
Syntheses and biological evaluations of highly functionalized hydroxamate containing and N-methylthio monobactams as anti-tuberculosis and β-lactamase inhibitory agents†‡
Mark W.
Majewski
a,
Kyle D.
Watson
a,
Sanghyun
Cho
b,
Patricia A.
Miller
a,
Scott G.
Franzblau
b and
Marvin J.
Miller
*a
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: mmiller1@nd.edu; Fax: +1 574 631 6652; Tel: +1 574 631 7571
bInstitute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, MIC 964, Rm. 412, IL 60612, USA
First published on 5th October 2015
Abstract
Both the resurgence of tuberculosis (TB) and antibiotic resistance continue to threaten modern healthcare and new means of combating pathogenic bacterial infections are needed. The syntheses of monobactams possessing hydroxamate and N-methylthio functionality are described, as well as their anti-TB, in vitro β-lactamase inhibitory, and general antimicrobial evaluations. A number of compounds exhibited significant anti-TB and β-lactamase inhibitory activity, with MIC values in the range of 25 to <0.19 μM against Mycobacteria tuberculosis (M. tb), and Ki values in the range of 25–0.03 μM against purified NDM-1 and VIM-1 lystate metallo β-lactamases. This work suggests that these scaffolds may serve as promising leads in developing new antibiotics and/or β-lactamase inhibitors.
Introduction
Over the past 20 years, the antibiotic depository has become less effective as a result of both antibiotic overuse and an increase in pathogenic bacterial resistance. Most notably, tuberculosis (TB) and the ESKAPE pathogens have posed great challenges in regards to effective treatments. TB is a highly infectious disease and the second major cause of death as the result of a bacterial infection worldwide.1 TB, caused by Mycobacterium tuberculosis (M. tb), typically targets the lungs and can be easily transmitted among individuals. On average, there is a new case of TB each second2 and, although most are latent infections, the active cases of TB continue to grow, approaching 10 million as of 2010.3 The ESKAPE bacteria refer to the six most common bacteria found in hospital settings (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter species) and pose a threat to health care due to the incredible rate at which these strains can become resistant to antibiotics. The aforementioned pathogens have been especially characterized by their remarkable ability to develop resistance to a myriad of antibiotics because of the many mechanisms developed: limited permeability, efflux pumps, and β-lactamases being the most well known.4 Among these defense mechanisms, the β-lactamases, notably class B metallo enzymes, have garnered considerable attention especially since there is no clinically available class B inhibitor.5 In an effort to help address this situation, we have recently become interested in developing hydroxamate-containing monocyclic β-lactams (monobactams) and substituted azetidinones with different peripheral functionality and evaluating their activity against M. tb and a panel of β-lactamase enzymes in vitro. Herein we report on the syntheses and biological assessments of these intriguing compounds.Among the major classes of antibacterial agents available, the β-lactam family continues to persist, still making up the majority of the antibiotic market today (Fig. 1).6 Although this family has undergone evolution in attempts to address microbial resistance, this class still remains a mainstay in the never ending microbial war. Classically, β-lactam antibiotics work by inhibiting the formation of peptidoglycan cross-linkages in the bacterial cell wall.7 Maintenance of this peptidoglycan layer is accomplished by transpeptidase enzymes. Here, the β-lactam acts as a mimic of the normal substrate for the enzymes during the cross-link formation, ultimately forming an acyl-enzyme intermediate that is sterically blocked, thus leading to inhibition.8
 | ||
| Fig. 1 Representative β-lactam antibiotics. | ||
Reports of β-lactam compounds with potent anti-TB activity, however, have been scarce. Certain classic β-lactams can exhibit anti-TB activity when administered in combination with clavulanate, a β-lactamase inhibitor (Fig. 2, 1).9,10 Furthermore, monobactam alkylthiols and halogen substituted aromatic monobactams have also demonstrated intrinsic activity (Fig. 2, 2–3).11,12 In general, β-lactams have not been widely used in TB therapy for three major reasons: issues with permeability of the cell wall of M. tb, the persistent threat of inactivation by β-lactamases, and poor activity in vivo.13
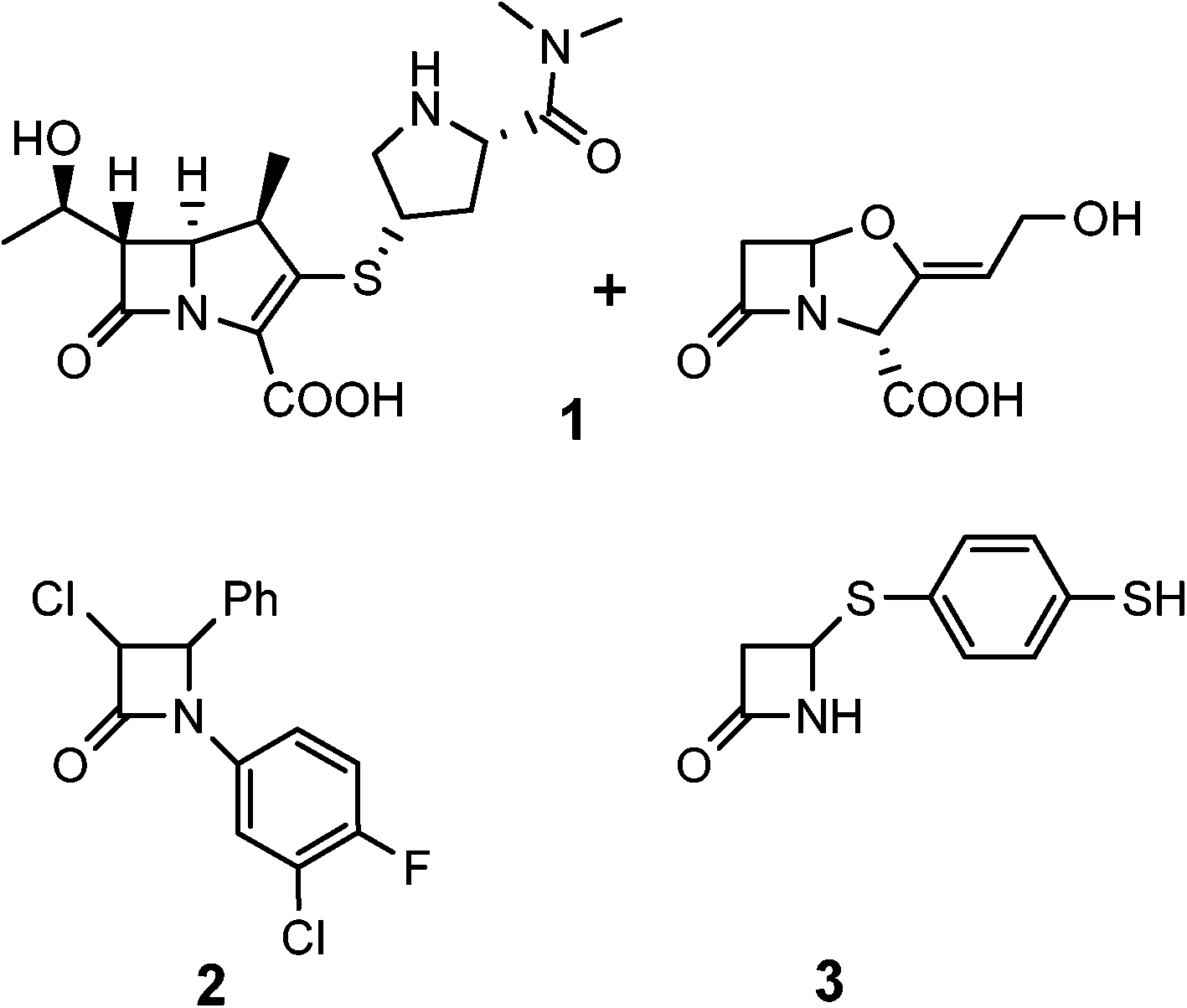 | ||
| Fig. 2 Some reported β-lactams with anti-TB activity. | ||
Although there are many treatments for TB (i.e. isoniazid, rifampin, etc.), there still remain many issues with these current regimens such as long treatment duration, drug toxicity, detrimental drug–drug interactions, and resistance.14 Over the last decade, however, a number of promising new compounds have been reported that have demonstrated potent activity against M. tb. These are highlighted by delamanid and bedaquiline, the latter recently approved for clinical use.15 Additionally, others such as nitroimidazoles (specifically PA-824),16 benzothiazinones (BTZs, specifically BTZ-043),17 imidazopyridines,18 and acrylic acid ethyl ethers19 all have recently been reported as potent anti-TB agents.
With the history of hydroxamates as useful enzyme inhibitors,20 it was of interest to see if the combination of hydroxamates with monobactams would lead to a new class of medicinal agents. Specifically, we were interested in the incorporation of more lipophilic hydroxamates and electronically varied aromatic rings into these monobactam scaffolds. As mentioned previously, one of the challenges in TB therapy is the penetrability of drugs, the result of the lipophilic cell wall of M. tb. Thus, we hypothesized that monobactams without its normal acidic sulfonate group at the nitrogen, in combination with lipophilic hydroxamates, including protected forms for extended use, and electron deficient aromatic rings may not only aid in passage through the thick cell wall of M. tb, but also potentiate activity. These features are summarized in Fig. 3. Our lab has already demonstrated that the incorporation of both hydroxamates and more lipophilic cyclic hydroxamates into cephalosporins led to potential new anti-microbial agents.21 Furthermore, the incorporation of alkylthio groups at the monobactam nitrogen has produced a fascinating new class of anti-cancer and anti-S. aureus agents.22 Herein we describe the syntheses of hydroxamate and N-methylthiol monobactams and their biological evaluations.
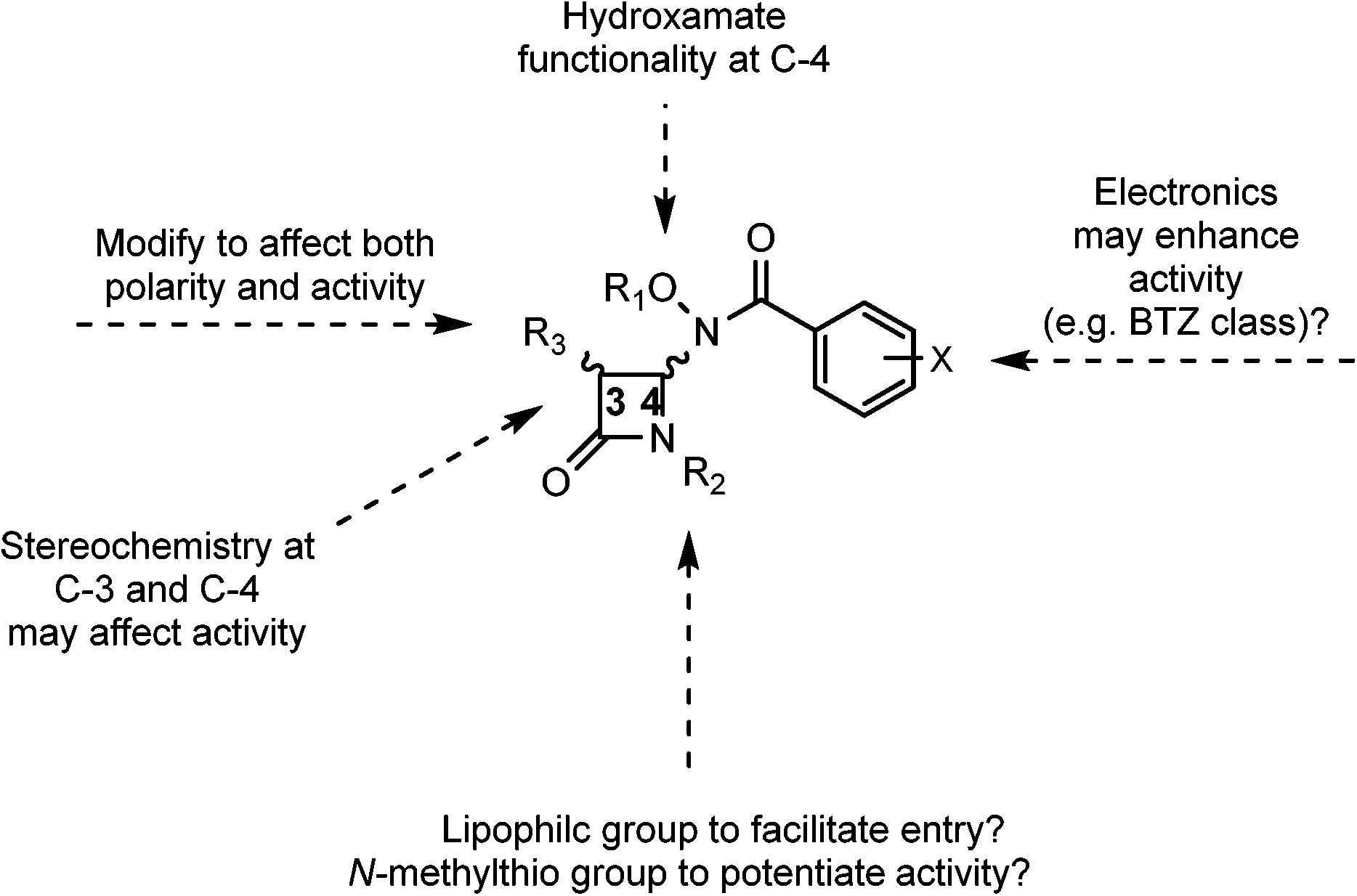 | ||
| Fig. 3 Monobactam key features. | ||
Results and discussion
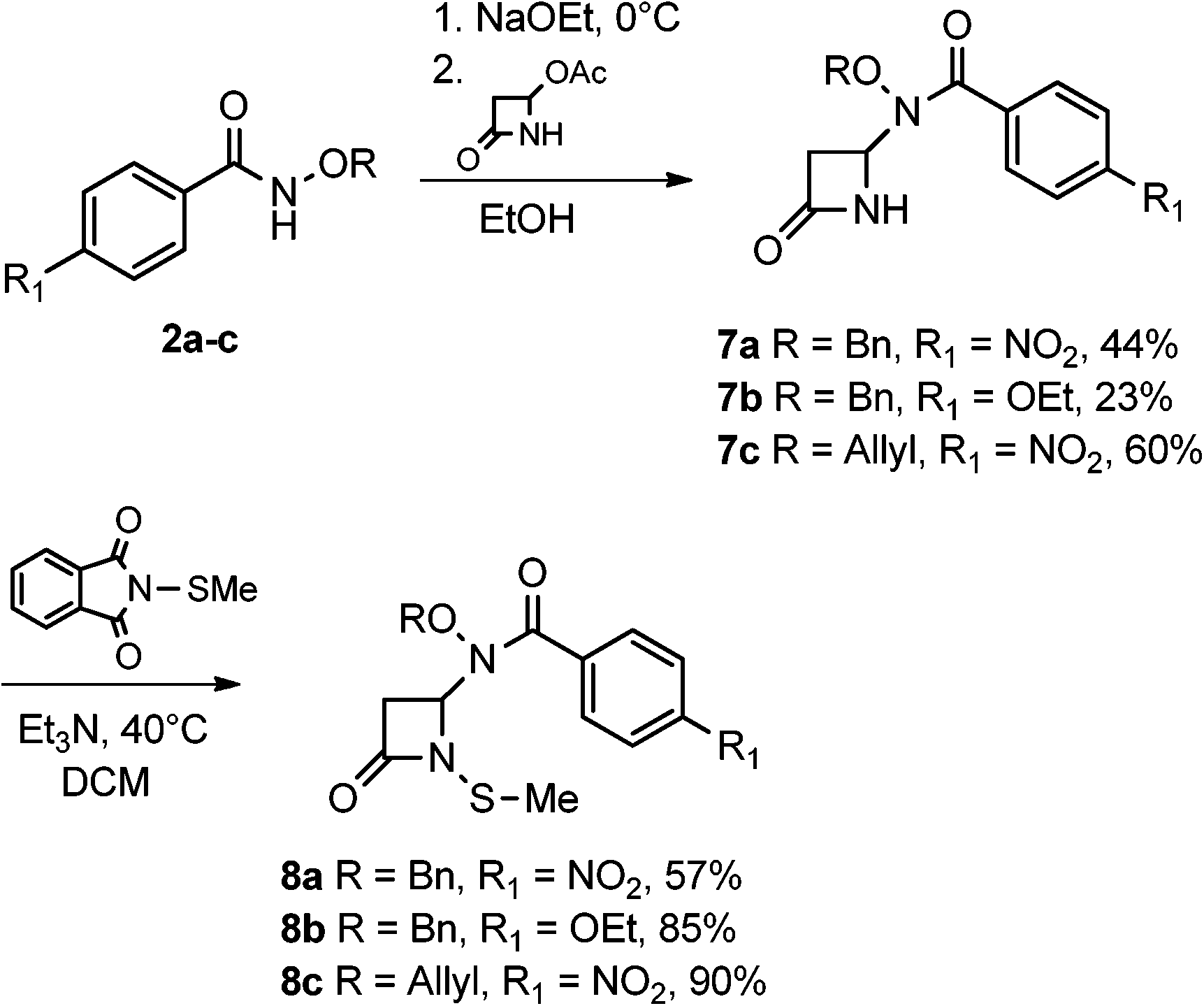
The syntheses and anti-TB evaluation of the necessary hydroxamate precursors are reported elsewhere.23 Once in hand, these hydroxamates were then reacted with a variety of azetidinones to give an initial set of monobactams, first those devoid of functionality at the C-3 position (Schemes 1–3). Hydroxamates 1–3 were added to 4-acetoxy-2-azetidinone, under similar conditions employed by Clauss24 with other nucleophiles, to give racemic monobactams 4, 7a–c, and 9a–b. Intermediate 4 was both N-thiolated with 2-(methylthio)isoindoline-1,3-dione25 and also separately silylated with TBSCl to give 5 and 6, respectively. Monobactams 7a–c and 9a–b were also N-thiolated to give 8a–c and 10a–b in good yields.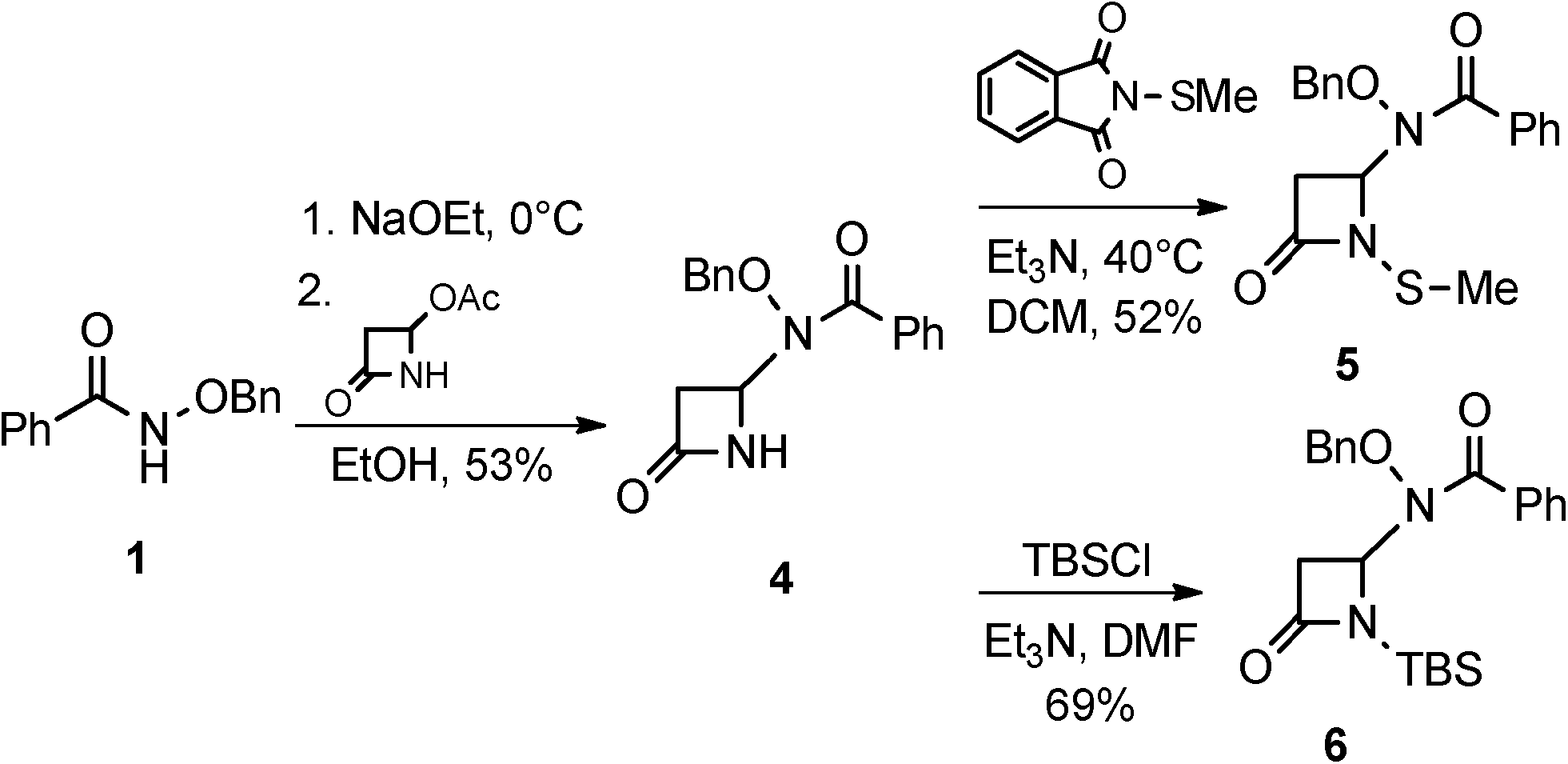 | ||
| Scheme 1 Racemic syntheses of 3-unsubstituted monobactams. | ||
 | ||
| Scheme 2 Racemic syntheses of monobactams with varied aromatic electronics. | ||
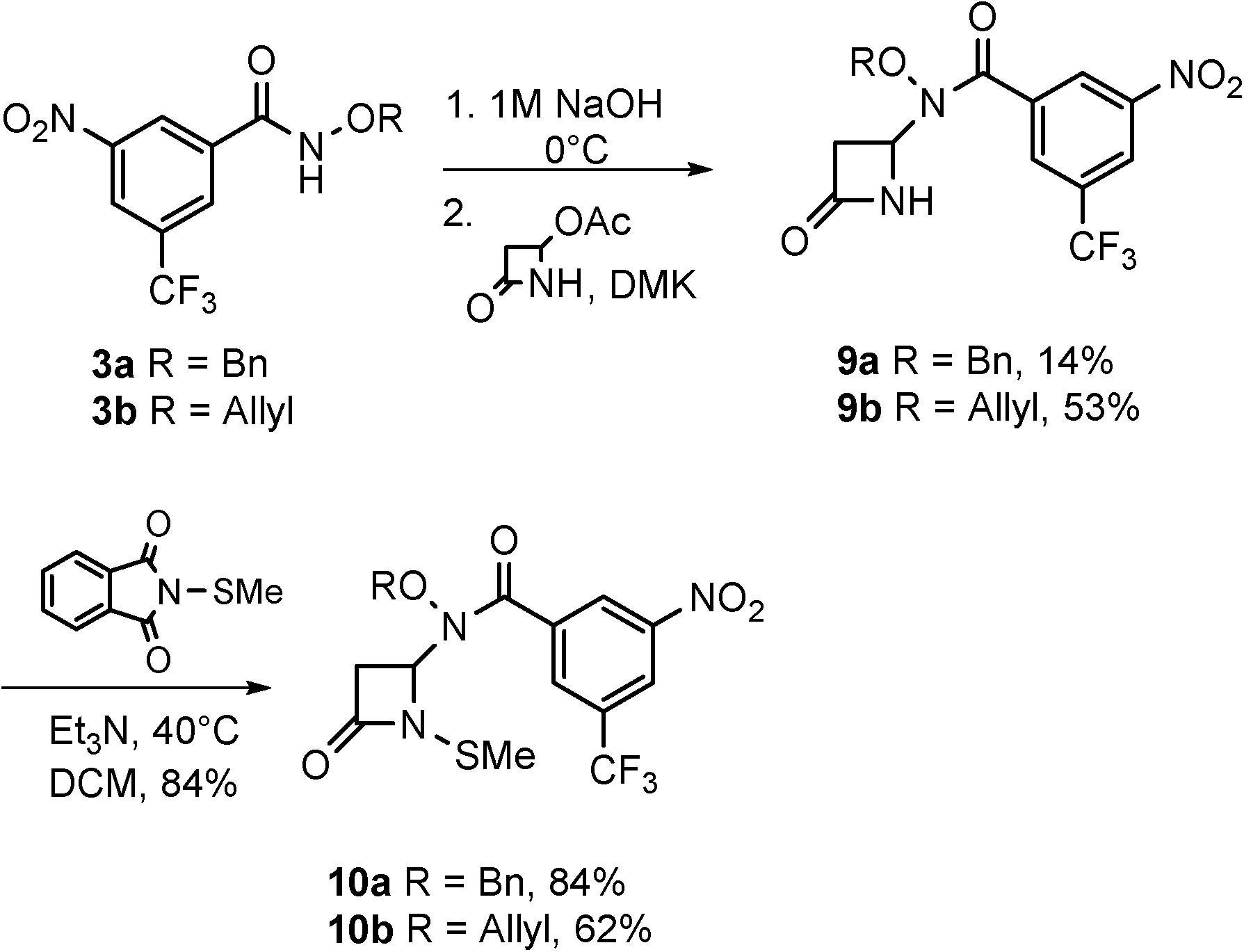 | ||
| Scheme 3 Racemic syntheses of monobactams with 3-nitro-5-(trifluoromethyl) aromatic group. | ||
We then turned our attention to synthesizing compounds that did possess established β-lactam sidechains, specifically, compounds with the hydroxyethyl, phenylacetamido, and phenoxyacetamido sidechains. Based on classic precedent, we hypothesized that these moieties may be needed to potentiate activity.26 As shown in Scheme 4, 1 was added to (2R,3R)-3-((R)-1-((tert-butyldimethylsilyl)oxy)ethyl)-4-oxoazetidin-2-yl acetate27 to give 11 in modest yield. The expected trans stereochemistry between the C-3 and C-4 hydrogens was verified using homo decoupling NMR experiments and the fact that β-lactam protons give distinctively different coupling values depending on whether they are cis or trans to each other.28 Next, was to remove the silyl protecting group. In most cases, a fluoride source, such as TBAF is employed for this conversion. However, in our hands none of the various sources of fluoride worked. Rather, stirring 11 in 1 M HCl with careful monitoring produced 12. Additionally, intermediate 11 was N-thiolated to give 13 and then also subjected to standard hydrogenolysis conditions to give hydroxamic acid 14.
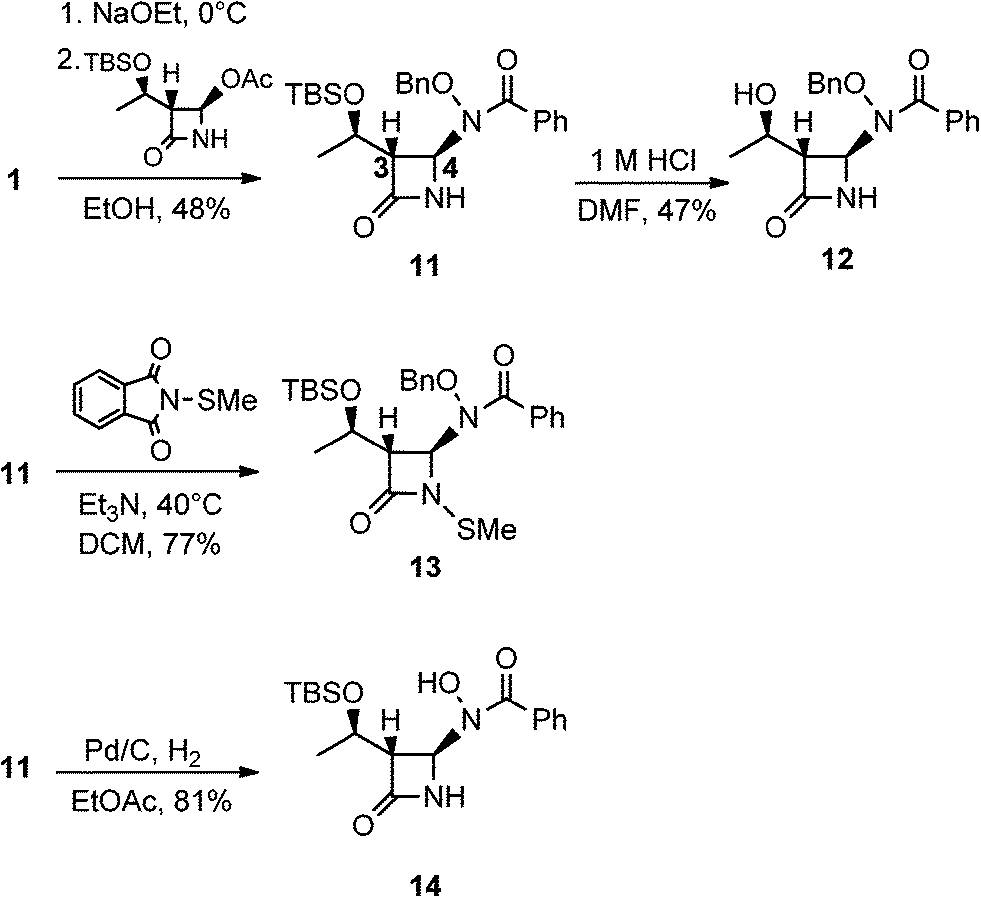 | ||
| Scheme 4 Syntheses of monobactams with a carbapenam side chain. | ||
Scheme 5 illustrates the syntheses of hydroxamate-containing monobactams with the phenylacetamido sidechain. This synthetic sequence started with the degradation of penicillin G29 to ultimately give the appropriately functionalized azetidinone. Thus, once generated from penicillin G, 15 was then subjected to ozonolysis and methanolysis to give key azetidinone 16. This intermediate was N-thiolated to give 17 and also reacted with hydroxamate 1 to give monobactam 18. Again, the expected trans stereochemistry was verified with NMR homo decupling analyses. Lastly, 18 was N-thiolated to yield product 19.
 | ||
| Scheme 5 Syntheses of monobactams with penicillin G side chain. | ||
Scheme 6 illustrates the preparation of hydroxamate containing monobactams containing the phenoxyacetamido, or penicillin V sidechain. The initial chemical transformations here are analogous to those in Scheme 5. Thus, penicillin V potassium salt was degraded with mercuric acetate in acetic acid, subjected to ozonolysis, and finally methanolysis to give key azetidinone 20. This azetidinone was then separately reacted with N-((tert-butyldimethylsilyl)oxy)benzamide and three additional hydroxamates to give 21 and highly functionalized monobactams 23a–c, respectively. To confirm the expected trans relationship between the C-3 and C-4 hydrogens, homo decoupling experiments were conducted on 23b. Lastly, N-thiolation of 23a–b yielded 24a–b.
 | ||
| Scheme 6 Syntheses of monobactams with penicillin V side chain. | ||
All compounds synthesized were then subjected to biological evaluations. Displayed in Table 1 are MIC data for select compounds against M. tb in the microplate alamar blue assay (MABA).30 As demonstrated, a number of compounds exhibited notable media-dependent activity against M. tb with differences in MIC values from different media being attributed to factors such as compound solubility, carbon source and media age.31 With regards to monobactams devoid of functionality at the C-3 position (Scheme 1), we saw that the more lipophilic and electron deficient the compound, generally the more active it was against M. tb. This observation can be highlighted by comparing the activities of compounds 5–10.
| Compound | 7H12a | GASb | Compound | 7H12a | GASb |
|---|---|---|---|---|---|
| a 7H12 = 7H9 medium + casitone, palmitic acid, albumin, and catalase. b GAS = glycerol-alanine salts medium. | |||||
| 5 | >50 | 17.86 | 14 | >50 | >50 |
| 6 | 49.20 | 0.28 | 17 | 47.65 | 10.67 |
| 7b | >100 | 49.32 | 18 | >100 | 6.05 |
| 8a | >100 | 24.23 | 19 | 47.33 | 11.56 |
| 8b | >100 | 34.78 | 23a | 5.93 | <0.195 |
| 9a | 44.72 | 30.27 | 24a | 6.00 | >50 |
| 10a | 23.38 | 5.92 | 24b | >100 | 64.14 |
| 11 | >100 | >100 | BTZ-043 | <0.004 | <0.004 |
| 12 | >100 | >100 | Rifampin | 0.05 | 0.04 |
| 13 | 10.26 | 0.753 | |||
Compounds that possessed hydroxyethyl functionality at C-3 led to some interesting observations. Based on the results for 3-unsubstituted compounds, it was surprising that 11 did not exhibit noticeable anti-TB activity, despite this compound possessing both lipophilic benzyl and TBS groups. Hypothesizing that the TBS group may need to be removed in order to activate the compound, we carried out the TBS removal (Scheme 4); however, 12 was inactive as well. This further solidified the notion that factors other than significant lipophilicity were crucial for the activity of these compounds. Furthermore, it was observed that once 11 was N-thiolated, the product obtained (13) demonstrated good activity against M. tb in both types of media utilized. In this case, it can be inferred that the methylthio substituent is important for activity with this set of compounds.
For compounds with penicillin G and V sidechains installed, we saw that having the phenoxyacetamido, or penicillin V sidechain, generated compounds with increased anti-TB activity. This can be readily illustrated by comparing the activities of 19 with 24a and 18 with 23a. Overall, it was most intriguing that such anti-TB activity was seen considering these monobactams lack a classic ionizable group normally assumed to be necessary for antibiotic activity. An interesting trend seen among all compounds evaluated was that N-thiolation often generated compounds with some measurable degree of anti-TB activity. First reported by Turos, N-alkylthio β-lactams have emerged as a new class of β-lactam scaffolds which have demonstrated excellent bacteriostatic activity against S. aureus and MRSA, in addition to anti-cancer and fungal activity. Turos has shown that these β-lactams exhibited their bacteriostatic activity against S. Aureus by in vivo transfer of the N-thiol group to coenzyme A, ultimately disrupting essential fatty acid biosynthesis.32 However, to our knowledge, there have not been any reports on the extension of N-thiolated β-lactams as anti-TB agents.
The second evaluation performed was the Kirby–Bauer antibacterial agar diffusion assay.33 It was seen that a number of compounds targeted Gram-positive bacteria, especially M. vaccae, with notable zones of inhibition in the range of 20–37 mm (see ESI‡). Based on the aforementioned anti-TB activity, this result was not unexpected since both M. tb and M. vaccae bacterial strains are from the same genus. However, it should be noted that a number of the most potent TB agents reported herein (6, 13, and 24a) were not very active in the agar studies. We also determined minimum inhibitory concentration (MIC) values for compounds of interest. Notably, both compounds 9a and 10b exhibited MICs of 6.25 μM against M. vaccae.
Lastly, select compounds were screened against a broad panel of β-lactamases in vitro at and using protocols established by Rempex pharmaceuticals. Our studies included enzymes from all four classes (A, B, C, and D). Here, synthesized compounds were incubated with purified enzyme (or lysate) for 10 min, then an enzyme substrate was added, and the optical density of the wells against time was measured and Ki values determined,34 with pertinent results illustrated in Table 2. For studies with metallo β-lactamase NDM-1, imipenem was utilized as the enzyme substrate while for all other enzymes, nitrocefin was employed. Overall, we observed that many of the monobactams acted as good β-lactamase inhibitors, with activity noted against a broad range of enzymes screened. Highlighted among these results was the broad spectrum of inhibition that compounds 8c and 19 exhibited. Azetidinone 8c, seen as one of the most active compounds in β-lactamase inhibition screenings showed inhibitory activity against CTX-M-14, NDM-1, and VIM-1 with calculated Ki values of 0.64, 0.03, and 22.7 μM, respectively. Compounds 6, 18, 19, and 24a were of outstanding interest, as a result of their activity against both M. tb and β-lactamases. With these encouraging results, we then tested all compounds for synergy with various β-lactam antibiotics against drug resistant, β-lactamase producing strains. However, interestingly we saw that the combinations were inactive.
| Enzyme | CTX-M-14 (purified) | Oxa-48 (purified) | NDM-1 (purified) | VIM-1 (lysate) |
|---|---|---|---|---|
| Class | A | D | B | B |
| Compound | K i (μM) | |||
| a Oxa-48: oxacillinase β-lactamase; NDM-1: New-Delhi metallo β-lactamase; VIM-1: Verona integron-encoded metallo β-lactamase. | ||||
| 6 | >160 | 1.72 | >160 | >160 |
| 7b | >160 | >160 | >160 | >160 |
| 7c | >160 | >160 | >160 | >160 |
| 8a | >160 | >160 | 0.97 | >160 |
| 8b | >160 | >160 | 0.07 | >160 |
| 8c | 0.64 | >160 | 0.03 | 22.7 |
| 9b | >160 | 1.93 | >160 | >160 |
| 10b | >160 | >160 | 0.04 | 59.27 |
| 11 | 0.25 | >160 | >160 | >160 |
| 12 | >160 | >160 | >160 | >160 |
| 13 | 0.40 | >160 | >160 | >160 |
| 14 | 0.32 | >160 | >160 | >160 |
| 18 | >160 | >160 | 5.54 | >160 |
| 19 | >160 | 1.95 | 0.07 | 2.86 |
| 20 | >160 | >160 | 5.87 | >160 |
| 21 | >160 | >160 | 0.96 | 31.2 |
| 23a | 0.05 | >160 | >160 | >160 |
| 24a | >160 | >160 | 0.06 | >160 |
| 24b | >160 | >160 | 0.15 | >160 |
When looking for trends from the data set in Table 2, it seemed apparent that N-thiolation was quite important in potentiating activity against the class B metallo β-lactamases. As ready examples, when comparing activities against class B enzymes, the conversion from 7b to 8b, 7c to 8c, and 23b (inactive, >160) to 24b all resulted in the conversion of an inactive compound to a potent inhibitor of NDM-1. The interesting exception was compound 13, whose activity was directed specifically at CTX-M-14 despite bearing the methylthio substituent. Further, silyl removal to give 12 resulted in an inactive compound.
Thiol containing compounds as inhibitors of β-lactamases are well established in the literature.35 For example, it has been shown that 9-(2-amidoethenylthio)-9-deoxy derivatives of clavulanic acid acted as good inhibitors of TEM-1 and Staphylococcal β-lactamases.36 Furthermore, mercaptoacetic acid thiol acid derivatives were reported as potent inhibitors of the B. cereus metallo enzyme.37 It was proposed that active substrates were hydrolyzed by the enzyme, releasing the thiol component which then formed a covalent bond with the active site cysteine. Another mercaptocarboxylate inhibitor; however, was shown to interact with IMP-1 metallo β-lactamase from P. aeruginosa through two key interactions: direct coordination of the sulfur to the Zn in the active site, and binding in the hydrophobic pocket.38 To the best of our knowledge, there are no reports of thiol-containing monobactams that act as potent and/or selective inhibitors of β-lactamases in the literature. Lastly, it was intriguing that none of the compounds in this report possessed an ionizable group, such as a carboxylate or –SO3, normally required for antibacterial and/or β-lactamase inhibitory activity. However based on the above precedent and discussions, it seems to not be required.
Conclusions
To close, we have synthesized a library of highly functionalized monobactams and have subjected them to antimicrobial testing. Many compounds exhibited notable anti-TB activity (MIC values 10.65 to <0.195 μM) and selective β-lactamase inhibitory activity (Ki values in the range 20 to 0.03 μM) against purified enzymes. Although at this stage of our studies the exact target(s) and/or general mode of action remain unknown, these compounds may serve as scaffolds against both TB and β-lactamases.Acknowledgements
We acknowledge the University of Notre Dame, and the NIH (NIH-2R01-AI054193 and NIH-T32GMO75782) for support of this work. We acknowledge Rempex Pharmaceuticals for providing β-lactamase enzymes for screening during a CBBI supported internship at Rempex by K. D. W. We acknowledge Nonka Sevova (Mass Spectrometry and Proteomics Facility, UND) for mass spectroscopic analyses, the ND Energy Materials Characterization Facility for use of IR instrumentation, and the Lizzadro NMR facilities for use of NMR instrumentation. M. W. M acknowledges an ECK Global Health Fellowship (2014-2015, UND). K. D. W. acknowledges a CBBI Fellowship (2013-2015, UND).References
- T. S. Chitre and K. G. Bothara, J. Chem. Pharm. Res., 2011, 3, 479–488 CAS.
- Tuberculosis World Health Organization, http://who.int/mediacentre/factsheets/fs104/en/index.html, Retrieved 12 November 2009, Fact sheet No 104, 2007.
- C. Dye and B. G. Williams, Science, 2010, 328, 856–861 CrossRef CAS PubMed.
- E. Peeters, H. J. Nelis and T. Coenye, J. Antimicrob. Chemother., 2009, 64, 801–809 CrossRef CAS PubMed.
- C. Bebrone, P. Lassaux, L. Vercheval, J. S. Sohier, A. Jehaes, E. Sauvage and M. Galleni, Drugs, 2010, 70, 651–679 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, 2nd edn, 2001, pp. 390–393 Search PubMed.
- L. I. Llarrull, S. A. Testero, J. F. Fisher and S. Mobashery, Curr. Opin. Microbiol., 2010, 13, 551–557 CrossRef CAS PubMed.
- J. E. Hugonnet, L. W. Tremblay, H. I. Boshoff, C. E. Barry and J. S. Blanchard, Science, 2009, 323, 1215–1218 CrossRef CAS PubMed.
- S. Solapure, N. Dinesh, R. Shandil, V. Ramachandran, S. Sharma, D. Bhattacharjee, S. Ganguly, J. Reddy, V. Ahuja, V. Panduga, M. Parab, K. G. Vishwas, N. Kumar, M. Balganesh and V. Balasubramanian, Antimicrob. Agents Chemother., 2013, 57, 2506–2510 CrossRef CAS PubMed.
- M. B. Kostova, C. J. Myers, T. N. Beck, B. J. Plotkin, J. M. Green, H. I. M. Boshoff, C. E. Barry, J. R. Deschamps and M. I. Konaklieva, Bioorg. Med. Chem., 2011, 19, 6842–6852 CrossRef CAS PubMed.
- B. M. Dinnimath, S. M. Hipparagi and M. Gowda, Int. J. Pharm. Technol., 2011, 3, 3792–3801 CAS.
- H. F. Chambers, J. Turner, G. F. Schecter, M. Kawamura and P. C. Hopewell, Antimicrob. Agents Chemother., 2005, 49, 2816–2821 CrossRef CAS PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Nat. Rev. Drug Discovery, 2013, 12, 388–404 CrossRef CAS PubMed.
- E. B. Wong, K. A. Cohen and W. R. Bishai, Trends Microbiol., 2013, 21, 493–501 CrossRef CAS PubMed.
- R. Singh, U. Manjunatha, H. I. M. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim, L. Zhang, S. Kang, T. H. Keller, J. Jiricek and C. E. Barry, III, Science, 2008, 322, 1392–1395 CrossRef CAS PubMed.
- V. Makarov, G. Manina, K. Mikusova, U. Möllmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, B. Silvia, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- G. C. Moraski, L. D. Markley, J. Cramer, P. A. Hipskind, H. Boshoff, M. A. Bailey, T. Alling, J. Ollinger, T. Parish and M. J. Miller, ACS Med. Chem. Lett., 2013, 4, 675–679 CrossRef CAS PubMed.
- M. S. Kabir, O. A. Namjoshi, R. Verma, R. Polanowski, S. M. Krueger, D. Sherman, M. A. Rott, W. R. Schwan, A. Monte and J. M. Cook, Bioorg. Med. Chem., 2010, 18, 4178–4186 CrossRef CAS PubMed.
- (a) M. Flipo, T. Beghyn, J. Charton, V. A. Leroux, B. P. Deprez and R. F. Deprez-Poulain, Bioorg. Med. Chem., 2007, 15, 63–76 CrossRef CAS PubMed; (b) J. J. Irwin, F. M. Raushel and B. K. Shoichet, Biochemistry, 2005, 44, 12316–12328 CrossRef CAS PubMed; (c) Y. M. Lin and M. J. Miller, J. Org. Chem., 1999, 64, 7451–7458 CrossRef CAS.
- M. J. Miller, G. Zhao, S. Vakulenko, S. Franzblau and U. Möllmann, Org. Biomol. Chem., 2006, 4, 4178–4185 CAS.
- E. Turos, Top. Heterocycl. Chem., 2013, 30, 147–182 CAS.
- M. W. Majewski, S. Cho, P. A. Miller, S. G. Franzblau and M. J. Miller, Bioorg. Med. Chem. Lett., 2015 DOI:10.1016/j.bmcl.2015.04.099.
- K. Clauss, D. Grimm and G. Prossel, Justus Liebigs Ann. Chem., 1974, 4, 539–560 Search PubMed.
- C. Cesario and M. J. Miller, J. Org. Chem., 2009, 74, 5730–5733 CrossRef CAS PubMed.
- M. I. Page and A. P. Laws, Tetrahedron, 2000, 56, 5631–5638 CrossRef CAS.
- Purchased from Ontario Chemicals, Unit 7A, 291 Woodlawn Road W. Guelph, Ontario, N1H7L6 Canada.
- P. Bevilacqua and J. L. Roberts, Synth. Commun., 1983, 13, 797–808 CrossRef CAS.
- E. Oh, Y. Y. Lee and Y. M. Goo, Arch. Pharm. Res., 1988, 11, 163–165 CrossRef CAS.
- (a) L. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004–1009 CAS; (b) J. J. De Voss, K. Rutter, B. G. Schroeder, H. Su, Y. Zhu and C. E. Barry 3rd, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1252–1257 CrossRef CAS PubMed; (c) S. H. Cho, S. Warit, B. Wan, C. H. Hwang, G. F. Pauli and S. G. Franzblau, Antimicrob. Agents Chemother., 2007, 51, 1380–1385 CrossRef CAS PubMed.
- S. G. Franzblau, M. A. DeGroote, S. H. Cho, K. Andries, E. Nuermberger, I. M. Orme, K. Mdluli, I. Angelo-Barturen, T. Dick, V. Dartois and A. J. Lenaerts, Tuberculosis, 2012, 92, 453–488 CrossRef CAS PubMed.
- (a) E. Turos, M. I. Konaklieva, R. X. F. Ren, H. Shi, J. Gonzalez, S. Dickey and D. Lim, Tetrahedron, 2000, 56, 5571–5578 CrossRef CAS; (b) B. Heldreth, T. E. Long, S. Jang, G. S. K. Reddy, E. Turos, S. Dickey and D. V. Lim, Bioorg. Med. Chem., 2006, 14, 3775–3784 CrossRef CAS PubMed; (c) K. D. Revell, B. Heldreth, T. E. Long, S. Jang and E. Turos, Bioorg. Med. Chem., 2007, 15, 2453–2467 CrossRef CAS PubMed.
- (a) A. W. Bauer, D. M. Perry and W. M. M. Kirby, Arch. Intern. Med., 1959, 104, 208–216 CrossRef CAS; (b) A. W. Bauer, W. M. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493–496 CAS.
- S. G. Waley, Biochemistry, 1982, 205, 631–633 CrossRef CAS.
- (a) S. Siemann, A. J. Clarke, T. Viswanatha and G. I. Dmitrienko, Biochemistry, 2003, 42, 1673–1683 CrossRef CAS PubMed; (b) W. Jin, Y. Arakawa, H. Yasuzawa, T. Taki, R. Hashiguchi, K. Mitsutani, A. Shoga, Y. Yamaguchi, H. Kurosaki, N. Shibata, M. Ohta and M. Goto, Biol. Pharm. Bull., 2004, 27, 851–856 CrossRef CAS PubMed; (c) A. Badarau, A. Llinas, A. P. Laws, C. Damblon and M. I. Page, Biochemistry, 2005, 44, 8578–8589 CrossRef CAS PubMed.
- G. Brooks, K. J. Coleman, S. Davies and P. A. J. Hunter, Antibiotics, 1988, 41, 892–898 CrossRef CAS.
- D. J. Payne, J. H. Bateson, B. C. Gasson, D. Proctor, T. Khushi, T. H. Farmer, D. A. Tolson, D. Bell, P. W. Skett, A. C. Marshall, R. Reid, L. Ghosez, Y. Combret and J. Marchand-Brynaert, Antimicrob. Agents Chemother., 1997, 41, 135–140 CAS.
- N. O. Concha, C. A. Janson, P. Rowling, S. Pearson, C. A. Cheever, B. P. Clarke, C. Lewis, M. Galleni, J. M. Frere, D. J. Payne, J. H. Bateson and S. S. Abdel-Meguid, Biochemistry, 2000, 39, 4288–4298 CrossRef CAS PubMed.
Footnotes |
| † This manuscript was written through contributions of all authors. M. W. M. synthesized and characterized all compounds and wrote both this report and the supporting information section. K. D. W. performed β-lactamase screenings, IR characterization, and assisted with decoupling experiments. S. C. performed the MABA assay, P. A. M. performed MIC and diffusion assays. All authors have given approval to the final version of the manuscript. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and characterization data. See DOI: 10.1039/c5md00340g |
| This journal is © The Royal Society of Chemistry 2016 |