
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A kinetic mechanism of repair of DNA containing α-anomeric deoxyadenosine by human apurinic/apyrimidinic endonuclease 1
N. A.
Timofeyeva
* and
O. S.
Fedorova
 *
*
Siberian Branch of the Russian Academy of Sciences, Institute of Chemical Biology and Fundamental Medicine, Novosibirsk 630090, Russia. E-mail: fedorova@niboch.nsc.ru; na_timof@niboch.nsc.ru
First published on 14th September 2016
Abstract
α-Anomers of 2′-deoxyadenosine (αdA) are major products of deoxyadenosine damage when DNA is γ-irradiated under anoxic conditions. Such lesions are a threat to genomic stability and are known to be processed by human apurinic/apyrimidinic endonuclease 1 (APE1). The aim of this study was to determine whether the α-anomeric structure enhances enzyme recognition. For this purpose, we analyzed the kinetic mechanism of αdA conversion by APE1 using a stopped-flow fluorescence technique. Our data reveals that the initial formation of the complex of APE1 with an αdA-containing substrate is followed by at least three conformational transitions in this complex that correspond to the induced fit leading to the formation of a catalytically competent complex. A local perturbation around the αdA lesion in the DNA duplex allows APE1 to avoid the initial conformational changes observed earlier in the case of the enzyme binding to an undamaged ligand, abasic-site-, tetrahydrofuran-, or 5,6-dihydrouridine-containing substrates. The αdA structure promotes recognition by the enzyme but dramatically impedes formation of the catalytically competent complex and hydrolysis of the 5′-phosphodiester bond. A step following the chemical reaction, possibly a release of the αdA-containing product, is rate-limiting for the overall enzymatic process, though an α-anomeric nucleotide at the 5′ terminus of the DNA nick accelerates dissociation of the enzyme–product complex. Our results show that the efficiency of αdA lesion conversion by APE1 is very low. Nonetheless, αdA repair by APE1 is probably a biologically relevant process.
1. Introduction
The human genome is continuously assaulted by various reactive species of either endogenous or exogenous origin; this process results in the formation of DNA lesions. The α-anomer of 2′-deoxyadenosine (αdA) is one of such lesions. It arises from deoxyadenosine exposed to γ-irradiation under anaerobic conditions.1 This compound is generated by abstraction of the anomeric hydrogen atom at C1′ by a free hydroxyl radical, which leads to epimerization at the C1′ position.2,3 αdA is found to be a major product of deoxyadenosine damage when poly(dA), poly(dA–dT), and salmon testis DNA are γ-irradiated under a nitrogen atmosphere.4 Formation of αdA lesions in human cells may have serious consequences because the eukaryotic nucleus is a poorly oxygenated intracellular compartment.5,6 Besides, tumor cells have adapted the ability to grow in hypoxic environments, and radiation therapy provides the conditions for such damage.7In order to sustain cellular genome integrity, DNA lesions must be repaired. The majority of damaged DNA bases are processed by the base excision repair (BER) pathway initiated by DNA glycosylases. Nevertheless, the αdA lesion is not repaired by DNA glycosylases/apurinic/apyrimidinic (AP) lyases. Instead, Escherichia coli endonuclease IV (Nfo), Saccharomyces cerevisiae AP endonuclease 1 (Apn1), and human AP endonuclease 1 (Ape1) directly incise the phosphodiester bond 5′ to the lesion in DNA, as part of the nucleotide incision repair (NIR) pathway.8–10
Because the repair pathway for the αdA lesions is conserved from E. coli to yeast and humans, it has been suggested that base lesions with the α-conformation with respect to the N-glycosidic bond may be biologically significant.10 The αdA lesion constitutes a moderate block to DNA replication catalyzed by Pol I in vitro, and the nucleotide insertion frequency opposite αdA is in the order T > C ≥ A ≫ G.11 Furthermore, αdA represents a moderate block to the replication of transfected M13 vectors containing αdA, where the replicating enzyme is Pol III. The ability of in vivo replication to pass through αdA was found to be ∼20% relative to the normal A base. αdA generates a single nucleotide deletion in vivo. These data suggest that αdA is potentially mutagenic.12
Probably, the structure of DNA containing an αdA lesion should develop preexisting deformations and/or be easily deformable for specific recognition by AP endonucleases. Several studies have been carried out to ascertain the possible influence of a single αdA inserted into regular oligonucleotide strands on their structures.
Initially,13 it was shown that duplexes containing a αdA/N pair (N = A, G, C, or T) in the middle do not undergo global conformational changes such as transitions to the A or Z forms of DNA. At the same time, the in vacuo energy minimization data suggested that precise positioning of the base and sugar–phosphate backbone deviates from the canonical one. The adenine ring protrudes in the minor groove, with the N6 amino group projecting well into that groove. For the duplexes containing an αdA/pyrimidine pair, displacement of the sugar–phosphate backbone from the canonical position is relatively minor. The αdA/T base pair was found to form two hydrogen bonds almost equivalent in distances and angles to the canonical A/T base pair, although the second hydrogen bond forms between atoms N6 of αdA and O2 of the opposing T. The movement of the N6 amino group into the minor groove is accompanied by a slight shift (inside the helix) of the opposing T. A minor displacement of the sugar–phosphate backbone occurs at the lesion site and a base immediately 5′ to αdA. The base-stacking interaction between αdA and a base located 5′ to this lesion is interrupted. The duplexes containing an αdA/purine pair show large distortion of the sugar–phosphate backbone. This distortion is not localized at the lesion site but extends to the surrounding sequences. The distortions in the duplex αdA/G produce a distinct kink and a bulge in the DNA backbone.13
Furthermore,14 molecular dynamics (MD) simulations of the structure and dynamics of a DNA duplex containing a single αdA residue in an aqueous solution showed the formation of a nonclassical αdA/T pair. In one simulation, the bases tended to mimic the classical Watson–Crick pair, and two stable hydrogen bonds were observed, though the second hydrogen bond was between N6 of αdA and O2 of the opposing T. In another simulation, the alignment of both bases involved did not allow for any effective hydrogen bonding, only in some periods of simulation did one hydrogen bond form between N6 of αdA and O2 of the opposing T. As portrayed earlier,13 it was shown that a single αdA/T base pair has a destabilizing effect mostly on its nearest neighbors, particularly with respect to sugar puckering and base stacking.14 Still, the duplex axis course was found to be less stable in the α-anomerized model than in the reference molecule. Those authors observed competition between effective base stacking of the modified residue and its stable alignment within the pair. The effective hydrogen bonding in the αdA/T base pair forces the modified residue to adopt a conformation that weakens its stacking capability. These findings are in contrast to the classical DNA duplex structures where both types of interactions (stacking and hydrogen bonding) cooperate in stabilizing their conformations.14
The nuclear magnetic resonance (NMR) solution studies15 established that the single αdA residue (within the αdA/T pair) flanked by cytosines (5′-CαdAC-3′) is intrahelical and is stacked in a reverse Watson–Crick fashion consistent with the slight decrease in thermostability. The base stacking interactions of αdA with its flanking nucleotides were found to be altered due to the change in chirality at C1′. The stacked αdA residue results in an 18° kink of the helical axis into the major groove. These changes are accompanied by enlargement of the minor groove 3′ to the modification. The conformation of the flanking base-paired segments was not altered strongly relative to a B-type conformation.15 A comparison of different flanking sequences revealed that the minor groove topology and kink are dependent on the sequence surrounding the αdA lesion.16
The cocrystal structures of Nfo, Apn1, or APE1 bound to a DNA duplex containing a single αdA nucleotide have not been obtained to date. An attempt to solve the crystal structure of the E. coli Nfo-H69A mutant bound to an intact duplex DNA containing an αdA/T base pair was successful at determining the high-resolution crystal structure of Nfo-H69A bound to a NIR product.17 The αdA/T base pair was shown to be well stacked in the DNA, and the phosphate group of αdA points toward the solvent. The αdA base, which is rotated 180° around the C1′–N9 axis (compared with an adenine base) makes modified Watson–Crick contacts. Its N6 atom interacts with the O2 atom of the opposing T. In contrast, the preceding base pair (C/G) is unpaired, with both bases being extrahelical.17
The kinked helical axis and the enlarged minor groove around the lesion revealed by NMR for αdA-containing DNA15 were also demonstrated in the crystal structures of an apurinic DNA substrate in complex with Endo IV18 or APE1.19,20 Thus, it was suggested that the α-anomeric structure promotes its recognition by the enzyme because of structural distortions that are already on the path toward their values in the subsequent complex. The presence of a kink and enlarged minor groove is expected to facilitate the enzymatic access both by reducing the energetic cost of forming the initial distortion and by further reducing the energetic cost of driving the nucleic acid into its final conformation.15
The objective of the present study was to determine whether the α-anomeric structure enhances enzyme recognition. To this end, we used the stopped-flow approach to study the conformational dynamics and the kinetic mechanism of the APE1 interaction with the αdA-containing substrates. Our data reveals that the αdA structure promotes recognition by the enzyme but dramatically impedes formation of the catalytically competent complex and hydrolysis of the 5′-phosphodiester bond.
2. Materials and methods
2.1. The enzyme and oligonucleotides
Human APE1 was overexpressed in the BL21 (DE3) pLysS E. coli cells by means of the pXC53 plasmid carrying the APE1 gene, and was purified as described elsewhere.21 The concentration of APE1 was determined by the Bradford assay using BSA as a standard. Oligodeoxyribonucleotides (ODNs; 5′ → 3′) d(ATGCACATCGTCTACATGCTTATGCAGTCA), d(ATGCACATCGTCTAACTGCTTATGCAGTCA), d(TGACTGCATA(αA)GCAXGTAGACGATGTGCAT) (X = T or NH2C6T, which is 5-[N-(trifluoroacetylaminohexyl)-3-acrylimido]-2′-deoxyuridine [Amino-Modifier C6 dT; Glen Research, USA], a modified thymine containing a hexamethylene amino linker on the C5 atom), d(TGACTGCATA(αA)GCAG(NH2C6T)TAGACGATGTGCAT) and d(TGACTGCATA) were synthesized in the Laboratory of Bionanotechnology (Institute of Chemical Biology and Fundamental Medicine, the Siberian Branch of the Russian Academy of Sciences, Russia) by the standard phosphoramidite method using monomers from Glen Research (USA) and were purified using anion exchange and reversed-phase high-performance liquid chromatography (HPLC). Oligodeoxyribonucleotides d(TGACTGCAT(2-aPu)(αA)GCATGTAGACGATGTGCAT) (where 2-aPu is 2-aminopurine: a highly fluorescent analog of adenine) and pd((αA)GCATGTAGACGATGTGCAT) were synthesized by Eurogentec (France) and kindly provided to us by Dr Alexander A. Ishchenko (CNRS UMR8200 Université Paris-Sud XI, Institut de Cancérologie Gustave Roussy, Villejuif, France). The concentration of each individual single-stranded (ss) ODN was determined via absorbance at 260 nm with the extinction coefficients calculated from the nearest-neighbor data for mono- and dinucleotides.222.2. Oligonucleotide labeling
A fluorophore, 5-(and 6-)carboxytetramethylrhodamine (TAMRA), was covalently attached to ODNs d(TGACTGCATA(αA)GCA(NH2C6T)GTAGACGATGTGCAT) and d(TGACTGCATA(αA)GCAG(NH2C6T)TAGACGATGTGCAT) at position 5 of the modified thymine heterocyclic base (NH2C6T) through a C6 amino linker. The ODN (10 optical units in 20 μL of H2O) was mixed with 150 μL of 0.1 M Na2B4O7 (pH 8.5). 5(6)-TAMRA succinimidyl ester [5(6)-TAMRA-SE; 4 mg] was dissolved in 64 μL of dimethyl formamide (DMF). The ODN solution was treated with 32 μL of 5(6)-TAMRA-SE in DMF. The reaction mixture was incubated with constant shaking for 1 h at room temperature. Then, 16 μL of 5(6)-TAMRA-SE in DMF was added, and the reaction mixture was incubated for one more hour. The incubation was continued for another 2 h after addition of the remaining 16 μL of 5(6)-TAMRA-SE in DMF. After that, the reaction mixture was incubated without shaking for 3 h at 4 °C, and precipitated with 2% LiClO4 in acetone. Purification of the reaction mixture to get rid of the free dye was performed using gel filtration chromatography. The TAMRA-labeled OND was separated from the unlabeled ODN using denaturing polyacrylamide gel electrophoresis (PAGE) in a 20% acrylamide gel containing 8 M urea. When necessary, the 5′ terminus of the ODN d(TGACTGCATA(αA)GCATGTAGACGATGTGCAT) was labeled with 32P according to the standard procedure23 using T4 polynucleotide kinase (20 U; SibEnzyme, Russia) and [γ-32P]ATP (Biosan, Russia). The oligonucleotide duplexes used in this study are presented in Fig. 1. | ||
| Fig. 1 Schematic presentation of various DNA duplexes used in this study. | ||
2.3. Kinetic fluorescence experiments
The kinetics of the APE1 interactions with the DNA substrates was recorded on an SX.20 stopped-flow spectrometer (Applied Photophysics, Ltd, UK). Fluorescence signals of either the protein tryptophan residues, the substrate TAMRA or the 2-aPu residues were detected. The dead time of the instrument was 1.04 ms. APE1 and αdA-containing DNA were rapidly mixed at 25 °C in a reaction buffer consisting of 20 mM HEPES/KOH pH 6.8, 50 mM KCl, and 0.01 mM MgCl2. The Trp fluorescence intensity of APE1 was recorded at 1 μM concentration of protein in the reaction chamber, and the 0.5–2.0 μM concentration of a double-stranded (ds) ODN substrate. The TAMRA fluorescence intensity and fluorescence anisotropy were measured at 1 μM substrate concentration in the reaction chamber, at an APE1 concentration of 1.0–3.0 μM. The 2-aPu fluorescence was measured at 1 or 3 μM substrate concentration in the reaction chamber, at 1.0–1.5 μM or 3.0–4.5 μM APE1, respectively. The emitted fluorescence of Trp in APE1 was detected through a long-pass Schott filter WG320 at λem > 320 nm with excitation at 281 nm. The emitted fluorescence of TAMRA was detected through a long-pass filter at λem > 580 nm with excitation at 550 nm (Lytkarino Optical Glass Factory, Russia). The emitted fluorescence of 2-aPu was detected through a long-pass Corion filter LG-370 at λem > 370 nm with excitation at 310 nm. The fluorescence was excited using the monochromator of the stopped-flow instrument. For anisotropy measurements, the light was polarized by means of calcite prisms for the incident beam and in front of each of the two photomultiplier detectors arranged in a T-configuration. The stopped-flow spectrometer was equipped with an open cuvette. Diffusion from the open cuvette could be a source of additional error in the kinetic parameters of very slow processes. To prevent such an error, 2-aPu fluorescence traces of the substrate upon its interaction with APE1 were also recorded using the Cary Eclipse spectrofluorometer (Agilent Technologies, USA) equipped with a closed cuvette. In the latter case, solutions of the enzyme and substrate were incubated separately at 25 °C for 10 min before manual mixing within the cuvette. The reaction was allowed to proceed at 25 °C in the reaction buffer mentioned above. The emitted fluorescence of 2-aPu was measured at 370 nm (λex = 310 nm). The excitation and emission wavelengths were adjusted using the monochromators of the Cary Eclipse instrument. At the start of the process, each fluorescence trace of Trp (t ≤ 10 s) or 2-aPu (t ≤ 200 s) represented the average of ≥4 experiments, and each TAMRA fluorescence anisotropy or fluorescence intensity trace (t < 1 s) represented the average of ≥12 experiments. For longer periods, no averaging was performed. The resulting kinetic traces were numerically integrated and fitted using nonlinear regression analysis in the DynaFit software (BioKin, USA).24 The initial regions of the kinetic traces were fitted to a kinetic scheme containing one bimolecular step of formation of the enzyme–substrate complex. Furthermore, consecutive spreading of the time slot with simultaneous elaboration of the kinetic mechanism was performed. The subsequent steps were first order (or pseudo-first order) and corresponded to isomerization of the enzyme–substrate complex, as they were observed prior to the catalytic step of hydrolysis of the substrate 5′-phosphodiester bond. Thus, a minimal kinetic scheme describing the empirical data was determined (Fig. 2). | ||
| Fig. 2 The minimal kinetic scheme derived from kinetic traces (both fluorescence and anisotropy of fluorescence). | ||
The kinetic parameters were obtained by numerical integration of the system of kinetic differential equations (eqn (1)) and the least-square global nonlinear fitting of the total fluorescence intensity (or fluorescence anisotropy) (F, total fluorescence intensity or fluorescence anisotropy; Fb, background fluorescence intensity or fluorescence anisotropy; fi, the coefficient of specific fluorescence [or specific anisotropy] for each discernible DNA conformer; [Si(t)], concentration of the conformer at any given time point t [i = 0 denotes free DNA; i > 0 means enzyme–DNA complexes]).25,26
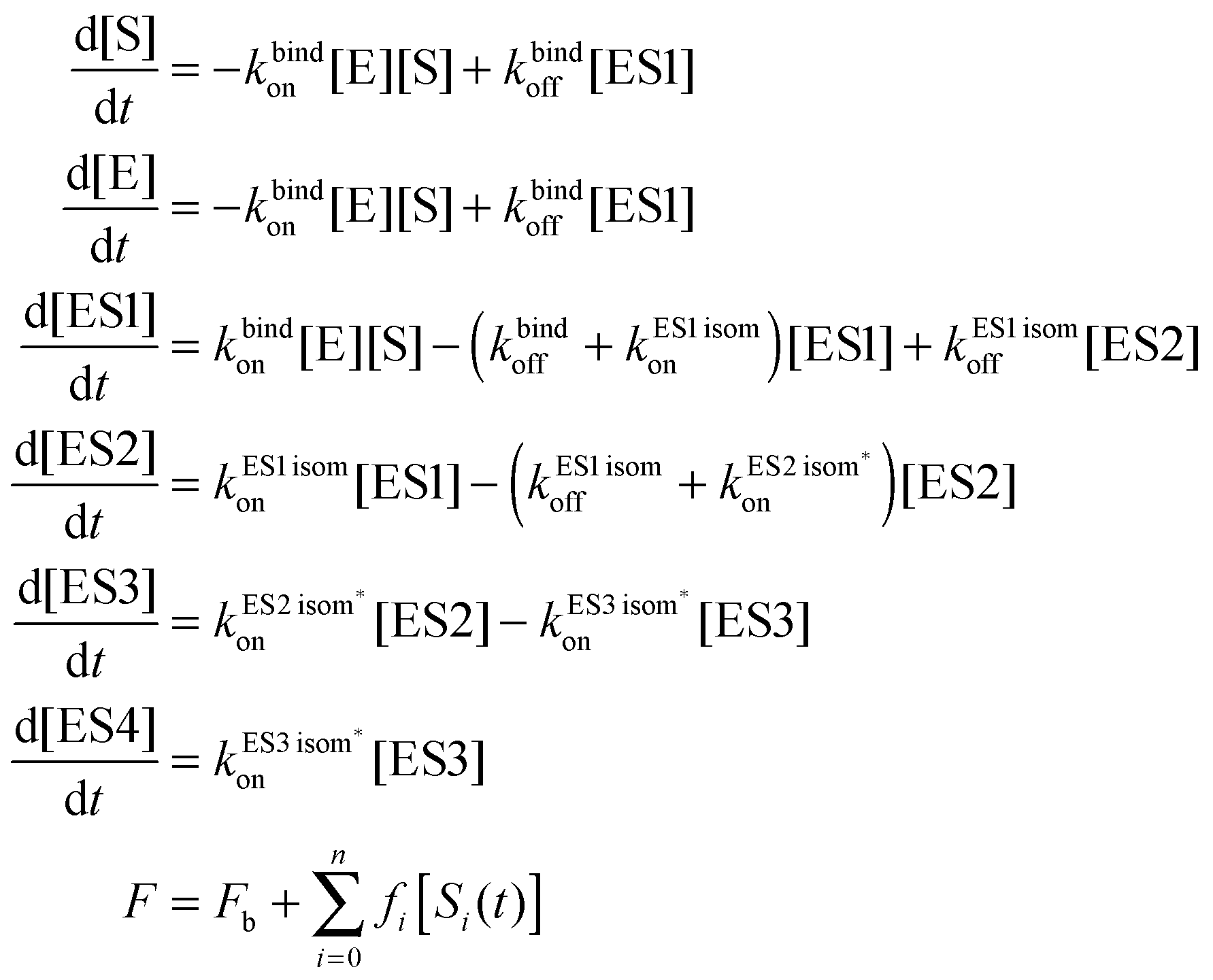 | (1) |
2.4. Steady-state fluorescence titration
To analyze the binding affinity of APE1 for an αdA-containing DNA substrate and for the product of this substrate cleavage, the enzyme was titrated in the reaction buffer (see above) under steady-state conditions with the αdA-substrate or with the ODNs reconstituting the products of substrate cleavage. The emitted fluorescence of Trp in the protein was measured at 330 nm (λex = 281 nm) using a Cary Eclipse spectrofluorometer (Agilent Technologies). The excitation and emission wavelengths were adjusted using the monochromators supplied with the instrument. Each point of the fluorescence titration curve was obtained by measurement of the fluorescence intensity of the separate solutions (80 μL) containing the enzyme (1 μM) and dsODN (0.5–20 μM). Each mixture of APE1 with the reaction product was incubated at 25 °C for 10 min before analysis. In the case of APE1 titration with an αdA-containing substrate, solutions of enzyme and substrate were incubated separately at 25 °C for 10 min before manual mixing in the cuvette. The time from the mixing to the fluorescence measuring was no more than 1 min. Within this period, no more than 1% of an αdA-containing substrate was cleaved, thus most of the substrate was unchanged. The fluorescence intensity values (F) in the presence of oligonucleotides were adjusted for the inner filter effect due to oligonucleotides' absorption at 281 nm as described elsewhere.27 The dissociation constants of the enzyme–substrate (or enzyme–product) complexes were calculated from the dependence of the enzyme's fluorescence intensity on the dsODN concentration according to the following equations: | (2) |
| F = fE × [E] + fEL × [EL] | (3) |
| fE = F0/[E]0; fEL = Flim/[E]0 | (4) |
 | (5) |
2.5. Incision assays of a [32P]-labeled αdA-substrate
Two series of time course experiments with cleavage of a 5′-[32P]-labeled αdA-containing substrate were conducted at 25 °C in the reaction buffer (see above). In one series, the reaction solution contained 1 μM APE1 and 0.75, 1.0, or 1.25 μM αdA-DNA, and in the other series, the reaction solution contained 20 μM APE1 and 0.5, 0.75, or 1.0 μM αdA-DNA. The enzyme was rapidly added to the 32P-labeled substrate. The reaction was quenched at designated intervals by addition of reaction mixture aliquots into thin-walled tubes that contained hot double-distilled water (these tubes were placed in boiling water beforehand). The tubes containing the reaction mixture aliquots were incubated for 2 min in boiling water and were then cooled to room temperature. To digest the insoluble enzyme–DNA complex that partially formed after the heating, the reaction mixture aliquots were treated with trypsin (APE1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trypsin = 12:1) in 50 mM NH4HCO3 at 37 °C for 16–18 h. The loading dye solution containing 8 M urea was added to the reaction mixture aliquots, which were then heated at 90 °C for 3 min. The substrates were separated from the products via denaturing PAGE in 20% acrylamide gels containing 7 M urea at 50 °C. The gels were dried, and the bands were visualized using a Molecular Imager FX phosphorimager (Bio-Rad, USA) and quantified in the Gel-Pro Analyzer software (Media Cybernetics, USA). Each kinetic curve was fitted separately to a one-exponential equation using the OriginPro software (OriginLab Corporation, USA). For the kinetic curves series obtained at 20 μM APE1, the weighted average of the observed kinetic constant (
:trypsin = 12:1) in 50 mM NH4HCO3 at 37 °C for 16–18 h. The loading dye solution containing 8 M urea was added to the reaction mixture aliquots, which were then heated at 90 °C for 3 min. The substrates were separated from the products via denaturing PAGE in 20% acrylamide gels containing 7 M urea at 50 °C. The gels were dried, and the bands were visualized using a Molecular Imager FX phosphorimager (Bio-Rad, USA) and quantified in the Gel-Pro Analyzer software (Media Cybernetics, USA). Each kinetic curve was fitted separately to a one-exponential equation using the OriginPro software (OriginLab Corporation, USA). For the kinetic curves series obtained at 20 μM APE1, the weighted average of the observed kinetic constant (![[k with combining macron]](https://www.rsc.org/images/entities/i_char_006b_0304.gif) ) was calculated as follows:
) was calculated as follows: | (6) |
(s()) was calculated as follows: | (7) |
3. Results
3.1. Equilibrium binding of APE1 to αdA-DNA
To determine the binding affinity of APE1 for a DNA substrate containing an αdA/T pair and for the product of cleavage of this substrate, the enzyme was titrated under steady-state conditions with the corresponding DNA. The equilibrium constant for dissociation of the APE1 complex with the αdA-containing substrate (αdA/T), KSd, was determined by recording the dependence of the enzyme fluorescence intensity on the substrate concentration (Fig. 3). The fitted value of KSd is equal to 3.3 ± 0.2 μM. Most of the substrate (∼99%) was unchanged under the conditions used in this experiment (see section Steady-state fluorescence titration in the Materials and methods section and Fig. 4), and therefore, this KSd value does reflect the interaction of APE1 with the substrate but not with the product. The equilibrium constant for dissociation of the complex of APE1 with the αdA-product (KPd) was determined by recording the dependence of the enzyme fluorescence intensity on the concentration of the product of αdA-substrate cleavage (Fig. 3). To reconstitute the reaction product, we used a nicked oligonucleotide duplex containing the αdA residue at the 5′ terminus at the site of the nick (Fig. 1). The fitted value of KPd is equal to 2.3 ± 0.2 μM, which is 1.5-fold lower than KSd. The comparison of the obtained KSd and KPd values showed that APE1 binds the αdA-containing product 1.5-fold more strongly than the αdA-containing substrate. According to these data, we believe that the product of αdA-containing substrate cleavage may inhibit APE1 competitively in the process of hydrolysis of the αdA-containing substrate. | ||
| Fig. 3 Equilibrium binding of APE1 to αdA-DNA. Fluorescence titration of 1 μM APE1 with the αdA/T-containing substrate (magenta squares) or with the ODNs reconstituting the products of substrate cleavage (blue triangles). a.u., arbitrary units. | ||
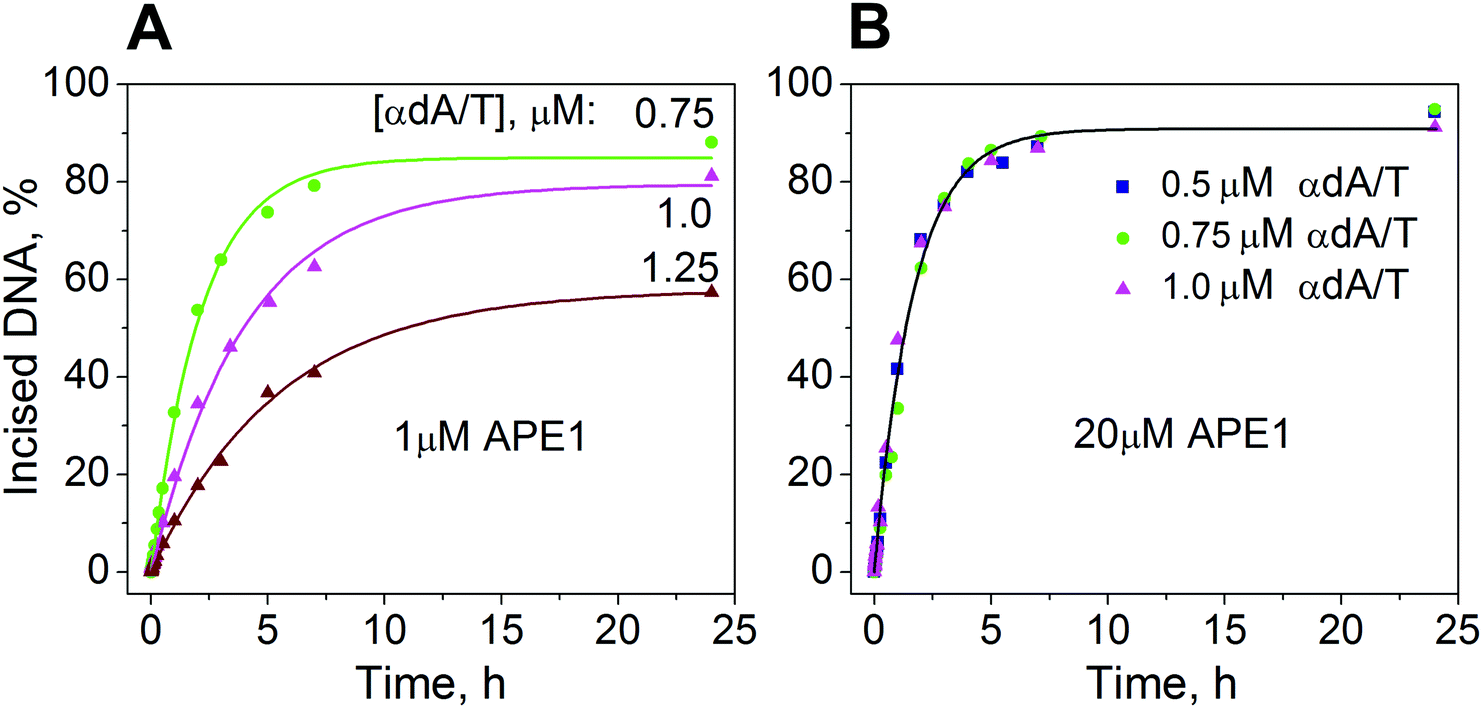 | ||
| Fig. 4 Incision assays of the [32P]-labeled αdA-containing substrate. Time courses of cleavage of a 5′-[32P]-αdA/T-containing substrate by APE1 [1 μM (A) or 20 μM (B)]. Concentrations of the substrate are indicated. | ||
3.2. Incision assays of the [32P]-labeled αdA-containing substrate
To determine the rate of accumulation of the incised product, PAGE was used to follow the time courses for product formation during processing of the 5′-[32P]-labeled αdA/T-containing substrate by APE1. Two series of time course experiments on product accumulation were performed. One series was conducted at the APE1 concentration of 1.0 μM and at 0.75, 1.0, or 1.25 μM αdA/T-containing substrate (Fig. 4A). Under these conditions, only ∼20% of the substrate was in complex with the enzyme as the KSd was equal to 3.3 μM. To process the substrate completely, APE1 should undergo multiple turnovers. Therefore, in our study, such concentrations were termed multiple-turnover conditions. The resulting time traces showed that the rate of product accumulation depends inversely on the initial concentration of the substrate. To determine the maximal rate of incision 5′ to the αdA lesion, a second series of experiments were conducted under the conditions where APE1 (20 μM) was present in excess over the substrate (0.5, 0.75, or 1 μM) (Fig. 4B). Under these conditions, ∼90% of the substrate was in complex with the enzyme. To process ∼90% of the substrate, APE1 had to undergo a single turnover; therefore, such concentrations were termed single-turnover conditions.APE1 was found to cleave ∼90% of the αdA-containing substrate within 7 h under single-turnover conditions (Fig. 4B). Under multiple-turnover conditions, this protein cleaved 40–80% of the αdA-containing substrate, depending on the substrate concentration, within the same period (Fig. 4A).
Previously,28 APE1 was shown to cleave more than 60% of a DHU-containing substrate within 35 h under multiple-turnover conditions. The time course of accumulation of the incised DHU-product showed a rapid burst of the product for periods of <20 s followed by a slow increase in the amount of the product. The burst amplitude was less than the initial concentration of the enzyme, although 100% of the APE1 molecules were shown to be capable of binding. We hypothesized that the decreased amplitude of the initial product accumulation means that the enzyme exists in two conformations that are in equilibrium. At the start, one part of the enzyme exists in the conformation that is energetically less favorable but more active for DHU-substrate cleavage. Another part of the enzyme exists in the conformation that is energetically more favorable but significantly less active for cleavage of this substrate.
In the present work, we did not observe a burst in the kinetic curves of accumulation of the αdA-containing product (Fig. 4). Each kinetic curve was fitted separately to a one-exponential equation.
Under single-turnover conditions (Fig. 4B), the rate of product accumulation did not depend on the initial concentration of the αdA-containing substrate. Probably, this rate was limited either by the rate of the enzymatic hydrolysis of the 5′-phosphodiester bond of the substrate or by the rate of conformational rearrangement (in the enzyme–substrate complex) that occurred prior to the incision reaction and led to the formation of the catalytically competent complex. The weighted average of the observed kinetic constant of product accumulation was (1.57 ± 0.04) × 10−4 s−1 for the series of kinetic curves obtained under single-turnover conditions (Table 1). The value of this constant was ∼5 orders of magnitude lower than the values of the rate constants for enzymatic hydrolysis of the 5′-phosphodiester bond in DHU- or AP site-containing substrates.25,28
| k bindon (M−1 s−1) × 10−8 | k bindoff (s−1) |
k
ES1isomon (s−1) |
k
ES1isomoff (s−1) |
k
ES2isomon (s−1) × 103 |
k
ES3isomon (s−1) × 104 |
k cut (s−1) × 104 | |
|---|---|---|---|---|---|---|---|
| The indicated variance data represent standard deviations of theoretically fitted plots from the stopped-flow curves. Actual error values also involve experimental errors, not exceeding 20%.a Data taken from another study.25b The average value taken from another study.28 | |||||||
| αdA(4TTAMRA)/T | |||||||
| Fluorescence anisotropy recording | 1.5 ± 0.2 | ≤1 | |||||
| Fluorescence recording | 17.2 ± 0.8 | 15.8 ± 0.4 | |||||
| αdA(5TTAMRA)/T | |||||||
| Fluorescence anisotropy recording | 1.6 ± 0.2 | ≤1 | |||||
| Fluorescence recording | 15.2 ± 0.6 | 17.8 ± 0.4 | 7.8 ± 0.1 | ||||
| (2-aPu)αdA/T | |||||||
| [αdA/T] = 3 μM | 1.4 ± 0.2 | ≤1 | 7.0 ± 0.2 {6.8 ± 0.1} | 5.53 ± 0.03 {5.11 ± 0.01} | |||
| {[αdA/T] = 1 μM} | |||||||
| Fluorescence recording | |||||||
| [32P]-αdA/T | |||||||
| [APE1] = 20 μM | 1.57 ± 0.04 | ||||||
|
{[APE1] = 1 μM
[αdA/T] = 0.75 μM = 1 μM = 1.25 μM} PAGE analysis |
{1.29 ± 0.05
0.70 ± 0.03 0.51 ± 0.02} |
||||||
| L-ligand | 4.4 × 10−3a |
12a | |||||
| DHU-substrate | 2.9 × 10−2a |
18a | 51 × 104b |
||||
| AP-substrate | 1.8a | 120a | 97 × 104a |
||||
Meanwhile, under multiple-turnover conditions, the rate of product accumulation was limited by a slow step that follows the chemical reaction. This step possibly corresponded to the enzyme release from the stable complex with a nicked αdA-containing product. An interesting outcome of this experiment was that the rate of product accumulation under multiple-turnover conditions decreased with increasing concentrations of the αdA-containing substrate (Fig. 4A). These data also indicated that the αdA-containing product inhibits APE1 competitively in the process of hydrolysis of the αdA-containing substrate. The values of the observed equilibrium kinetic constants of product accumulation were (1.29 ± 0.05) × 10−4 s−1, (0.70 ± 0.03) × 10−4 s−1 and (0.51 ± 0.02) × 10−4 s−1 for reactions with 0.75, 1.0, or 1.25 μM αdA-containing substrate, respectively (Table 1). These values were lower than the value of the kinetic constant of product accumulation under single-turnover conditions (1.57 ± 0.04) × 10−4 s−1. Thus, the APE1 release from the complex with the αdA-containing product seems to be limiting the overall enzymatic process of hydrolysis of the αdA-containing substrate and appears to determine its rate under the steady-state conditions.
3.3. Invariance of Trp fluorescence intensity during the interaction of APE1 with an αdA-containing substrate
To determine the kinetic mechanism of APE1 interaction with an αdA-containing substrate, an effort was made to record the conformational changes in the enzyme using a stopped-flow approach with detection of intrinsic tryptophan (Trp) fluorescence. We expected that the binding of the αdA-containing substrate would result in changes in intrinsic APE1 fluorescence because of conformational rearrangements in the enzyme. Unexpectedly, no significant changes in protein fluorescence intensity were observed during the interaction of APE1 with the αdA-containing substrate (data not shown). In contrast, earlier we observed25 considerable changes in Trp fluorescence intensity during APE1 interaction with substrates containing an AP site, (3-hydroxytetrahydrofuran-2-yl)methyl phosphate (F, tetrahydrofuran), or DHU. These changes reflected the formation of enzyme–substrate complexes and subsequent conformational rearrangements of these complexes. Moreover, the kinetics of APE1 binding to an undamaged DNA ligand (L) showed two phases of changing Trp fluorescence.25 Thus, our results revealed that local perturbation around the αdA lesion in the DNA duplex allows APE1 to avoid the conformational changes observed earlier in the case of the enzyme binding to an undamaged ligand L containing a nonspecific β-anomeric nucleoside and specific substrates containing AP, F, or DHU sites.3.4. Anisotropy and fluorescence changes of TAMRA-labeled αdA-containing substrates due to APE1 action
Because we could not register APE1 interactions with the αdA-containing substrate by detecting the intrinsic tryptophan fluorescence intensity, we decided to carry out the stopped-flow experiments at increasing APE1 concentrations using fluorescently labelled DNA molecules. To monitor the formation of the enzyme–substrate complex, we attached TAMRA to the DNA duplex and registered changes in TAMRA fluorescence intensity and anisotropy during the enzymatic process. TAMRA was covalently attached via an amino linker to position 5 of a thymine that was located in the αdA-containing DNA strand and displaced either three (αdA(4TTAMRA)/T) or four (αdA(5TTAMRA)/T) nucleotides in the 3′ direction from αdA (Fig. 1). The fluorophore at these positions was probably located next to the protein molecule when the latter was bound to the αdA lesion.When APE1 and the TAMRA-containing substrate approached each other, the fluorophore mobility became restricted, thus resulting in an increase in the fluorescence anisotropy signal. Indeed, when TAMRA fluorescence anisotropy was monitored by the stopped-flow method, an increase in the signal (with a subsequent plateau) was observed in the time range from 0 to ∼20 ms (Fig. 5). This increase in the fluorescence anisotropy signal likely reflected the formation of an initial enzyme–substrate complex (ES1). Such initial binding most likely fits a single binding equilibrium. Fitting of the experimental data yielded the forward rate constant for the initial binding of APE1 to the αdA/T-containing substrate (kbindon, Table 1). At the same time, we could not determine the precise value of the reverse rate constant (kbindoff) presumably because of a strong shift of the equilibrium toward the enzyme–substrate complex. We successfully determined only the upper limit of the reverse rate constant kbindoff ≤ 1 s−1.
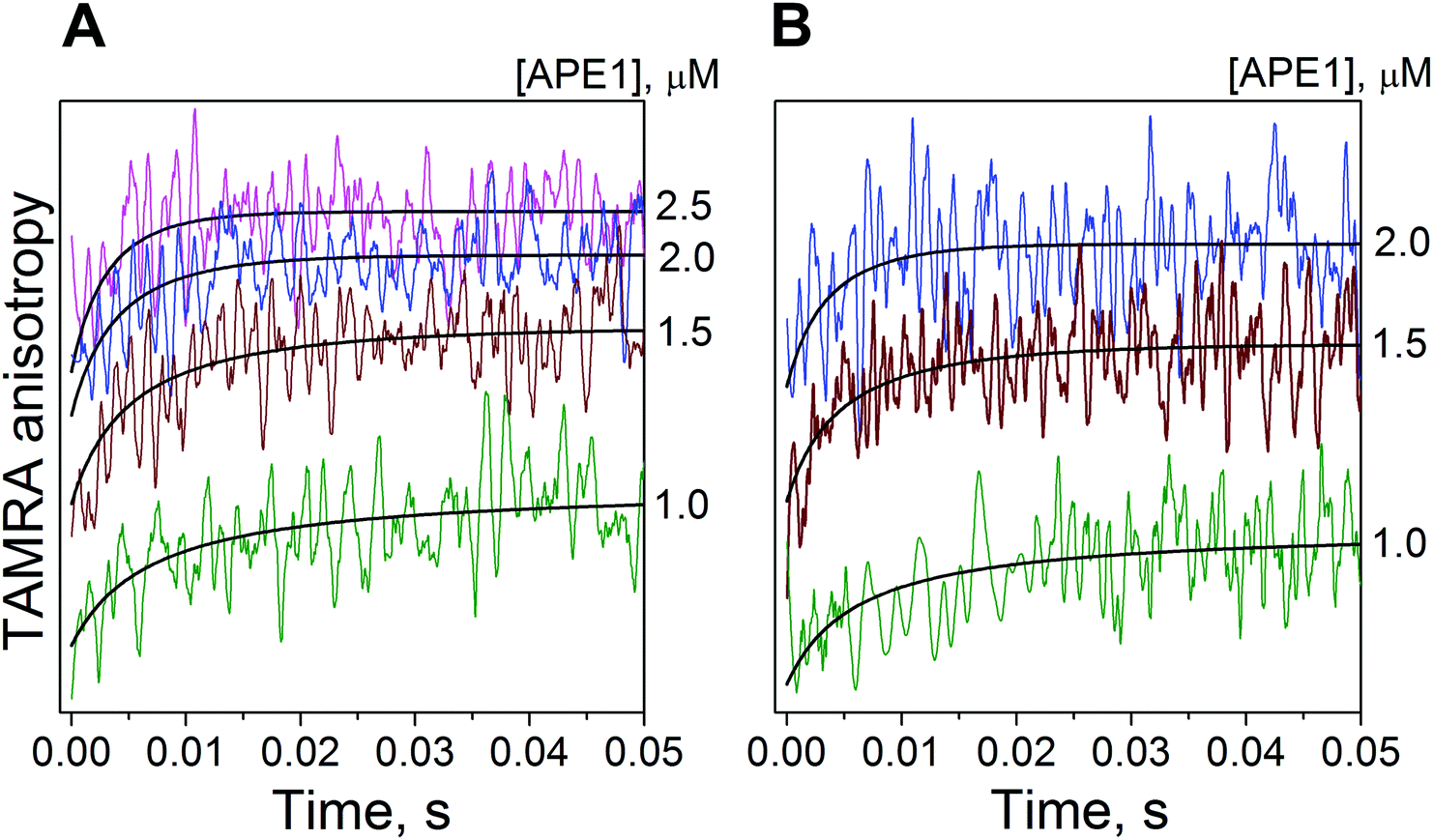 | ||
| Fig. 5 Anisotropy changes of TAMRA-labeled αdA-containing substrates due to APE1 action. TAMRA anisotropy traces of substrates [αdA(4TTAMRA)/T] (A) or [αdA(5TTAMRA)/T] (B) during their interaction with APE1. The smooth curves are the result of a fitting procedure. The concentrations of the substrates are 1 μM. The concentrations of APE1 are indicated next to the right axis. | ||
Kinetic traces of TAMRA fluorescence showed an increase in fluorescence intensity within the time range from 0 to ∼60 ms with a plateau after 60 ms (Fig. 6A and B). This increase likely reflected both formation of an initial enzyme–substrate complex (ES1) and isomerization of the initial complex to yield the second DNA–protein complex (ES2) because formation of the initial complex alone was shown above to take place within 20 ms. Such two-stage binding in all likelihood can be described by two reversible steps, but TAMRA fluorescence kinetic traces (Fig. 6A and B) did not allow us to divide these two steps. Therefore, when processing fluorescence kinetic traces (Fig. 6A and B), we assumed the kinetic constants of the initial enzyme–substrate complex formation (kbindon, kbindoff) to be equal to the corresponding rate constants obtained during fitting of the fluorescence anisotropy kinetic traces (Fig. 5). Fitting the fluorescence traces yielded the rate constants of initial enzyme–substrate complex isomerization (kES1isomon, kES1isomoff, Table 1).
 | ||
| Fig. 6 Fluorescence changes of TAMRA-labeled αdA-containing substrates due to APE1 action. TAMRA fluorescence traces of substrates [αdA(4TTAMRA)/T] (A) or [αdA(5TTAMRA)/T] (B and C) during their interaction with APE1. The smooth curves are the result of a fitting procedure. The concentrations of the substrates are 1 μM. The concentrations of APE1 are indicated next to the right axis. | ||
The slow increase in the TAMRA fluorescence intensity up to ∼900 s with a plateau was observed in the fluorescence kinetic traces during APE1 interaction with an αA(5TTAMRA)/T-containing substrate (Fig. 6C). This increase probably corresponded to the second slow change in the conformation of the αdA-containing substrate in complex with the protein because APE1 cleaved no more than 10% of the 5′-[32P]-labeled αdA-containing substrate during 900 s under both single-turnover (see Fig. 4B) and multiple-turnover conditions (see Fig. 4A). Probably, this second conformational change of the αdA-containing substrate was also reversible. We were able to observe only equilibrium accumulation of the third protein complex with DNA (ES3; because this process proceeded very slowly), and we could reliably determine only the rate of accumulation of complex ES3 (kES2isomon, Table 1).
3.5. Fluorescence changes of the 2-aminopurine-labeled αdA-containing substrate due to APE1 action
The dynamics of conformational rearrangements in the αdA-containing DNA substrate during the interaction with APE1 were also recorded by detecting changes in the fluorescence intensity of the 2-aminopurine residue (2-aPu, a highly fluorescent analog of adenine) incorporated into the DNA strand at the 5′-side of the damaged site. Within a DNA strand, the fluorescence of 2-aPu is quenched due to aromatic stacking with neighboring DNA bases.29 The stacking of 2-aPu with aromatic residues of proteins also leads to the quenching of its fluorescence.30 Disruption of 2-aPu aromatic stacking increases fluorescence intensity.31 Owing to conformational changes of DNA in complex with APE1, one can expect the changes of the fluorescence intensity of 2-aPu residues located in the enzyme-binding site.During the interaction of 1 μM substrate (2-aPu)αdA/T with APE1 (1.0, 1.25, or 1.5 μM), the traces of 2-aPu fluorescence showed a two-phase character (Fig. 7A). In the first phase, the 2-aPu fluorescence intensity showed an increase for periods <1000 s. This increase coincided with the slow increase in TAMRA fluorescence intensity in the kinetic traces presented in Fig. 6C. A further increase in 2-aPu fluorescence intensity for periods of >1000 s corresponded to the second phase (Fig. 7A). The increase in concentrations of the substrate (2-aPu)αdA/T (3 μM) and APE1 (3.0, 3.75, or 4.5 μM) allowed us to detect changes in 2-aPu fluorescence intensity at the start; this period reflected the step of initial enzyme–substrate complex formation (Fig. 7B). Fitting of the experimental data (Fig. 7B) yielded the forward rate constant value of the initial binding of APE1 with the αdA/T-substrate (Table 1). The reverse rate constant kbindoff was estimated to be ≤1 s−1 as in the case of traces of TAMRA fluorescence anisotropy. The rapid increase in 2-aPu fluorescence intensity at the initial times was followed by a slow two-phase increase in the fluorescence signal. These two slow phases were analogous to the slow phases of 2-aPu fluorescence intensity increase in the kinetic traces obtained at lower concentrations of the reactants (Fig. 7A). Slow phases in the time courses for both series (Fig. 7A and B) probably reflected two slow conformational changes of the enzyme–substrate complex. Most likely, both steps of the slow conformational changes were reversible, but fitting of the fluorescence traces (Fig. 7) yielded only the rate constant values of the equilibrium accumulation of complexes ES3 (kES2isomon, Table 1) and ES4 (kES3isomon, Table 1).
 | ||
| Fig. 7 2-Aminopurine fluorescence traces of substrate (2-aPu)αdA/T during its interaction with APE1. Traces were recorded by means of an SX.20 stopped-flow spectrometer. The smooth curves are the result of a fitting procedure. The concentrations of the substrates are 1 μM (A) or 3 μM (B). The concentrations of APE1 are indicated next to the right axis. | ||
We also used a spectrofluorometer equipped with a closed cuvette to obtain the 2-aminopurine fluorescence traces of substrate (2-aPu)αdA/T during its interaction with APE1 (Fig. 8). Fitting of these fluorescence traces yielded the rate constant of the equilibrium accumulation of complex ES4 (kES3isomon = (6.17 ± 0.05) × 10−4 s−1), which is in good agreement with the corresponding rate constant obtained by fitting of the stopped-flow kinetic traces (Table 1).
 | ||
| Fig. 8 2-Aminopurine fluorescence traces of substrate (2-aPu)αdA/T (1 μM) during its interaction with APE1. The traces were recorded using a Cary Eclipse spectrofluorometer. The smooth curves are the result of a fitting procedure. The concentrations of APE1 are indicated next to the right axis. | ||
4. Discussion
Taken together, our results allowed us to propose a minimal kinetic mechanism of the conversion of an αdA site in the DNA substrate by APE1 (Fig. 9). As follows from the data described above, APE1 rapidly generates the initial enzyme–substrate complex ES1. This complex undergoes one rapid (ES2) and at least two slow conformational transitions with the formation of complexes ES2 and ES3, ES4, respectively. These conformational transitions correspond to the induced fit of the enzyme active site and its rearrangement into the catalytically competent conformation (complex ES4). After that, the catalytic chemical step of hydrolysis of the substrate 5′-phosphodiester bond takes place, resulting in formation of the APE1 complex with a nicked product EP. After that, the process of APE1 release from the enzyme–product complex occurs and is characterized by the dissociation constant KPd. This terminal dissociation step likely limits the overall process of enzymatic hydrolysis of an αdA-containing substrate. Therefore, the product of cleavage of an αdA-substrate inhibits APE1 competitively in the process of conversion of αdA-containing DNA.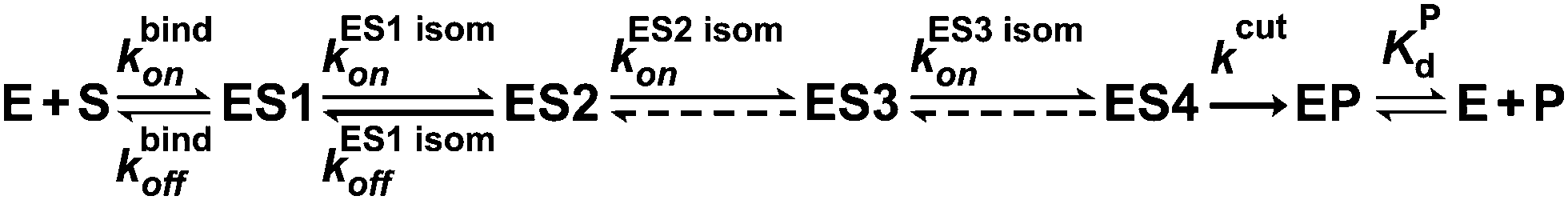 | ||
| Fig. 9 A minimal kinetic scheme describing conversion of an αdA-containing DNA substrate by APE1. E, APE1; S, DNA substrate; ESi, enzyme–substrate complexes; EP, the complex of APE1 with the DNA product; P, DNA product. | ||
In accordance with the concept of induced fit, the enzyme in complex with the substrate undergoes conformational changes leading to a thermodynamically favorable substrate-bound conformation.32,33 The analysis of the cocrystal structure of human APE1 bound to abasic DNA showed that the AP site is very tightly packed in the enzyme active site.20 Therefore, the enzyme existing in the optimal conformation for interacting with its major substrate, the AP site, does not have to bind the αdA lesion in its own active site. The co-crystal structure of human APE1 bound to αdA-containing DNA has not been obtained yet, but it is clear that APE1 should undergo the conformational changes in its active site region to incise the DNA sugar–phosphate backbone on the 5′ side of such a structurally noncognate lesion as αdA.
In our previous study, by monitoring changes in the Trp fluorescence intensity, we showed that APE1 undergoes conformational changes in complex with substrates containing DHU, an AP site, or tetrahydrofuran (F).25 Moreover, we have shown that the formation of an APE1 complex with undamaged DNA is also followed by conformational changes in the enzyme. Obviously, the latter conformational changes do not lead to the formation of the catalytically competent complex. Thus, moving along the DNA during a search for specific sites, the enzyme changes its own conformation, probably continuously trying to form a catalytically competent complex. APE1 successfully forms such a complex only when interacting with DNA that contains lesions that are substrates for the enzyme.
On the other hand, in the present study, we demonstrated that the Trp fluorescence intensity does not change when APE1 interacts with a DNA substrate containing an αdA/T base pair. The structural NMR studies established that in solution the αdA/T pair resulted in an 18° kink of the DNA helical axis and an enlargement of the minor groove around the lesion.15
At the same time, structural data have shown that the APE1-bound AP-containing DNA bends by ∼35° and its minor groove widens by ∼2 Å around the lesion.20 The APE1 stabilization of such an abasic DNA conformation is reached due to the interaction of the substrate with amino acid residues emanating from four loops and from one α-helix of the protein molecule. Thus, during the initial steps of the binding, APE1 is most likely to bend the DNA and widen its minor groove. Because the DNA substrate containing the αdA residue is initially distorted as necessary, the conformational changes in the APE1 molecule, previously observed in the cases of the enzyme binding with L-ligand, AP-, F-, and DHU-containing substrates, are absent. It should be pointed out that one cannot rule out the existence of APE1 conformational rearrangements that occur without changes in Trp fluorescence intensity during the enzyme initial binding to the αdA-containing substrate.
We successfully registered formation of an initial APE1 complex, ES1, with the αdA-containing DNA substrate by detecting fluorescence anisotropy changes in the TAMRA attached to the DNA. The fluorophore is covalently attached to the thymine residue that is located in the αdA-containing DNA strand and dislodges either three (αA(4TTAMRA)) or four (αA(5TTAMRA)) nucleotides in the 3′ direction from αdA. The TAMRA residue at these positions is probably located next to the protein binding site.
The forward rate constant of the initial specific binding of APE1 to the αdA-containing DNA substrate (kbindon) is significantly higher than the corresponding constants for the undamaged DNA L-ligand25 and the DHU-containing substrate25 (Table 1). On the other hand, for the binding of APE1 with the αdA-substrate in the buffer optimal for the NIR pathway, the value of kbindon almost coincides with the corresponding value for binding of APE1 to the AP-substrate in the buffer optimal for the BER pathway25 (Table 1). The value of the reverse rate constant for the initial specific binding of APE1 to the αdA-containing DNA substrate is estimated to be ≤1 s−1. This rate constant is lower than the corresponding reverse rate constants measured earlier for the L-ligand, and DHU- or AP-containing DNA substrates25 (Table 1).
Thus, these results indicate that in the case of APE1 interaction with αdA-containing DNA, the equilibrium of the initial binding step is shifted toward formation of the enzyme–substrate complex much more strongly than in the cases of the L-ligand or DHU- and AP-substrates. We believe that such an equilibrium shift of the initial binding step may ensure efficient recognition of the αdA lesion by APE1.
In this work, we have shown that the initial complex of the enzyme with the αdA-substrate (ES1) undergoes a rapid conformational rearrangement (into ES2) followed by at least two slow conformational transitions (into ES3 and ES4). Equilibria of these slow conformational changes are most likely shifted backward strongly; this situation probably explains the relatively high value of the equilibrium constant for dissociation of the APE1 complex with the αdA-containing substrate (KSd = 3.3 ± 0.2 μM).
Therefore, our data suggest that during the APE1 interaction with the αdA-containing DNA substrate, formation of the catalytically competent complex occurs through mutual conformational changes (induced fit) in the enzyme–substrate complex. One can see that the process of induced fit is extremely slow, much slower than in the case of APE1 interaction with abasic DNA.25 In addition, during processing of the DHU-containing substrate, the conformational transitions leading to the formation of the catalytically competent complex take place significantly faster than during the processing of the αdA-substrate by APE1.28 It has been proposed15 that the kink and enlargement of the minor groove of an αdA-containing substrate facilitate the enzymatic access both by reducing the energetic costs of distortion of the initial DNA helix and by further driving of the nucleic acid into its final conformation. Our data proves that the α-anomeric structure of the substrate does enhance recognition by the enzyme but dramatically impedes formation of the catalytically competent complex.
The formation of the catalytically competent enzyme complex with the αdA-containing substrate is followed by hydrolysis of the 5′-phosphodiester bond of the substrate. The maximal rate of accumulation of the incised product was observed under single-turnover conditions. The rate constant of the chemical step is very low ((1.57 ± 0.04) × 10−4 s−1, Table 1). This rate is ∼5 orders of magnitude lower than the rate constants of the APE1 hydrolytic cleavage of substrates containing a DHU or AP site25,28 (Table 1). Nevertheless, the rate-limiting step of the overall enzymatic process is likely the APE1 release from the complex with an αdA-containing product. This conclusion follows from determination of the rates of αdA-product accumulation under multiple-turnover conditions. The observed kinetic constants of αdA-product accumulation are (1.29 ± 0.05) × 10−4 s−1, (0.70 ± 0.03) × 10−4 s−1 and (0.51 ± 0.02) × 10−4 s−1 for αdA-substrate concentrations 0.75, 1.0, and 1.25 μM, respectively, i.e., they depend inversely on the substrate concentrations (Table 1). In our previous study, we also demonstrated that the APE1 complex with the product of cleavage of a DHU-containing substrate is stable, and that the enzyme release from this complex limits the overall enzymatic process.28 The value of the rate constant for the limiting step of enzyme release from the complex with the DHU-containing product calculated from the time course of [32P]-labeled product accumulation is (1.3 ± 0.3) × 10−5 s−1.28 Apparently, the existence of a tight complex of APE1 with products of substrate cleavage ensures coordinated binding of substrates to other proteins involved in a repair process and the transfer of substrates from one enzyme to another. Such coordination may protect cells from cytotoxic and mutagenic effects of intermediate repair products such as DNA single-strand breaks. Meanwhile, our data show that enzyme dissociation from the complex with the αdA-containing product takes place at least fourfold faster than from the complex with the DHU-containing product. Thus, an α-anomeric structure at the 5′ terminus at the site of the DNA nick likely accelerates dissociation of the enzyme–product complex.
The comparison of the obtained values of equilibrium dissociation constants revealed that APE1 binds to the αdA-containing product 1.5-fold more strongly than the αdA-containing substrate. Thus, the αdA-containing product may inhibit APE1 competitively in the process of hydrolysis of the αdA-containing substrate. Earlier,28 we demonstrated that the product of cleavage of a substrate containing 5,6-dihydrouridine (DHU) can also inhibit this enzyme competitively. The values of equilibrium dissociation constants obtained for interactions of APE1 with a DHU-containing substrate (KS(DHU)d = 1.3 μM) and DHU-containing product (KP(DHU)d = 1.4 μM) were almost identical, indicating that APE1 showed similar affinity for the DHU-containing substrate and for the product. According to our data, the lower stability of the complex of APE1 with the αdA/T-containing substrate—in comparison to the enzyme complex with the αdA-containing product—may be explained by greater perturbation of the structure of the αdA-containing substrate as compared to the structure of the αdA-containing nicked product. Our results revealed that APE1 binds products of the cleavage of DNA containing αdA (KPd = 2.3 μM) or tetrahydrofuran (KP(F)d = 2.1 μM25), an abasic-site analog, with almost the same affinity in an identical buffer. At the same time, the binding affinity of APE1 for the αdA-containing product is a little weaker than for the DHU-containing product (KP(DHU)d = 1.4 μM25).
The αdA lesion is not repaired by DNA-glycosylases/AP-lyases but rather by endonucleases during the nucleotide incision repair (NIR) pathway. This repair pathway is conserved from E. coli to yeast and humans. Our in vitro experiments revealed that the human AP endonuclease APE1 can effectively recognize the αdA-containing substrate. APE1 incises this substrate significantly more slowly than substrates containing DHU or an AP site, but is released faster from the complex with the αdA-containing reaction product than from a complex with a reaction product containing DHU. All these data indicate that the repair of αdA via the NIR pathway should have biological significance.
Acknowledgements
This work was supported by a grant from the Russian Academy of Sciences “Molecular and cellular biology” (6.11) to NAT, OSF. Data analysis was specifically funded by a grant from the Russian Science Foundation (14-14-00063) to OSF.References
- N. Mariaggi, R. Teoule, J. Cadet, H. Dickie and E. Hugues, Radiat. Res., 1979, 79, 431–438 CrossRef CAS
.
- A. Bonicel, N. Mariaggi, E. Hughes and R. Teoule, Radiat. Res., 1980, 83, 19–26 CrossRef CAS PubMed
-
J. A. Raleigh, A. F. Fuciarelli and C. R. Kulatunga, Anticarcinogenesis and Radiation Protection, Plenum Press, New York, 1987, pp. 33–39 Search PubMed
- K. B. Lesiak and K. T. Wheeler, Radiat. Res., 1990, 121, 328–337 CrossRef CAS PubMed
- H. Joenje, Mutat. Res., 1989, 219, 193–208 CAS
- T. Lindahl, Nature, 1993, 362, 709–715 CrossRef CAS PubMed
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed
- H. Ide, K. Tedzuka, H. Shimzu, Y. Kimura, A. A. Purmal, S. S. Wallace and Y. W. Kow, Biochemistry, 1994, 33, 7842–7847 CrossRef CAS PubMed
- L. Gros, A. A. Ishchenko, H. Ide, R. H. Elder and M. K. Saparbaev, Nucleic Acids Res., 2004, 32, 73–81 CrossRef CAS PubMed
- A. A. Ishchenko, H. Ide, D. Ramotar, G. Nevinsky and M. Saparbaev, Biochemistry, 2004, 43, 15210–15216 CrossRef CAS PubMed
- H. Ide, T. Yamaoka and Y. Kimura, Biochemistry, 1994, 33, 7127–7133 CrossRef CAS PubMed
- H. Shimizu, R. Yagi, Y. Kimura, K. Makino, H. Terato, Y. Ohyama and H. Ide, Nucleic Acids Res., 1997, 25, 597–603 CrossRef CAS PubMed
- H. Ide, H. Shimizu, Y. Kimura, S. Sakamoto, K. Makino, M. Glackin, S. S. Wallace, H. Nakamuta, M. Sasaki and N. Sugimoto, Biochemistry, 1995, 34, 6947–6955 CrossRef CAS PubMed
- L. Bielecki and R. W. Adamiak, Acta Biochim. Pol., 2001, 48, 103–111 CAS
- J. M. Aramini, S. H. Cleaver, R. T. Pon, R. P. Cunningham and M. W. Germann, J. Mol. Biol., 2004, 338, 77–91 CrossRef CAS PubMed
- C. N. Johnson, A. M. Spring, S. Desai, R. P. Cunningham and M. W. Germann, J. Mol. Biol., 2012, 416, 425–437 CrossRef CAS PubMed
- A. Mazouzi, A. Vigouroux, B. Aikeshev, P. J. Brooks, M. K. Saparbaev, S. Morera and A. A. Ishchenko, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3071–E3080 CrossRef CAS PubMed
- D. J. Hosfield, Y. Guan, B. J. Haas, R. P. Cunningham and J. A. Tainer, Cell, 1999, 98, 397–408 CrossRef CAS PubMed
- C. D. Mol, D. J. Hosfield and J. A. Tainer, Mutat. Res., 2000, 460, 211–229 CAS
- C. D. Mol, T. Izumi, S. Mitra and J. A. Tainer, Nature, 2000, 403, 451–456 CrossRef CAS PubMed
- L. Y. Kanazhevskaya, V. V. Koval, D. O. Zharkov, P. R. Strauss and O. S. Fedorova, Biochemistry, 2010, 49, 6451–6461 CrossRef CAS PubMed
-
G. D. Fasman, Handbook of Biochemistry and Molecular Biology – Nucleic Acids, GRC Press, Cleveland, 1975, vol. 1 Search PubMed
-
J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor NY, 3rd edn, 2001 Search PubMed
- P. Kuzmic, Anal. Biochem., 1996, 237, 260–273 CrossRef CAS PubMed
- N. A. Timofeyeva, V. V. Koval, D. G. Knorre, D. O. Zharkov, M. K. Saparbaev, A. A. Ishchenko and O. S. Fedorova, J. Biomol. Struct. Dyn., 2009, 26, 637–652 CAS
- M. V. Lukina, A. V. Popov, V. V. Koval, Y. N. Vorobjev, O. S. Fedorova and D. O. Zharkov, J. Biol. Chem., 2013, 288, 28936–28947 CrossRef CAS PubMed
- N. A. Timofeyeva, V. V. Koval, A. A. Ishchenko, M. K. Saparbaev and O. S. Fedorova, PLoS One, 2011, 6, e24063 CAS
- N. A. Timofeyeva, V. V. Koval, A. A. Ishchenko, M. K. Saparbaev and O. S. Fedorova, Biochemistry (Moscow), 2011, 76, 273–281 CrossRef CAS
- E. L. Rachofsky, R. Osman and J. B. Ross, Biochemistry, 2001, 40, 946–956 CrossRef CAS PubMed
- C. A. Dunlap and M. D. Tsai, Biochemistry, 2002, 41, 11226–11235 CrossRef CAS PubMed
- T. M. Nordlund, S. Andersson, L. Nilsson, R. Rigler, A. Gräslund and L. W. McLaughlin, Biochemistry, 1989, 28, 9095–9103 CrossRef CAS PubMed
- D. E. Koshland Jr., Adv. Enzymol. Relat. Subj. Biochem., 1960, 22, 45–97 CAS
- G. G. Hammes, Y.-C. Chang and T. G. Oas, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13737–13741 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |