-mPEG (lane 1), epoxy-mPEG (lane 2), carbonate-mPEG (lane 3) and TsT-mPEG (lane 4). (Marker (M) is a PageRuler pre-stained protein ladder; each lane was loaded with 20 μg of the material; the gel was stained with Coomassie brilliant blue G). | ||
| Sample | Molecular weight/kDa | Remaining amines | Protein activity/% | mPEG chains | |
|---|---|---|---|---|---|
| SDS | MALDI | ||||
| LYZ(TFP-mPEG) | 21–24 | 20–24 | 1.17 | 60 | 4 |
| LYZ(epoxy-mPEG) | 23–130 | 30–34 | 1.32 | 80 | 9 |
| LYZ(carbonate-mPEG) | 26–36 | 26–30 | 0.47 | 62 | 7 |
| LYZ(TsT-mPEG) | 36–55 | 33–40 | 0.04 | 13 | 10–12 |
The lysozyme modification with epoxy-mPEG results in higher molecular weights of 30–34 kDa (SDS, MALDI-ToF MS) corresponding to around 9 attached PEG chains. Apparently, the higher reactivity of the epoxide compared to active esters, leads to the reaction with additional nucleophilic groups on the protein surface, for example any of the three available tyrosines.
The modification of LYZ with carbonate-mPEG leads to narrowly defined products similar to the TFP-mPEG modification, but with slightly higher molecular weights around 26–30 kDa (see also Fig. 3). This indicates that around 7 mPEG chains per protein were introduced.
The reaction with TsT-mPEG yields products with a molecular weight of 33–40 kDa (SDS, MALDI-ToF MS). Since one TsT molecule carries two 2 kDa mPEG chains, 5–6 TsT molecules were introduced onto the protein surface, leading to around 10–12 mPEG chains overall. This represents the highest PEG modification compared to the other used mPEGs. Similarly, the fluorescamine assay confirms the high surface shielding by indicating that no free amines are available.
After modification, all proteins were soluble in organic solvents and used later for the preparation of nanoparticles.
In addition to increasing the lipophilicity of the proteins, we also tested for the structural integrity and remaining enzymatic activity after PEGylation. It was the initial aim to perform a mild surface modification without harming the native three-dimensional structure. Therefore, we performed a fluorescence assay with the synthetic lysozyme substrate 4-methylumbelliferyl-β-D-N,N′,N′′ triacetylchitotrioside to determine the enzymatic activity of the PEGylated samples compared to native LYZ (see the ESI‡). Epoxy-mPEG modified lysozyme showed the highest remaining activity of 80%, followed by carbonate-mPEG and TFP-mPEG modified LYZ with an activity of 60–62% compared to the native enzyme. Apparently, the active site of lysozyme is not significantly affected by the surface PEGylation. Most likely, the reduced activity can be attributed to the shielding effects of the PEG chains, restricting the access of the trisaccharide substrate to the active site. In comparison, the modification with TsT-mPEG leads to a significant loss of activity (19%, similar to a previous report with mono-mPEG functionalized TsT29). This is in accordance with the high amount of PEGylation seen in SDS and MALDI-ToF MS experiments, indicating both high shielding and a possible modification of amino acids close to the active site.
In addition, CD spectroscopy was performed to analyze the protein conformation after PEGylation (Fig. 4B and ESI‡). For all modifications, the CD spectra are very similar to that of native lysozyme, suggesting that most secondary structure elements were preserved and no or only minimal unfolding or denaturation occurred.
 | ||
| Fig. 4 Protein activity assay for native LYZ and surface modified samples (detailed results are given in Table 1 and ESI‡) (A); CD spectra indicate no loss of secondary structure elements after PEGylation (B); after the formation of nanoparticles, the NTA measurements for all protein particles result in average diameters of around 100–130 nm (C) (as an example, epoxy-mPEG modified LYZ is shown here). TEM images of the particles demonstrate that individual proteins (small black dots within the particle) assemble into spherical particles. | ||
Nanoparticle preparation
As previously shown, a single-emulsion based method was used for the formulation of non-toxic, stable nanoparticles.29 The high surface PEGylation renders the proteins fully soluble in organic solvents and an emulsion-based oil-in-water (o/w) solvent evaporation method for the formation of nanoparticles is possible. This lipophilic switch of the proteins has the advantage that no denaturation steps are necessary for the formation of a hydrophobic material as part of the nanoparticle formation. Also, the native conformation of the biomacromolecule can be retained and no crosslinking agents to stabilize the resulting nanoparticles are necessary.For this, the PEGylated proteins were dissolved in dichloromethane and then added to a PBS buffer (pH 7.4). This biphasic system was sonicated, resulting in a nano-emulsion. After the evaporation of the organic solvent, protein nanoparticles with an average diameter of around 100–130 nm formed (see the ESI‡). TEM images of the particles show that individual proteins (dLYZ = 3.5 nm for native lysozyme; dLYZ = 17 nm after PEGylation) assemble into spherical particles. Each nanoparticle contains, depending on the diameter, around 250–450 modified proteins (Fig. 4C).
The nanoparticles show a zeta potential in the range of −13 to −19 mV (see the ESI‡). The unmodified lysozyme has a positive zeta potential, whereas the PEG-modified proteins have a slightly negative potential due to the functionalization of the positively charged amino acids on the surface of the proteins. The negative zeta potential of the final nanoparticles is, in particular, beneficial for a stable particle suspension since it prevents aggregation and unwanted nonspecific interactions e.g. with blood proteins.46,47 Stored at 4 °C in the PBS buffer, the nanoparticle suspensions are stable over prolonged time (min. 2 months) (see the ESI‡).
Conclusions
We evaluated four different methods for a high density PEGylation of proteins. For this, linear PEG chains were end-group activated to react with nucleophilic amino acids on the surface of lysozyme in order to increase their lipophilicity. The solubility in organic solvents allows the application of an emulsion-based solvent evaporation method for the formation of nanoparticles. Using the modified proteins as particle materials, it is possible to form stable nanoparticles in the size range of 100–130 nm without crosslinking or denaturation steps. The resulting nanoparticle properties (size, surface potential, particle stability and toxicity) meet all the requirements necessary for therapeutic applications as delivery platform.We could show that the efficiency and extent of protein modification significantly vary among the different PEGylation methods. The lowest amount of surface modification was achieved with TFP-activated mPEGs (4 chains), whereas epoxy-mPEG and carbonate-mPEG resulted in a higher amount with around 7–9 mPEG chains per protein. The most PEG chains (10–12) can be introduced with TsT-mPEG, partly since each protein reactive TsT group carries two PEG chains. The different reactivities and modification efficiencies are important to consider when different sized proteins and enzymes are modified. However, in our case of the relatively small lysozyme (14.3 kDa), all methods led to the desired protein modifications that are soluble in organic solvents. Therefore, secondary criteria are gaining importance in the selection of the best PEGylation method. When looking at the initial synthesis of the activated mPEG chain, both TFP and epoxy end groups demonstrate the easiest, quickest and cleanest synthesis routes. Since the activation with epichlorohydrin requires standard –OH terminated mPEG (compared to a carboxylic acid for TFP activation), the epoxy activation should be preferred. Similarly, when looking at changes in structure and activity of the protein, mild and non-destructive surface PEGylations are preferable. Again, the modification with epoxy-mPEG is the method of choice, due to the relatively minor loss of catalytic activity (20% less, compared to native lysozyme).
In summary, the presented modification methods have the potential to be universally applied for any protein of interest, in order to increase its lipophilic character. The new results extend the current toolkit of possible methods for the high-surface PEGylation of proteins and can advance the development of new and innovative protein-based materials. This is in particular interesting for the production of catalytically active materials or biocatalysts in organic solvents.
Experimental
Materials and methods
All chemicals and reagents are commercially available and were used directly without further purification. 1H and 13C NMR spectra were recorded at 300 MHz or 600 MHz by using a Bruker NMR spectrometer. FT-IR spectra were recorded with a Nicolet Avatar 330-IR ATR-Unit from Thermo Electron Corporation. Size-exclusion chromatography was performed on an Agilent 1100 Series using PSS (Polymer Standards Service) as the standard. SDS-PAGE was performed as described elsewhere48 using a 15% polyacrylamide gel (Rothiphorese® 30 gel mix). The gel was stained with Coomassie Brilliant Blue G and a PageRuler pre-stained protein ladder (10–170 kDa) was used as the marker. MALDI-ToF MS measurements were performed with a Shimadzu Axima CFR MALDI-ToF mass spectrometer, equipped with a nitrogen laser delivering 3 ns laser pulses at 337 nm. CD spectra were recorded on a J-815 (JASCO). Nanoparticle Tracking Analysis (NTA) was performed on a NanoSight LM 14 equipped with a green laser (532 nm). Zeta potential measurements were performed on a Zetasizer Nano ZS instrument (Malvern).General procedure for mPEG activation
For the activation of methoxypolyethylene glycol (2000 g mol−1; Sigma-Aldrich, St. Louis), either cyanuric chloride (TsT), epichlorohydrin (epoxy) or p-nitrophenyl chloroformate (carbonate) was used. To obtain the tetrafluorophenol (TFP) active ester, α-methoxy-ω-carboxy PEG (2000 g mol−1; Rapp Polymer, Germany) was used. Detailed descriptions of the reaction conditions, yields and analytical data are given in the ESI.‡Modification of LYZ with TFP-mPEG
Lysozyme (15.0 mg, 1.0 μmol) was dissolved in 3 mL of 0.1 M carbonate buffer (pH 8) and combined with TFP-mPEG (48.4 mg, 22.0 μmol). The mixture was stirred at room temperature for 48 h. The product was purified by dialysis (Slide-A-Lyzer® dialysis cassettes, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO) against water for 3 days and was then lyophilized.
000 MWCO) against water for 3 days and was then lyophilized.
Modification of LYZ with epoxy-mPEG
Lysozyme (15.0 mg, 1.0 μmol) was dissolved in 3 mL of 0.1 M carbonate buffer (pH 8) and combined with epoxy-mPEG (44.0 mg, 22.0 μmol). The mixture was stirred at room temperature for 48 h. The product was purified by dialysis (Slide-A-Lyzer® dialysis cassettes, 10000 MWCO) against water for 3 days and was then lyophilized.
Modification of LYZ with carbonate-mPEG
Lysozyme (15.0 mg, 1.0 μmol) was dissolved in 3 mL of 0.1 M carbonate buffer (pH 8) and combined with carbonate-mPEG (44.0 mg, 22.0 μmol). The mixture was stirred at room temperature for 48 h. The product was purified by dialysis (Slide-A-Lyzer® dialysis cassettes, 10000 MWCO) against water for 7 days and was then lyophilized.
Modification of LYZ with TsT-mPEG
Lysozyme (5.0 mg, 0.3 μmol) was dissolved in 1 mL of 0.1 M borate buffer (pH 10) and combined with activated TsT-mPEG (195.70 mg, 48.9 μmol). After 2 h at 40 °C, the reaction was stopped with 2 mL of the phosphate buffer (pH 6). The excess of mPEG was removed with Microsep™ centrifugal devices (MWCO 30 kDa, PALL Corporation) and the resulting solution was lyophilized.Fluorescamine assay
The average number of free amino groups on the LYZ surface (after PEGylation) was determined by conducting a fluorescamine assay. All protein samples were dissolved in PBS (pH 7.4) at a concentration of 2 mg mL−1 and native lysozyme at a concentration of 0.2 mg mL−1. Hexylamine was used as the external standard in the concentration range of 19–40 μg mL−1 and was prepared similarly. Then, 125 μL of PBS (pH 7.4) was pipetted into each well of a 96-black-well-microplate (flat bottom). 25 μL of each sample, 25 μL of PBS (blank) or 25 μL of the hexylamine standard were added in triplicate to each well. Finally, 50 μL of 0.3 mg mL−1 fluorescamine solution (in acetone) was added, then mixed and measured immediately. For all measurements, the excitation wavelength was set to 380 nm, while the emission wavelength was set to 460 nm.Protein activity assay
PEGylated LYZ was dissolved in 0.1 M phosphate buffer (pH 5.2) and diluted to a final protein concentration of 2 μM. A solution of 4-methylumbelliferyl-β-D-N,N′,N′′ triacetylchitotrioside ((GlcNAc)3MeU) (20 μM in the same buffer) was preheated to 42 °C for 5 min. 200 μL of each solution were combined and further incubated at 42 °C. Every 30 min, 50 μL of each sample were transferred to 300 μL of 0.5 M glycine buffer (pH 12.0) to stop the catalytic activity of the protein and enhance the fluorescence intensity of methylumbelliferone. A solution of native lysozyme (2 μM), treated under the same conditions, was used as a reference. For all measurements, the excitation wavelength was set to 380 nm, while the emission wavelength was set to 460 nm.Nanoparticle preparation
A single-emulsion solvent evaporation method29 was applied for the preparation of nanoparticles using the PEGylated LYZ samples. LYZ(TFP-mPEG), LYZ(epoxy-mPEG), LYZ(carbonate-mPEG) and LYZ(TsT-mPEG) were dissolved at a concentration of 2.5 mg mL−1 in ice cold dichloromethane (DCM, 0.5 mL) and added to 2.5 mL of an ice cold PBS (pH 7.4) buffer. The mixture was sonicated for 45 s on ice, using an ultrasonicator (Bandelin Ultrasonic Homogenisator Sonoplus UW 70, Germany). The emulsion was stirred in a well-ventilated hood overnight for the evaporation of DCM.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) as part of the collaborative research center SFB 1066 (Project A2). We thank Monika Schmelzer of the working group of Prof. Dr. H. Frey (JGU Mainz, Germany) for the SEC measurements. Additionally, we thank Prof. Dr. P. Besenius, Prof. Dr. T. Efferth, Prof. Dr. B. Epe, and Prof. Dr. T. Schirmeister (JGU Mainz, Germany) for the kind permission to use some of their analytical equipment.References
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 157, 168–182 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- D. E. Borchmann, T. P. Carberry and M. Weck, Macromol. Rapid Commun., 2014, 35, 27–43 CrossRef CAS PubMed.
- M. A. Gauthier and H. A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- L. S. Witus and M. B. Francis, Acc. Chem. Res., 2011, 44, 774–783 CrossRef CAS PubMed.
- Y. Wu, D. Y. Ng, S. L. Kuan and T. Weil, Biomater. Sci., 2015, 3, 214–230 RSC.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- D. Pfister and M. Morbidelli, J. Controlled Release, 2014, 180, 134–149 CrossRef CAS PubMed.
- C. J. Fee and J. M. Van Alstine, Chem. Eng. Sci., 2006, 61, 924–939 CrossRef CAS.
- M. Barz, R. Luxenhofer, R. Zentel and M. J. Vicent, Polym. Chem., 2011, 2, 1900–1918 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442–1448 RSC.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- C. Mattos and D. Ringe, Curr. Opin. Struct. Biol., 2001, 11, 761–764 CrossRef CAS PubMed.
- Y. Inada, K. Takahashi, T. Yoshimoto, A. Ajima, A. Matsushima and Y. Saito, Trends Biotechnol., 1986, 4, 190–194 CrossRef CAS.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- E. Muller, D. Josic, T. Schroder and A. Moosmann, J. Chromatogr. A, 2010, 1217, 4696–4703 CrossRef PubMed.
- K. Mayolo-Deloisa, M. E. Lienqueo, B. Andrews, M. Rito-Palomares and J. A. Asenjo, J. Chromatogr. A, 2012, 1242, 11–16 CrossRef CAS PubMed.
- B. Castillo, R. J. Sola, A. Ferrer, G. Barletta and K. Griebenow, Biotechnol. Bioeng., 2008, 99, 9–17 CrossRef CAS PubMed.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS PubMed.
- L. Yang, F. Cui, D. Cun, A. Tao, K. Shi and W. Lin, Int. J. Pharm., 2007, 340, 163–172 CrossRef CAS PubMed.
- K. Langer, S. Balthasar, V. Vogel, N. Dinauer, H. von Briesen and D. Schubert, Int. J. Pharm., 2003, 257, 169–180 CrossRef CAS PubMed.
- S. Yu, P. Yao, M. Jiang and G. Zhang, Biopolymers, 2006, 83, 148–158 CrossRef CAS PubMed.
- S. H. Lee, D. Heng, W. K. Ng, H.-K. Chan and R. B. H. Tan, Int. J. Pharm., 2011, 403, 192–200 CrossRef CAS PubMed.
- M. Fach, L. Radi and P. R. Wich, J. Am. Chem. Soc., 2016 DOI:10.1021/jacs.6b06243.
- E. M. Bachelder, T. T. Beaudette, K. E. Broaders, J. Dashe and J. M. Frechet, J. Am. Chem. Soc., 2008, 130, 10494–10495 CrossRef CAS PubMed.
- R. Silva, H. Ferreira and A. Cavaco-Paulo, Biomacromolecules, 2011, 12, 3353–3368 CrossRef CAS PubMed.
- I. Sole, C. Solans, A. Maestro, C. Gonzalez and J. M. Gutierrez, J. Colloid Interface Sci., 2012, 376, 133–139 CrossRef CAS PubMed.
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- L. Zhang, C. R. Robertson, B. R. Green, T. H. Pruess, H. S. White and G. Bulaj, J. Med. Biochem., 2009, 52, 1310–1316 CAS.
- S.-J. Sung, S. H. Min, K. Y. Cho, S. Lee, Y.-J. Min, Y. I. Yeom and J.-K. Park, Biol. Pharm. Bull., 2003, 26, 492–500 CAS.
- R. Qi, Y. Gao, Y. Tang, R.-R. He, T.-L. Liu, Y. He, S. Sun, B.-Y. Li, Y.-B. Li and G. Liu, AAPS J., 2009, 11, 395–405 CrossRef CAS PubMed.
- K. Ono, Y. Kai, H. Maeda, F. Samizo, K. Sakurai, H. Nishimura and Y. Inada, J. Biomater. Sci., Polym. Ed., 1991, 2, 61–65 CrossRef CAS PubMed.
- K. R. Gee, E. A. Archer and H. C. Kang, Tetrahedron Lett., 1999, 40, 1471–1474 CrossRef CAS.
- M. R. Lockett, M. F. Phillips, J. L. Jarecki, D. Peelen and L. M. Smith, Langmuir, 2008, 24, 69–75 CrossRef CAS PubMed.
- J. Lim and E. E. Simanek, Adv. Drug Delivery Rev., 2012, 64, 826–835 CrossRef CAS PubMed.
- D. P. Blackall, J. K. Armstrong, H. J. Meiselman and T. C. Fisher, Blood, 2001, 97, 551–556 CrossRef CAS PubMed.
- H. Li, H. Zhou, S. Krieger, J. J. Parry, J. J. Whittenberg, A. V. Desai, B. E. Rogers, P. J. Kenis and D. E. Reichert, Bioconjugate Chem., 2014, 25, 761–772 CrossRef CAS PubMed.
- Z. N. Li, Z. F. Yuan, G. Y. Mu, M. Hu, L. J. Cao, Y. L. Zhang and M. X. Ge, Exp. Ther. Med., 2015, 10, 889–894 CAS.
- R. Banerjee, N. J. Pace, D. R. Brown and E. Weerapana, J. Am. Chem. Soc., 2013, 135, 2497–2500 CrossRef CAS PubMed.
- T. Wongpinyochit, P. Uhlmann, A. J. Urquhart and F. P. Seib, Biomacromolecules, 2015, 16, 3712–3722 CrossRef CAS PubMed.
- S. Das and A. Chaudhury, AAPS PharmSciTech, 2011, 12, 62–76 CrossRef CAS PubMed.
- C. Freitas and R. H. Muller, Int. J. Pharm., 1998, 168, 221–229 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interest. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00475f |
| This journal is © The Royal Society of Chemistry 2016 |