
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of the transmembrane domain in SNARE protein mediated membrane fusion: peptide nucleic acid/peptide model systems†
Jan-Dirk
Wehland
a,
Antonina S.
Lygina
a,
Pawan
Kumar
a,
Samit
Guha
a,
Barbara E.
Hubrich
a,
Reinhard
Jahn
b and
Ulf
Diederichsen
*a
aInstitute of Organic and Biomolecular Chemistry, Georg-August-University of Göttingen, Tammannstr. 2, 37077 Göttingen, Germany. E-mail: udieder@gwdg.de; Fax: +49-551-39-22944
bDepartment of Neurobiology, Max-Planck-Institute of Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany
First published on 14th June 2016
Abstract
Fusion of synaptic vesicles with the presynaptic plasma membrane is mediated by Soluble NSF (N-ethylmaleimide-sensitive factor) Attachment Protein Receptor proteins also known as SNAREs. The backbone of this essential process is the assembly of SNAREs from opposite membranes into tight four helix bundles forcing membranes in close proximity. With model systems resembling SNAREs with reduced complexity we aim to understand how these proteins work at the molecular level. Here, peptide nucleic acids (PNAs) are used as excellent candidates for mimicking the SNARE recognition motif by forming well-characterized duplex structures. Hybridization between complementary PNA strands anchored in liposomes through native transmembrane domains (TMDs) induces the merger of the outer leaflets of the participating vesicles but not of the inner leaflets. A series of PNA/peptide hybrids differing in the length of TMDs and charges at the C-terminal end is presented. Interestingly, mixing of both outer and inner leaflets is seen for TMDs containing an amide in place of the natural carboxylic acid at the C-terminal end. Charged side chains at the C-terminal end of the TMDs are shown to have a negative impact on the mixing of liposomes. The length of the TMDs is vital for fusion as with the use of shortened TMDs, fusion was completely prevented.
Introduction
Membrane fusion is an indispensable event in all forms of life. Understanding of the fundamental biophysical principles and the molecular mechanisms governing this process will lead to a better understanding of naturally occurring processes and will also open new opportunities in the field of drug delivery and tools for gene transfection. Intracellular fusion is mediated by specific molecular recognition of SNARE (Soluble NSF (N-ethylmaleimide-sensitive factor) Attachment Protein Receptor) proteins. Most of these proteins contain a C-terminal transmembrane domain (TMD) connected by a short linker to the recognition sequence assembling in the latter SNARE complex. In neuronal exocytosis, fusion of two membranes is facilitated by the interaction of the key SNARE proteins syntaxin-1A and SNAP-25 residing in the plasma membrane and synaptobrevin 2 in the vesicle membrane.1,2 Several mechanisms of SNARE mediated membrane fusion have been discussed but none of them is universally accepted.3,4 In particular, structural and functional requirements of TMDs in the fusion process are not precisely understood.5,6 A number of studies has suggested the necessity of TMDs for membrane fusion,7–13 but at what stage and how exactly TMDs may influence the fusion process is still a question to answer. The presence of continuous helices including the SNARE motif and the TMD/linker domain in the cis-SNARE complex makes a pitch for their involvement in the late stage events.2 Removal of amino acids from the C-terminal end has been shown to arrest the fusion at the hemifusion stage.7 Besides, insertions/deletions in the C-terminal half of synaptobrevin 2 TMDs have been shown to be detrimental for neurosecretion while mutations in the N-terminal half are better tolerated.9 Simulation studies have also suggested the participation of the C-terminal end of the TMDs in the fusion pore opening.11 Additionally, movement of the C-terminal end of the synaptobrevin 2 TMD from water to the membrane interface has been linked to the fusion pore opening.10 The addition of amino acids with charged side chains at the C-terminal end increases the energetic barrier for this movement, thereby having a negative impact on the mixing of liposomes.14 Thus, a critical role for the C-terminal half of the TMDs and in particular the last amino acids in membrane fusion has been indicated. However, to the best of our knowledge no study has been conducted with TMDs having a neutral C-terminal end, which could reflect the influence of charges at this position on the fusion process.Artificial fusogenic peptide model systems allow for simplifying the complexity of interactions in natural systems. Furthermore, they provide opportunities for systematically varying the chemical structure and composition in order to understand the fusion mechanism in detail. The reduction of molecular complexity and elucidation of important elements in the fusion process provide a detailed mechanistic insight at the molecular level, and furthermore, offer a chance to design artificial fusion systems with prescribed properties.15–17 Commonly, model systems consist of a recognition unit linked to a membrane anchor. A wide range of recognition units based on highly specific molecular interactions including peptide coiled-coils,18–24 DNA,25–29 peptide nucleic acids (PNAs),30,31 small molecule recognition,32,33 hydrogen bonding34,35 and diol-boronic acid mediated recognition36–38 has been used. Predominantly, lipids were applied as membrane anchors;15 peptide transmembrane domains are still the exception for artificial fusion-mediating constructs.18,30,31
We introduced a model system that is of reduced complexity compared to native SNAREs taking advantage of specific recognition between complementary strands of PNA. Native TMDs and linker regions of syntaxin-1A and synaptobrevin 2 were used as membrane anchors and the SNARE recognition motif was replaced with an artificial recognition unit consisting of PNA (Fig. 1). As reported previously, PNA/TMD constructs reconstituted in liposomes were capable of mediating the fusion of the outer membrane leaflets leading to the docking or hemifusion stage. However, SNARE protein like fusion of both leaflets was prevented.30 For the TMD/linker domains native SNARE peptide sequences were used. This allows for studying their possible contribution in the fusion process by systematically introducing structural modifications in an otherwise well-defined construct. In this study, we especially focus on the impact of charge and/or nature of amino acids at the C-termini of TMDs. Various TMD–PNA chimeras differing in the length of TMDs, or in charges at the C-terminal end are reported and the effect of these modifications on the fusion of model membranes is discussed. A transition from hemifusion to full fusion is accomplished by switching the charged carboxylic acid C-terminal ends (like SNAREs) to neutral amide C-termini.
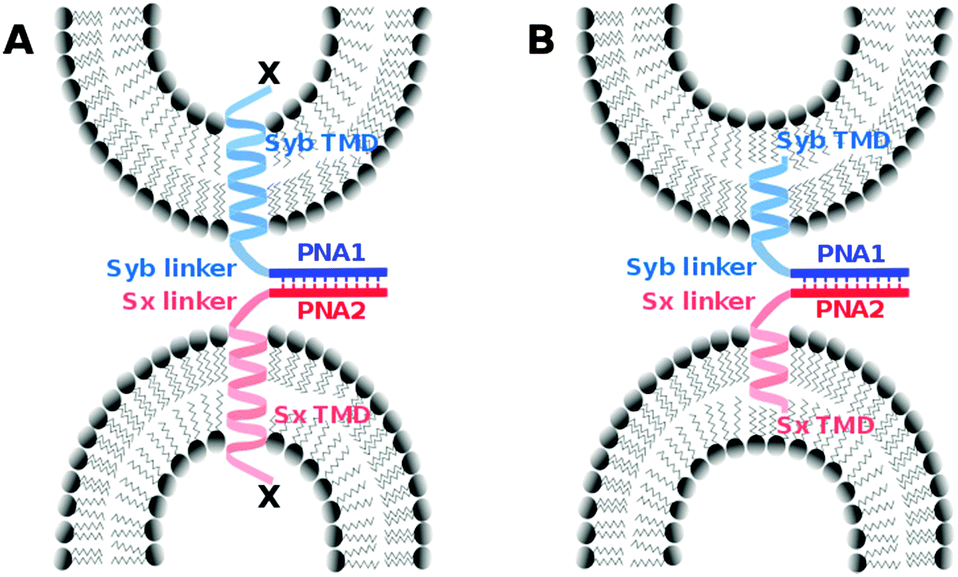 | ||
| Fig. 1 PNA/peptide model systems containing the native linker and TMD peptide sequence of synaptobrevin 2 (light blue) or syntaxin-1A (light red) as well as PNA oligomers (dark blue and dark red) as artificial recognition units with parallel strand orientation. (A) Variation of C-terminal amino acids (X) to investigate the influence of terminal charges on the fusion process. (B) PNA/peptide constructs with shortened TMDs. | ||
Results and discussion
Design and synthesis of various PNA/TMD hybrids
The design of SNARE protein mimics is based on the TMD and linker regions of native syntaxin-1A and synaptobrevin 2, whereas a PNA–PNA double strand replaces the tetrameric SNARE motif. The sequences for the PNA recognition unit were chosen so that the duplexes form selectively in a parallel manner with reasonable stability. PNA1 (gtagatcact) and PNA2 (catctagtga) form a parallel duplex with a stability of Tm = 45 °C.30,39 In analogy to the natural SNARE complex the parallel PNA duplex ensures the orientation of both TMD/linker units on the same side of the recognition duplex. The N-terminal ends of TMD/linker domains of native synaptobrevin 2 and syntaxin-1A were extended with PNA1 and PNA2, respectively. For control experiments, syntaxin-1A TMD/linker was also linked to PNA1. The peptides were assembled by automated microwave-assisted Fmoc solid phase peptide synthesis. Subsequent assembly of PNA recognition units was performed manually (see the ESI† for synthetic details) to obtain the designed PNA/peptide model systems with various C-terminal modifications (Table 1). The integrity of the PNA/peptide hybrids was determined by ESI-HRMS.| Hybrid | Sequence |
|---|---|
| PNA1 = H2N-gtagatcact; PNA2 = H2N-catctagtga; Syb = natural TMD/linker region of synaptobrevin 2; Sx = natural TMD/linker region of syntaxin-1A. Terminations: capital letters indicate exchange of natural C-terminal amino acids T (Syb) and G (Sx) for K (SybK and SxK) and E (SybE and SxE). Termination “a” indicates exchange of a carboxylate group (Syb, SybK, SybE, Sx, SxK and SxE) for an amide group (Syba, SybKa, SybEa Sxa, SxKa, SxEa). Termination “sh” indicates TMDs C-terminally shortened by 14 amino acids in the case of synaptobrevin 2 (Sybsh) and 16 amino acids in the case of syntaxin-1A (Sxsh), both with C-terminal amide groups. | |
| PNA1/Syb | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFST-COOH |
| PNA2/Sx | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFG-COOH |
| PNA1/Sx | PNA1-KYQSKARRKKIMIIICCVILGIIIASTIGGIFG-COOH |
| PNA1/Syba | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFST-CONH2 |
| PNA2/Sxa | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFG-CONH2 |
| PNA1/Sxa | PNA1-KYQSKARRKKIMIIICCVILGIIIASTIGGIFG-CONH2 |
| PNA1/SybK | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFSK-COOH |
| PNA2/SxK | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFK-COOH |
| PNA1/SxK | PNA1-KYQSKARRKKIMIIICCVILGIIIASTIGGIFK-COOH |
| PNA1/SybE | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFSE-COOH |
| PNA2/SxE | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFE-COOH |
| PNA1/SxE | PNA1-KYQSKARRKKIMIIICCVILGIIIASTIGGIFE-COOH |
| PNA1/SybKa | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFSK-CONH2 |
| PNA2/SxKa | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFK-CONH2 |
| PNA1/SxKa | PNA1-KYQSKARRKKIMIIICCVILGIIIASTIGGIFK-CONH2 |
| PNA1/SybEa | PNA1-KRKYWWKNLKMMIILGVICAIILIIIIVYFSE-CONH2 |
| PNA2/SxEa | PNA2-KYQSKARRKKIMIIICCVILGIIIASTIGGIFE-CONH2 |
| PNA1/Sybsh | PNA1-KRKYWWKNLKMMIILGVI-CONH2 |
| PNA2/Sxsh | PNA2-KYQSKARRKKIMIIICC-CONH2 |
PNA/TMD hybrids in membranes
Vesicles were prepared by using a ternary mixture of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and cholesterol (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:25 mol%). For the labeled vesicles, DOPE lipids modified with nitrobenzoxadiazole (NBD-DOPE) and lissamine rhodamine (Rh-DOPE) were used additionally (each 1.5 mol%) and the DOPE content was reduced accordingly (22 mol%). Synthesized PNA/peptide hybrids were incorporated in membranes in the vesicle preparation process (ESI†) yielding a peptide/lipid molar ratio of 1:200. Large unilamellar vesicles (LUVs) with a size of (100 ± 20) nm decorated with PNA/peptide hybrids were obtained by the standard extrusion procedure.40 Extrusion and the following fusion assays were performed in buffer solution (20 mM HEPES, 100 mM KCl, 1 mM EDTA, 1 mM DTT, pH = 7.4).
:25:25 mol%). For the labeled vesicles, DOPE lipids modified with nitrobenzoxadiazole (NBD-DOPE) and lissamine rhodamine (Rh-DOPE) were used additionally (each 1.5 mol%) and the DOPE content was reduced accordingly (22 mol%). Synthesized PNA/peptide hybrids were incorporated in membranes in the vesicle preparation process (ESI†) yielding a peptide/lipid molar ratio of 1:200. Large unilamellar vesicles (LUVs) with a size of (100 ± 20) nm decorated with PNA/peptide hybrids were obtained by the standard extrusion procedure.40 Extrusion and the following fusion assays were performed in buffer solution (20 mM HEPES, 100 mM KCl, 1 mM EDTA, 1 mM DTT, pH = 7.4).
The statistical distribution of PNA/peptide hybrids between outer and inner leaflets was determined. PNA1/Syb and PNA1/Sx were each labeled with an NBD fluorophore at the N-terminus. They were incorporated in liposomes and NBD emission was recorded. The vesicles were then treated with dithionite (S2O42−) which is known to selectively reduce NBD fluorescence only at the outer leaflet.41 For both labeled conjugates a loss in NBD emission of about 60% was recorded after dithionite treatment (Fig. 2). Thus, about 40% of PNA/TMD hybrids were directed towards the vesicles' interior concerning the recognition units.
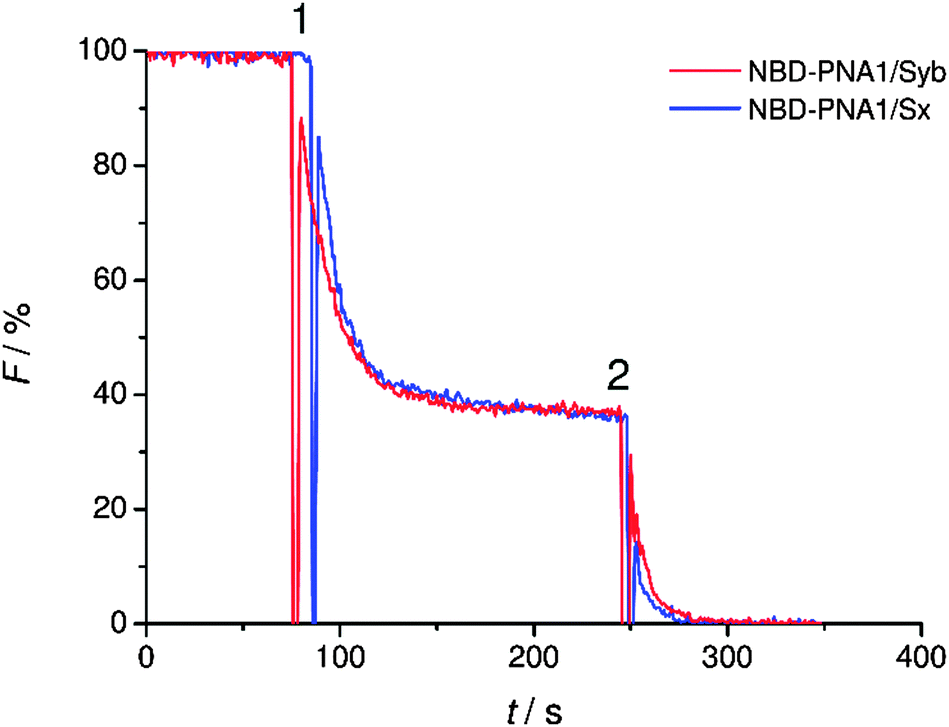 | ||
| Fig. 2 Fluorescence change after treatment of lipid vesicles containing NBD-labeled PNA1/Syb or PNA1/Sx with sodium dithionite (1) and complete elimination of fluorescence after destruction of the vesicles with Triton X-100 (2). | ||
Fusion activity of synthesized PNA/peptide hybrids
The capability of PNA/peptide hybrids to induce the fusion of liposomes was tested by employing a FRET based lipid mixing assay.42 Hybrids consisting of the PNA1 recognition unit and synaptobrevin 2 TMD/linker were anchored onto liposomes containing both donor (NBD) and acceptor (Rh) dyes. Hybrids containing the PNA2 recognition unit and syntaxin-1A TMD/linker were present in vesicles without any dyes. Prior to fusion, an efficient FRET effect was noticed due to the close distance between donor and acceptor dyes. Upon membrane fusion with unlabeled vesicles, the mixing of lipids goes along with an increase in the distance of dyes and consequently a decrease in FRET efficiency. This results in an increase of NBD emission. Notably, the change in FRET is not observed if vesicles aggregate without fusion. For differentiating between hemifusion and full fusion, lipid mixing assays were carried out without and with dithionite treated labeled vesicles to check for mixing of the inner leaflets. For further differentiation, content mixing assays using sulforhodamine B (SRB) loaded vesicles were performed (Fig. S1, ESI†). Control experiments were performed by using PNA1 based hybrids in both vesicle populations.Fusion activity of hybrids carrying a neutral C-terminus
TMDs of native SNARE proteins contain a carboxylic acid C-terminally, thereby providing a negative charge under physiological pH. As previously shown, PNA/peptide hybrids consisting of natural TMDs (PNA1/Syb and PNA2/Sx) failed to induce the mixing of the inner leaflets of interacting liposomes30 most likely arresting the hemifusion stage (Fig. S2, ESI†). Regarding the hypothesis of Lindau et al. a zipper-like recognition process forming the SNARE motif is followed by further zippering into the linker region, thereby pulling the TMDs into the membrane lipid bilayer.14 This hypothesis indicates the active participation of negatively charged C-termini in the fusion process and an interdependence of TMD C-termini with the recognition process. The PNA double strand recognition motif differs from the SNARE motif as it provides a duplex topology that is clearly separated from the peptide topology of the linker and TMD. Even in case PNA–PNA recognition is based on a zipper mechanism,43 zippering will stop after the PNA duplex is formed not transferring any further forces towards the membrane inserted TMD helix. This would explain why PNA/peptide mediated membrane fusion gets arrested at the hemifusion stage. This hypothesis is further supported by comparable SNARE analogs consisting of a continuous peptide strand of the TMD/linker region and a coiled-coil recognition unit.18 For these peptide-only SNARE models full fusion of liposomes was provided in agreement with the option for further zippering within a continuous topology. With the PNA based SNARE analogs PNA1/Syb and PNA2/Sx transfer of energy by zippering into the linker region is interrupted because of the topology change. Movement of the C-terminus of synaptobrevin 2 TMD from water to the membrane interface is predicted to affect fusion pore opening.10,14 This movement requires energy and is less favorable in case the terminal amino acid residues contain polar side chains and the C-terminus is negatively charged.Following the hypothesis of interdependence of recognition motif zippering and TMD retraction into the membrane, PNA/peptide constructs were investigated that contain the PNA recognition unit and at the same time an amide at the C-terminal end (PNA1/Syba and PNA2/Sxa). By replacing the negatively charged termini in PNA/peptide chimeras PNA1/Syb and PNA2/Sx by neutral ends the penetration of TMDs into the lipid bilayer should be assisted. The energy barrier for relocation of the TMD C-terminus into the lipid bilayer gets reduced by charge removal and the fusion process should be facilitated. Assays for vesicle fusion mediated by PNA/peptide chimera PNA1/Syba and PNA2/Sxa with neutral C-termini indeed provided a significant increase in NBD emission (Fig. 3). The increase was almost two fold higher compared to the emission obtained for constructs PNA1/Syb and PNA2/Sx containing negatively charged carboxylic acid C-termini (Fig. 3A). Moreover, efficient lipid mixing was noticed for vesicles pretreated with sodium dithionite indicating the fusion of both leaflets (Fig. 3B). No increase in NBD emission was recorded in the control experiment using non-complementary PNA/peptide hybrids (Fig. 3, PNA1/Syba and PNA1/Sxa). The result indicates the involvement of the C-termini of TMDs in the fusion process supporting the hypothesis of amplifying and expediting membrane fusion by removing charges at the C-termini of TMDs.
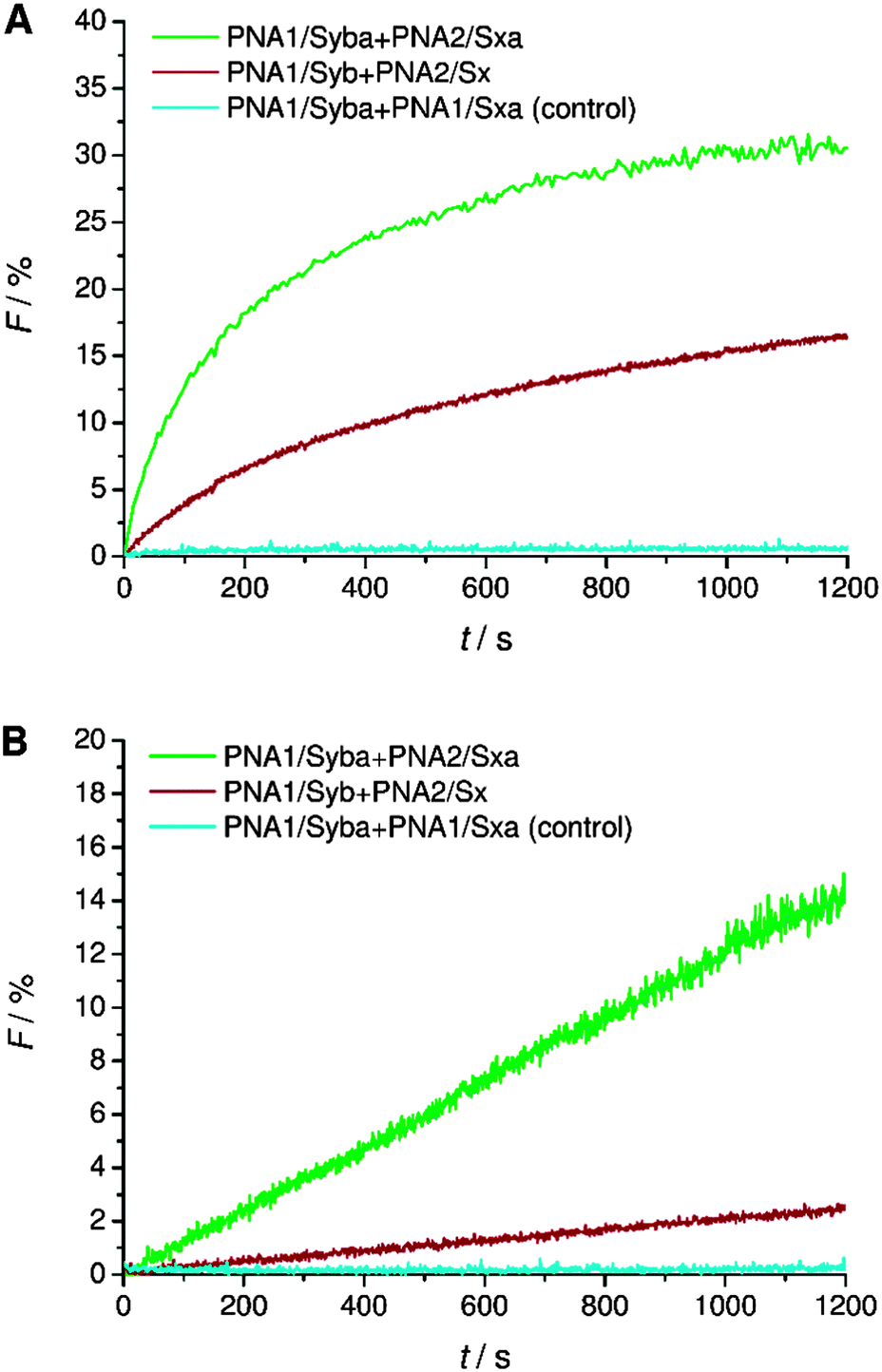 | ||
| Fig. 3 Lipid mixing assays for vesicles containing PNA/peptide hybrids bearing a negatively charged or neutral C-terminal end. (A) Total lipid mixing (both leaflets are labeled). (B) Inner leaflet mixing (donor dye in outer leaflets is reduced by dithionite treatment and therefore inactive): an increase of donor emission only takes place if inner leaflets undergo lipid mixing. For sequences of PNA/peptide chimera see Table 1. | ||
Fusion activity of hybrids containing charged side chains at the C-terminal amino acid
A systematic investigation of the influence of C-terminal charges in the PNA/peptide hybrid mediated fusion process was provided by a combination of carboxylic acid and amide termini with charged amino acid side chains of the terminal amino acids. Lysine or glutamic acid was introduced at the TMD C-termini for investigating the charge influence on liposome fusion. Mutation of the C-terminal residues maintains the effective length of the TMDs in contrast to the literature report where synaptobrevin 2 TMD was extended with charged residues.14 Lysine at the C-terminal end of the TMDs potentially can neutralize the negative charge of the terminus by forming a zwitterion. Liposomes decorated with hybrids containing lysine at the C-terminal end (PNA1/SybK and PNA2/SxK) showed a noticeable higher tendency to fuse than hybrids with unmodified natural TMDs (PNA1/Syb and PNA2/Sx) (Fig. 4A). Furthermore, mixing of inner leaflets was also noticed with vesicles pretreated with dithionite ions (Fig. 4B). Thus, zwitterionic arrangement at the C-terminal ends promotes the merger of both leaflets probably by reducing the net charge at the C-termini. In contrast, the presence of glutamate at the C-terminal ends of the TMDs renders vesicles non-fusogenic as no change in NBD emission was observed for liposomes carrying the hybrids PNA1/SybE and PNA2/SxE (Fig. 4). This could be due to an increase in the net charge at the C-terminal end and/or due to interactions of the negatively charged side chains with the surrounding lipids.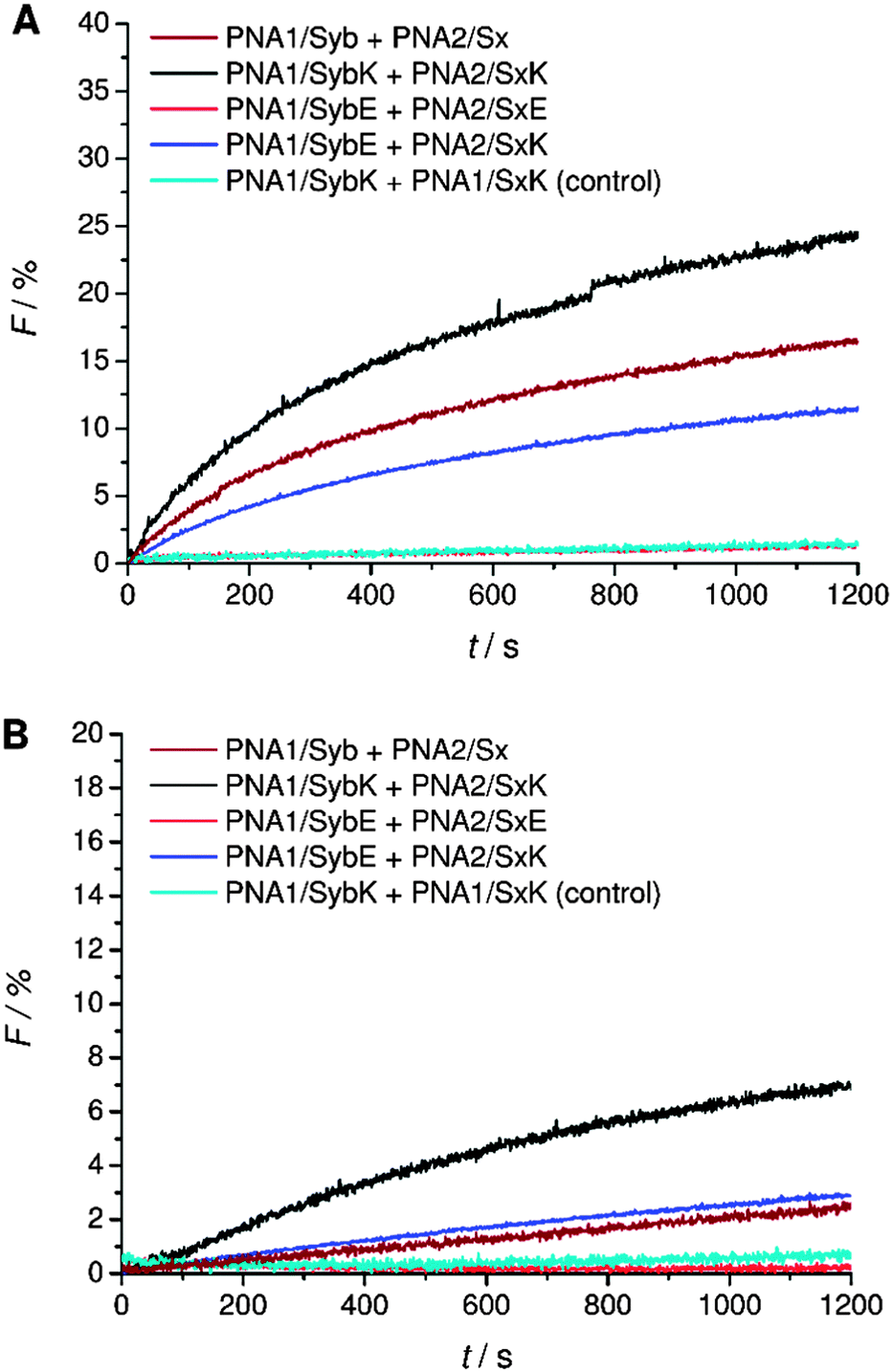 | ||
| Fig. 4 Lipid mixing assays for vesicles containing PNA/peptide hybrids with lysine or glutamate at the negatively charged carboxylic acid C-terminus. (A) Total lipid mixing. (B) Inner leaflet mixing. For sequences of PNA/peptide chimera see Table 1. | ||
Next, C-terminal amide model systems containing positively charged lysine or anionic glutamate as the C-terminal amino acid were studied (PNA1/SybKa, PNA2/SxKa, PNA1/SybEa, PNA2/SxEa). These TMDs carry charges at the side chain of the terminal amino acid allowing the modulation of the terminal charge scenario and to see if the charges need to be localized at the terminus or only in the TMD C-terminal region. The ability to induce liposome fusion dropped significantly with the introduction of positively charged side chains at otherwise neutral C-termini (Fig. 5, compare PNA1/Syba and PNA2/Sxa with PNA1/SybKa and PNA2/SxKa). Furthermore, with these constructs no noticeable mixing of inner leaflets was observed (Fig. 5B). In contrast, lysine at the C-terminus of TMDs with a carboxylate group (PNA1/SybK and PNA2/SxK) promoted liposome mixing (Fig. 4). A combination of a negatively charged C-terminus and a positively charged side chain is beneficial for fusion. Amide modified glutamate at the C-terminal end (hybrids PNA1/SybEa and PNA2/SxEa) provides negative charge through the side chain. No mixing of liposomes was noticed when vesicles containing hybrid PNA1/SybEa were mixed with those carrying hybrid PNA2/SxEa (Fig. 5). A similar observation was made for hybrids (PNA1/SybE, PNA2/SxE) containing glutamate at the C-terminal end of the natural TMDs (Fig. 4). These results strongly suggest that the negatively charged side chain of glutamate inhibits the mixing of liposomes. Furthermore, the possibility of interactions between the C-termini of two TMDs through side chains was tested.44 Just a slightly lower tendency for fusion was recorded for vesicles containing hybrid PNA1/SybE (C-terminal glutamic acid) when mixed with vesicles bearing PNA2/SxK (C-terminal lysine) compared to hybrids carrying TMDs with natural C-termini (PNA1/Syb and PNA2/Sx, Fig. 4). A combination of the positive impact of lysine and the negative impact of glutamate at the C-termini seems to be effective. Thus, the influence of the possible electrostatic interactions between oppositely charged side chains on fusion was not observed. This could be due to the involvement of the positively charged side chain of lysine in a zwitterionic arrangement with the carboxylic acid C-terminus. The situation becomes clearer by using hybrids (PNA1/SybEa and PNA2/SxKa) carrying charges only on the side chain. Here a zwitterionic structure is avoided by using the neutral amide at the C-terminus. The efficiency of constructs PNA1/SybEa and PNA2/SxKa for inducing fusion was lower than for hybrids with an uncharged amide at the C-terminal end (PNA1/Syba and PNA2/Sxa) (Fig. 4) indicating a less pronounced impact of charged C-terminal amino acid side chains on the fusion process. Moreover, only a small mixing of inner leaflets was recorded for vesicles containing hybrids PNA1/SybEa and PNA2/SxKa (Fig. 5B). It can be argued that any interaction between side chains is only possible after further zippering into the TMD/linker regions because of the resulting spatial proximity of side chains. However, this is avoided by topology differences between PNA units and peptide TMDs of SNARE mimics.
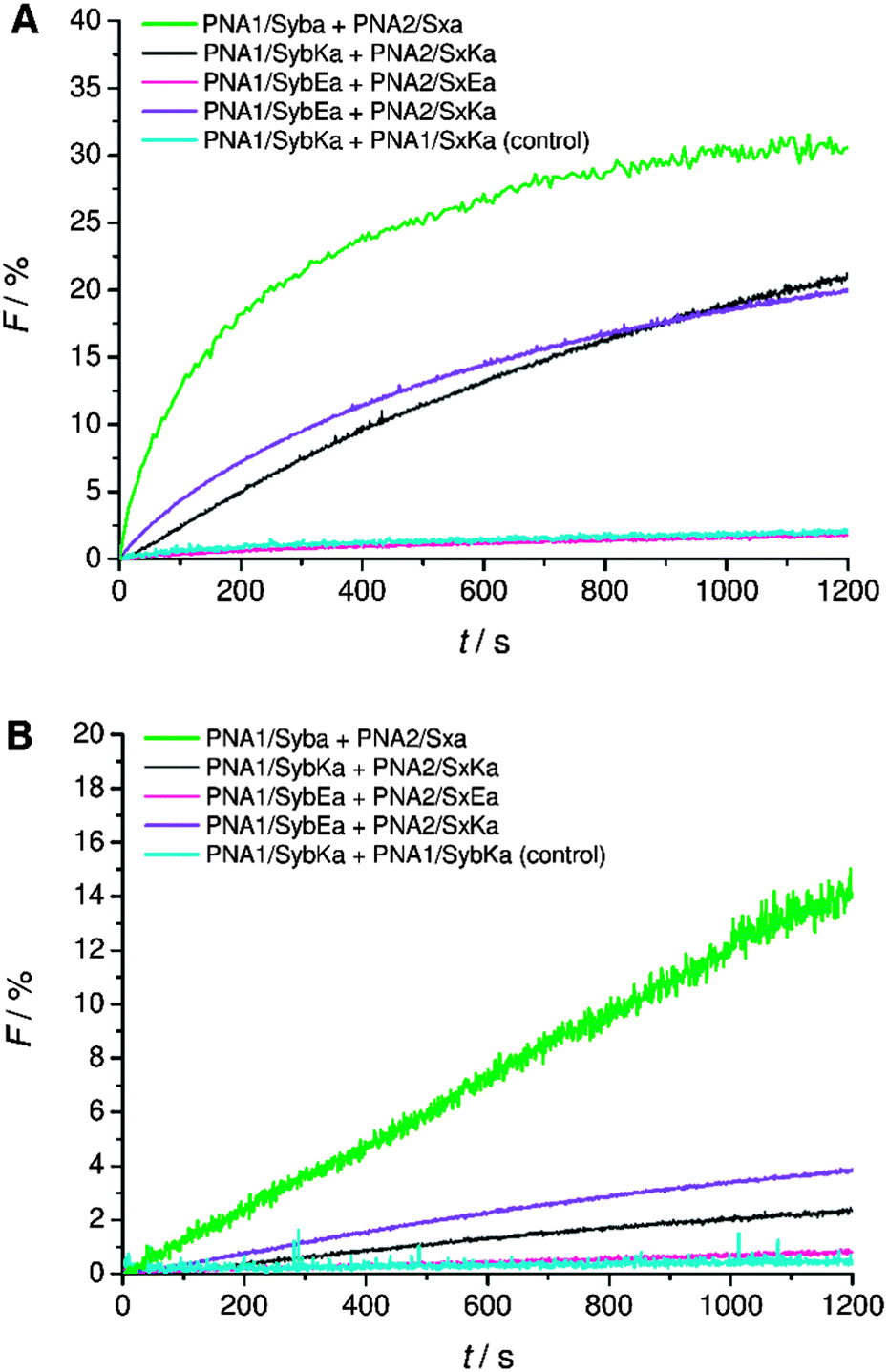 | ||
| Fig. 5 Lipid mixing assays for vesicles containing PNA/peptide hybrids with lysine or glutamate at the neutral amide C-terminus. (A) Total lipid mixing. (B) Inner leaflet mixing. For sequences of PNA/peptide chimera see Table 1. | ||
Fusion activity of hybrids containing shortened TMDs
Finally, the effect of reducing the length of TMDs on fusion was tested. For lipid-anchored SNAREs45 and the transmembrane deletion mutant of v-SNARE (Snc2p)7 it has been shown that the immersion depth of the hydrophobic anchor is an important determinant of the membrane fusion. The modified constructs (PNA1/Sybsh and PNA2/Sxsh) with shortened TMDs containing a neutral C-terminus were prepared. The lipid mixing efficiency with the shortened hybrid (PNA1/Sybsh or PNA2/Sxsh) was found to be significantly lower than that for the model system bearing full length TMDs (PNA1/Syba or PNA2/Sxa, Fig. 6). Interestingly, no fusion was noticed when both TMDs were shortened (Fig. 6). From lipid mixing experiments with these model systems we conclude on the contribution and recognition of the full-length TMDs in the fusion process as proposed for the native SNARE mediated fusion mechanism.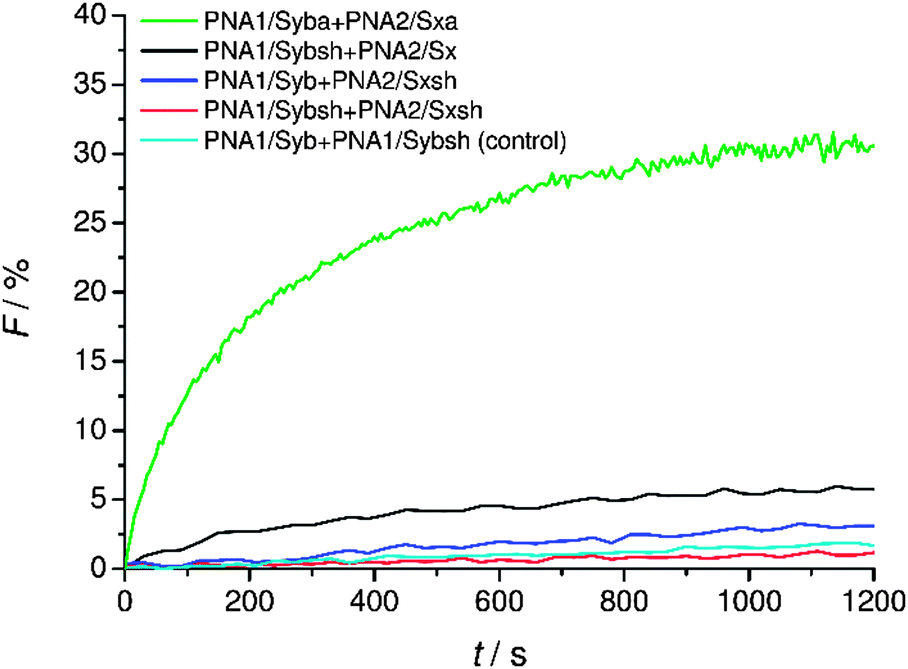 | ||
| Fig. 6 Lipid mixing assays for vesicles containing PNA/peptide hybrids with shortened TMDs. For sequences of PNA/peptide chimeras see Table 1. | ||
Conclusions
Novel simplified SNARE-mimicking systems consisting of TMD/linker segments of the natural membrane-bound SNARE proteins (syntaxin-1A and synaptobrevin 2) and peptide nucleic acid (PNA) recognition motifs were designed and synthesized. The prepared hybrids were incorporated into liposomes and tested for their capability to induce fusion. The role of charge at the C-terminal end of TMDs was studied by either replacing the natural carboxylic acid terminus with an uncharged amide and/or by introducing charged residues at the C-terminal amino acid side chain. Since PNA recognition motifs are topologically separated from the TMD/linker segments, the zippering proposed for the native SNARE recognition process is restricted to PNA double strand formation in the case of PNA/peptide hybrids. Here, dragging of the TMDs into the lipid bilayer is not possible as it is proposed for native SNARE proteins. Therefore, PNA/peptide SNARE analogs with a natural carboxylate C-terminus only lead to hemifusion. By changing the C-terminus from the negatively charged to the neutral stage, full fusion is recovered likely by facilitating penetration of TMDs into the lipid bilayer.Referred to the native SNARE protein fusion mechanism the results obtained by these model systems indicate the involvement of the C-terminus in late stages of the fusion process. An interdependence between the zippered formation of the SNARE recognition motif, further zippering up to linker regions, thereby dragging the TMD into the lipid bilayer, and participation of the C-terminal end of the TMDs in the reorientation of lipids in the fusion process of the inner leaflet is supported by the PNA/peptide SNARE analog model systems.
Acknowledgements
Generous support from the Deutsche Forschungsgemeinschaft DFG (SFB 803, project B5 ‘Construction of novel hybrid biooligomers as artificial SNAREs to dissect the mechanism of SNAREs in membrane docking and fusion’) is gratefully acknowledged. P. K. and S. G. acknowledge the Alexander von Humboldt foundation, Germany, for financial assistance.Notes and references
- A. T. Brunger, K. Weninger, M. Bowen and S. Chu, Annu. Rev. Biochem., 2009, 78, 903–928 CrossRef CAS PubMed.
- A. Stein, G. Weber, M. C. Wahl and R. Jahn, Nature, 2009, 460, 525–528 CrossRef CAS PubMed.
- W. Liu and V. Parpura, Sci. World J., 2010, 10, 1258–1268 CrossRef CAS PubMed.
- J. Rizo and J. Xu, Annu. Rev. Biophys., 2015, 44, 339–367 CrossRef CAS PubMed.
- D. Langosch, M. Hofmann and C. Ungermann, Cell. Mol. Life Sci., 2007, 64, 850–864 CrossRef CAS PubMed.
- J. Nikolaus and A. Herrmann, Biol. Chem., 2012, 393, 1231–1245 CrossRef CAS PubMed.
- Y. Xu, F. Zhang, Z. Su, J. A. McNew and Y.-K. Shin, Nat. Struct. Mol. Biol., 2005, 12, 417–422 Search PubMed.
- F. Deak, O.-H. Shin, E. T. Kavalali and T. C. Sudhof, J. Neurosci., 2006, 26, 6668–6676 CrossRef CAS PubMed.
- E. Fdez, M. Martinez-Salvador, M. Beard, P. Woodman and S. Hilfiker, J. Cell Sci., 2010, 123, 2473–2480 CrossRef CAS PubMed.
- Q. Fang and M. Lindau, Physiology, 2014, 29, 278–285 CrossRef CAS PubMed.
- H. J. Risselada, C. Kutzner and H. Grubmüller, ChemBioChem, 2011, 12, 1049–1055 CrossRef CAS PubMed.
- C.-W. Chang and M. B. Jackson, Biophys. J., 2015, 109, 76–84 CrossRef CAS PubMed.
- L. van Keimpema and T. Kroon, J. Neurosci., 2015, 35, 11459–11461 CrossRef CAS PubMed.
- A. N. Ngatchoua, K. Kislera, Q. Fang, A. M. Walter, Y. Zhao, D. Bruns, J. B. Sørensen and M. Lindau, PNAS, 2010, 107, 18463–18468 CrossRef PubMed.
- H. R. Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- M. Ma and D. Bong, Acc. Chem. Res., 2013, 46, 2988–2997 CrossRef CAS PubMed.
- P. Kumar, S. Guha and U. Diederichsen, J. Pept. Sci., 2015, 21, 621–629 CrossRef CAS PubMed.
- K. Meyenberg, A. S. Lygina, G. van den Bogaart, R. Jahn and U. Diederichsen, Chem. Commun., 2011, 47, 9405 RSC.
- H. Robson Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef CAS PubMed.
- T. Zheng, J. Voskuhl, F. Versluis, H. R. Zope, I. Tomatsu, H. R. Marsden and A. Kros, Chem. Commun., 2013, 49, 3649 RSC.
- H. Robson Marsden, A. V. Korobko, T. Zheng, J. Voskuhl and A. Kros, Biomater. Sci., 2013, 1, 1046 RSC.
- F. Versluis, J. Voskuhl, B. van Kolck, H. Zope, M. Bremmer, T. Albregtse and A. Kros, J. Am. Chem. Soc., 2013, 135, 8057–8062 CrossRef CAS PubMed.
- F. Versluis, J. Voskuhl, J. Vos, H. Friedrich, B. J. Ravoo, P. H. H. Bomans, M. C. A. Stuart, N. A. J. M. Sommerdijk and A. Kros, Soft Matter, 2014, 10, 9746–9751 RSC.
- A. Kashiwada, K. Matsuda, T. Mizuno and T. Tanaka, Chem. – Eur. J., 2008, 14, 7343–7350 CrossRef CAS PubMed.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584–9585 CrossRef CAS PubMed.
- G. Stengel, L. Simonsson, R. A. Campbell and F. Höök, J. Phys. Chem. B, 2008, 112, 8264–8274 CrossRef CAS PubMed.
- L. Simonsson, P. Jönsson, G. Stengel and F. Höök, ChemPhysChem, 2010, 11, 1011–1017 CrossRef CAS PubMed.
- Y.-H. M. Chan, B. van Lengerich and S. G. Boxer, Biointerphases, 2008, 3, FA17 CrossRef CAS PubMed.
- B. van Lengerich, R. J. Rawle, P. M. Bendix and S. G. Boxer, Biophys. J., 2013, 105, 409–419 CrossRef CAS PubMed.
- A. S. Lygina, K. Meyenberg, R. Jahn and U. Diederichsen, Angew. Chem., Int. Ed., 2011, 50, 8597–8601 CrossRef CAS PubMed.
- M. Sadek, D. Berndt, D. Milovanovic, R. Jahn and U. Diederichsen, ChemBioChem, 2016, 17, 479–485 CrossRef CAS PubMed.
- Y. Gong, Y. Luo and D. Bong, J. Am. Chem. Soc., 2006, 128, 14430–14431 CrossRef CAS PubMed.
- Y. Gong, M. Ma, Y. Luo and D. Bong, J. Am. Chem. Soc., 2008, 130, 6196–6205 CrossRef CAS PubMed.
- M. Ma, A. Paredes and D. Bong, J. Am. Chem. Soc., 2008, 130, 14456–14458 CrossRef CAS PubMed.
- M. Ma, Y. Gong and D. Bong, J. Am. Chem. Soc., 2009, 131, 16919–16926 CrossRef CAS PubMed.
- A. Kashiwada, M. Tsuboi and K. Matsuda, Chem. Commun., 2009, 695–697 RSC.
- A. Kashiwada, M. Tsuboi, T. Mizuno, T. Nagasaki and K. Matsuda, Soft Matter, 2009, 5, 4719–4725 RSC.
- A. Kashiwada, I. Yamane, M. Tsuboi, S. Ando and K. Matsuda, Langmuir, 2012, 28, 2299–2305 CrossRef CAS PubMed.
- S. Guha, J. Graf, B. Göricke and U. Diederichsen, J. Pept. Sci., 2013, 19, 415–422 CrossRef CAS PubMed.
- R. C. MacDonald, R. I. MacDonald, B. P. M. Menco, K. Takeshita, N. K. Subbarao and L. Hu, Biochim. Biophys. Acta, Biomembr., 1991, 1061, 297–303 CrossRef CAS.
- J. C. McIntyre and R. G. Sleight, Biochemistry, 1991, 30, 11819–11827 CrossRef CAS PubMed.
- D. K. Struck, D. Hoekstra and R. E. Pagano, Biochemistry, 1981, 20, 4093–4099 CrossRef CAS PubMed.
- S. Pensato, M. Renda, F. Leccia, M. Saviano, L. D. D'Andrea, C. Pedone, P. V. Pedone and A. Romanelli, Biopolymers, 2010, 93, 434–441 CrossRef CAS PubMed.
- H. J. Risselada and H. Grubmüller, Curr. Opin. Struct. Biol., 2012, 22, 187–196 CrossRef CAS PubMed.
- J. A. McNew, T. Weber, F. Parlati, R. J. Johnston, T. J. Melia, T. H. Söllner and J. E. Rothman, J. Cell Biol., 2000, 150, 105–118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00294c |
| This journal is © The Royal Society of Chemistry 2016 |