
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of triazole-based peptidomimetics of a PSD-95 PDZ domain inhibitor†‡
Anders
Bach
*,
Thomas B.
Pedersen
and
Kristian
Strømgaard
Department of Drug Design and Pharmacology, University of Copenhagen, Universitetsparken 2, DK-2100 Copenhagen, Denmark. E-mail: anders.bach@sund.ku.dk; Tel: +45 3533 6242
First published on 18th January 2016
Abstract
PSD-95 PDZ domains are biologically important and promising drug targets. Here, we discover a triazole-based peptidomimetic, 10, by ‘click chemistry’. Compound 10 inhibits the PDZ2/GluN2B interaction with affinity similar to tripeptide SAV and better than current small-molecules. Thus, 10 represents a new class of PSD-95 PDZ inhibitors.
1. Introduction
Postsynaptic density protein-95 (PSD-95) is a scaffolding protein found in neuronal synapses.1 PSD-95 contains three postsynaptic density protein-95/disks-large/zonula occludens-1 (PDZ) domains, which interact with the N-methyl-D-aspartate (NMDA) receptor and signalling proteins such as neuronal nitric oxide synthase (nNOS).2 Compounds targeting PDZ1 and PDZ2 of PSD-95 have shown promising effects in animal models of ischemic stroke,3–5 pain6–8 and neurodegenerative diseases,9 as they can disconnect disease-related excessive NMDA receptor activity from harmful down-stream mechanisms.3,10 PSD-95 is particularly promising as a drug target in relation to ischemic stroke, and peptide-based inhibitors are being pursued as drug candidates in preclinical and clinical trials.4,11PDZ domains are highly abundant in the human proteome and are often found in scaffolding proteins where they mediate protein–protein interactions and form signalling networks of key importance to cellular biology.12,13 In the canonical binding mode PDZ domains bind the C-terminal peptide part (4–7 amino acids) of its interaction partner (Fig. 1A and B). The PDZ binding pocket is created by the βB strand and αB helix of the PDZ domain and is narrow and shallow and thus well suited to recognize peptides.14–16 A small hydrophobic sub-pocket binds the side chain of the C-terminal amino acid (position P0, typically a Val, Leu or Ile) and serves as an anchor point for the interaction with peptide. The C-terminal carboxylic acid of the peptide ligand is crucial for binding17 and is accommodated by the conserved PDZ ‘GLGF’ loop.18 Peptides binding to PDZ domains form an anti-parallel β-sheet with the PDZ βB strand via amide backbone hydrogen bonds.14,18,19 Structure–activity relationship (SAR) investigations of both peptide and protein backbone have revealed that not all amide atoms of the peptide engage directly with the PDZ domain. Especially, the P−1 amino acid does not interact with PDZ, but seem to act more as a spacer that ensures the right positioning of the essential P0 and P−2 residues (Fig. 1A and B).18–20
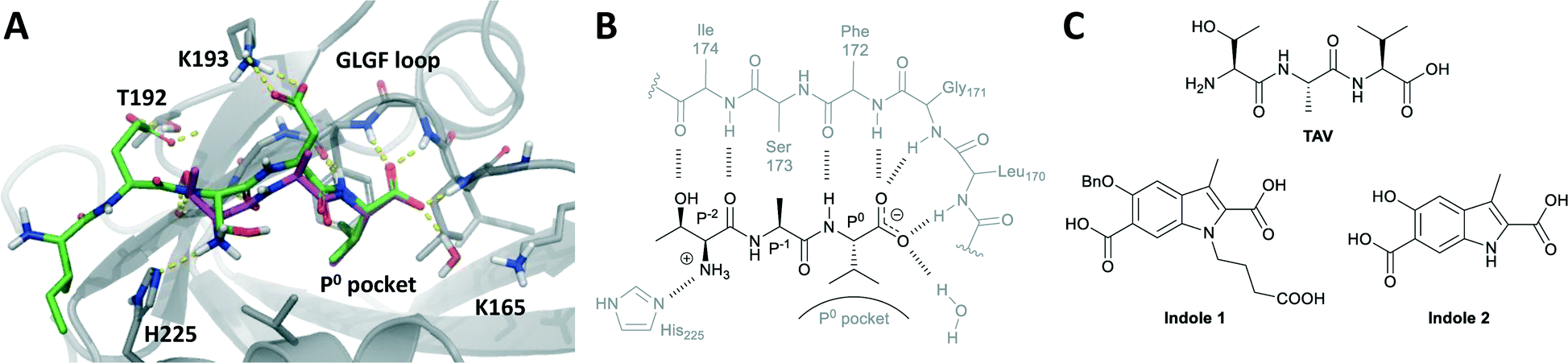 | ||
| Fig. 1 (A) pentapeptide IESDV (green), derived from the GluN2B sequence, and tripeptide TAV (magenta) docked into PSD-95 PDZ2. (B) Schematic 2D plot of TAV–PDZ2 interactions. (C) Reference compounds used herein: tripeptide TAV and indole-based small-molecules. | ||
Truncation studies of peptide ligands targeting PSD-95 have shown that pentapeptides display affinities similar to longer peptides, while tetra- and tripeptides show a stepwise reduction in affinity.15 pentapeptides have subsequently been derivatized into dimeric inhibitors with nanomolar potency.4,21 Likewise, the affinity of tetrapeptides has been increased about 20-fold by N-alkylations and stability can be enhanced by replacing amides with thioamides.15,20 Also, cyclization and expansion of the P−1 side chain of hexapeptides have led to peptidomimetic structures with increased affinities.22,23
Developing small-molecules targeting PDZ domains have shown to be difficult. Only few small-molecule inhibitors have been reported and affinities are in the low micromolar range in best cases.13 Until recently, no small-molecule PSD-95 PDZ inhibitors existed. However, Vogrig et al. reported a series of indole-based compounds (Fig. 1C) able to interact with PSD-95 PDZ domains (1H–15N heteronuclear single-quantum coherence, HSQC, NMR), inhibit the PSD-95-PDZ/5-HT2A receptor interaction (pull-down) and relieve pain in hyperalgesic rats.24
Herein, we pursue a peptidomimetic strategy introducing a rigid scaffold into a short PSD-95 peptide ligand with the potential of improving affinity, selectivity, and stability and provide novel chemotypes targeting PSD-95. We used the tripeptide Thr-Ala-Val (TAV) as starting point (Fig. 1A–C), which binds PDZ1 and PDZ2 with medium affinity (PDZ2: Ki ∼ 40 μM). TAV was identified from a peptide SAR study of the GluN2B C-terminal revealing that P0 Val is essential for affinity, P−1 Asp could be replaced with Ala, and Thr instead of Ser in P−2 leads to increased affinity.15 As TAV is small and reasonably potent it is an attractive starting point for converting peptides into more small-molecule-like compounds. Here, we prepared azide and alkyne analogues of the corresponding amino acids and dipeptides of TAV, which then served as building blocks for subsequent copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reactions thereby producing a series of 1,2,3-triazole containing analogues of TAV.25,26 Triazole resembles amide in planarity, size and electronic properties and has previously been used as an amide bioisoster resulting in potent and protease stable compounds.27–30 Thus, here we systematically investigated triazole as a potential bioisoster in PSD-95 PDZ domain peptide inhibitors and the possibility to rigidify the peptide backbone by implementing this small and aromatic ring. This strategy led to the identification of a new triazole-containing peptidomimetic against the PDZ2 domain of PSD-95.
2. Results and discussion
2.1. Design and synthesis
In the design process we first fragmentized TAV into the amino acid and dipeptide moieties (Thr, Thr-Ala, Ala-Val and Val) and replaced the resulting amines with azide groups and carbonyls with methyl azides to generate the four azide building blocks (1–4, Fig. 2). Likewise, carbonyls of peptide fragments were replaced with acetylene to get alkyne 5 and 6. Additionally, alkynes 7–8 were made where the acetylene replaces the central part of P−1 Ala and the important P0 and P−2 (Val and Thr) amino acids were preserved (Fig. 2). For 8, an alcohol group needed to replace the intended carbonyl in order to provide a water-stable building block. For both 7 and 8 it was envisioned that the carbonyl/alcohol could facilitate hydrogen bond interactions with the PDZ backbone. After preparing the building blocks, the strategy was to react the four azides (1–4) with the four alkynes (5–8) applying the CuAAC reaction so that the corresponding triazole structures (9–18) contained at least one Val and carboxylic acid group intended to bind into the PDZ domain P0 pocket (Fig. 2). This systematic strategy allows for a thorough investigation of the idea of introducing a rigid scaffold into the TAV backbone.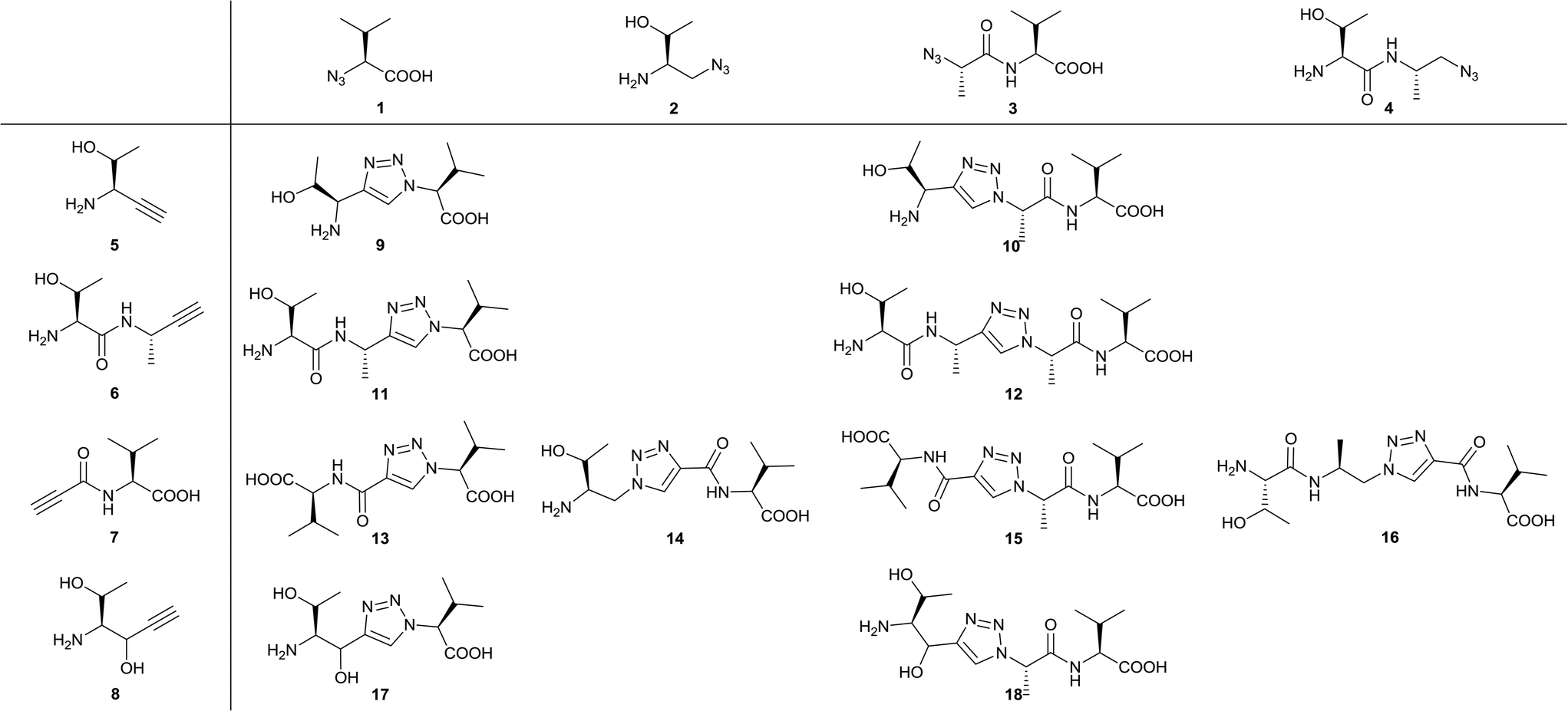 | ||
| Fig. 2 Azides 1–4 and alkynes 5–8 are combined into triazole-based peptidomimetic structures 9–18 all bearing a C-terminal Val and carboxylic acid. | ||
The synthesis of azides (1–4) and alkynes (5–8) were carried out in satisfactorily yields generally following standard procedures (Schemes 1 and 2). Azide 1 was synthesized in one step from L-valine in a diazo transfer reaction mediated by freshly prepared triflyl azide according to literature procedure.31,32 To generate azide 2, 19 (prepared by reduction of Boc- and tert-butyl protected L-threonine33) was mesylated on the alcohol group to provide intermediate 20 followed by a nucleophilic substitution reaction by treatment of sodium azide34 to get 21. Azide 2 was then obtained under mild deprotection conditions applying HCl in dioxane (Scheme 1). Azide 3 was synthesized by a solid-phase procedure starting from a Wang resin preloaded with Fmoc-protected L-valine (22). After deprotecting the Fmoc, (S)-2-azidopropanoic acid31 was coupled to the free valine amine to get resin-bound intermediate 23. The desired azide 3 was obtained by cleaving the resin under standard conditions with trifluoroacetic acid (TFA) (Scheme 1). Azide 4 was synthesized by coupling 24 (ref. 35) with Fmoc- and tert-butyl protected L-threonine using HBTU followed by deprotection under mild acidic conditions (Scheme 1).
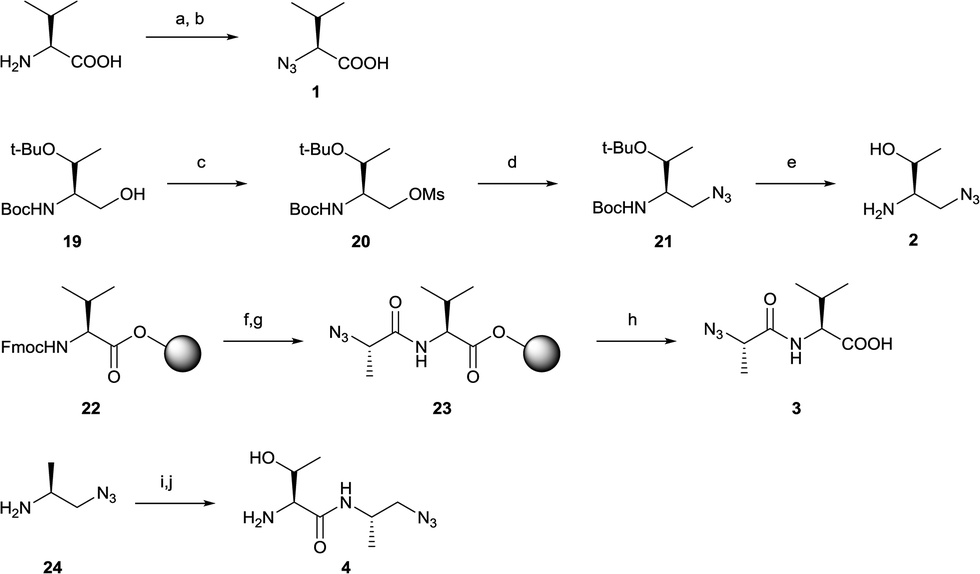 | ||
| Scheme 1 Synthesis of azides 1–4. Reagents and conditions: (a) Tf2O, NaN3, H2O, CH2Cl2, 0 °C, 2 h; (b) CuSO4, K2CO3, H2O, CH3OH, CH2Cl2, rt, overnight; (c) MsCl, Et3N, CH2Cl2, 0 °C to rt, 4 h; (d) NaN3, DMF, 50 °C, 18 h; (e) 4 M HCl in dioxan, rt, 1.5 h; (f) 20% piperidine in DMF; (g) (S)-2-azidopropanoic acid, HATU, HOAt, collidine, DMF, rt, 3 h; (h) TFA/tips/H2O (90/5/5), rt, 2 h; (i) Boc-Thr(tBu)-OH, HBTU, DIPEA, DMF, rt, 12 h; (j) 4 M HCl in dioxan, rt, 1.5 h. | ||
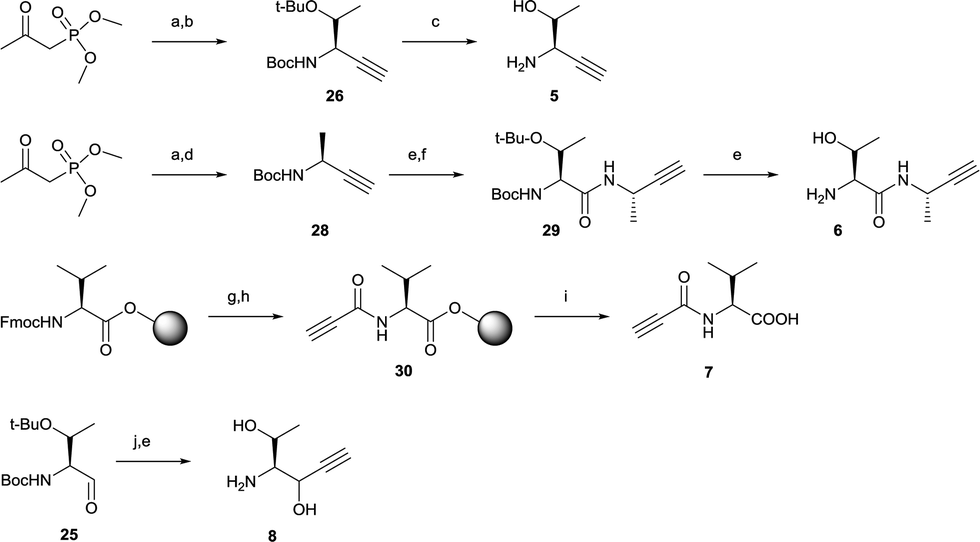 | ||
Scheme 2 Synthesis of alkynes 5–8. Reagents and conditions: (a) p-TsN3, K2CO3, CH3CN, rt, 2 h; (b) Boc-Thr(tBu)-CHO (25), CH3OH, rt, overnight; (c) (CH3)3Sil, CH2Cl2, rt, 10 min; (d) Boc-Ala-CHO (27), CH3OH, rt, overnight; (e) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 (1:1), 0 °C; (f) Boc-Thr(tBu)-OH, HATU, DIPEA, DMF, rt, 3 h; (g) 20% piperidine in DMF; (h) propiolic acid, EEDQ, DMF, rt, 2 h; (i) TFA:CH2Cl2:H2O (47.5:47.5:5), rt, 2 h; (j) ethynylmagnesium chloride, THF, 0 °C, 1 h. :CH2Cl2 (1:1), 0 °C; (f) Boc-Thr(tBu)-OH, HATU, DIPEA, DMF, rt, 3 h; (g) 20% piperidine in DMF; (h) propiolic acid, EEDQ, DMF, rt, 2 h; (i) TFA:CH2Cl2:H2O (47.5:47.5:5), rt, 2 h; (j) ethynylmagnesium chloride, THF, 0 °C, 1 h. | ||
Next, alkyne building blocks (5–8) were synthesized. To prepare 5, the Boc- and tert-butyl protected L-threonine was first converted to the corresponding aldehyde Boc-Thr(tBu)-CHO (25).33 This aldehyde was then reacted with freshly in situ prepared Bestmann–Ohira reagent (dimethyl diazo-2-oxopropylphosphonate),36 and the resulting intermediate alkyne 26 was readily deprotected in iodotrimethylsilane to generate 5 (Scheme 2).37 Likewise, the synthesis of alkyne 6 started with aldehyde 27 (Boc-Ala-CHO38) and reaction with the Bestmann–Ohira reagent to generate 28 (Scheme 2). After deprotecting the Boc this intermediate was coupled with Boc-Thr(tBu)-OH followed by another deprotection to facilitate alkyne 6. Alkyne 7 was generated by a solid-phase strategy where propiolic acid was coupled to resin-bound L-valine using N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ)25,39 to get intermediate 30 followed by cleavage from the resin (Scheme 2). Alkyne 8 was synthesized by a Grignard reaction where ethynylmagnesium chloride was added to the carbonyl of aldehyde 25 followed by deprotection with TFA (Scheme 2).
The triazole-based peptidomimetic structures 9–18 were then synthesized by standard CuAAC reaction conditions using a 1:1 ratio of alkyne and azide, 0.4 eq. CuSO4 and 2 eq. sodium L-ascorbate in water/tert-butanol at room temperature (ESI‡). For 11–18, the reactions completed within 30 minutes and compounds were obtained in reasonably to fine yields (>30%) after HPLC purification. Triazoles 9 and 10, both based on alkyne 5, needed modified conditions to give satisfactorily yields (66–67%). For 9, heating to 55 °C for 18 hours was applied, while for 10, acetonitrile (20%) replaced tert-butanol, more sodium L-ascorbate (6 eq.) was added together with DIPEA and reaction time was prolonged to 65 hours at room temperature. Solutions of all tested compounds were prepared in deuterated PBS buffer and concentrations accurately determined by qNMR prior to assay tests.
2.2. Biological evaluation
Triazole-based compounds 9–18 were tested in a fluorescence polarization (FP) assay in a heterologous competition mode.15,20,21 Increasing concentrations of test substance were added to a fixed concentration of PDZ protein and fluorescent peptide probes, which were prepared by Cy5-labeling 11-mer peptides corresponding to the C-termini of the GluN2B subunit of NMDA receptors or CRIPT.15 We first tested 9–18 against PDZ2 of PSD-95 together with the control tripeptides TAV and the weaker binding tripeptide SAV.15 To compare the triazole compounds with current leading small-molecule PSD-95 PDZ inhibitors we synthesized Indole 1 and 2 (Fig. 1C) according to literature24 and tested them in the FP assay.Among 9–18, compound 10 showed robust inhibition of the PDZ2/probe interaction with a Ki of 289 μM (Fig. 3). This affinity is ∼8-fold lower than the TAV tripeptide (Ki = 38 μM) from which it was designed, but within the same affinity-range as SAV (Ki = 229 μM). Triazoles 11 and 13 showed some sign of inhibition (Fig. 3), but affinities were too low to reliably determine the Ki values. Surprisingly, Indole 1 only demonstrated marginal inhibitory activity towards PDZ2, and a Ki value could not be determined due to a high bottom plateau. Indole 2 was found more potent and gave a credible inhibition curve and a Ki value of 530 μM towards PDZ2 (Fig. 3).
 | ||
| Fig. 3 FP assay: triazoles and reference compounds tested for affinity to PDZ1 and PDZ2 of PSD-95. Cy5-GluN2B is used as probe at 50 nM; [PDZ2] = 3.75 μM; [PDZ1] = 15 μM. For PDZ2, the following Ki values were determined: 38 ± 9 μM (TAV), 229 ± 15 μM (SAV), 530 ± 96 μM (Indole 2) and 289 ± 41 μM (10). Ki values for Indole 1, 11 and 13 are >500 μM. For PDZ1, the following Ki values were determined: 74 ± 8 μM (TAV) and 480 ± 160 μM (SAV). Error bars indicate SEM based on three individual measurements. | ||
Next, we tested the active triazole 10 and reference compounds towards PDZ1 and PDZ3 of PSD-95 by FP in a similar way as for PDZ2. From previous studies it is known that PDZ1 recognizes the same peptide sequences as PDZ2 although generally with a lower affinity than PDZ2; while the selectivity profile for PDZ3 is very different from PDZ1 and PDZ2.15,16 This was reflected in our results, as TAV and SAV demonstrated higher Ki values towards PDZ1 (74 and 480 μM, respectively) than for PDZ2. For 10 and Indole 1 and 2 some inhibitory activity towards PDZ1 is observed, but their weaker affinities prevented Ki determination (estimated Ki > 1 mM). For PDZ3 we observed no activity for 10 or the two indoles (data not shown).
Overall, these FP data demonstrate that the novel triazole-based peptidomimetic structure 10 binds PDZ2 of PSD-95 with Ki value of 289 μM and can fully displace the PDZ2/probe interaction. The affinity to PDZ1 was much weaker (Ki > 500 μM) and no activity to PDZ3 was observed. Compared to tripeptides, 10 is equipotent to SAV and 8-fold weaker than TAV. On the other hand, 10 is several fold more potent than Indole 1 and about 2-fold more potent than Indole 2. Vogrig et al.24 demonstrated that Indole 1 inhibits the PSD-95-PDZ1/5-HT2A interaction with an IC50 of 190 μM in an in vitro pull-down assay. Indole 2 was not tested in this assay, as it was estimated to be a weak binder based on 1H–15N HSQC NMR. Also, Indole 1 showed analgesic effects in a rat neuropathic pain model. However, based on our FP data these reported pharmacological effects might be mediated by other functions than a specific inhibition of PSD-95 PDZ domains.
In order to exclude any unconventional mechanism or artefactual events of the novel triazole compound we tested 10 by isothermal titration calorimetry (ITC). The ligands, TAV and 10, were titrated into PDZ2 under low c value conditions. The c value corresponds to the ratio of the receptor concentration and the dissociation constant and should normally be 10–100 to get high-quality data.40,41 However, for weak interactions this is not practical obtainable and instead experiments are run at low c values (often <1).41 In this alternative setup it is important that ligand concentration is several fold higher than protein concentration and that the concentration of ligand and receptor are precisely known (as stoichiometry information is lost). When properly designed the integrated heats form a hyperbolic graph from which the affinity can be reliable extracted. TAV titrated into PDZ2 resulted in substantial signal, and when integrated heats were fitted to a one-site binding model Kd was determined to 101 μM (Fig. 4A). For 10, the measured heats were smaller but still resulted in a typical and reliable low c value thermogram and Kd of 598 μM (Fig. 4B). The relative affinities between TAV and 10 agree very well for FP and ITC, although the Kd values suggested by ITC were 2–2.5 fold higher than the FP Ki values. However, this is considered an acceptable margin considering the low affinities and the concomitant experimental challenges.41 Overall, the ITC measurements confirm the FP data and corroborate that 10 binds specifically to the PDZ2 binding pocket strongly enough to produce measurable heat in ITC and displace a peptide probe in FP.
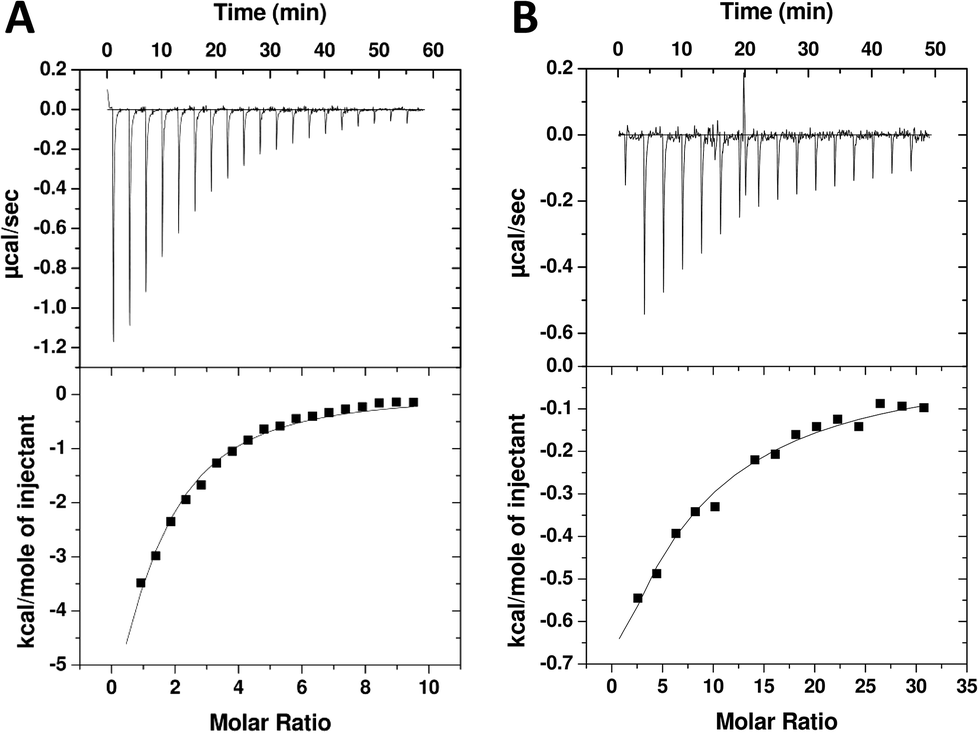 | ||
| Fig. 4 Representative ITC data for TAV titrated into 50 μM PDZ2 (A) and 10 titrated into 35 μM PDZ2 (B). The syringe was loaded with 2305 μM TAV or 5140 μM 10. Results for TAV/PDZ2: Kd = 101 ± 19 μM; ΔH = −15.0 ± 0.99 kcal mol−1, −TΔS = 9.48 ± 1.1 kcal mol−1. Results for 10/PDZ2: Kd = 598 ± 136 μM; ΔH = −11.8 ± 3.1 kcal mol−1, −TΔS = 7.42 ± 3.2 kcal mol−1. All values are average ± SD based on three experiments. | ||
2.3. Computational evaluation
In order to explore how 10 could interact with PSD-95 PDZ2 we performed computational docking studies using Glide and a homology model of PDZ2 based on the crystal structure of PDZ3 in complex with the CRIPT peptide (1BE9),14 as previously described.15,20,21 When docking penta- and tetrapeptides into this model these are positioned in the same conformation as CRIPT in PDZ3 (see Fig. 1) and with an excellent agreement among the highest ranking docking poses. For TAV the suggested conformations also align very well with experimentally determined peptide-PDZ structures, e.g. Val is positioned in the P0 pocket and backbone interactions to PDZ are apparent (Fig. 1 and 5). The N-terminal amino acid, Thr in TAV, shows a higher degree of conformational variation among the poses, but most conformations suggest that Thr's hydroxyl group interacts with the β-strand (βB) of PDZ while the N-terminal amine engages with His225 on the α-helix (B) as illustrated in Fig. 1 and 5. Docking poses of 10 suggest a binding mode very similar to TAV (Fig. 5). The P0 Val side chain binds in the hydrophobic P0 pocket of PDZ and the carboxylic acid of 10 interacts with the GLGF loop in all top 10 poses. Additionally, both 10 and TAV donate their P0 amide hydrogen to the PDZ backbone. Interestingly, in the top ranking pose of 10 (Fig. 5), the N3 triazole nitrogen accepts a hydrogen atom from a PDZ backbone amide bond similar to the carbonyl group of TAV's P−2 amide illustrating the amide isosteric properties of triazole. Also, the top pose of 10 suggests that the P−2 Thr-residue binds PDZ in the same way as seen for TAV (Fig. 5). However, generally the poses of 10 agree less well in the P−2 position compared to TAV, and for example other poses of 10 suggest that its amine does not interact with PDZ2 at all. Perhaps this variation in the P−2 region reflects less tight interactions in that part of the molecule and could perhaps explain why TAV is more potent than 10. Overall, this computational analysis illustrates that 10 possesses suitable proportions and electronic properties allowing it to fit into the PDZ pocket, and that compound 10 is likely to bind PDZ2 in an similar mode as the tripeptide TAV.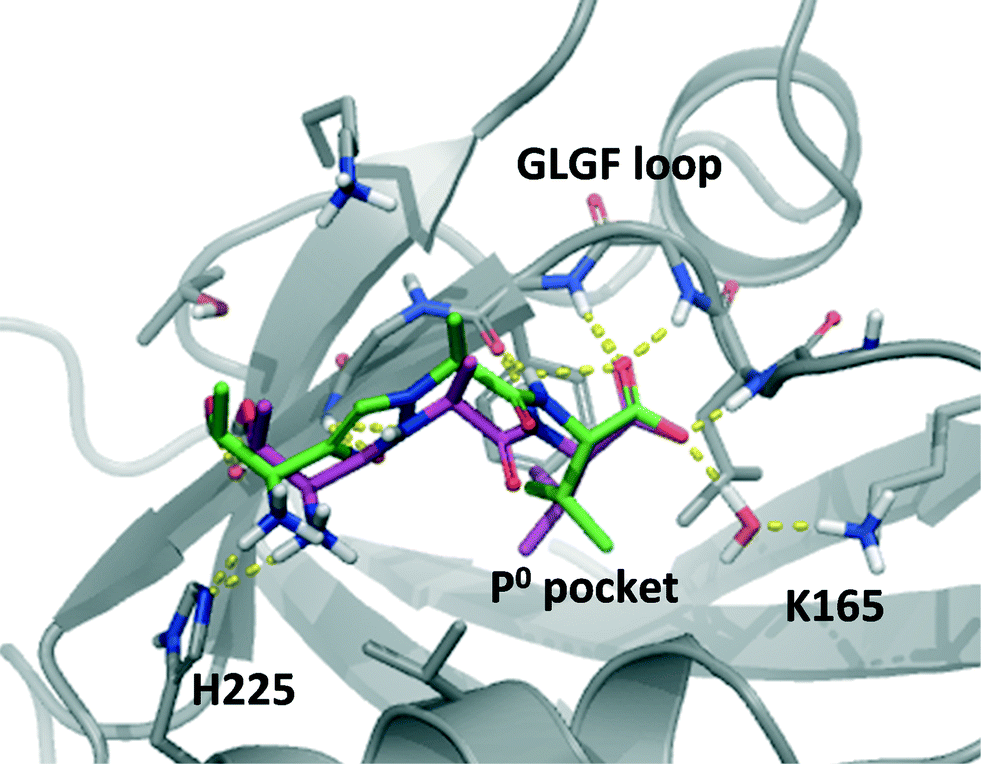 | ||
| Fig. 5 Compound 10 (green) and TAV (magenta) docked into PSD-95 PDZ2. The highest ranking poses are shown. | ||
3. Conclusions
In this study we have pursued a peptidomimetic strategy where we rigidify the original peptide backbone by introducing a small triazole group. The feasibility of the CuAAC reaction is exploited to make several close analogues in order to provide a thorough investigation of the strategy. The short tripeptide TAV was used as starting point for the design of triazole-based compounds. Azide and alkyne building blocks 1–8 were synthesized in satisfactorily yields and so were the corresponding triazole-based peptidomimetic structures 9–18. Compound 10 fully inhibited the PDZ2/peptide probe interaction with a Ki value of 289 μM. This affinity is lower than for TAV, but similar to the corresponding SAV tripeptide and two to several fold better than seen for the recently published indole-based small-molecule PSD-95 PDZ inhibitors (Indole 1–2). ITC studies of 10 show that its interaction with PDZ2 produces heat and gives a thermogram typical for a true, but low-affinity, binder. Finally, computational docking studies suggest that 10 binds PDZ2 in a binding mode very similar to TAV.This study illustrates the challenges of generating non-peptide based PDZ inhibitors with reasonable potencies. When deviating from the peptide structure ligand-affinity is often lost, and as seen for the triazole-series even subtle changes dramatically affect affinity. Also, the affinities of reported indole small-molecules were found to be low in our hands. Still, based on our ‘click chemistry’ strategy we discovered compound 10, which represents a novel class of PSD-95 PDZ inhibitors. It demonstrated reliable inhibitory activity in our well-established FP and ITC assays and could inspire future efforts in the field of PDZ domain medicinal chemistry.
Notes and references
- M. Sheng and C. C. Hoogenraad, Annu. Rev. Biochem., 2007, 76, 823 CrossRef CAS PubMed.
- K. S. Christopherson, B. J. Hillier, W. A. Lim and D. S. Bredt, J. Biol. Chem., 1999, 274, 27467 CrossRef CAS PubMed.
- M. Aarts, Y. Liu, L. Liu, S. Besshoh, M. Arundine, J. W. Gurd, Y. T. Wang, M. W. Salter and M. Tymianski, Science, 2002, 298, 846 CrossRef CAS PubMed.
- A. Bach, B. H. Clausen, M. Møller, B. Vestergaard, C. N. Chi, A. Round, P. L. Sørensen, K. B. Nissen, J. S. Kastrup, M. Gajhede, P. Jemth, A. S. Kristensen, P. Lundström, K. L. Lambertsen and K. Strømgaard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3317 CrossRef CAS PubMed.
- D. J. Cook, L. Teves and M. Tymianski, Nature, 2012, 483, 213 CrossRef CAS PubMed.
- F. Tao, Q. Su and R. A. Johns, Mol. Ther., 2008, 16, 1776 CrossRef CAS PubMed.
- B. W. LeBlanc, M. Iwata, A. P. Mallon, C. N. Rupasinghe, D. J. Goebel, J. Marshall, M. R. Spaller and C. Y. Saab, Neuroscience, 2010, 167, 490 CrossRef CAS PubMed.
- R. D'Mello, F. Marchand, S. Pezet, S. B. McMahon and A. H. Dickenson, Mol. Ther., 2011, 19, 1780 CrossRef PubMed.
- L. M. Ittner, Y. D. Ke, F. Delerue, M. Bi, A. Gladbach, J. van Eersel, H. Wolfing, B. C. Chieng, M. J. Christie, I. A. Napier, A. Eckert, M. Staufenbiel, E. Hardeman and J. Gotz, Cell, 2010, 142, 387 CrossRef CAS PubMed.
- F. X. Soriano, M. A. Martel, S. Papadia, A. Vaslin, P. Baxter, C. Rickman, J. Forder, M. Tymianski, R. Duncan, M. Aarts, P. Clarke, D. J. Wyllie and G. E. Hardingham, J. Neurosci., 2008, 28, 10696 CrossRef CAS PubMed.
- M. D. Hill, R. H. Martin, D. Mikulis, J. H. Wong, F. L. Silver, K. G. Terbrugge, G. Milot, W. M. Clark, R. L. Macdonald, M. E. Kelly, M. Boulton, I. Fleetwood, C. McDougall, T. Gunnarsson, M. Chow, C. Lum, R. Dodd, J. Poublanc, T. Krings, A. M. Demchuk, M. Goyal, R. Anderson, J. Bishop, D. Garman, M. Tymianski and ENACT trial investigators, Lancet Neurol., 2012, 11, 942 CrossRef CAS PubMed.
- J. Schultz, R. R. Copley, T. Doerks, C. P. Ponting and P. Bork, Nucleic Acids Res., 2000, 28, 231 CrossRef CAS PubMed.
- C. N. Chi, A. Bach, K. Strømgaard, S. Gianni and P. Jemth, BioFactors, 2012, 38, 338 CrossRef CAS PubMed.
- D. A. Doyle, A. Lee, J. Lewis, E. Kim, M. Sheng and R. MacKinnon, Cell, 1996, 85, 1067 CrossRef CAS PubMed.
- A. Bach, C. N. Chi, T. B. Olsen, S. W. Pedersen, M. U. Røder, G. F. Pang, R. P. Clausen, P. Jemth and K. Strømgaard, J. Med. Chem., 2008, 51, 6450 CrossRef CAS PubMed.
- M. A. Stiffler, J. R. Chen, V. P. Grantcharova, Y. Lei, D. Fuchs, J. E. Allen, L. A. Zaslavskaia and G. MacBeath, Science, 2007, 317, 364 CrossRef CAS PubMed.
- B. Z. Harris, F. W. Lau, N. Fujii, R. K. Guy and W. A. Lim, Biochemistry, 2003, 42, 2797 CrossRef CAS PubMed.
- S. W. Pedersen, S. B. Pedersen, L. Anker, G. Hultqvist, A. S. Kristensen, P. Jemth and K. Strømgaard, Nat. Commun., 2014, 5, 3215 Search PubMed.
- J. N. Eildal, G. Hultqvist, T. Balle, N. Stuhr-Hansen, S. Padrah, S. Gianni, K. Strømgaard and P. Jemth, J. Am. Chem. Soc., 2013, 135, 12998 CrossRef CAS PubMed.
- A. Bach, J. N. Eildal, N. Stuhr-Hansen, R. Deeskamp, M. Gottschalk, S. W. Pedersen, A. S. Kristensen and K. Strømgaard, J. Med. Chem., 2011, 54, 1333 CrossRef CAS PubMed.
- A. Bach, C. N. Chi, G. F. Pang, L. Olsen, A. S. Kristensen, P. Jemth and K. Strømgaard, Angew. Chem., Int. Ed., 2009, 48, 9685 CrossRef CAS PubMed.
- A. Piserchio, G. D. Salinas, T. Li, J. Marshall, M. R. Spaller and D. F. Mierke, Chem. Biol., 2004, 11, 469 CrossRef CAS PubMed.
- D. G. Udugamasooriya, S. C. Sharma and M. R. Spaller, ChemBioChem, 2008, 9, 1587 CrossRef CAS PubMed.
- A. Vogrig, L. Dorr, N. Bouzidi, B. Boucherle, A. S. Wattiez, E. Cassier, G. Vallon, I. Ripoche, I. Abrunhosa-Thomas, P. Marin, L. Nauton, V. Thery, C. Courteix, L. Y. Lian and S. Ducki, ACS Chem. Biol., 2013, 8, 2209 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674 RSC.
- A. Trabocchi and A. Guarna, in Peptidomimetics in Organic and Medicinal Chemistry, John Wiley & Sons, Ltd, 2014, ch. 5, pp. 99–121 Search PubMed.
- A. Brik, J. Alexandratos, Y. C. Lin, J. H. Elder, A. J. Olson, A. Wlodawer, D. S. Goodsell and C. H. Wong, ChemBioChem, 2005, 6, 1167 CrossRef CAS PubMed.
- I. E. Valverde, A. Bauman, C. A. Kluba, S. Vomstein, M. A. Walter and T. L. Mindt, Angew. Chem., Int. Ed., 2013, 52, 8957 CrossRef CAS PubMed.
- J. T. Lundquist and J. C. Pelletier, Org. Lett., 2001, 3, 781 CrossRef CAS PubMed.
- H. Johansson and D. S. Pedersen, Eur. J. Org. Chem., 2012, 4267 CrossRef.
- A. Bach and K. Strømgaard, Synthesis, 2011, 807 CAS.
- N. B. Kondekar and P. Kumar, Synth. Commun., 2011, 41, 1301 CrossRef CAS.
- S. Mosca, F. Wojcik and L. Hartmann, Macromol. Rapid Commun., 2011, 32, 197 CrossRef CAS PubMed.
- G. J. Roth, B. Leipold, S. G. Müller and H. J. Bestmann, Synthesis, 2004, 640 CAS.
- G. Reginato, A. Mordini, F. Messina, A. Degl'Innocenti and G. Poli, Tetrahedron, 1996, 52, 10985 CrossRef CAS.
- P. K. Chakravarty, W. J. Greenlee, W. H. Parsons, A. A. Patchett, P. Combs, A. Roth, R. D. Busch and T. N. Mellin, J. Med. Chem., 1989, 32, 1886 CrossRef CAS PubMed.
- N. Umezawa, N. Matsumoto, S. Iwama, N. Kato and T. Higuchi, Bioorg. Med. Chem., 2010, 18, 6340 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L. N. Lin, Anal. Biochem., 1989, 179, 131 CrossRef CAS PubMed.
- W. B. Turnbull and A. H. Daranas, J. Am. Chem. Soc., 2003, 125, 14859 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Synthesis of compounds and experimental description of assays and docking. See DOI: 10.1039/c5md00445d |
| This journal is © The Royal Society of Chemistry 2016 |