
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinView: a visual comparative sequence analysis tool for integrated kinome research
Daniel Ian
McSkimming
a,
Shima
Dastgheib
b,
Timothy R.
Baffi
c,
Dominic P.
Byrne
d,
Samantha
Ferries
d,
Steven Thomas
Scott
e,
Alexandra C.
Newton
c,
Claire E.
Eyers
 d,
Krzysztof J.
Kochut
b,
Patrick A.
Eyers
d and
Natarajan
Kannan
*ae
d,
Krzysztof J.
Kochut
b,
Patrick A.
Eyers
d and
Natarajan
Kannan
*ae
aInstitute of Bioinformatics, University of Georgia, Athens, GA 30602, USA. E-mail: nkannan@uga.edu
bDepartment of Computer Science, University of Georgia, Athens, GA 30602, USA
cDepartment of Pharmacology, University of California at San Diego, La Jolla, CA 92093, USA
dDepartment of Biochemistry, Institute of Integrative Biology, University of Liverpool, Liverpool, UK
eDepartment of Biochemistry & Molecular Biology, University of Georgia, Athens, GA 30602, USA
First published on 5th October 2016
Abstract
Multiple sequence alignments (MSAs) are a fundamental analysis tool used throughout biology to investigate relationships between protein sequence, structure, function, evolutionary history, and patterns of disease-associated variants. However, their widespread application in systems biology research is currently hindered by the lack of user-friendly tools to simultaneously visualize, manipulate and query the information conceptualized in large sequence alignments, and the challenges in integrating MSAs with multiple orthogonal data such as cancer variants and post-translational modifications, which are often stored in heterogeneous data sources and formats. Here, we present the Multiple Sequence Alignment Ontology (MSAOnt), which represents a profile or consensus alignment in an ontological format. Subsets of the alignment are easily selected through the SPARQL Protocol and RDF Query Language for downstream statistical analysis or visualization. We have also created the Kinome Viewer (KinView), an interactive integrative visualization that places eukaryotic protein kinase cancer variants in the context of natural sequence variation and experimentally determined post-translational modifications, which play central roles in the regulation of cellular signaling pathways. Using KinView, we identified differential phosphorylation patterns between tyrosine and serine/threonine kinases in the activation segment, a major kinase regulatory region that is often mutated in proliferative diseases. We discuss cancer variants that disrupt phosphorylation sites in the activation segment, and show how KinView can be used as a comparative tool to identify differences and similarities in natural variation, cancer variants and post-translational modifications between kinase groups, families and subfamilies. Based on KinView comparisons, we identify and experimentally characterize a regulatory tyrosine (Y177PLK4) in the PLK4 C-terminal activation segment region termed the P+1 loop. To further demonstrate the application of KinView in hypothesis generation and testing, we formulate and validate a hypothesis explaining a novel predicted loss-of-function variant (D523NPKCβ) in the regulatory spine of PKCβ, a recently identified tumor suppressor kinase. KinView provides a novel, extensible interface for performing comparative analyses between subsets of kinases and for integrating multiple types of residue specific annotations in user friendly formats.
Background
Multiple sequence alignments (MSAs) are a fundamental analysis tool used throughout comparative biology to investigate the relationships connecting protein sequence, structure, function, and evolutionary history. The patterns of conservation and variation in MSAs reflect functional similarities and differences between aligned sequences and often serve as a conceptual starting point for generating testable hypotheses regarding protein functions. MSAs are fundamental for structure prediction,1–3 domain identification, molecular evolution,4–6 and phylogenetic analysis.7–9 Recent studies have also employed MSAs to predict the structural and functional impact of disease mutations,10,11 and for distinguishing disease variants from silent variants.12–14 In general, disease variants target moderate to highly conserved residues, while silent variants remain uniformly distributed with respect to residue conservation.12,15 Thus accurate estimation of conservation in MSAs is critical for distinguishing disease-associated (‘driver’) from silent variants.Residues involved in regulatory functions such as post-translational modifications (PTMs),16–18 (reviewed in ref. 19) can be identified by examining the patterns of conservation and variation in orthologous sequences. Phosphorylation, in particular, is a prevalent type of PTM that regulates protein functions through covalent modification of hydroxyl groups on targeted serine, threonine or tyrosine residues. Although large-scale proteomic studies have identified thousands of PTMs in protein domains, the functional significance of many of these PTMs are largely unknown, though analysis of PTMs in the context of conserved and variable residues in MSAs have provided new insights into speciation and functional specialization in signaling proteins.20,21 More recently, PTMs have emerged as a valuable source of information for identifying mutations associated with complex disease signaling22–24 and identifying novel drug targets.25 For example, cancer mutations can rewire signaling pathways by removing conserved phosphorylation sites or by introducing new phosphorylation sites.24,26 To systematically identify and characterize such mutations, however, signaling and cancer variants need to be integrated and analyzed in the context of PTMs and evolutionary patterns.
Integrative analysis of cancer, PTM and evolutionary data, however, is a challenge because of the size and disparity of these data sources and formats. Consider, for example, the eukaryotic protein kinases (ePKs), a large family of proteins associated with cellular regulation and disease that encode the complex phosphorylation events found in diverse signaling networks. Evolutionary data on ePKs is encoded in thousands of sequences from diverse organisms, and extracting this data requires construction and mining of large MSAs. Likewise, information on kinase PTMs and cancer variants are stored in databases such as dbPTM27 and COSMIC,28,29 respectively, and syntactic differences in the file formats of these data sources pose unique challenges in integrative data mining. Simple queries such as “what mutations alter conserved phosphorylation sites in the protein kinase domain”, currently require a time consuming (and error-prone) procedure involving retrieval of information from different data sources (Fig. 1A) and post-processing data through customized programs and scripts. Estimating conservation of amino acid residues from MSAs is especially challenging, because protein kinases have diversified into major groups, families and sub-families during the course of evolution and accurate estimation of conservation requires comparisons of sequences across various hierarchical categories.30–34 Finally, inconsistencies in MSAs due to the alignment method of choice, input sequences, and parameters pose additional challenges in data interpretation, reproducibility and sharing.
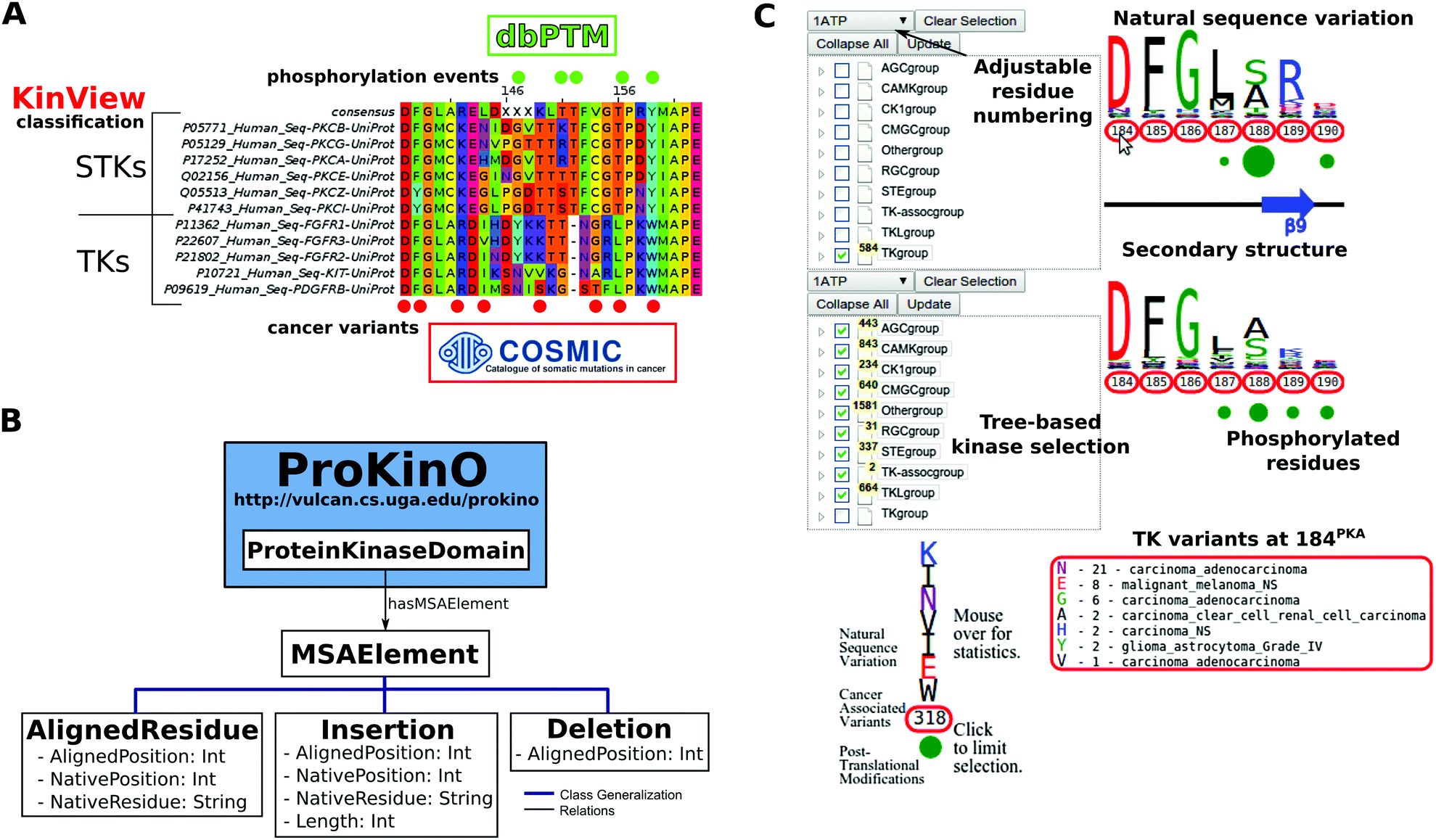 | ||
| Fig. 1 (A) Example benefits of integrative analysis. Placing cancer variants in the context of natural sequence variation and post-translational modifications provides mechanistic insight as to how cancer variants can disrupt functionally significant residues, like phosphorylation sites, to deregulate kinase catalytic activity and rewire signaling networks. Using the classification of the human kinome allows us to consider differences between the kinase groups, families and subfamilies. (B) MSAOnt schema. MSAOnt consists of three main classes: AlignedResidue, Insertion and Deletion. Using these three classes, we can fully represent an alignment of sequences to a profile or consensus. It is connected to the ProKinO ProteinKinaseDomain class through the hasMSAElement relation. (C) The KinView interface. The interface is divided horizontally into top and bottom regions. Each region has an associated tree structure to select the kinase group, family, subfamily or domains of interest. The pull-down menu above the tree adjusts the residue numbering to match any Human kinase UniProt numbering. After clicking ‘Update’, the natural sequence variation of the selected kinases is displayed using a weblogo. Red circumscribed residue numbers show positions with cancer associated variants, while green circles show positions with experimentally validated post-translational modifications (PTMs). The secondary structure is displayed between the top and bottom regions. Detailed information is displayed by hovering the mouse over a residue number (cancer variants) or green circle (PTMs). | ||
Ontologies have emerged as a powerful tool for addressing the data integration challenge. For example, the Gene Ontology35 and the Protein Ontology (PRO)94 have served as vehicles of knowledge for the biological community for nearly two decades, and domain ontologies, such as the Protein Kinase Ontology (ProKinO), have captured knowledge specific to the protein kinase domain.36,37 While such domain-specific ontologies have enabled integrative mining of data and generated testable hypotheses for functional studies, they do not conceptualize information related to the patterns of conservation and variation in MSAs. To significantly enhance the application of ProKinO in systems biology analysis, we report the Multiple Sequence Alignment Ontology (MSAOnt). The advantage of representing protein kinase sequence alignments in the form of an ontology is that it (i) enables queries to examine the patterns of conservation and variation at each position within the protein kinase domain, (ii) allows rapid and consistent comparisons of protein kinase sequences based on their evolutionary grouping (group, family, sub-family etc.) and (iii) provides a framework for data sharing, annotation and reproducibility within the kinase community. MSAOnt is integrated with ProKinO37,38 and currently contains information on kinase sequences from 15 organisms (Fig. 1B).
We have also created an accompanying integrative visualization tool, termed KinView, which enables experts and novices to perform comparative analyses of cancer variants in the context of natural sequence variation and post-translational modifications across evolutionary groups of kinases (Fig. 1C). The integration of these orthogonal types of data provides new insight into functional effects of cancer variants and provides testable hypotheses for the vast numbers of experimental studies that contribute towards a biochemical, biophysical and mathematical understanding of kinome-responsive biosystems. As a new example, we identify and experimentally characterize a conserved tyrosine in the activation loop of the master centriole-regulating kinase Polo Like Kinase 4 (PLK4) and demonstrate its likely role in regulating kinase activity through dimerization and auto-phosphorylation. Likewise, we identify and experimentally characterize a novel loss-of-function mutation in the F-helix of the tumor suppressor Protein Kinase C Beta (PKCβ). These state-of-the art ontology and visualization tools are freely available online through the ProKinO browser at http://vulcan.cs.uga.edu/prokino. A demo video showing basic KinView usage is available at http://https://www.youtube.com/watch?v=sLHC0yevAJo.
Results and discussion
The development of MSAOnt and KinView provides a framework for integrating and relating evolutionary, disease variant and PTM data on kinases in an interactive and visually appealing way, which has the potential to support a scientific community that increasingly relies on evolutionary and disease-relevant changes in structure and function to understand protein kinase signaling and its biological importance. Below, we highlight the application of KinView in knowledge discovery and hypothesis generation, by identifying several highly conserved phosphoacceptor sites in the activation segment (lying between the DFG and APE motifs) of STKs and experimentally characterizing a conserved tyrosine in the PLK4 P+1 loop that is associated with kinase regulation through autophosphorylation-dependent activation. We also demonstrate the application of KinView through correlative analysis of cancer, PTM, and evolutionary data in Fibroblast Growth Factor Receptor (FGFR) and Platelet-Derived Growth Factor Receptor (PDGFR) kinases. Finally, we use KinView to identify kinases with cancer variants at the very highly conserved F-helix aspartate residue and experimentally validate a predicted loss of function mutation in the putative tumor suppressor kinase PKCβ.KinView based comparison of PTK and STK activation segments reveal divergent patterns of phosphorylation
The activation segment is a functionally important flexible region of the catalytic domain that is post-translationally modified in diverse kinases. Modification of serine, threonine or tyrosine residues in the activation segment, typically through ‘upstream’ kinase-mediated phosphorylation, reversibly regulates catalytic activity in the majority of Protein Tyrosine Kinases (PTKs) and Serine/Threonine Kinases (STKs).39–42 Since the activation segment is one of the most variable regions of the kinase domain, we used KinView to visualize the patterns of conservation and variation between PTKs and STKs, and observe correlations between publicly available cancer variants, PTMs and natural sequence variation. A snapshot of the PTK and STK KinView comparison is shown (Fig. 2A). Nearly every residue in the activation segment is mutated in at-least one cancer type in both PTKs and STKs, as indicated in red circumscribing the residue numbers. In contrast, visualization of the PTMs, shown in green circles below the residue numbers, indicates interesting differences in the phosphorylation patterns. In PTKs, the major phosphorylation sites occur in the N-terminal activation segment, most commonly at residues prior to 194PKA. In contrast, STKs are phosphorylated across the entire activation segment, with major (sometimes combinatorial) phosphorylation sites described at conserved residues equivalent to T197PKA, T201PKA and Y204PKA.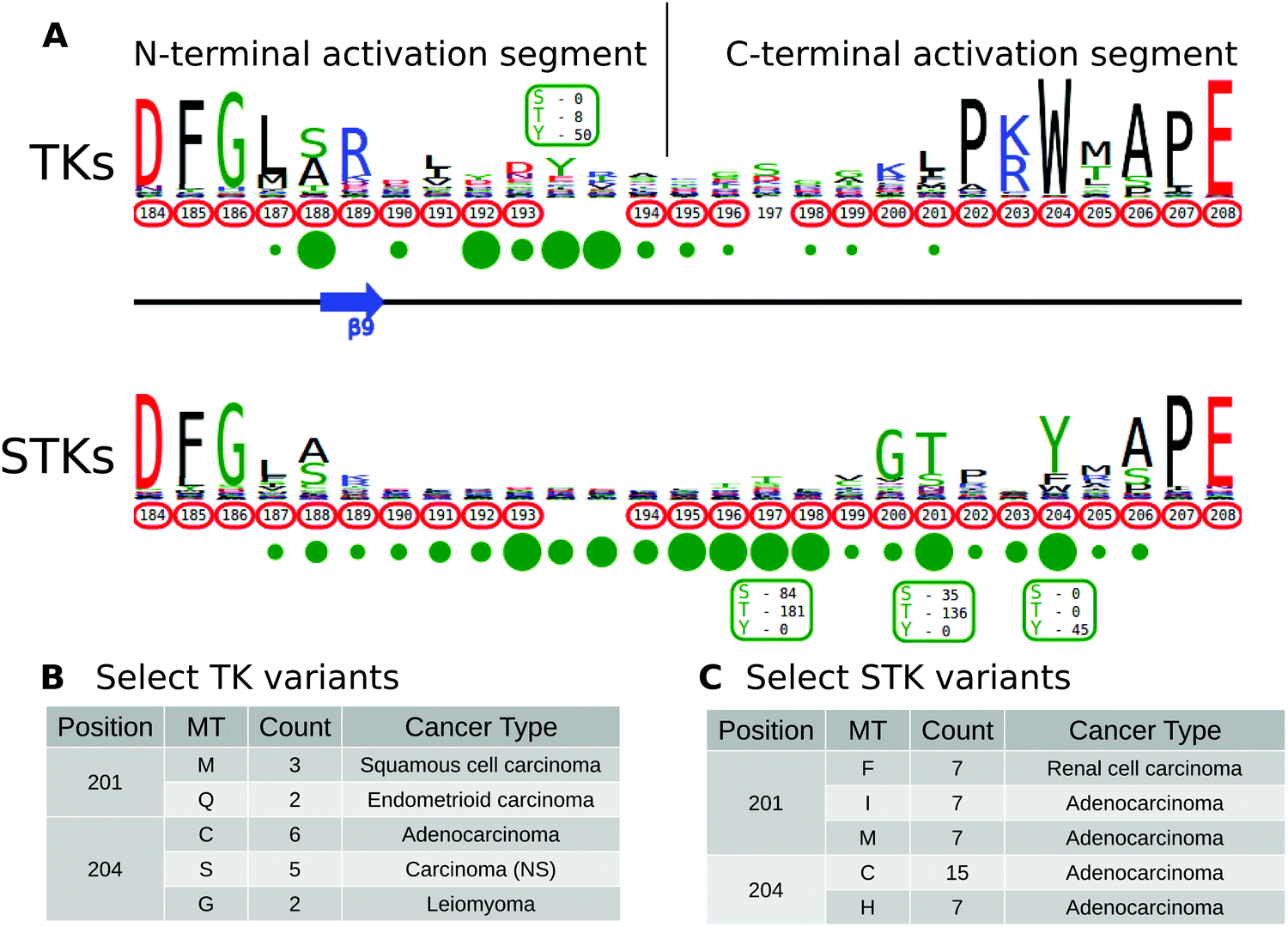 | ||
| Fig. 2 (A) KinView comparison of PTK and STK activation segments. The natural sequence variation is displayed in a weblogo format, with cancer variants represented with red circumscribed residue numbers and post-translational modifications represented with green circles. Major phosphorylation sites have the residue type and number of experimentally validated phosphorylation events displayed. Note, the lack of residue numbers below two columns reflect a deletion in PKA relative to the alignment profiles. (B) A subset of cancer associated variants in PTKs. Here, we show the common variants effecting positions 201PKA and 204PKA in PTKs. The mutant type (MT), count and most commonly associated cancer type are shown. (C) A subset of cancer associated variants in STKs. Here, we show the common variants effecting positions 201PKA and 204PKA in STKs. The mutant type (MT), count and most commonly associated cancer type are shown. | ||
While phosphorylation hotspots in the activation segment (from 194PKA to 198PKA) are expected based on decades of experimental data,43 the frequent conservation and modification of T201PKA and Y204PKA in STKs is rather surprising given the comparative lack of attention paid to studying this region amongst the ePKs. T201PKA and Y204PKA are located in the substrate binding (P+1) pocket of the activation segment, and therefore phosphorylation of these residues is predicted to alter kinase activity and/or modulate substrate binding. Consistent with this view, T387CHK2 (201PKA) is a major auto-phosphorylation site in CHK2 associated with cell cycle checkpoint regulation.44,45 Likewise, phosphorylation of the equivalent residue in COT/Tpl2, T290COT (201PKA), regulates catalytic activity and is required for degradation by the proteasome.46 In the Microtubule Affinity Regulating Kinase 2 (MARK2), phosphorylation of T212MARK2 (201PKA) by GSK3β kinase is reported to lead to inactivation.47 The functional significance of phosphorylation of a conserved Tyr, Y204PKA, has also been validated experimentally in several STKs that are regulated by ‘upstream’ kinases. For example, the phosphorylation of Y315AKT1 (204PKA), as well as Y326AKT1 (215PKA), is necessary for the activation of AKT1 by Src.48–50 This modification is distinct from the PDK1-mediated T-loop (T308AKT1) and mTORC2-mediated hydrophobic (S473AKT1) sites of Ser/Thr phosphorylation induced by insulin and growth factor downstream of PI3 kinases.51 IKKβ and PKCδ are also phosphorylated at the equivalent Tyr residue by Src and in H2O2 treated cells, respectively.52,53 Interestingly, in the Extracellular Signal-Regulated Kinase 1 (ERK1), phosphorylation of either T207ERK1 (201PKA) or Y210ERK1 (204PKA) reduces kinase activity.54 The preponderance of T201PKA and Y204PKA phosphorylation in STKs and the observed divergence in PTKs (where the Tyr is replaced by non-phosphorylatable hydrophobic residues) suggest that regulation by modification of P+1 pocket residues is a selective feature of STKs.
Eleven kinase domains have been crystallized with phosphorylated P+1 pocket residues covering three STKs: the mitotic checkpoint serine/threonine-protein kinase Bub1, the dual specificity protein kinase CLK1 and the dual specificity protein kinase TTK/MPS1. While P+1 pocket phosphorylation has been shown to increase the affinity of Bub1 towards its histone substrate,55,56 none of the crystallized activation segments adopt a canonical active conformation.55–58 Indeed, the potent inhibitory effect of T686 mutations in the P+1 pocket of TTK/MPS1 are very well-documented in cells, and is an established method to inactivate TTK/MPS1.59 Moreover, X-ray structure of an inactive (non-phosphorylated) T686A TTK/MPS1 P+1 loop mutant demonstrate that it also adopts an inactive conformation.60,61 These observations have implications both for understanding the subset of protein kinases that are regulated by substrate-driven dimerization prior to autophosphorylation and activation, and for interpreting the large number of cancer variants observed in the activation segment (see below).
KinView based comparison of cancer variation in PTK and STK activation segments
Having identified the P+1 pocket as a novel evolutionary-conserved phosphorylation site in the STKs, we next sought to compare and contrast reported cancer variants observed in the activation segment using the KinView software. To identify changes occurring at equivalent positions without the use of MSAOnt and KinView currently requires identifying the native residue number corresponding to T201PKA and Y204PKA for each kinase, and subsequently filtering the COSMIC database to retrieve variants occurring at the equivalent positions. This is challenging, especially since amino acid numbering can vary between different databases, leading to confusion and the need for timely contextual analysis to confirm which amino acid is functionally implicated. However, using KinView, cancer variants mapping to positions T201PKA and Y204PKA can be readily identified and quantified by simply hovering the mouse over the red circumscribed residue number (Fig. 2B and C). Interestingly, cancer variants at 201PKA in STKs favor mutation to non-phosphorylated phenylalanine, isoleucine and methionine while those at 204PKA favor cysteine and histidine more strongly than hydrophobic residues (Fig. 2C). Indeed, PTKs naturally conserve hydrophobic residues at 201PKA and 204PKA. As both T201PKA and Y204PKA are frequently phosphorylated in STKs, one mechanism that tumor cells might utilize to deregulate these kinases is to abolish the possibility of phosphorylation through acceptor site mutation. Consistent with this view, the T387ACHK2 (201PKA) mutation in CHK2 abolishes catalytic activity.44,45 The equivalent mutation in NEK6, T210ANEK6 (201PKA), not only decreases phosphorylation of STAT3, a NEK6 substrate, but also decreases cell proliferation and oncogenic transformation in mouse epidermal cells.62 Likewise, CHK2 harbors a recurrent Y390CCHK2 (204PKA) deleterious mutation in breast cancer that impairs p53 activation and DNA damage response,63 and the corresponding Y188FIKKβ (204PKA) in IKKβ is also associated with decreased kinase activity.52Prediction and characterization of a putative phosphorylation site (Y204PKA) in PLK4
The Ser/Thr protein kinase PLK4 is a master controller of centriole duplication, and PLK4 mutations are associated with human cancer-associated pathologies.64 PLK4 catalytic activity is tightly regulated by autophosphorylation,65 and several lines of evidence suggest an in trans-mediated mechanism of autoregulation in cells that is also likely to be important for maintenance of genomic stability.66–68 Activated PLK4 regulates the phosphorylation of an expanding family of centriolar proteins, which control centrosome duplication and ciliogenesis. PLK4 autoactivation relieves PLK4 Polo box-mediated autoinhibition in cis, which helps promote dimerization,69 and in turn generates a PLK4 phosphodegron, which induces rapid PLK4 degradation through the ubiquitin proteasome pathway.70,71 The phosphorylation of a conserved activation loop residue (T170PLK4) is associated with PLK4 catalytic activity in transfected human cells,72 and several Ser/Thr residues in the activation segment (equivalent to human T170PLK4, T174PLK4 and S179PLK4) are the product of PLK4 autophosphorylation.69,71 Interestingly, the PLK4 activation segment also contains a highly conserved Tyr residue (Y177PLK4), at the equivalent position to Y204PKA, which we hypothesized might contribute to PLK4 regulation (Fig. 3A). To investigate the effects of Y177PLK4 mutations, we expressed four truncated human PLK4 catalytic domain (PLK4 1–269) proteins in bacteria, a simple model system for analyzing enzymatic and inhibitor parameters in PLK4.73 Y177FPLK4 PLK4 cannot be phosphorylated, and maintains a hydrophobic residue in the P+1 motif, whereas Y177EPLK4 PLK4 is designed to mimic a phosphorylated residue due to the constitutive negative charge on the acidic glutamate side chain. As shown in Fig. 3B, WT and Y177FPLK4 PLK4 exhibited similar electrophoretic mobility after separation by SDS-PAGE. In contrast, the Y177EPLK4 substitution behaved like a ‘kinase-dead’ D154APLK4 PLK4 mutant, since electrophoretic mobility was increased, consistent with decreased PLK4 phosphorylation. Comprehensive tandem MS/MS analysis confirmed the presence of phosphorylated Y177PLK4 in trypsinized preparations of PLK4 (1–269), suggesting that this was a bona fide phosphorylation site (Fig. 3C). In addition, we found Y177PLK4 accompanying T170PLK4 phosphorylation in the same tryptic phosphopeptide, proving that phosphorylation on T-loop (T170PLK4) and P+1 loop (Y177PLK4) residues can occur simultaneously (Fig. 3D). Phosphorylation at Y177PLK4 was previously identified in recombinant human PLK4,74 but peptide fragmentation spectra, or the effects of mutations at this P+1 loop site were not reported.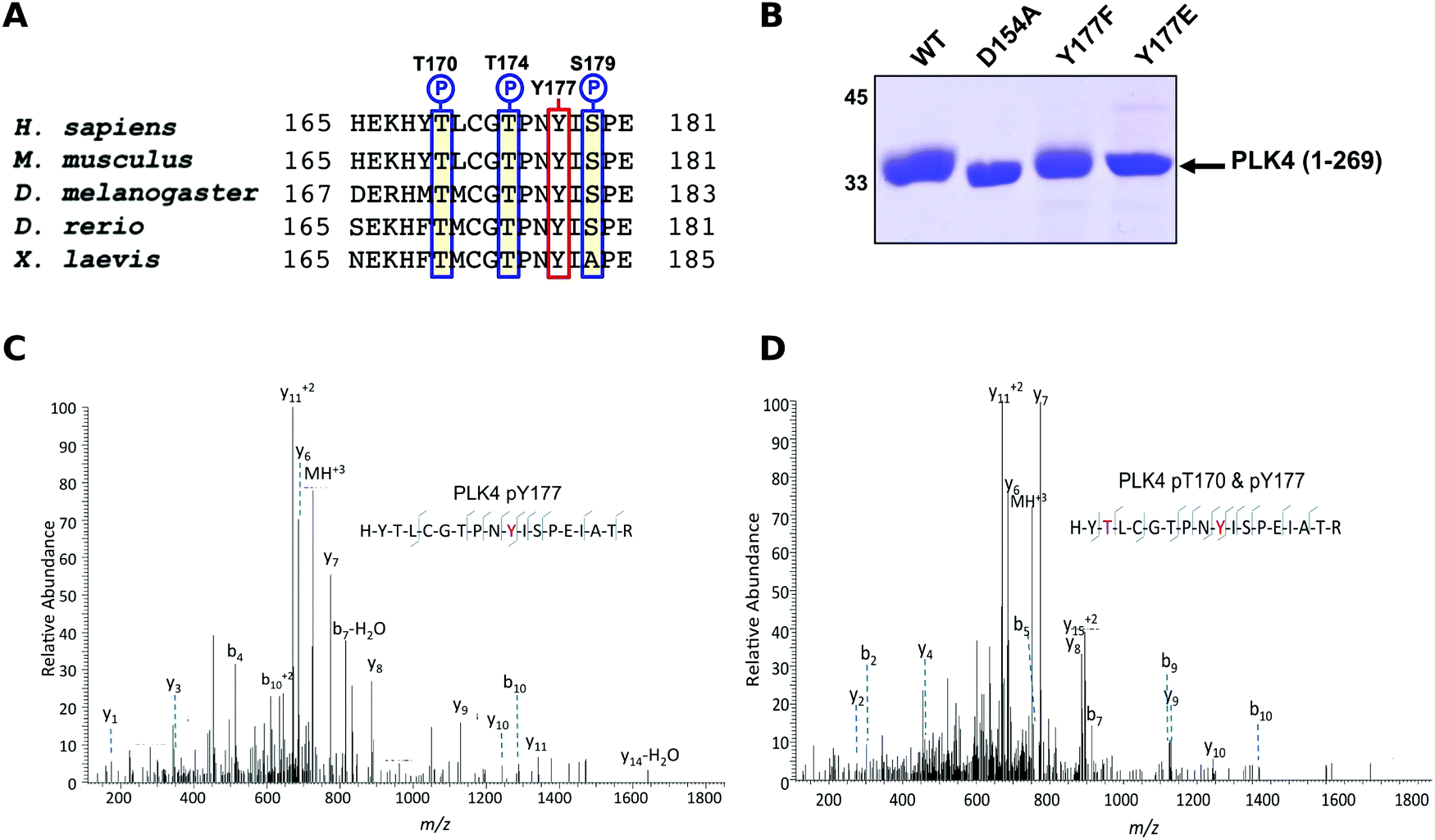 | ||
| Fig. 3 (A) Sequence alignment of model eukaryotic PLK4 activation segments, omitting the ‘DFG’ motif, and ending at the ‘APE’ motif. Conserved Ser/Thr amino acids are outlined in blue, and conserved Y170PLK4 is outlined in red. (B) Coomassie blue staining of 3 μg of PLK4 proteins separated by SDS-PAGE. (C) Collision dissociation product ion tandem mass spectra identifying phosphorylation of T177PLK4 or (D) dual phosphorylation of T170PLK4 and Y177PLK4 in the same PLK4 phosphopeptide. Matched product ions are indicated. | ||
To assess whether Y177PLK4 mutations influenced PLK4 folding, stability or ATP binding, we assessed purified proteins by DSF and thermal denaturation (Table 1). DSF is a standard technique for analysis of catalytically active and inactive protein kinases and their interaction with ligands.34,75 We found that PLK4 Y177EPLK4, Y177FPLK4 and D164APLK4 substitutions exhibited a narrow range of Tm values for unfolding, between 35.8 °C (Y177EPLK4, least stable) and 38.0 °C (WT PLK4, most stable) (Table 1). In addition, the two Tyr mutants exhibited no defects in ATP binding when compared to WT PLK4, whereas the kinase-dead D154APLK4 mutation, which indirectly prevents ATP binding by blocking divalent cation binding,76 was unable to induce PLK4 stabilization. Next, we evaluated the effects of Y177PLK4 substitutions on PLK4 inhibitor binding. We found that the potent ATP-competitive PLK4 inhibitors VX-68073 and staurosporine77 induced thermal stabilization amongst all three PLK4 proteins, whereas a negative control PLK1 inhibitor BI253677 did not. These data establish that mutations at Y177PLK4 do not profoundly affect ATP or small molecule binding susceptibility in the nucleotide-binding site. We next evaluated the activity and phosphorylation status of Y177PLK4 substitutions, including the critical T170PLK4 activation site of autophosphorylation (Fig. 3A). To accomplish this, we generated a new polyclonal phosphospecific pT170PLK4 antibody and confirmed its phosphospecificity at this site using purified phosphorylated WT and D154APLK4 PLK4 proteins (Fig. 4A). As shown in Fig. 4B, a kinetic autophosphorylation assay demonstrated essentially identical incorporation into PLK4 by WT and Y177FPLK4 mutants, proving that conservation of an aromatic side chain did not affect autophosphorylation activity. In contrast, the PLK4 phosphomimic Y177EPLK4 exhibited less than 5% of the activity of WT or Y177FPLK4 PLK4, and this residual activity was completely inhibited by the PLK4 inhibitor VX-680. Interestingly, the Y177EPLK4 mutant did not contain any detectable Thr 170PLK4 phosphorylation when assessed with the phosphospecific antibody, suggesting that trans autophosphorylation had been abolished in this mutant, consistent with a role for the Y177PLK4 P+1 residue in regulating substrate binding. These data suggest the potential importance of a Tyr or Phe residue in the P+1 loop for promotion of substrate phosphorylation in PLK4, since mutation to Glu prevents autophosphorylation and completely abolishes PLK4 activity. Multiple Ser/Thr kinases that autoactivate in bacteria contain a conserved hydrophobic residue in the P+1 loop. The best understood of these is PKA,78 and experimental evidence suggests that an aromatic amino acid (Phe or Tyr) is needed for formation of an allosterically-connected conformational network, RI-subunit interaction and peptide substrate binding. Mutation of Y204APKA induces a 30-fold decrease in the rate of enzyme turnover, although the effects of phosphomimetics or installment of phosphotyrosine at Y204PKA were not reported.78 Our study suggests that prevention of autoactivation (presumably through substrate blockade) can be achieved in PLK4 by the simple introduction of a negative charge in the P+1 loop, as might occur reversibly should the Tyr side chain also become phosphorylated in vivo. We also find that phosphorylated Y177PLK4 co-exists alongside phosphorylated T170PLK4 PLK4 in vitro, and it will be interesting to evaluate how Y177PLK4 phosphorylation affects the activity of T170PLK4 phosphorylated PLK4, although the resistance of pY177PLK4 in PLK4 to dephosphorylation makes this experiment challenging.74 However, our finding that non-phosphorylatable Y177FPLK4 still maintained normal autophosphorylation compared to WT PLK4 suggests the presence of very low levels of pY177PLK4 in our WT PLK4 preparations. It will be important to attempt the converse experiment by increasing the levels of pY177PLK4 and evaluating effects on substrate binding. These data might focus analysis of other Ser/Thr or dual-specificity protein kinases that are regulated by autophosphorylation, and it will be particularly interesting to observe the cellular effects of phosphomimics or tyrosine phosphorylation in the P+1 loops of PKA, Aurora kinases,79 GSK380 and DYRKs.81
| Protein | T m/°C | ΔTm/°C | ||||||
|---|---|---|---|---|---|---|---|---|
| +ATP:Mn2+ | +10 μM VX-680 | +100 μM VX-680 | +10 μM staurosporine | +100 μM staurosporine | +10 μM BI2536 | +100 μM BI2536 | ||
| PLK4 (1–269) | 38.03 ± 0.10 | 2.16 ± 0.23 | 6.06 ± 0.27 | 12.55 ± 1.74 | 11.00 ± 0.13 | 16.46 ± 0.15 | −0.94 ± 0.57 | −1.09 ± 0.57 |
| PLK4 Y177E | 35.82 ± 0.01 | 2.98 ± 0.11 | 7.73 ± 0.16 | 13.62 ± 4.82 | 12.76 ± 0.33 | 17.79 ± 0.32 | −0.82 ± 0.57 | −0.75 ± 0.19 |
| PLK4 Y177F | 36.06 ± 0.07 | 2.81 ± 0.04 | 8.25 ± 0.76 | 15.94 ± 0.13 | 13.37 ± 0.95 | 18.14 ± 0.68 | 0.36 ± 1.36 | −0.37 ± 0.82 |
| PLK4 D154A | 37.49 ± 0.26 | 0.37 ± 0.16 | 8.73 ± 0.06 | 14.13 ± 0.41 | 11.97 ± 0.02 | 17.50 ± 0.18 | 0.45 ± 0.22 | −0.20 ± 0.9 |
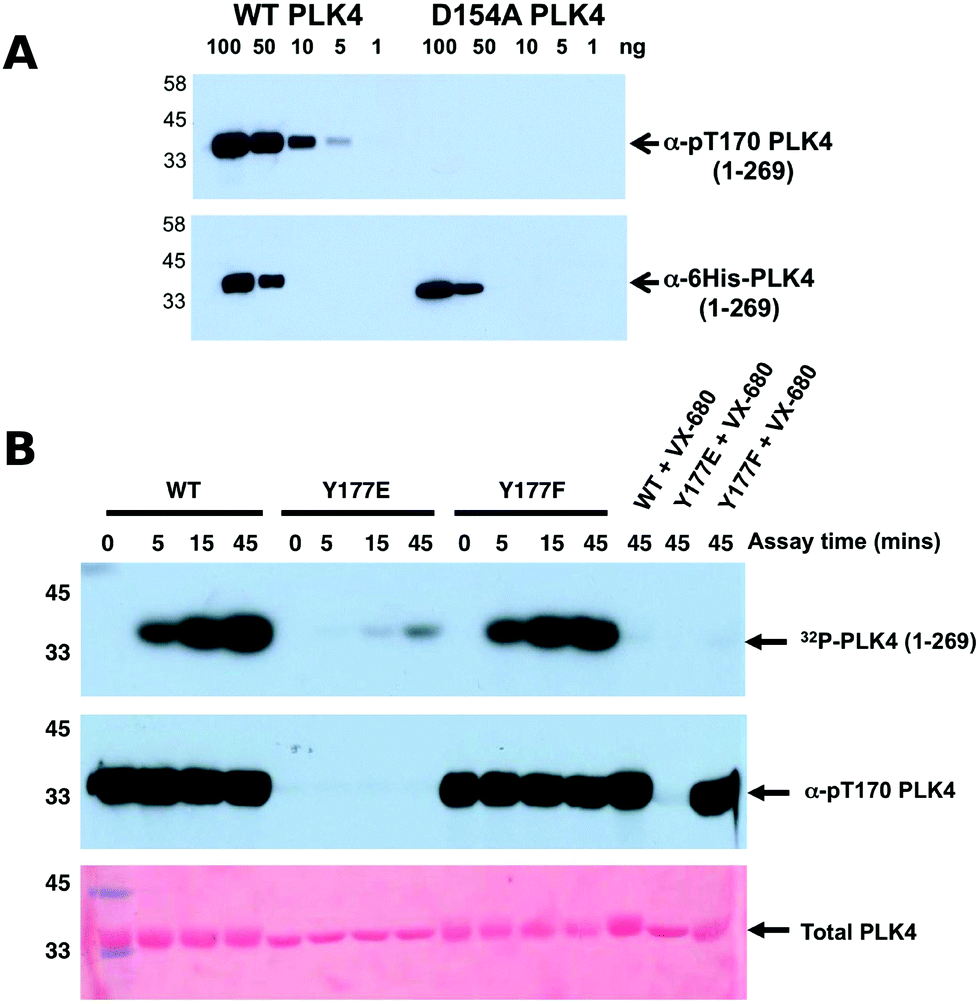 | ||
| Fig. 4 (A) Wild type PLK4 (1–269) or D154APLK4 mutant PLK4 (1–296) were serially diluted and the indicated amounts (ng) were separated by SDS PAGE. Proteins were transferred to a nitrocellulose membrane and probed with rabbit pT170PLK4 PLK4 or 6His antibodies. Antibody binding was visualised using goat anti-rabbit secondary antibodies attached to HRP by ECL. (B) PLK4 Y177EPLK4 mutation greatly reduces 32P incorporation (autophosphorylation) into PLK4 (top panel) and lacks detectable T170PLK4 phosphorylation before or after ATP addition when probed with pT170PLK4 PLK4 antibody (middle panel). Equal loading of proteins was confirmed by staining the membrane with Ponceau S (bottom panel). | ||
Comparisons of PTMs, natural and disease variants in the activation loop of FGFR and PDGFR
KinView enables comparative analysis of any subset of kinases, including comparisons of closely related families that are similar in sequence, but differ in their mechanisms of regulation. PDGFR and FGFR families within the PTK group are one such example. Both FGFR and PDGFR are frequently mutated in cancers, and while some of these mutations have been well-studied,82,83 the structural/functional impacts of many others are largely unknown. We employed KinView to perform a comparative analysis of these two families by selecting the FGFR family on the top half of KinView and the PDGFR family on the bottom. As these families are closely related, there is less natural sequence variation between them, noted by similarities in their corresponding weblogos. The C-terminal activation segment is fairly well conserved between these families, though striking differences can be observed in the N-terminal segment (Fig. 5A). Position S855PDGFRβ (193PKA) is a highly conserved phosphorylation site in the activation segment of PDGFR family members, displayed in KinView through both the maximal height of serine (S) at position 855 and the green circle below. The S855PDGFRβ equivalent residue in the FGFR family, 652FGFR1 (193PKA), is a highly conserved aspartate, which mimics the negative charge of a phosphorylated serine. Biochemical studies on PDGFR and FGFR have shown distinct roles for S855PDGFRβ and D652FGFR1, respectively. In PDGFR, phosphorylation of S855PDGFRβ (193PKA) by Ck1-γ2 acts as a negative regulator of PDGFRβ activity,84 while in FGFR, D652FGFR1 (193PKA) contributes to substrate recognition and the formation of the trans-phosphorylating FGFR dimer.85 These functions are presumably altered by conserved cancer variants observed at these positions (Fig. 5C).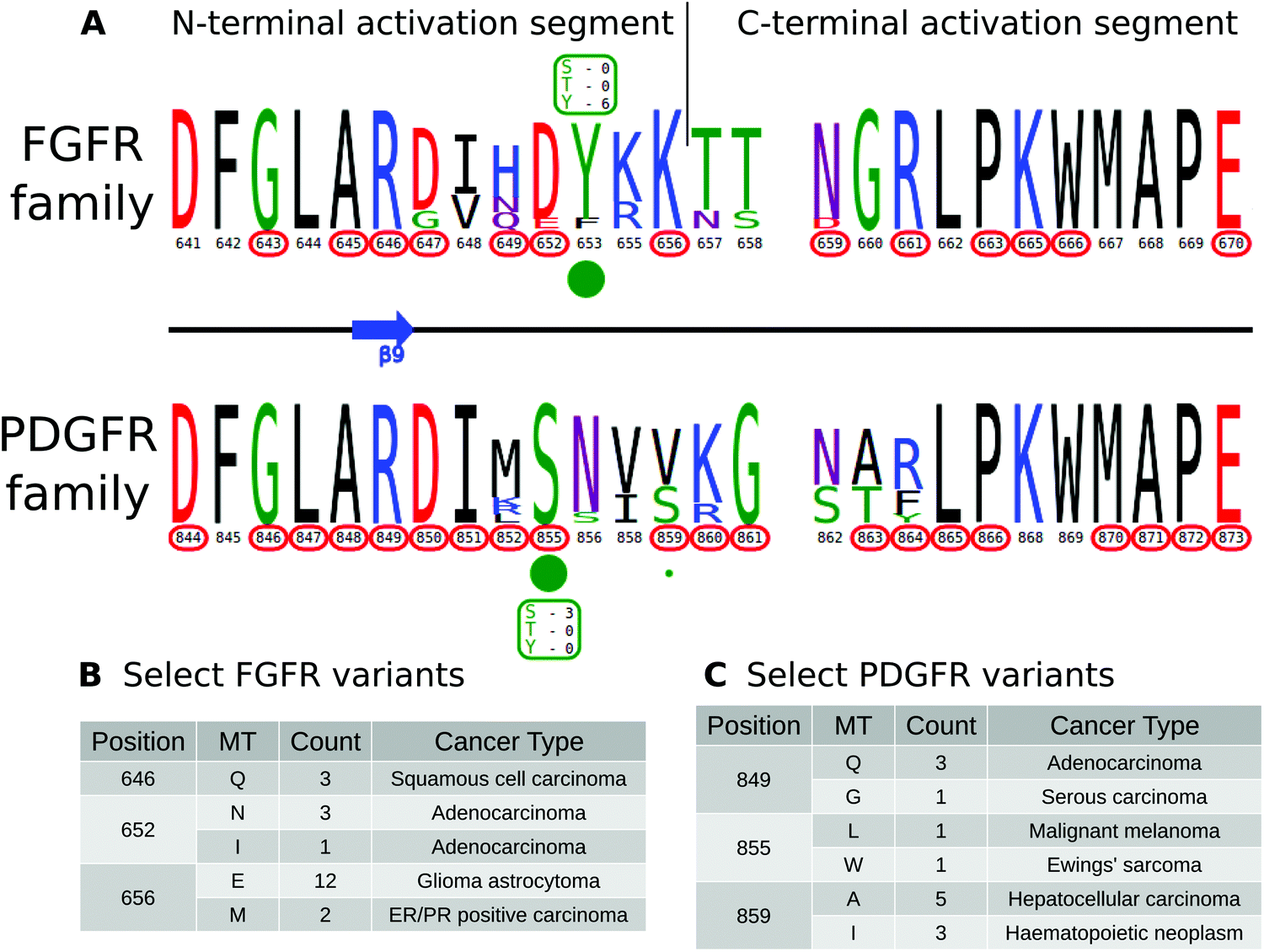 | ||
| Fig. 5 (A) KinView comparison of FGFR family and PDGFR family activation segments. The natural sequence variation is displayed in a weblogo format, with cancer variants represented with red circumscribed residue numbers and post-translational modifications represented with green circles. Major phosphorylation sites have the residue type and number of experimentally validated phosphorylation events displayed. (B) A subset of cancer associated variants in FGFR family. Here, we show the common variants effecting positions 646FGFR1, 652FGFR1 and 656FGFR1 in the FGFR family. The mutant type (MT), count and most commonly associated cancer type are shown. Note the high frequency of K656EFGFR1 variants, which structurally mimic the phosphorylation of the preceding tyrosine. (C) A subset of cancer associated variants in PDGFR family. Here, we show the common variants effecting positions 849PDGFRβ, 855PDGFRβ and 859PDGFRβ in the PDGFR family. The mutant type (MT), count and most commonly associated cancer type are shown. | ||
KinView clearly shows the differing patterns of cancer variation between the FGFR and PDGFR families. The FGFR family, for example, is more sparsely altered than the PDGFR family, with approximately half as many residues (10 vs. 19, respectively) mutated in cancers (Fig. 5A). Using KinView, we see that there is only a single N-terminal activation segment residue, R646, which is both universally conserved and mutated across both families. This arginine (R849PDGFRβ) is located at the DFG+3 position and is highly conserved throughout receptor tyrosine kinases (RTKs). In available FGFR crystal structures, R646FGFR1 coordinates with the phosphorylated residue in the activation loop and is associated with allosteric regulation of the kinase domain.86 Thus mutation of R646FGFR1 in squamous cell and adenocarcinomas (Fig. 5B and C), is predicted to alter allosteric regulation of FGFR2 activity.
KinView based hypothesis generation and experimental validation identifies a novel loss of function mutation in PKCβ
Conserved non-catalytic residues are often mutated in cancers and KinView based mining of such sites can provide new testable hypotheses for functional studies. By observing the letter height in the ePK alignment, for example, we can see that D220PKA, located in the R-spine stabilizing F-helix, is highly conserved across all ePKs. Some 97% of the kinases in ProKinO conserve an aspartate at 220PKA, which is even more highly conserved than the catalytically relevant HRD and DFG aspartate residues found at positions 166PKA (89.17%) and 184PKA (91.58%), respectively. D220PKA is highly mutated to an uncharged asparagine in a variety of cancers, particularly malignant melanoma. KinView-based identification of kinases that harbor the D220NPKA mutation revealed 45 kinases, 8 of which belong to the AGC family (Fig. 6A). One kinase that harbors the D220NPKA mutation is PKCβ, an AGC kinase member of the conventional PKC subfamily. Although members of the PKC family of kinases have recently been shown to function as tumor suppressors through characterization of cancer variants,87 the functional impact of D523NPKCβ (D220NPKA) has not been established. Since D220PKA anchors the regulatory spine to the αF-helix and plays a critical role in kinase functions,88,89 we predicted that the D523NPKCβ would decrease PKC activity. To test this hypothesis, we first transfected mCherry tagged wild-type or D523NPKCβ PKCβII in COS7 cells and examined its phosphorylation state, a marker of correct processing and an event that requires the catalytic function of the enzyme.90 Western blot analysis revealed that the phosphorylation of the mutant PKCβII was impaired: the total PKCβII migrated primarily as a faster mobility band after SDS-PAGE (indicated by dash) that corresponds to unphosphorylated protein, and this species was not recognized by phospho-specific antibodies to any of the three priming phosphorylation events in the activation loop, the C-tail turn and hydrophobic motifs (Fig. 6B). Importantly, this mutant was inactive in cells. By using a genetically-encoded fluorescence based reporter, C Kinase Activity Reporter, CKAR91 to measure agonist-evoked activity in real time in live cells, we found that whereas overexpressed wild-type PKCβII caused an increase in reporter read-out (blue) relative to untransfected cells (yellow), the D523NPKCβ mutant did not (Fig. 6C). Treatment with phorbol ester, to maximally activate PKC, resulted in some activity above endogenous levels for the D523NPKCβ, likely resulting from the small amount of phosphorylated enzyme. These data strongly support the hypothesis that the D523NPKCβ allele is loss-of-function.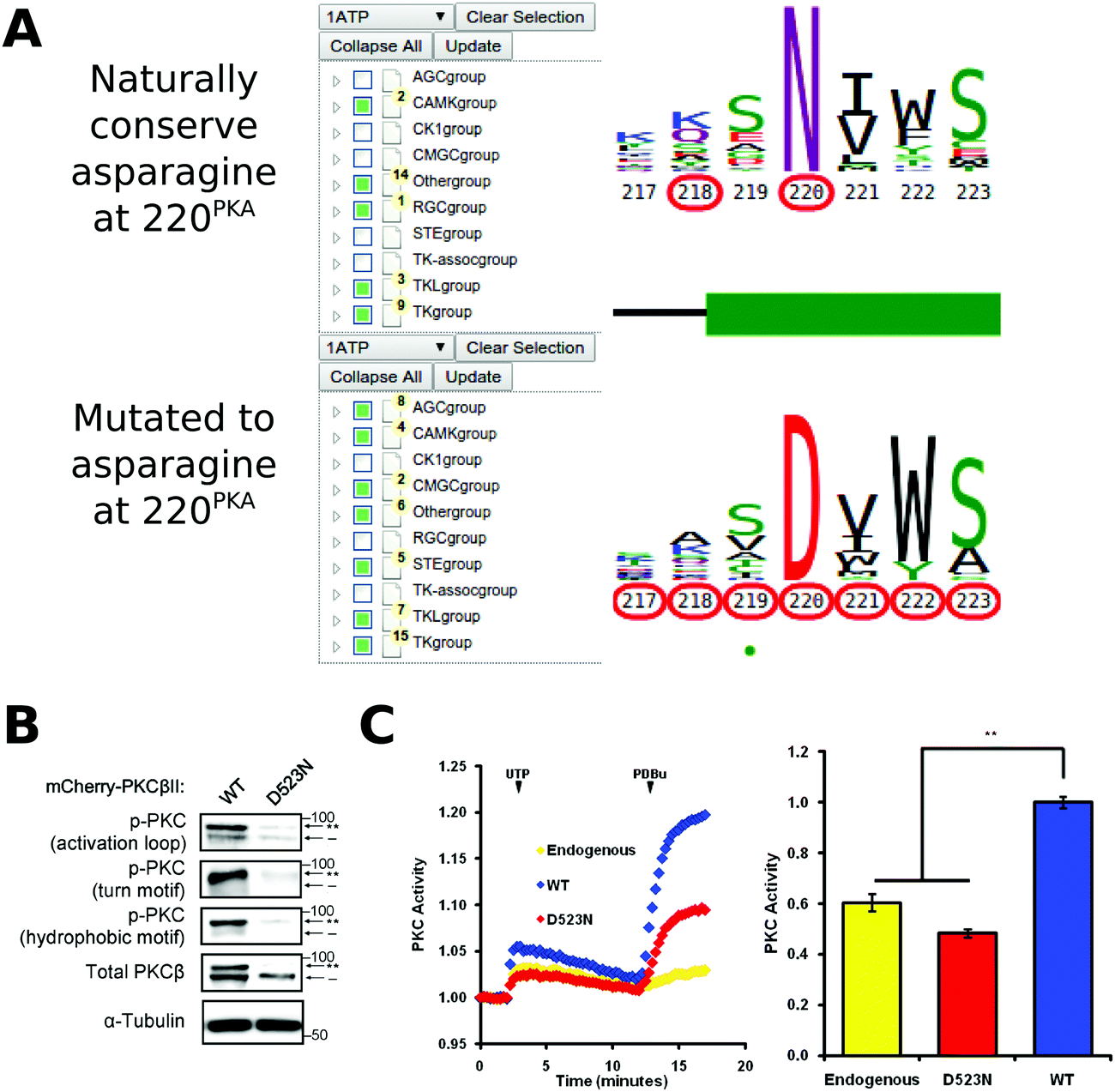 | ||
| Fig. 6 (A) KinView selection of kinases that naturally conserve an asparagine at position 220PKA (top) and those that have mutations to asparagine at position 220PKA (bottom). Note that the AGC group does not naturally conserve an asparagine at 220PKA among the 15 organisms included in ProKinO. (B) Impaired phosphorylation of D523NPKCβ PKCβII variant. The mutation to asparagine decreases the priming phosphorylations, which require the activity of PKC, in the activation loop, the C-tail turn motif and the C-tail hydrophobic motif. (C) Normalized FRET ratio changes showing PKC activity from COS7 cells co-expressing CKAR and mCherry-tagged PKCβII mutant stimulated with 100 μM UTP followed by 200 nM PDBu. Graph on the right shows the signaling output resulting from UTP stimulation (see Methods), quantified and normalized to WT PKC activity. **p < 0.01 as compared with WT, using a repeated-measures one-way ANOVA. | ||
Conclusion
We have described the formalization of multiple sequence alignments in an ontological format, which allows for diverse and novel methods of interrogating a sequence alignment. It is important to recognize that we are not describing new methods for generating an alignment, but rather encouraging their sharing and reuse by any researcher with an interest in protein kinases, disease-associated mutations and the systems analysis of cellular signaling. With the MSAOnt population tool, researchers can take any multiple sequence alignment and generate a public ontology that is available through standard web interfaces. As many gene families are currently described using MSAs, we expect these tools to encourage and simplify the process of generating a domain-specific ontology.The accuracy of MSAOnt data, and any subsequent analysis, is entirely dependent on the quality of the alignment from which it is created. Although we have used highly curated alignments, the possibility of misalignments in highly variable loop regions cannot be entirely ruled out. Such misalignments can obscure correlative analysis of cancer and natural variants using KinView and the resulting patterns from such variable regions should be interpreted with caution. The current analysis has been performed with kinases from 15 species and limited to the modification of residues by phosphorylation, which is a central mechanism through which protein kinases are regulated enzymatically. Consistent with this description, we present mass spectrometric evidence that the P+1 loop Tyr residue in PLK4, whose diverse biological effects are driven through Ser/Thr-dependent autophosphorylation, is also phosphorylated. Indeed, the stoichiometric introduction of a negative charge at the P+1 loop Tyr induces complete inactivation of PLK4 expressed in bacteria, driven through blockade of autoactivation by autophosphorylation at the activating T170PLK4 residue. Given that PLK4 activation occurs through an in trans mechanism in cells,69,71 it is tempting to speculate that covalent modification of the equivalent Tyr residue might be important as an additional regulatory mechanism in Ser/Thr kinases that are regulated through Ser/Thr (or Tyr) autophosphorylation in the activation loop. Indeed, this mode of regulation has been suggested for the AGC kinase AKT as a novel mechanism for fine-tuning other phosphorylation events lying downstream of PDK1 and mTORC2,50,51 and could also be relevant in dual-specificity kinases that undergo initial activation through autophosphorylation on the N-terminal activation loop, such as DYRK and GSK3β. Extending the analysis beyond the 15 species and inclusion of other types of PTMs such as ubiquitination and glycosylation could provide additional insights into disease and natural variants in the kinase domain relevant to catalytic regulation or complex formation.
The visualization tools we have developed will encourage the frequent construction and comparison of weblogos, a graphical interface for simultaneously considering both the relative frequency of amino acids and the information content contained in a position. They are also extensible and can incorporate any type of residue-level annotation stored in a compatible ontology, although we have initially focused on placing cancer variant data in the context of natural sequence variation and post-translational modifications, as way to generate experimentally testable hypotheses concerning potential effects on catalysis. As an example, we use KinView to identify and subsequently experimentally validate a suspected loss of function variant in PKCβ.
Finally, KinView provides a novel visual method for performing integrative comparative analyses between kinase groups, families and subfamilies. KinView is fundamentally different from other graphical visualization tools, like the molecular evolutionary genetics analysis (MEGA) toolkit,92,93 as it does not rank order variants or provide mutation-impact scores. Instead, KinView lets the user make informed decisions by providing an interactive visualization framework for correlating natural, disease and PTM data all in one place. Although we focused our analysis on a subset of comparisons, tens of thousands of comparisons can be performed using KinView and the kinase domain evolutionary hierarchy. Thus, KinView-based comparisons spanning the entire kinase domain and incorporation of structural visualization such as JMoL or PyMoL will be critical in predicting and testing the functional impact of cancer variants. Finally, to expand the scope of KinView and ProKinO in cancer kinome mining, integration with other ontologies such as the Protein Ontology94 will be essential.
Methods
The MSA ontology
The MSA Ontology (MSAOnt) provides a simple schema for relating a set of sequences to a profile or consensus sequence (Fig. 1B). The idea of representing multiple sequence alignments as an ontology has been previously proposed in the multiple alignment ontology (MAO).95 However, there are fundamental differences between MAO and MSAOnt. MSAOnt is a minimalist representation of the components of a profile or consensus alignment, and thus contains classes to handle insertions and deletions as single objects. MAO, in contrast, places equal significance on each residue position, even in insertion regions where only a handful of possibly thousands of sequences may be represented, which can drastically increase the number of instances when considering large protein families, like the eukaryotic protein kinases. The remaining sequences would need to instantiate a deletion character (‘–’) at each of the insertion positions, increasing the size of our ePK alignment from 241 residues to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 789. Although the MAO is reported to be available through the Open Biomedical Ontology (OBO) Foundry96 and hosted on a separate website (http://bips.u-strasbg.fr/LBGI/MAO/mao.html), OBO lists the ontology as deprecated and the published website no longer exists. Further, there is no software available which generates a populated instance of MAO.
789. Although the MAO is reported to be available through the Open Biomedical Ontology (OBO) Foundry96 and hosted on a separate website (http://bips.u-strasbg.fr/LBGI/MAO/mao.html), OBO lists the ontology as deprecated and the published website no longer exists. Further, there is no software available which generates a populated instance of MAO.
In MSAOnt, subclasses of the MSAElement class provide the standard three alignment elements: AlignedResidue, Insertion and Deletion. The AlignedResidue class describes an individual residue, providing the position and amino acid found in the native sequence as well as the position to which it is aligned. Instances of the Deletion class contain a single data property, capturing the aligned position deleted in the given sequence. Finally, the Insertion class contains four data properties: one describing the aligned position prior to the insertion, the native sequence of the insertion segment, the native position of the first amino acid in the insertion and insertion length.
As the schema of MSAOnt is intended to be populated with instances, we also created an open-source Python program for populating MSAOnt given an MSA in CMA or aligned FASTA formats. The CMA format97 distinguishes insertion segments using lowercase letters, while aligned residues are formatted in uppercase. As aligned FASTAs don't include this information, we infer it using the percentage of sequences lacking a residue at a given position. If it is greater than p%, we consider sequences with residues at that position to have an insertion relative to the consensus and no AlignedResidue instances are generated. Instead, a single Insertion element is instantiated. In contrast, if it is less than p%, sequences with no residue at that position have a Deletion element instantiated, while the remaining sequences are described using AlignedResidue instances. We have parameterized p and set a default value of 25. The output consists of the instantiated MSAOnt as an RDF graph, serialized to an output RDF file. This file can then be loaded into a triple store, such as Virtuoso, Jena or TopBraid, accessed through the associated SPARQL endpoint, easily shared with others, and integrated with existing ontologies. It can also be loaded and queried programmatically on a local machine, using an ontology software package like RDFlib.98
By semantically linking instances of the MSAElement class to the sequence in which they are contained, MSAOnt enables novel modes of querying MSA information. While some questions, such as “what is the amino acid distribution at position Y?”, can be answered in a straightforward manner by parsing or visualizing a standard MSA, MSAOnt provides a method for identifying sequences with desired characteristics. For example, sequences containing a specified amino acid at the gatekeeper position (121PKA), an aligned residue position in the kinase domain critical for inhibitor design and drug resistance, can be readily identified by querying MSAOnt, while performing the same task without MSAOnt would require writing specialized software to parse aligned sequences.
Incorporation of MSAOnt in ProKinO
The Protein Kinase Ontology (ProKinO) is a kinase-centric knowledgebase that integrates data from multiple manually curated information sources. It is located at http://vulcan.cs.uga.edu/prokino and provides a wealth of data on kinases, including somatic cancer related variants (COSMIC),28 reactions and pathways in which they participate (Reactome),99,100 post-translational modifications (dbPTM),27 available sequences and crystal structures (KinBase, RCSB),101,102 general functional information (UniProt),103 as well as internally generated and literature derived motif data. Conceptually, we separated the idea of sequence and protein domain into two classes: the Sequence class, which provides full protein sequences for a kinase, and the ProteinKinaseDomain class, which is a representation of the kinase domain alone. Some kinases, like the Jak family, contain multiple kinase domains, which we have labeled pk1 and pk2, following the convention in UniProt.Previously, we loosely incorporated some MSA information into ProKinO by providing a PKA equivalent position for cancer variants and functionally relevant motifs, but the ProteinKinaseDomain class serves as an ideal anchor for the MSAElement instances, which describe our alignment of the kinase domain. Thus, for incorporation in ProKinO, we modified the MSAOnt schema to relate MSAElements to the ProteinKinaseDomain class in ProKinO. Further, to reduce the complexity of querying, the ProKinO namespace subsumes that of MSAOnt.
Visualization
When the number of aligned sequences is large, it can be difficult to visualize the entire alignment and accurately estimate conserved regions. Instead, we can collapse any number of aligned sequences to a single weblogo image that captures both the conservation and relative frequency of amino acids at each aligned position.104 The total height of a column is determined by its information content, while the height of each letter within a column is determined by the relative frequency of the amino acid it represents. Many weblogo visualization tools are available, both through the command line and web server interfaces, but they typically require a FASTA formatted input file containing the aligned sequences to be visualized. This prerequisite requires manual manipulation of sequence files, which complicates and devalues the use of weblogos for comparative analyses. For example, we can easily provide a FASTA file containing the full kinome alignment and generate its corresponding weblogo. If we then want to see how conservation among protein tyrosine kinases (PTKs) differs, we would first need to identify and extract the sequences of the PTKs and generate a new image. To narrow our focus to a specific family, another round of sequence identification and extraction is necessary. Instead of round after round of FASTA manipulation, we sought to leverage ProKinO's SPARQL endpoint to extract exactly those sequences we wish displayed.SGVizler is a JavaScript wrapper developed to visualize the results of SPARQL queries in a web browser.105 It supports many of the charts available through Google Chart Tools, but can also generate complex visualizations through the manual manipulation of scalable vector graphics elements. To this end, we extended SGVizler by adding a new type of visualization: the weblogo, for visualizing the patterns of conservation and variation in MSAs.104 The SGVizler weblogo accepts the results of an aggregate SPARQL query containing three columns of data: a numeric representation of the aligned position, a string representing amino acids present and, lastly, their quantity. The conservation and relative frequencies are then calculated from these counts and displayed in a weblogo format, with amino acids colored by their biochemical properties. By altering the query, we can select sequence subsets for display.
KinView
To further exemplify the utility of MSAOnt and provide a tool for the rapid comparison of kinase sequences and residue level annotations, we created KinView, a web-based ProKinO specific kinase viewer written in JavaScript (Fig. 1C). The display is split horizontally, allowing two sets of sequences to be simultaneously displayed. For each set, we utilize the hierarchical kinase domain classifications in ProKinO to allow the graphical selection of specific domain sequences, via a tree-based structure on the left side of the browser. We can update the residue numbering to match any human kinase UniProt sequence, using a pull-down menu above the tree structure, though the default numbering is from the mouse PKA crystal structure 1atp. Every position in our alignment profiles is displayed, with insertions and deletions relative to the profile dependent on the numbering chosen. Insertions are displayed as two consecutive aligned columns with non-consecutive numbering. For example, epidermal growth factor receptor (EGFR) has an insertion relative to our profiles at native positions 752EGFR and 753EGFR in the β3-αC loop. Deletions, on the other hand, are displayed as aligned columns with no residue number shown beneath. In the activation segment, for example, PKA has a deletion relative to the profiles between residues 193PKA and 194PKA (Fig. 2A). Upon loading, the top and bottom display a weblogo of the entire ePK alignment. Hovering the mouse over the weblogo displays the relative frequency of each amino acid at that position. Residues with associated cancer variants have their positions outlined in red, while the information is hidden until the mouse hovers over a particular residue number. Upon hovering, the cancer variants found at that position and the cancer type in which they are most commonly found, in the selected sequences, are displayed at the bottom. Similarly, residues with experimentally validated PTMs have an associated green circle, whose radius provides a rough measure of the number of modifications observed at a given position. Hovering the mouse over the circle displays the exact number of kinases with experimentally validated modifications mapping to the position, which are normalized by kinase to remove multiplicities that arise when a modification is reported in more than one study. Secondary structure information is displayed between the logos, with β-strands denoted as blue arrows and α-helices as green rectangles.We can also limit the results in several ways. By clicking on the weblogo, an amino acid selection interface appears. After clicking on and submitting the desired amino acids, MSAOnt is queried to identify in which kinases the selected amino acids are naturally conserved. Similarly, by clicking on the residue number or the green PTM circle, we can identify which kinases have selected mutant types or modified residues, respectively, at that position. After submission, the tree-based list of kinases is correspondingly updated, the weblogo is redrawn and the variant and PTM data is updated. For example, to identify which kinases naturally conserve a tyrosine at 204PKA, we can simply click on the weblogo above position 204, which displays a table of the residues naturally conserved at that position. By selecting ‘Y’ and clicking ‘Submit’, an MSAOnt query is submitted and the selection tree, natural sequence variation, cancer variants and post-translational modifications are updated to include information about kinases with Y204PKA. Similarly, if we want to identify kinases with experimentally validated phosphorylation events at 204PKA, we click on the green circle below position 204 and select the modified residue type. Again, the selection tree and integrated data is updated to reflect only those kinases that are known to be phosphorylated at 204PKA. KinView has been successfully tested on the Chrome, Firefox and Safari web browsers.
Protein expression, antibodies and reagents
The pan anti-phospho-PKC activation loop antibody (p500) was described previously.106 The anti-phospho-PKCα/βII turn motif (T638/641; 9375S) and pan anti-phospho-PKC hydrophobic motif (βII S660; 9371S) antibodies were purchased from Cell Signaling. Anti-PKCβ antibody was purchased from BD Transduction Laboratories. The anti-α-tubulin (T6074) antibody was from Sigma. Phorbol 12,13-dibutyrate (PDBu) and uridine-5-triphosphate (UTP) were purchased from Sigma-Aldrich. 6His-N-terminally tagged human PLK4 (amino acids 1–269) was expressed and purified as described previously107 and affinity purified using Ni-NTA agarose. Proteins were eluted from beads by incubation with 0.5 M imidazole. Described PLK4 mutations were introduced using standard PCR-based site-directed mutagenesis protocols and confirmed by sequencing the PLK4 1–269 coding region. pT170 PLK4 phosphospecific antibody was raised in rabbits against a phosphorylated PLK4 T-loop consensus peptide, and purified using standard affinity protocols prior to storage at −20 °C59. The PLK4 inhibitors VX680 and staurosporine and the PLK1 inhibitor BI2536 were employed after dilution from 50 mM DMSO stocks stored at −80 °C79.Kinase assays and analysis of PLK4 autophosphorylation
The kinetics of PLK4 autophosphorylation was measured by two methods. Initially 2 μg (∼60 pmol) of WT, Y177E, and Y177F PLK4 (1–269) recombinant protein in 50 mM Tris, pH 7.4, 100 mM NaCl, 1 mM DTT and 100 mM imidazole were assayed at 30 °C in the presence of 200 μM ATP (2 μCi 32P ATP per assay) and 10 mM MgCl2. VX-680 (10 μM) was pre-incubated prior to ATP addition, or 1% (v/v) DMSO was included as solvent control. The reactions were terminated at the indicated time points by denaturation in SDS sample buffer prior to separation by SDS-PAGE and transfer to nitrocellulose membranes. 32P-incorporation into PLK4 (autophosphorylation) was detected by autoradiography. The total amount of PLK4 loaded was detected by Ponceau S staining. In addition, samples were immunoblotted with purified pT170 PLK4 antibody (1:1000 dilution) and phosphorylation at T170 was detected with a rabbit HRP-conjugated secondary antibody by ECL.
Differential scanning fluorimetry (DSF)
The fluorescence emission of Sypro Orange dye (1:4000 final dilution from stock) was measured to construct thermal denaturation profiles for individual PLK4 proteins (4 μM) and evaluated as described using the Boltzmann equation (Mohanty et al., 2016) in the presence of 1 mM ATP complexed with 10 mM MgCl2 or the indicated concentrations of kinase inhibitor (4% (v/v) final DMSO concentration). Control samples all contained 4% DMSO as solvent control. Denaturation assays were performed using a StepOnePlus RT-PCR machine employing a thermal ramp between 25–95 °C (0.3 °C step per data point). Mean ΔTm values ± SD from duplicate experiments were calculated by subtracting the control Tm value from the Tm value measured in the presence of ligand.
Cell culture, transfection, and immunoblotting
All cells were maintained in DMEM (Corning) containing 10% fetal bovine serum (Atlanta Biologicals) and penicillin/streptomycin (Corning) at 37 °C, in 5% CO2. Transient transfection of COS7 was carried out using the FuGENE 6 transfection reagent (Roche) for 24 h. Cells were lysed in 50 mM Tris, pH 7.4, 1% Triton X-100, 50 mM NaF, 10 mM Na4P2O7, 100 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, and 50 nM okadaic acid. Whole cell lysates were analyzed by SDS-PAGE and immunoblotting via chemiluminescence on a FluorChemQ imaging system (Alpha Innotech).FRET imaging and analysis
Cells were imaged as described previously.108 For activity measurements, cells were co-transfected with the indicated mCherry-tagged PKC and CKAR. UTP-stimulated PKC activity traces were quantified by area under the curve and normalized to WT PKC activity. Data represent the average of three independent experiments. Comparisons for UTP-stimulated activity were made using a repeated-measures one-way ANOVA followed by a post hoc Dunnett's multiple comparison test. **p < 0.01 as compared with the WT group. All statistical tests were performed using Prism software version 6.0e for Mac (Graphpad software).Plasmid constructs
The C Kinase Activity Reporter (CKAR)91 was previously described. pENTR clones of DNA encoding human PKCβII were from the Ultimate Human ORF Library (Life Technologies). These were N-terminally tagged with mCherry via Gateway cloning (Life Technologies) into pDEST-mCherry, which was generated from pcDNA3 (Life Technologies) with mCherry DNA (gift from Roger Tsien) subcloned into the HindIII and EcoRV sites and blunt ligation of the Reading Frame Cassette A (Life Technologies) into the EcoRV site of pcDNA3. PLK4 (1–269) was cloned into pET30 Ek/LIC and expressed in E. coli strain BL21(DE3) pLysS, as described.107PLK4 digestion and mass spectrometry
.raw files were converted to .mgf files in Proteome Discoverer. HCD and ETHCD spectra were separated according to ETD reaction time (<39 ms selects HCD spectra) generating two separate .mgf files. Using an in-house built Perl script, the two .mgf files were merged and searched using MASCOT (2.1) against the E. coli IPI database (24/03/15; 4551 sequences) with the sequence of human PLK4 (1–269) included. Parameters were set as follows: MS1 tolerance of 10 ppm; MS/MS mass tolerance of 0.6 Da; carbamidomethylation of cysteine was set as a fixed modification; phosphorylation of serine and threonine, phosphorylation of tyrosine and oxidation of methionine were set as variable modifications. The tandem MS data for the identified phosphopeptides were interrogated manually and confirmed.
Funding
Funding for NK from the National Science Foundation (MCB-1149106) and National Institutes of Health (GM114409-01) is acknowledged. This work was supported by National Institutes of Health grant GM3154- (to ACN), North West Cancer Research grants CR1037 and CR1088 (to PAE), and Biotechnology and Biological Sciences Research Council project grant (BB/M012557/1) and DTP PhD studentship (to CEE).Availability of data and materials
Requests for PLK4 constructs and pT170 phosphospecific antibody should be directed to PAE. MSAOnt population software is available at http://https://github.com/esbg/msaont (DOI: http://10.5281/zenodo.50804). KinView is available through the ProKinO browser at http://vulcan.cs.uga.edu/prokino.Competing interests
The authors declare that they have no competing interests.References
- D. T. Jones, J. Mol. Biol., 1999, 292, 195–202 CrossRef CAS PubMed.
- J. A. Cuff and G. J. Barton, Proteins, 2000, 40, 502–511 CrossRef CAS.
- J. A. Cuff, M. E. Clamp, A. S. Siddiqui, M. Finlay and G. J. Barton, Bioinformatics, 1998, 14, 892–893 CrossRef CAS PubMed.
- S. K. Hanks and A. M. Quinn, Methods Enzymol., 1991, 200, 38–62 CAS.
- C. P. Ponting, J. Schultz, F. Milpetz and P. Bork, Nucleic Acids Res., 1999, 27, 229–232 CrossRef CAS PubMed.
- N. Furnham, N. L. Dawson, S. A. Rahman, J. M. Thornton and C. A. Orengo, J. Mol. Biol., 2016, 428, 253–267 CrossRef CAS PubMed.
- M. Gouy, S. Guindon and O. Gascuel, Mol. Biol. Evol., 2010, 27, 221–224 CrossRef CAS PubMed.
- A. J. Drummond and A. Rambaut, BMC Evol. Biol., 2007, 7, 214 CrossRef PubMed.
- H. A. Schmidt, K. Strimmer, M. Vingron and A. von Haeseler, Bioinformatics, 2002, 18, 502–504 CrossRef CAS PubMed.
- I. A. Adzhubei, S. Schmidt, L. Peshkin, V. E. Ramensky, A. Gerasimova, P. Bork, A. S. Kondrashov and S. R. Sunyaev, Nat. Methods, 2010, 7, 248–249 CrossRef CAS PubMed.
- I. Adzhubei, D. M. Jordan and S. R. Sunyaev, Current Protocols in Human Genetics, 2013, ch. 7, unit 7.20 Search PubMed.
- M. P. Miller and S. Kumar, Hum. Mol. Genet., 2001, 10, 2319–2328 CrossRef CAS PubMed.
- M. P. Miller, J. D. Parker, S. W. Rissing and S. Kumar, Ann. Hum. Genet., 2003, 67, 567–579 CrossRef CAS PubMed.
- S. Kumar, M. Sanderford, V. E. Gray, J. Ye and L. Liu, Nat. Methods, 2012, 9, 855–856 CrossRef CAS PubMed.
- R. Notaro, A. Afolayan and L. Luzzatto, FASEB J., 2000, 14, 485–494 CAS.
- P. Beltrao, V. Albanèse, L. R. Kenner, D. L. Swaney, A. Burlingame, J. Villén, W. A. Lim, J. S. Fraser, J. Frydman and N. J. Krogan, Cell, 2012, 150, 413–425 CrossRef CAS PubMed.
- C. R. Landry, E. D. Levy and S. W. Michnick, Trends Genet., 2009, 25, 193–197 CrossRef CAS PubMed.
- A. N. Nguyen Ba and A. M. Moses, Mol. Biol. Evol., 2010, 27, 2027–2037 CrossRef CAS PubMed.
- P. Beltrao, P. Bork, N. J. Krogan and V. van Noort, Mol. Syst. Biol., 2013, 9, 714 CrossRef PubMed.
- P. Beltrao, J. C. Trinidad, D. Fiedler, A. Roguev, W. A. Lim, K. M. Shokat, A. L. Burlingame and N. J. Krogan, PLoS Biol., 2009, 7, e1000134 Search PubMed.
- J. R. Johnson, S. D. Santos, T. Johnson, U. Pieper, M. Strumillo, O. Wagih, A. Sali, N. J. Krogan and P. Beltrao, PLoS Comput. Biol., 2015, 11, e1004362 Search PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- N. Blom, S. Gammeltoft and S. Brunak, J. Mol. Biol., 1999, 294, 1351–1362 CrossRef CAS PubMed.
- C. S. Tan, B. Bodenmiller, A. Pasculescu, M. Jovanovic, M. O. Hengartner, C. Jørgensen, G. D. Bader, R. Aebersold, T. Pawson and R. Linding, Sci. Signaling, 2009, 2, ra39 CrossRef PubMed.
- X. D. Zhang, J. Song, P. Bork and X. M. Zhao, Sci. Rep., 2016, 6, 20558 CrossRef CAS PubMed.
- S. Sunyaev, V. Ramensky, I. Koch, W. Lathe, A. S. Kondrashov and P. Bork, Hum. Mol. Genet., 2001, 10, 591–597 CrossRef CAS PubMed.
- T. Y. Lee, H. D. Huang, J. H. Hung, H. Y. Huang, Y. S. Yang and T. H. Wang, Nucleic Acids Res., 2006, 34, D622–D627 CrossRef CAS PubMed.
- S. A. Forbes, D. Beare, P. Gunasekaran, K. Leung, N. Bindal, H. Boutselakis, M. Ding, S. Bamford, C. Cole, S. Ward, C. Y. Kok, M. Jia, T. De, J. W. Teague, M. R. Stratton, U. McDermott and P. J. Campbell, Nucleic Acids Res., 2015, 43, D805–D811 CrossRef PubMed.
- S. Bamford, E. Dawson, S. Forbes, J. Clements, R. Pettett, A. Dogan, A. Flanagan, J. Teague, P. A. Futreal, M. R. Stratton and R. Wooster, Br. J. Cancer, 2004, 91, 355–358 CAS.
- S. K. Hanks and T. Hunter, FASEB J., 1995, 9, 576–596 CAS.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- N. Kannan, N. Haste, S. S. Taylor and A. F. Neuwald, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1272–1277 CrossRef CAS PubMed.
- H. S. Meharena, P. Chang, M. M. Keshwani, K. Oruganty, A. K. Nene, N. Kannan, S. S. Taylor and A. P. Kornev, PLoS Biol., 2013, 11, e1001680 CAS.
- S. Mohanty, E. J. Kennedy, F. W. Herberg, R. Hui, S. S. Taylor, G. Langsley and N. Kannan, Biochim. Biophys. Acta, 2015, 1854, 1575–1585 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- G. Gosal, K. J. Kochut and N. Kannan, PLoS One, 2011, 6, e28782 CAS.
- D. I. McSkimming, S. Dastgheib, E. Talevich, A. Narayanan, S. Katiyar, S. S. Taylor, K. Kochut and N. Kannan, Hum. Mutat., 2015, 36, 175–186 CrossRef CAS PubMed.
- G. Gosal, K. J. Kochut and N. Kannan, PLoS One, 2011, 6, e28782 CAS.
- L. N. Johnson, M. E. Noble and D. J. Owen, Cell, 1996, 85, 149–158 CrossRef CAS PubMed.
- S. S. Taylor, J. Yang, J. Wu, N. M. Haste, E. Radzio-Andzelm and G. Anand, Biochim. Biophys. Acta, 2004, 1697, 259–269 CrossRef CAS PubMed.
- M. Huse and J. Kuriyan, Cell, 2002, 109, 275–282 CrossRef CAS PubMed.
- B. Nolen, C. Y. Yun, C. F. Wong, J. A. McCammon, X. D. Fu and G. Ghosh, Nat. Struct. Biol., 2001, 8, 176–183 CrossRef CAS PubMed.
- J. A. Endicott, M. E. Noble and L. N. Johnson, Annu. Rev. Biochem., 2012, 81, 587–613 CrossRef CAS PubMed.
- C. H. Lee and J. H. Chung, J. Biol. Chem., 2001, 276, 30537–30541 CrossRef CAS PubMed.
- G. Buscemi, P. Perego, N. Carenini, M. Nakanishi, L. Chessa, J. Chen, K. Khanna and D. Delia, Oncogene, 2004, 23, 7691–7700 CrossRef CAS PubMed.
- J. Cho and P. N. Tsichlis, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2350–2355 CrossRef CAS PubMed.
- T. Timm, K. Balusamy, X. Li, J. Biernat, E. Mandelkow and E. M. Mandelkow, J. Biol. Chem., 2008, 283, 18873–18882 CrossRef CAS PubMed.
- T. Jiang and Y. Qiu, J. Biol. Chem., 2003, 278, 15789–15793 CrossRef CAS PubMed.
- H. S. Jung, D. W. Kim, Y. S. Jo, H. K. Chung, J. H. Song, J. S. Park, K. C. Park, S. H. Park, J. H. Hwang, K. W. Jo and M. Shong, Mol. Endocrinol., 2005, 19, 2748–2759 CrossRef CAS PubMed.
- R. Chen, O. Kim, J. Yang, K. Sato, K. M. Eisenmann, J. McCarthy, H. Chen and Y. Qiu, J. Biol. Chem., 2001, 276, 31858–31862 CrossRef CAS PubMed.
- L. R. Pearce, D. Komander and D. R. Alessi, Nat. Rev. Mol. Cell Biol., 2010, 11, 9–22 CrossRef CAS PubMed.
- W. C. Huang, J. J. Chen and C. C. Chen, J. Biol. Chem., 2003, 278, 9944–9952 CrossRef CAS PubMed.
- H. Konishi, M. Tanaka, Y. Takemura, H. Matsuzaki, Y. Ono, U. Kikkawa and Y. Nishizuka, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11233–11237 CrossRef CAS.
- S. Lai and S. Pelech, Mol. Biol. Cell, 2016, 27, 1040–1050 CrossRef CAS PubMed.
- C. Breit, T. Bange, A. Petrovic, J. R. Weir, F. Müller, D. Vogt and A. Musacchio, PLoS One, 2015, 10, e0144673 Search PubMed.
- Z. Lin, L. Jia, D. R. Tomchick, X. Luo and H. Yu, Structure, 2014, 22, 1616–1627 CrossRef CAS PubMed.
- O. Fedorov, K. Huber, A. Eisenreich, P. Filippakopoulos, O. King, A. N. Bullock, D. Szklarczyk, L. J. Jensen, D. Fabbro, J. Trappe, U. Rauch, F. Bracher and S. Knapp, Chem. Biol., 2011, 18, 67–76 CrossRef CAS PubMed.
- S. Naud, I. M. Westwood, A. Faisal, P. Sheldrake, V. Bavetsias, B. Atrash, K. M. Cheung, M. Liu, A. Hayes, J. Schmitt, A. Wood, V. Choi, K. Boxall, G. Mak, M. Gurden, M. Valenti, A. de Haven Brandon, A. Henley, R. Baker, C. McAndrew, B. Matijssen, R. Burke, S. Hoelder, S. A. Eccles, F. I. Raynaud, S. Linardopoulos, R. L. van Montfort and J. Blagg, J. Med. Chem., 2013, 56, 10045–10065 CrossRef CAS PubMed.
- R. K. Tyler, M. L. Chu, H. Johnson, E. A. McKenzie, S. J. Gaskell and P. A. Eyers, Biochem. J., 2009, 417, 173–181 CrossRef CAS PubMed.
- M. L. Chu, L. M. Chavas, K. T. Douglas, P. A. Eyers and L. Tabernero, J. Biol. Chem., 2008, 283, 21495–21500 CrossRef CAS PubMed.
- M. L. Chu, Z. Lang, L. M. Chavas, J. Neres, O. S. Fedorova, L. Tabernero, M. Cherry, D. H. Williams, K. T. Douglas and P. A. Eyers, Biochemistry, 2010, 49, 1689–1701 CrossRef CAS PubMed.
- Y. J. Jeon, K. Y. Lee, Y. Y. Cho, A. Pugliese, H. G. Kim, C. H. Jeong, A. M. Bode and Z. Dong, J. Biol. Chem., 2010, 285, 28126–28133 CrossRef CAS PubMed.
- N. Wang, H. Ding, C. Liu, X. Li, L. Wei, J. Yu, M. Liu, M. Ying, W. Gao, H. Jiang and Y. Wang, Oncogene, 2015, 34, 5198–5205 CrossRef CAS PubMed.
- C. A. Martin, I. Ahmad, A. Klingseisen, M. S. Hussain, L. S. Bicknell, A. Leitch, G. Nürnberg, M. R. Toliat, J. E. Murray, D. Hunt, F. Khan, Z. Ali, S. Tinschert, J. Ding, C. Keith, M. E. Harley, P. Heyn, R. Müller, I. Hoffmann, V. C. Daire, H. Dollfus, L. Dupuis, A. Bashamboo, K. McElreavey, A. Kariminejad, R. Mendoza-Londono, A. T. Moore, A. Saggar, C. Schlechter, R. Weleber, H. Thiele, J. Altmüller, W. Höhne, M. E. Hurles, A. A. Noegel, S. M. Baig, P. Nürnberg and A. P. Jackson, Nat. Genet., 2014, 46, 1283–1292 CrossRef CAS PubMed.
- T. C. Moyer, K. M. Clutario, B. G. Lambrus, V. Daggubati and A. J. Holland, J. Cell Biol., 2015, 209, 863–878 CrossRef CAS PubMed.
- G. Guderian, J. Westendorf, A. Uldschmid and E. A. Nigg, J. Cell Sci., 2010, 123, 2163–2169 CrossRef CAS PubMed.
- C. A. Lopes, S. C. Jana, I. Cunha-Ferreira, S. Zitouni, I. Bento, P. Duarte, S. Gilberto, F. Freixo, A. Guerrero, M. Francia, M. Lince-Faria, J. Carneiro and M. Bettencourt-Dias, Dev. Cell, 2015, 35, 222–235 CrossRef CAS PubMed.
- A. J. Holland, D. Fachinetti, Q. Zhu, M. Bauer, I. M. Verma, E. A. Nigg and D. W. Cleveland, Genes Dev., 2012, 26, 2684–2689 CrossRef CAS PubMed.
- J. E. Klebba, D. W. Buster, T. A. McLamarrah, N. M. Rusan and G. C. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E657–E666 CrossRef CAS PubMed.
- I. Cunha-Ferreira, A. Rodrigues-Martins, I. Bento, M. Riparbelli, W. Zhang, E. Laue, G. Callaini, D. M. Glover and M. Bettencourt-Dias, Curr. Biol., 2009, 19, 43–49 CrossRef CAS PubMed.
- J. E. Klebba, D. W. Buster, A. L. Nguyen, S. Swatkoski, M. Gucek, N. M. Rusan and G. C. Rogers, Curr. Biol., 2013, 23, 2255–2261 CrossRef CAS PubMed.
- T. Nakamura, H. Saito and M. Takekawa, Nat. Commun., 2013, 4, 1775 CrossRef PubMed.
- D. A. Sloane, M. Z. Trikic, M. L. Chu, M. B. Lamers, C. S. Mason, I. Mueller, W. J. Savory, D. H. Williams and P. A. Eyers, ACS Chem. Biol., 2010, 5, 563–576 CrossRef CAS PubMed.
- A. Shrestha, G. Hamilton, E. O'Neill, S. Knapp and J. M. Elkins, Protein Expression Purif., 2012, 81, 136–143 CrossRef CAS PubMed.
- J. M. Murphy, Q. Zhang, S. N. Young, M. L. Reese, F. P. Bailey, P. A. Eyers, D. Ungureanu, H. Hammaren, O. Silvennoinen, L. N. Varghese, K. Chen, A. Tripaydonis, N. Jura, K. Fukuda, J. Qin, Z. Nimchuk, M. B. Mudgett, S. Elowe, C. L. Gee, L. Liu, R. J. Daly, G. Manning, J. J. Babon and I. S. Lucet, Biochem. J., 2014, 457, 323–334 CrossRef CAS PubMed.
- V. Reiterer, P. A. Eyers and H. Farhan, Trends Cell Biol., 2014, 24, 489–505 CrossRef CAS PubMed.
- E. F. Johnson, K. D. Stewart, K. W. Woods, V. L. Giranda and Y. Luo, Biochemistry, 2007, 46, 9551–9563 CrossRef CAS PubMed.
- M. J. Moore, J. A. Adams and S. S. Taylor, J. Biol. Chem., 2003, 278, 10613–10618 CrossRef CAS PubMed.
- P. J. Scutt, M. L. Chu, D. A. Sloane, M. Cherry, C. R. Bignell, D. H. Williams and P. A. Eyers, J. Biol. Chem., 2009, 284, 15880–15893 CrossRef CAS PubMed.
- P. A. Lochhead, R. Kinstrie, G. Sibbet, T. Rawjee, N. Morrice and V. Cleghon, Mol. Cell, 2006, 24, 627–633 CrossRef CAS PubMed.
- R. Kinstrie, N. Luebbering, D. Miranda-Saavedra, G. Sibbet, J. Han, P. A. Lochhead and V. Cleghon, Sci. Signaling, 2010, 3, ra16 CrossRef PubMed.
- H. Chen, Z. Huang, K. Dutta, S. Blais, T. A. Neubert, X. Li, D. Cowburn, N. J. Traaseth and M. Mohammadi, Cell Rep., 2013, 4, 376–384 CrossRef CAS PubMed.
- Z. Huang, H. Chen, S. Blais, T. A. Neubert, X. Li and M. Mohammadi, Structure, 2013, 21, 1889–1896 CrossRef CAS PubMed.
- E. B. Bioukar, N. C. Marricco, D. Zuo and L. Larose, J. Biol. Chem., 1999, 274, 21457–21463 CrossRef CAS PubMed.
- Y. Kobashigawa, S. Amano, M. Yokogawa, H. Kumeta, H. Morioka, M. Inouye, J. Schlessinger and F. Inagaki, Genes Cells, 2015, 20, 860–870 CrossRef CAS PubMed.
- H. Chen, J. Ma, W. Li, A. V. Eliseenkova, C. Xu, T. A. Neubert, W. T. Miller and M. Mohammadi, Mol. Cell, 2007, 27, 717–730 CrossRef CAS PubMed.
- C. E. Antal, A. M. Hudson, E. Kang, C. Zanca, C. Wirth, N. L. Stephenson, E. W. Trotter, L. L. Gallegos, C. J. Miller, F. B. Furnari, T. Hunter, J. Brognard and A. C. Newton, Cell, 2015, 160, 489–502 CrossRef CAS PubMed.
- K. Oruganty, N. S. Talathi, Z. A. Wood and N. Kannan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 924–929 CrossRef CAS PubMed.
- E. M. Lisabeth, C. Fernandez and E. B. Pasquale, Biochemistry, 2012, 51, 1464–1475 CrossRef CAS PubMed.
- A. C. Newton, Am. J. Physiol.: Endocrinol. Metab., 2010, 298, E395–E402 CrossRef CAS PubMed.
- J. D. Violin, J. Zhang, R. Y. Tsien and A. C. Newton, J. Cell Biol., 2003, 161, 899–909 CrossRef CAS PubMed.
- S. Kumar, G. Stecher and K. Tamura, Mol. Biol. Evol., 2016, 33, 1870–1874 CrossRef CAS PubMed.
- G. Stecher, L. Liu, M. Sanderford, D. Peterson, K. Tamura and S. Kumar, Bioinformatics, 2014, 30, 1305–1307 CrossRef CAS PubMed.
- D. A. Natale, C. N. Arighi, W. C. Barker, J. A. Blake, C. J. Bult, M. Caudy, H. J. Drabkin, P. D'Eustachio, A. V. Evsikov, H. Huang, J. Nchoutmboube, N. V. Roberts, B. Smith, J. Zhang and C. H. Wu, Nucleic Acids Res., 2011, 39, D539–D545 CrossRef CAS PubMed.
- J. D. Thompson, S. R. Holbrook, K. Katoh, P. Koehl, D. Moras, E. Westhof and O. Poch, Nucleic Acids Res., 2005, 33, 4164–4171 CrossRef CAS PubMed.
- B. Smith, M. Ashburner, C. Rosse, J. Bard, W. Bug, W. Ceusters, L. J. Goldberg, K. Eilbeck, A. Ireland, C. J. Mungall, N. Leontis, P. Rocca-Serra, A. Ruttenberg, S. A. Sansone, R. H. Scheuermann, N. Shah, P. L. Whetzel, S. Lewis and O. Consortium, Nat. Biotechnol., 2007, 25, 1251–1255 CrossRef CAS PubMed.
- N. Kannan and A. F. Neuwald, Protein Sci., 2004, 13, 2059–2077 CrossRef CAS PubMed.
- D. Krech, RDFlib: A Python Library for Working with RDF [Computer software], 2006, http://github.com/RDFLib/rdflib Search PubMed.
- D. Croft, A. F. Mundo, R. Haw, M. Milacic, J. Weiser, G. Wu, M. Caudy, P. Garapati, M. Gillespie, M. R. Kamdar, B. Jassal, S. Jupe, L. Matthews, B. May, S. Palatnik, K. Rothfels, V. Shamovsky, H. Song, M. Williams, E. Birney, H. Hermjakob, L. Stein and P. D'Eustachio, Nucleic Acids Res., 2014, 42, D472–D477 CrossRef CAS PubMed.
- M. Milacic, R. Haw, K. Rothfels, G. Wu, D. Croft, H. Hermjakob, P. D'Eustachio and L. Stein, Cancers, 2012, 4, 1180–1211 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Y. Zhai, M. J. Chen and G. Manning, KinBase, http://kinase.com/web/current/kinbase/.
- U. Consortium, Nucleic Acids Res., 2015, 43, D204–D212 CrossRef PubMed.
- T. D. Schneider and R. M. Stephens, Nucleic Acids Res., 1990, 18, 6097–6100 CrossRef CAS PubMed.
- M. G. Skjæveland, The Semantic Web: ESWC 2012 Satellite Events, Springer, 2012, pp. 361–365 Search PubMed.
- E. M. Dutil, A. Toker and A. C. Newton, Curr. Biol., 1998, 8, 1366–1375 CrossRef CAS PubMed.
- S. Atasoy, B. Glocker, S. Giannarou, D. Mateus, A. Meining, G. Z. Yang and N. Navab, Med. Image Comput. Comput. Assist. Interv., 2009, 12, 499–506 Search PubMed.
- L. L. Gallegos, M. T. Kunkel and A. C. Newton, J. Biol. Chem., 2006, 281, 30947–30956 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |