
Arginine-linked neomycin B dimers: synthesis, rRNA binding, and resistance enzyme activity†‡
Yi
Jin
a,
Derrick
Watkins
b,
Natalya N.
Degtyareva
b,
Keith D.
Green
c,
Meredith N.
Spano
b,
Sylvie
Garneau-Tsodikova
*c and
Dev P.
Arya
*ab
aClemson University, Department of Chemistry, Clemson, SC 29634, USA. E-mail: dparya@clemson.edu
bNUBAD, LLC, Greenville, SC 29605, USA
cUniversity of Kentucky, Department of Pharmaceutical Sciences, 789 South Limestone Street, Lexington, KY 40536-0596, USA. E-mail: sylviegtsodikova@uky.edu; Fax: 859 257 7585; Tel: 859 218 1686
First published on 3rd November 2015
Abstract
The nucleotides comprising the ribosomal decoding center are highly conserved, as they are important for maintaining translational fidelity. The bacterial A-site has a small base variation as compared with the human analogue, allowing aminoglycoside (AG) antibiotics to selectively bind within this region of the ribosome and negatively affect microbial protein synthesis. Here, by using a fluorescence displacement screening assay, we demonstrate that neomycin B (NEO) dimers connected by L-arginine-containing linkers of varying length and composition bind with higher affinity to model A-site RNAs compared to NEO, with IC50 values ranging from ~40–70 nM, and that a certain range of linker lengths demonstrates a clear preference for the bacterial A-site RNA over the human analogue. Furthermore, AG-modifying enzymes (AMEs), such as AG O-phosphotransferases, which are responsible for conferring antibiotic resistance in many types of infectious bacteria, demonstrate markedly reduced activity against several of the L-arginine-linked NEO dimers in vitro. The antimicrobial activity of these dimers against several bacterial strains is weaker than that of the parent NEO.
Introduction
The growing problem of antibiotic resistance presents an urgent need for the development of new therapeutics to treat bacterial infections. One class of commonly prescribed broad-spectrum antibiotics, the aminoglycosides (AGs), combat Gram-positive and Gram-negative bacteria by selectively binding bacterial ribosomes and inhibiting protein synthesis.1,2 A particularly vulnerable site for antibiotic targeting is the decoding center of the small ribosomal subunit, which is responsible for matching the correct aminoacyl-transfer (t)RNA with the three-nucleotide codon of the messenger (m)RNA. The binding of AGs to the aminoacyl (A)-site of the ribosome interferes with this recognition process, which results in mistranslation of the mRNA.3–6Bacterial AG antibiotic resistance is commonly conferred via AG-modifying enzymes (AMEs), namely AG O-phosphotransferases (APHs), AG N-acetyltransferases (AACs), and AG O-nucleotidyltransferases (ANTs), which render their substrate molecules unable to bind at their respective ribosomal sites of action.7–10 Resistant bacterial strains carry plasmids containing genes for these enzymes, and any forthcoming AG antibiotics must be able to bypass this troublesome resistance mechanism.
Therefore, when developing novel AGs, it is crucial that (i) they be highly selective for the bacterial A-site over the human A-site counterpart, as mutations in human mitochondrial ribosomes that result in higher affinity for these ligands often lead to drug toxicity,11,12 and (ii) they evade the action of the AMEs responsible for the majority of resistance to these drugs. In this work, we have combined the two aforementioned molecular design strategies in order to produce novel compounds that are both poor substrates for AMEs and display increased affinities for their RNA targets over their unmodified parent AG. We report the synthesis and screening of a series of neomycin B (NEO) dimers that are joined by L-arginine-containing linkers of varying lengths. NEO dimers with triazole, urea, and thiourea linkages have been shown to be poor substrates for certain AMEs,13 and both NEO dimers and L-arginine-conjugated AGs have been reported to increase ligand affinity for nucleic acids as compared with their unconjugated counterparts.14–21 Other AG homo- and hetero-dimers have also been reported to display promise in enhancing RNA binding, improving antibiotic activity, and/or resisting the action of AMEs.22–29 Our novel dimers display higher affinities for both human and bacterial RNA A-site model constructs than does NEO, as shown by a fluorescence displacement assay, and also show slight binding preferences among the bacterial and human constructs used in this study. Significantly, AMEs display very different activity for these L-arginine-linked NEO dimer substrates than they do for NEO, and notably, these dimers are very poor substrates especially for the AG O-phosphotransferases APH(2′′)-Ia and APH(3′)-Ia. Despite these collective differences, the dimers' antimicrobial activities are comparable to those of NEO.
Results and discussion
Synthesis of L-arginine NEO dimers
We successfully achieved the synthesis of nine novel triazole-linked L-arginine-NEO dimers (compounds 7–15) by using a high-yielding robust synthetic “click chemistry” approach followed by standard 9-fluorenylmethoxycarbonyl (Fmoc)-based solid-phase peptide synthesis (Fig. 1). Our initial focus was to synthesize the terminal dialkyne linker 4 in order to achieve triazole-linked NEO dimers via click chemistry. To this end, we synthesized compound 4 in two steps from ethyl 3,5-dibromobenzoate (2) via Sonogashira–Hagihara cross-coupling followed by removal of the 2-hydroxyisopropyl group of compound 3 to generate free terminal alkynes in molecule 4 (Fig. 1A). We synthesized the Boc-protected NEO azide from commercially available NEO in three steps30 and further coupled it with the dialkyne compound 4 to successfully produce the triazole-linked NEO dimer 5via click chemistry (Fig. 1A). The presence of a free carboxyl functional group in 5 proved to be extremely beneficial for further modifications using solid-phase peptide chemistry. To this end, we rapidly generated a library of L-arginine-NEO dimers 7–15 with varying chain lengths by using Fmoc-based solid-phase peptide chemistry (Fig. 1B and S1B‡). Compound 5 was further deprotected to obtain NEO dimer 6 with a free carboxyl group (Fig. S1A‡), which was used as a control molecule for the binding study. Detailed experimental procedures and characterization data (NMR spectra and HRMS/MALDI-TOF MS; Fig. S2–S23‡) for compounds 6–15 are presented in the ESI.‡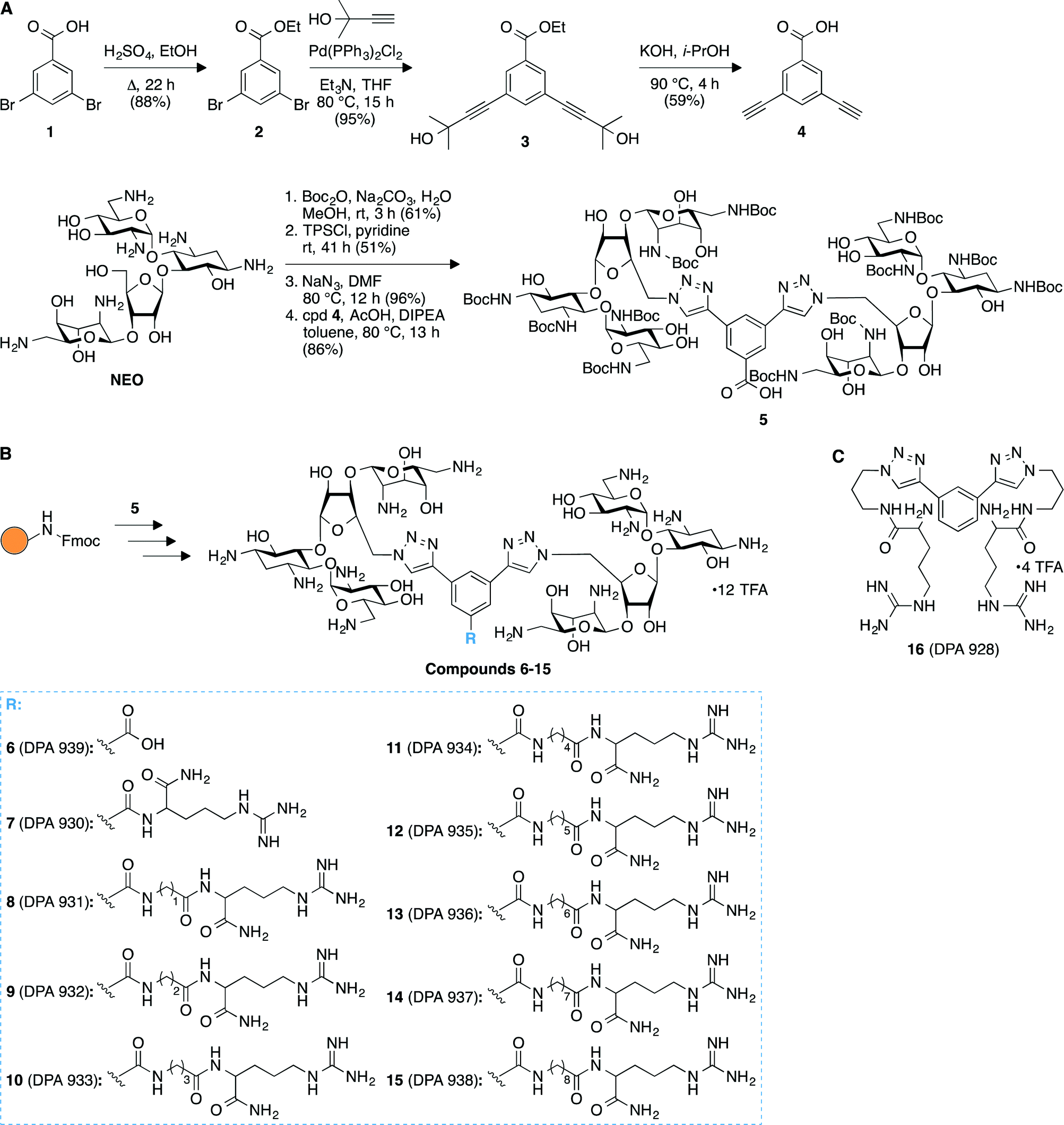 | ||
| Fig. 1 Synthetic schemes for the preparation of A. compound 5, and B. arginine-linked NEO dimers 6–15. C. Structure of compound 16 used as a control in binding studies to model A-sites. | ||
Screening of compounds 6–16 for binding to model A-sites
In order to establish relative affinity of the L-arginine-linked NEO dimers for human and bacterial ribosomal A-sites, we assessed dimers 6–15, NEO, and a control molecule 16 (Fig. 1C), along with a 27-base model A-site RNA oligomers representing E. coli and human rRNA sequences by using a fluorescence displacement assay (Fig. 2).31–33 In this ligand binding assay, F-NEO, a conjugate of NEO and fluorescein, serves as a fluorescent reporter of binding when a ligand displaces it.34 Fluorescence emission intensity of F-NEO is decreased when it is bound within the major groove of RNA, analogously to non-conjugated NEO, as confirmed by docking experiments. Upon displacement from the RNA target oligomer by a competitive binder, the fluorescence of F-NEO increases, thereby reporting on ligand binding.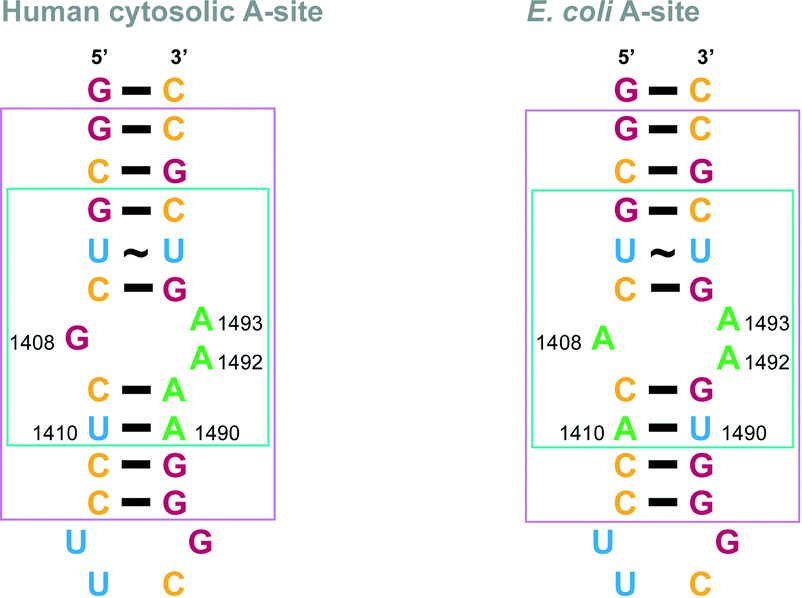 | ||
| Fig. 2 rRNA A-site constructs. Nucleotides implicated in AG binding are shown in a turquoise box in the E. coli and human A-site structures. Nucleotides that match part of the crystal structures used for modelling in Fig. 5 are shown in a pink box in the E. coli and human A-site structures. Nucleotides are colored as follow: A = green, G = red, C = yellow, and U = blue. | ||
The dissociation constants (Kd) between the F-NEO probe and A-sites measured by direct titrations were 4 ± 1 nM and 23 ± 3 nM for E. coli and human A-sites, respectively. The approximate four-to-six-fold difference between affinities of NEO to E. coli and human A-sites has been previously documented.35 We assessed the percentage of displacement of F-NEO by compounds 6–16 by comparison of fluorescence emission of F-NEO (Fig. 3) in the presence of model A-site RNAs and again upon addition of a single concentration of the tested compounds. In order to better distinguish between NEO-like binders to model A-site RNAs, the ligand concentration used in competitive binding experiments was approximately equal to the NEO IC50, where the IC50 concentration is defined as a concentration at which 50% of F-NEO is displaced. No increase in emission due to the displacement of the probe was observed for control molecule 16, indicating that it did not bind appreciably to either of the A-site RNAs. All NEO dimers, 6–15, demonstrated stronger binding preferences for both A-sites as compared with NEO. This result is consistent with previous findings demonstrating that the presence of a single positively-charged arginine enhances the binding ability of NEO dimers to RNAs as compared with NEO alone.30
 | ||
| Fig. 3 Bar graph showing the percentage of displacement of F-NEO by compounds 6–16. NEO is shown as a control. | ||
We also observed that the linker length affected the binding affinities of the L-arginine-linked NEO dimers to the A-site RNAs as shown by IC50 measurements (Table 1 and Fig. S24–S25‡). Compounds 13 and 14, with 6 and 7 consecutive carbons within their linkers, respectively, demonstrated the strongest affinity for the E. coli A-site of all the compounds tested. IC50 values were also measured for human A-site. Measured IC50 values of these compounds for the human A-site were similar to those measured for the E. coli A-site (Table 1). The affinities of compounds 7, 11, 12, and 13 were higher for the E. coli A-site than for the human homologue, indicating that the consecutive carbons chains within the arginine linkers have an optimum length of 4–6 carbons, and that the presence of this carbon chain assists in distinction between two A-sites. Since the reported IC50 values were determined by F-NEO displacement and are therefore relative to F-NEO affinity for each RNA, and since F-NEO has a ~5.8-fold larger affinity for the E. coli versus the human A-site RNA, we can report that all of the L-arginine-linked NEO dimers have a higher affinity for the E. coli A-site than does NEO, and most likely preferences for one A-site for another are actually larger than the reported IC50 values imply. Therefore, we have calculated selectivity factor that indicates the preferential affinity ratio of each compound for the E. coli A-site as compared with the human A-site. The affinity of NEO for the E. coli A-site is five times stronger than for the human homologue.
| Compound | E. coli A-site | Human A-site | Selectivity factor |
|---|---|---|---|
| NEO | 87 ± 6 | 76 ± 13 | 5.0 |
| 6 | 51 ± 6 | 50 ± 5 | 5.6 |
| 7 | 55 ± 1 | 71 ± 1 | 7.4 |
| 8 | 62 ± 2 | 58 ± 2 | 5.3 |
| 9 | 70 ± 3 | 58 ± 1 | 4.7 |
| 10 | 56 ± 8 | 52 ± 3 | 5.3 |
| 11 | 52 ± 2 | 70 ± 8 | 7.4 |
| 12 | 54 ± 4 | 68 ± 6 | 7.2 |
| 13 | 44 ± 2 | 56 ± 1 | 7.3 |
| 14 | 48 ± 2 | 43 ± 1 | 5.2 |
| 15 | 60 ± 3 | 62 ± 3 | 5.9 |
Antibacterial activity
Having studied the binding of our compounds to model A-sites, we next investigated our L-arginine-linked NEO dimers 6–15 for their inhibition of antimicrobial growth against three bacterial strains: Bacillus cereus ATCC 11778, Staphylococcus epidermidis ATCC 12228, and Staphylococcus aureus ATCC 25923. The minimum inhibitory concentration (MIC) values for dimers 6–15 and NEO, which served as a control, are summarized in Table 2. Due to the large molecular weight of our compounds (ranging from 2818 to 3284 g mol−1) we provide the MIC values in μM, which we feel is best suited for larger molecules. However, as it is standard practice to provide MIC values in μg mL−1 in microbiology, we also included these values into parentheses in Table 2. A range is reported for the concentrations at which a growth inflection point was not observed. While most of the NEO dimers showed little activity against S. epidermidis and S. aureus, all dimers had a profound inhibitory effect on the growth of the spore-forming B. cereus, demonstrating the selectivity of our compounds towards certain bacterial strains. Interestingly, compound 6, which does not contain arginine, showed better inhibition of S. aureus and S. epidermidis than the L-arginine-containing NEO dimers 7–15. All dimers showed promising activity (MIC values ranging from 0.8–13 μM) against B. cereus, with dimers 8 and 9 being the best with MIC values of 1.6 μM and 0.8 μM, respectively. As observed previously, the positive charge of the L-arginine group in the linker is likely to assist with bacterial membrane penetration and binding to the negatively charged RNA.| Compound | B. cereus ATCC 11778 | S. epidermidis ATCC 12228 | S. aureus ATCC 25923 |
|---|---|---|---|
| NEO | ≤0.1 (≤0.1) | 0.2 (0.2) | 0.2 (0.2) |
| 6 | 3–6 (9–18) | 13 (35) | 13 (35) |
| 7 | 3–6. (10–19) | >50 (>154) | >50 (>154) |
| 8 | 1.6 (5) | >50 (>157) | 50 (157) |
| 9 | 0.8 (2.5) | 50 (158) | 50 (158) |
| 10 | 1.6–3 (5–10) | >50 (>159) | >50 (>159) |
| 11 | 3–13 (10–40) | >50 (>159) | >50 (>159) |
| 12 | 1.6–3 (5–10) | >50 (>160) | >50 (>160) |
| 13 | 3 (10) | >50 (>161) | >50 (>161) |
| 14 | 3–6 (10–19) | 25 (81) | >50 (>161) |
| 15 | 3–6 (10–20) | 50 (164) | >50 (>164) |
Aminoglycoside-modifying enzyme (AME) activity assays
Since the alteration of AG functional groups by AMEs is the most common mechanism of bacterial AG resistance, we finally investigated our NEO dimers 6–15 as potential substrates of N-acetyltransferase (AAC) and O-phosphotransferase (APH) resistance enzymes (Fig. 4). For the AACs, we selected AAC(2′)-Ic from Mycobacterium tuberculosis,36–38 AAC(3)-IV from Escherichia coli,39,40 and AAC(6′)-Ie from the bifunctional AAC(6′)-Ie/APH(2′′)-Ia from S. aureus,39,41–43 which are known to acetylate NEO at the 2′-, 3, and 6′-position, respectively. We also elected to test against Eis, a unique AAC found in various bacterial strains, which is known to multi-acetylate AGs at a variety of positions.38,44–48 For the APHs, we selected APH(2′′)-Ia and APH(3′)-Ia,13,49 which commonly phosphorylate NEO. The activity of the enzymes for each compound was normalized to that of NEO. It is important to note that we did not test our NEO dimers 6–15 against ANTs due to the facts that (i) most ANTs modify streptomycin or spectinomycin,7 and (ii) the bacterial strains against which these compounds were tested in this study are not known to contain ANTs that modify NEO.13 | ||
| Fig. 4 Bar graph showing the relative initial rates of the listed AMEs with NEO and L-arginine-linked NEO dimers 6–15. All rates were normalized to the parent NEO. | ||
Interestingly, the AAC(3)-IV enzyme that acetylates the 3-amino group of NEO was found to be similarly or more active against certain dimers (6 and 8–12) than NEO. Its activity was found to be inversely proportional to linker length, and gradually decreased with increasing linker length for L-arginine-containing linkers. Compound 7 (n = 0), was modified by AAC(3)-IV at approximately 30% of the rate when compared to the NEO modification.
All other AMEs tested demonstrated much lower enzymatic activity towards compounds 6–15, which were found to be poorer substrates for these enzymes than NEO. The most likely explanation for these positive results is that steric hindrance from the bulky second NEO and/or the increase in molecular dynamics afforded by the flexible linker prevents proper and/or stable binding of the NEO group at the active site of the enzyme. In support of this argument, compound 15 with longest L-arginine tail is the least susceptible substrate for the AMEs tested.
Overall, the O-phosphotransferases APH(2′′)-Ia and APH(3′)-Ia, as well as AAC(6′)-Ie/APH-(2′′)-Ia showed greatly reduced activity against the dimers. Since phosphorylation of AG functional groups by APH(3′)-Ia is one of the most commonly encountered AG antibiotic resistance mechanisms,10 these results indicate that our L-arginine-linked dimers are a promising avenue for future development as antimicrobial therapeutics.
A computer generated model of dimer 11 bound to the two RNA A-sites
Docking experiments using Auto Dock were performed with dimer 11, as it showed a significant difference in binding between the two A-sites. These docking experiments were performed to offer a potential explanation of our experimental findings. Indeed, different binding modes were observed when we docked 11 with E. coli as well as human RNA A-sites. In the 11-E. coli RNA A-site complex, NEO rings interact in the same region of the RNA (Fig. 5, please see pink boxed region), as established for AG (NEO) binding with E. coli RNA A-site (Fig. 2). However, in the binding of 11 with human RNA A-site, NEO rings interact with RNA bases away from adenines A1490–A1493 (Fig. 5). The difference in binding sites may also cause the observed difference in binding site of the L-arginine containing side chain of 11 (Fig. 5). Together, these two factors likely contribute to the seven-fold difference in binding of 11 with E. coli RNA A-site as compared to human RNA A-site.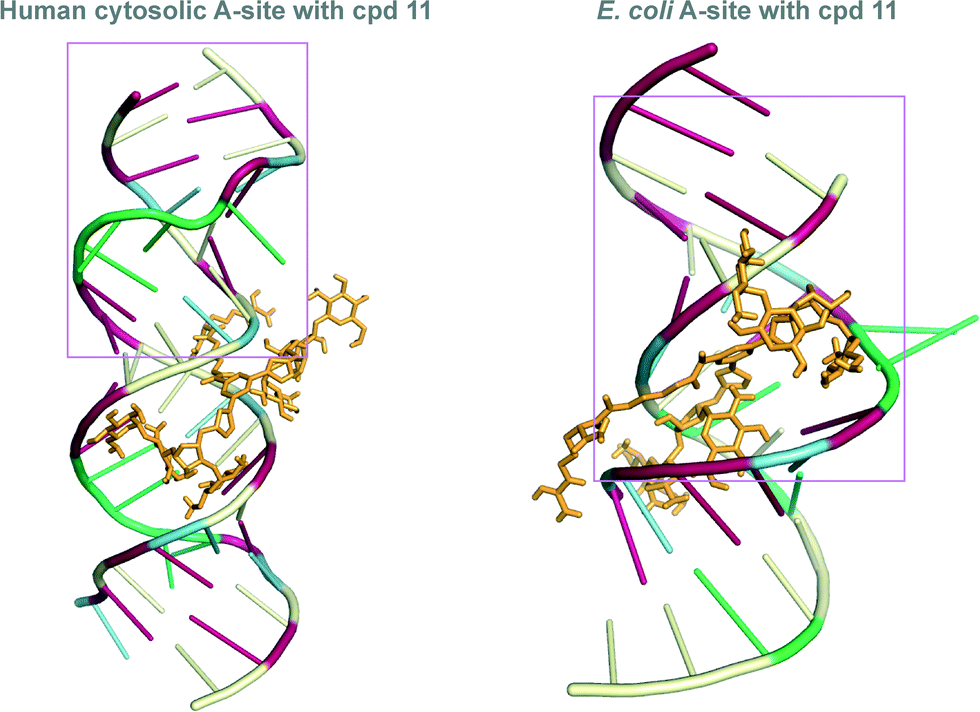 | ||
| Fig. 5 Docked structures of the L-arginine-linked NEO dimer 11 (depicted as orange stick) with the E. coli (right) and human RNA (left) A-sites. The nucleoside bases are colored as follow: A = green, G = red, C = pale yellow, and U = blue. The pink box represent the residues that are identical to those presented in Fig. 2. | ||
Conclusions
In summary, we have designed a series of NEO dimers with L-arginine-containing linkers of varying lengths to target the ribosomal A-site. These dimers displayed higher affinity for both human and bacterial RNA A-site model constructs than the parent NEO does, as shown by a fluorescence displacement assay. These compounds also showed slight binding preferences among the bacterial and human A-site model constructs, with a carbon linker length of 4–6 being optimal for E. coli A-site preference over the human sequence. The dimers displayed antimicrobial effects that were weaker than their parent AG, NEO. It is interesting to note that dimer 6 also displayed significant antibacterial activity. Significantly, AMEs showed very different activity for the arginine-linked NEO dimer substrates than they do for NEO. As compared with NEO, our dimers are very poor substrates for several AMEs, especially for the phosphotransferases APH(2′′)-Ia and APH(3′)-Ia, as well as for the AAC(6′)-Ie/APH-(2′′)-Ia. Our L-arginine-linked NEO dimers therefore present a potential avenue for the development of therapeutics to combat AG antibiotic-resistant infections.Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants 2R42GM097917 (D. P. A.) and AI090048 (S. G.-T.) and by startup funds from the College of Pharmacy at the University of Kentucky (S. G.-T.). We thank Dr. Sayantan Bhaduri for assistance with the description of the synthetic scheme. We thank Dr. Souvik Sur for modelling.Notes and references
- J. L. Houghton, K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 880–902 CrossRef CAS PubMed.
- M. Y. Fosso, Y. Li and S. Garneau-Tsodikova, Med. Chem. Commun., 2014, 5, 1075–1091 RSC.
- D. Moazed and H. F. Noller, J. Mol. Biol., 1990, 211, 135–145 CrossRef CAS PubMed.
- D. Moazed and H. F. Noller, Nature, 1987, 327, 389–394 CrossRef CAS PubMed.
- E. A. De Stasio, D. Moazed, H. F. Noller and A. E. Dahlberg, EMBO J., 1989, 8, 1213–1216 CAS.
- C.-H. Wong, M. Hendrix, E. S. Priestley and W. A. Greenberg, Chem. Biol., 1998, 5, 397–406 CrossRef CAS PubMed.
- M. S. Ramirez and M. E. Tolmasky, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- C. A. Smith and E. N. Baker, Curr. Drug Targets: Infect. Disord., 2002, 2, 143–160 CrossRef CAS.
- K. J. Labby and S. Garneau-Tsodikova, Future Med. Chem., 2013, 5, 1285–1309 CrossRef CAS PubMed.
- S. Garneau-Tsodikova and K. J. Labby, MedChemComm, 2015 10.1039/C5MD00344J.
- S. Hong, K. A. Harris, K. D. Fanning, K. L. Sarachan, K. M. Frohlich and P. F. Agris, J. Biol. Chem., 2015, 290, 19273–19286 CrossRef CAS PubMed.
- S. Hong, K. Fanning, K. Harris, K. Frohlich and P. Agris, FASEB J., 2015, 29, 575.520 Search PubMed.
- D. Watkins, S. Kumar, K. D. Green, D. P. Arya and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 3899–3905 CrossRef CAS PubMed.
- A. Litovchick, A. G. Evdokimov and A. Lapidot, Biochemistry, 2000, 39, 2838–2852 CrossRef CAS PubMed.
- A. Litovchick, A. G. Evdokimov and A. Lapidot, FEBS Lett., 1999, 445, 73–79 CrossRef CAS PubMed.
- A. Berchanski and A. Lapidot, Bioconjugate Chem., 2008, 19, 1896–1906 CrossRef CAS PubMed.
- C. Cabrera, A. Gutierrez, J. Barretina, J. Blanco, A. Litovchick, A. Lapidot, B. Clotet and J. A. Este, Antiviral Res., 2002, 53, 1–8 CrossRef CAS PubMed.
- S. Kumar, L. Xue and D. P. Arya, J. Am. Chem. Soc., 2011, 133, 7361–7375 CrossRef CAS PubMed.
- S. Kumar, P. Kellish, W. E. Robinson Jr., D. Wang, D. H. Appella and D. P. Arya, Biochemistry, 2012, 51, 2331–2347 CrossRef CAS PubMed.
- S. Kumar, M. N. Spano and D. P. Arya, Bioorg. Med. Chem., 2015, 23, 3105–3109 CrossRef CAS PubMed.
- A. King, D. Watkins, S. Kumar, N. Ranjan, C. Gong, J. Whitlock and D. P. Arya, Antimicrob. Agents Chemother., 2013, 57, 4717–4726 CrossRef CAS PubMed.
- K. Michael, H. Wang and Y. Tor, Bioorg. Med. Chem., 1999, 7, 1361–1371 CrossRef CAS PubMed.
- S. J. Sucheck, A. L. Wong, K. M. Koeller, D. D. Boehr, K.-A. Draker, P. Sears, G. D. Wright and C.-H. Wong, J. Am. Chem. Soc., 2000, 122, 5230–5231 CrossRef CAS.
- F. Agnelli, S. J. Sucheck, K. A. Marby, D. Rabuka, S. L. Yao, P. S. Sears, F. S. Liang and C. H. Wong, Angew. Chem., Int. Ed., 2004, 43, 1562–1566 CrossRef CAS PubMed.
- N. W. Luedtke, Q. Liu and Y. Tor, Biochemistry, 2003, 42, 11391–11403 CrossRef CAS PubMed.
- A. Bodlenner, A. Alix, J. M. Weibel, P. Pale, E. Ennifar, J. C. Paillart, P. Walter, R. Marquet and P. Dumas, Org. Lett., 2007, 9, 4415–4418 CrossRef CAS PubMed.
- A. G. Santana, A. Batisda, T. M. Del Campo, J. L. Asensio and J. Revuelta, Synlett, 2011, 2, 219–222 Search PubMed.
- S. Hanessian, J. P. Maianti, R. D. Matias, L. A. Feeney and E. S. Armstrong, Org. Lett., 2011, 13, 6476–6479 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, K. D. Green, K. J. Labby, M. Feldman, S. Garneau-Tsodikova and M. Fridman, J. Med. Chem., 2013, 56, 5613–5625 CrossRef CAS PubMed.
- L. Jiang, D. Watkins, Y. Jin, C. Gong, A. King, A. Z. Washington, K. D. Green, S. Garneau-Tsodikova, A. K. Oyelere and D. P. Arya, ACS Chem. Biol., 2015, 10, 1278–1289 CrossRef CAS PubMed.
- B. Francois, R. J. Russell, J. B. Murray, F. Aboul-ela, B. Masquida, Q. Vicens and E. Westhof, Nucleic Acids Res., 2005, 33, 5677–5690 CrossRef CAS PubMed.
- M. I. Recht, S. Douthwaite, K. D. Dahlquist and J. D. Puglisi, J. Mol. Biol., 1999, 286, 33–43 CrossRef CAS PubMed.
- M. I. Recht and J. D. Puglisi, Antimicrob. Agents Chemother., 2001, 45, 2414–2419 CrossRef CAS PubMed.
- D. Watkins, F. A. Norris, S. Kumar and D. P. Arya, Anal. Biochem., 2013, 434, 300–307 CrossRef CAS PubMed.
- D. H. Ryu and R. R. Rando, Bioorg. Med. Chem., 2001, 9, 2601–2608 CrossRef CAS PubMed.
- J. A. Ainsa, E. Perez, V. Pelicic, F. X. Berthet, B. Gicquel and C. Martin, Mol. Microbiol., 1997, 24, 431–441 CAS.
- M. W. Vetting, S. S. Hegde, F. Javid-Majd, J. S. Blanchard and S. L. Roderick, Nat. Struct. Biol., 2002, 9, 653–658 CrossRef CAS PubMed.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- K. D. Green, W. Chen, J. L. Houghton, M. Fridman and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 119–126 CrossRef CAS PubMed.
- M. L. Magalhaes and J. S. Blanchard, Biochemistry, 2005, 44, 16275–16283 CrossRef CAS PubMed.
- D. M. Daigle, D. W. Hughes and G. D. Wright, Chem. Biol., 1999, 6, 99–110 CrossRef CAS PubMed.
- D. D. Boehr, D. M. Daigle and G. D. Wright, Biochemistry, 2004, 43, 9846–9855 CrossRef CAS PubMed.
- S. J. Caldwell and A. M. Berghuis, Antimicrob. Agents Chemother., 2012, 56, 1899–1906 CrossRef CAS PubMed.
- K. D. Green, T. Biswas, C. Chang, R. Wu, W. Chen, B. K. Janes, D. Chalupska, P. Gornicki, P. C. Hanna, O. V. Tsodikov, A. Joachimiak and S. Garneau-Tsodikova, Biochemistry, 2015, 54, 3197–3206 CrossRef CAS PubMed.
- K. D. Green, R. E. Pricer, M. N. Stewart and S. Garneau-Tsodikova, ACS Infect. Dis., 2015, 1, 272–283 CrossRef CAS.
- O. V. Tsodikov, K. D. Green and S. Garneau-Tsodikova, PLoS One, 2014, 9, e92370 Search PubMed.
- J. L. Houghton, T. Biswas, W. Chen, O. V. Tsodikov and S. Garneau-Tsodikova, ChemBioChem, 2013, 14, 2127–2135 CrossRef CAS PubMed.
- R. E. Pricer, J. L. Houghton, K. D. Green, A. S. Mayhoub and S. Garneau-Tsodikova, Mol. BioSyst., 2012, 8, 3305–3313 RSC.
- P. J. Stogios, P. Spanogiannopoulos, E. Evdokimova, O. Egorova, T. Shakya, N. Todorovic, A. Capretta, G. D. Wright and A. Savchenko, Biochem. J., 2013, 454, 191–200 CrossRef CAS PubMed.
Footnotes |
| † DPA conceived and designed the dimer compounds and RNA binding experiments. YJ synthesized the dimers. KDG, under SGT supervision, performed MIC values determination against bacterial strains and experiments against AMEs. NND assisted with manuscript preparation. NND and DW performed RNA screening assays. MNS, SGT, and DPA wrote the manuscript. SGT prepared all figures. |
| ‡ Electronic supplementary information (ESI) available: A figure showing the preparation of compounds 6–15 (Fig. S1) is presented. Experimental procedures and images of spectroscopic characterization of all newly synthesized compounds (Fig. S2–S23) and of IC50 plots (Fig. S24 and S25) are also provided. See DOI: 10.1039/c5md00427f |
| This journal is © The Royal Society of Chemistry 2016 |