
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Delivery of siRNA using ternary complexes containing branched cationic peptides: the role of peptide sequence, branching and targeting†
Laila
Kudsiova
a,
Katharina
Welser‡
b,
Frederick
Campbell§
b,
Atefeh
Mohammadi
b,
Natalie
Dawson
b,
Lili
Cui
a,
Helen C.
Hailes
b,
M. Jayne
Lawrence
a and
Alethea B.
Tabor
*b
aInstitute of Pharmaceutical Science, King's College London, Franklin–Wilkins Building, 150 Stamford Street, Waterloo Campus, London SE1 9NH, UK
bDepartment of Chemistry, University College London, Christopher Ingold Laboratories, 20, Gordon Street, London WC1H 0AJ, UK. E-mail: a.b.tabor@ucl.ac.uk; Fax: +44(0) 20 7679 7463; Tel: +44(0) 20 7679 4695
First published on 22nd January 2016
Abstract
Ternary nanocomplexes, composed of bifunctional cationic peptides, lipids and siRNA, as delivery vehicles for siRNA have been investigated. The study is the first to determine the optimal sequence and architecture of the bifunctional cationic peptide used for siRNA packaging and delivery using lipopolyplexes. Specifically three series of cationic peptides of differing sequence, degrees of branching and cell-targeting sequences were co-formulated with siRNA and vesicles prepared from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the cationic lipid DOTMA and the helper lipid, DOPE. The level of siRNA knockdown achieved in the human alveolar cell line, A549-luc cells, in both reduced serum and in serum supplemented media was evaluated, and the results correlated to the nanocomplex structure (established using a range of physico-chemical tools, namely small angle neutron scattering, transmission electron microscopy, dynamic light scattering and zeta potential measurement); the conformational properties of each component (circular dichroism); the degree of protection of the siRNA in the lipopolyplex (using gel shift assays) and to the cellular uptake, localisation and toxicity of the nanocomplexes (confocal microscopy). Although the size, charge, structure and stability of the various lipopolyplexes were broadly similar, it was clear that lipopolyplexes formulated from branched peptides containing His-Lys sequences perform best as siRNA delivery agents in serum, with protection of the siRNA in serum balanced against efficient release of the siRNA into the cytoplasm of the cell.
:1 molar ratio of the cationic lipid DOTMA and the helper lipid, DOPE. The level of siRNA knockdown achieved in the human alveolar cell line, A549-luc cells, in both reduced serum and in serum supplemented media was evaluated, and the results correlated to the nanocomplex structure (established using a range of physico-chemical tools, namely small angle neutron scattering, transmission electron microscopy, dynamic light scattering and zeta potential measurement); the conformational properties of each component (circular dichroism); the degree of protection of the siRNA in the lipopolyplex (using gel shift assays) and to the cellular uptake, localisation and toxicity of the nanocomplexes (confocal microscopy). Although the size, charge, structure and stability of the various lipopolyplexes were broadly similar, it was clear that lipopolyplexes formulated from branched peptides containing His-Lys sequences perform best as siRNA delivery agents in serum, with protection of the siRNA in serum balanced against efficient release of the siRNA into the cytoplasm of the cell.
Introduction
Small interfering RNA (siRNA) can be used to knock down a wide range of protein targets in viral and cancer cells, raising the possibility that siRNA can be used as a therapy for the treatment of cancers and viral infections. In order for siRNA to be successfully administered in vivo for therapeutic purposes, it must be packaged into nanoparticles in such a way that it is not only protected from enzymatic degradation and aggregation by serum proteins but also targeted to the cell type of interest. Once the target cell has been reached, the siRNA-carrying nanoparticle must then be efficiently internalised, generally via endocytosis.1,2 Although many of the strategies that have been used for packaging and cellular delivery of DNA3 have the potential to be adapted for the delivery of siRNA, siRNA differs from DNA in several crucial respects.4 Firstly, the 21 nucleotide base pairs of siRNA are significantly smaller in size than the plasmid DNA usually used for gene delivery, so it is likely that siRNA containing nanoparticle formulations will contain more than one siRNA molecule. In addition, whereas plasmid DNA can be highly condensed by complexation with cationic lipids or polymers to form lipoplexes or polyplexes, siRNA (21 base pairs) is unlikely to be significantly condensed, preferring instead to behave as a rigid rod. Finally, although both nanoparticles carrying siRNA and plasmid DNA must both escape from the endosome, plasmid DNA has to reach the nucleus in order for gene expression to occur, whereas siRNA has the advantage of only having to be released into the cytoplasm where it will be loaded onto the multi-protein RNA-inducing silencing complex (RISC) thereby initiating the process of sequence-specific degradation of the mRNA targets.4Cationic polymers, which can complex negatively charged siRNA into nanosized particles, have been successfully used for siRNA delivery in vitro.1–4 Nanoparticles formulated with cationic dendrimers such as poly(amido amine) (PAMAM) have been particularly effective in delivering siRNA for gene silencing. However, very few siRNA delivery systems are currently in pre-clinical studies or in clinical trials.2 One major drawback for clinical applications of cationic dendrimer-containing formulations is their toxicity. Cationic dendrimers are cytotoxic due to their interactions with biological membranes as well as their induced haemolytic and haematological toxicity.5 Nanoparticles formulated from siRNA and cationic peptides (either linear or branched) may therefore be an attractive alternative for siRNA delivery, because of their reduced toxicity and their potential biodegradability.
Two approaches to the design of cationic peptide sequences for the complexation and delivery of siRNA to cells have been reported, exploiting either Arg-rich, or mixed His/Lys, sequences of cationic residues. The first approach was based on the observation that cell-penetrating peptides (CPP) are frequently Arg-rich sequences. It is believed that nanoparticles incorporating CPP sequences can translocate directly into cells through the cell membrane, or enter cells via endocytosis.6 Indeed non-covalent nanocomplexes of siRNA with7 (L-Arg)9 or with8 (L-Arg)8 have been successfully used to deliver siRNA into cells, with the siRNA localised at the perinuclear region after release from the complex. After uptake of these nanocomplexes into the cells via endocytosis, the CPP may also disrupt the endosomal membrane, allowing the siRNA to be released into the cytoplasm. Kumar et al. used a chimaeric peptide, consisting of a 29-residue sequence of the rabies virus glycoprotein (RVG) linked to a (D-Arg)9 sequence, to bind and deliver siRNA to neuronal cells in vitro.9 The same complex of chimaeric peptide and siRNA was also used to successfully deliver siRNA to macrophages,10 while a CD7-specific single chain antibody conjugated to (D-Arg)9 was used to selectively deliver siRNA to T cells.11 In these formulations, it was believed that as the unnatural D-Arg residues are not recognised by enzymes or receptors, these cationic peptides cannot act as CPP and are resistant to serum proteases in vivo.11
The second approach was based on the hypothesis that nanocomplexes containing His-rich peptide sequences are able to act as a “proton sponge” and thus to neutralise the acidity of the interior of the endosome. This is believed to lead to swelling and rupture of the endosome with subsequent release of the nanocomplexes into the cytoplasm.12 Mixson and co-workers studied highly branched peptide carriers, with different repeating sequences of His and Lys residues in the branches, and targeted with an integrin-binding RGD sequence at the C-terminus, as reagents for the delivery of siRNA.13 Significantly, the level of gene knockdown achieved with the best peptide carrier was comparable to or better than the ‘gold standards’, Lipofectamine 2000 or Oligofectamine but without their associated toxicity.13 It was also found that branched peptide carriers based on sequences such as (HHHK)4 that resulted in the best level of siRNA delivery, were significantly different from those peptide sequences previously optimised by this group for successful plasmid DNA delivery. These siRNA/(HHHK)4 peptide carrier nanocomplexes were also shown to be effective for systemic delivery in vivo.14 PEG-modified versions of these nanocomplexes with cRGD targeted ligands showed good pharmacokinetics and tumor-selective siRNA delivery in vivo.15,16 Linear, amphipathic histidine-rich peptide sequences, which also contain Lys, Ala and Leu residues, have also been reported to be effective for siRNA delivery,17 and disulfide-linked polymers of Cys-His-Lys peptides18 have been used to deliver a broad range of nucleic acids, including siRNA.19
The gene delivery properties of ternary nanocomplexes, known as lipopolyplexes, composed of lipid, peptide and DNA, have recently been investigated, using: a bifunctional peptide with a (Lys)16 domain to bind and condense DNA and a cyclic peptide targeting domain that partially protrudes from the nanocomplex and mediates cell-specific receptor-mediated endocytosis of the vector;20 and a 1:1 mixture of a cationic lipid, structurally related to N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), and the neutral lipid, dioleylphosphatidylethanolamine (DOPE) (which can enhance liposome fusion with the endosomal membrane, leading to endosomal escape21). This vector has been optimised for a number of applications, including the development of a peptide targeting sequence for gene delivery to respiratory cells.22 Shielding lipids containing short ethylene glycol (EG) sequences,23–25 and peptides that are cleaved within the endosome25 to give vectors that are stable in the systemic circulation but disassemble once internalized,25,26 have been incorporated. The effect of lipid chain geometry on the structure and transfection properties of lipopolyplexes and lipoplexes (binary complexes of lipid and DNA) has also been investigated.27,28 Most recently, the effect of cationic residue sequence and branching on the level of plasmid DNA deliver achieved with lipopolyplexes has been studied, with branched Arg-rich peptides and linear Lys-rich peptides being most effective.29
In this work, the effects of varying the sequence of cationic residues and the degree of branching on the packaging and delivery of siRNA in DOTMA:DOPE:peptide:siRNA lipopolyplexes (LPRs) have been studied. In light of the various studies on polyplex delivery of siRNA reported by other groups, we have for the first time carried out a detailed comparison of whether Lys, His, L-Arg, D-Arg or mixed His/Lys sequences would be most effective in mediating siRNA delivery, in both peptide:siRNA polyplexes (PR) and LPR formulations. We have also investigated the effect of peptide sequence and structure on the overall structure, siRNA protection, and cytoplasmic release of the nanocomplex, and are able to relate this to the effectiveness of the LPRs as siRNA delivery vehicles.
Results
Peptide design
A comprehensive set of linear (B0), singly (B1) and doubly-branched polycationic peptides (B2) has been studied to explore the effect of both cationic sequence and peptide structure on the complexation of siRNA and the gene silencing efficiency of LPRs. To test the LPRs in a human alveolar cell line, the peptide sequences included a targeting sequence, CYGLPHKFC (sequence [Y]) which was selected by phage display to bind human airway epithelial cells and mediate good DNA transfection efficiency.22 The targeting [Y] and polycationic sequences are linked by either a non-cleavable spacer sequence (GAGA (L2)) or a cleavable sequence (RVRRGA (L1)). L1 is recognised by the endosomal enzyme furin, and peptides containing this sequence have been previously demonstrated to enhance DNA transfection by LPD complexes.25,26Three groups of peptides were synthesised for formulation and testing. In Group 1 we aimed to compare the effects of Lys, Arg, D-Arg and His residues in peptides of the same length and degree of branching (Fig. 1). In order to determine whether or not the targeting sequence plays a major role in cellular uptake a control, sK6B1-L1-[Y], with a scrambled version of the [Y] sequence, was prepared. The Group 2 peptides are designed to compare the effects of linear, singly and doubly branched peptides on LPR knockdown, and a second control peptide, K12, (devoid of a targeting sequence) has also been included to investigate the role of the targeting sequence. Four peptides (Group 3) based on the alternating HK sequences recently reported by Mixson and co-workers,13 were also studied. In this paper, repeating HHHK sequences in 4-branched and 8-branched peptides were found to be particularly effective in siRNA delivery, and therefore the singly-branched peptides (H3K)4B1-L1-[Y] and (KH3)4B1-L1-[Y] were prepared. In contrast, Mixson and co-workers reported that HHK sequences were less effective for siRNA delivery but gave better pDNA transfection, and the peptide (HHK)4B1-L1-[Y] was therefore also studied. As a further comparison, a fourth peptide based on a repeating HHHHK sequence, (H4K)3B1-L1-[Y] was synthesised. All LPRs were prepared using vesicles comprising of a 1:1 molar ratio of cationic lipid, DOTMA, and neutral helper lipid, DOPE, (Lipofectin®). For the purposes of comparison the biophysical and transfection properties of PRs, prepared by complexing the various peptides with siRNA, were investigated.
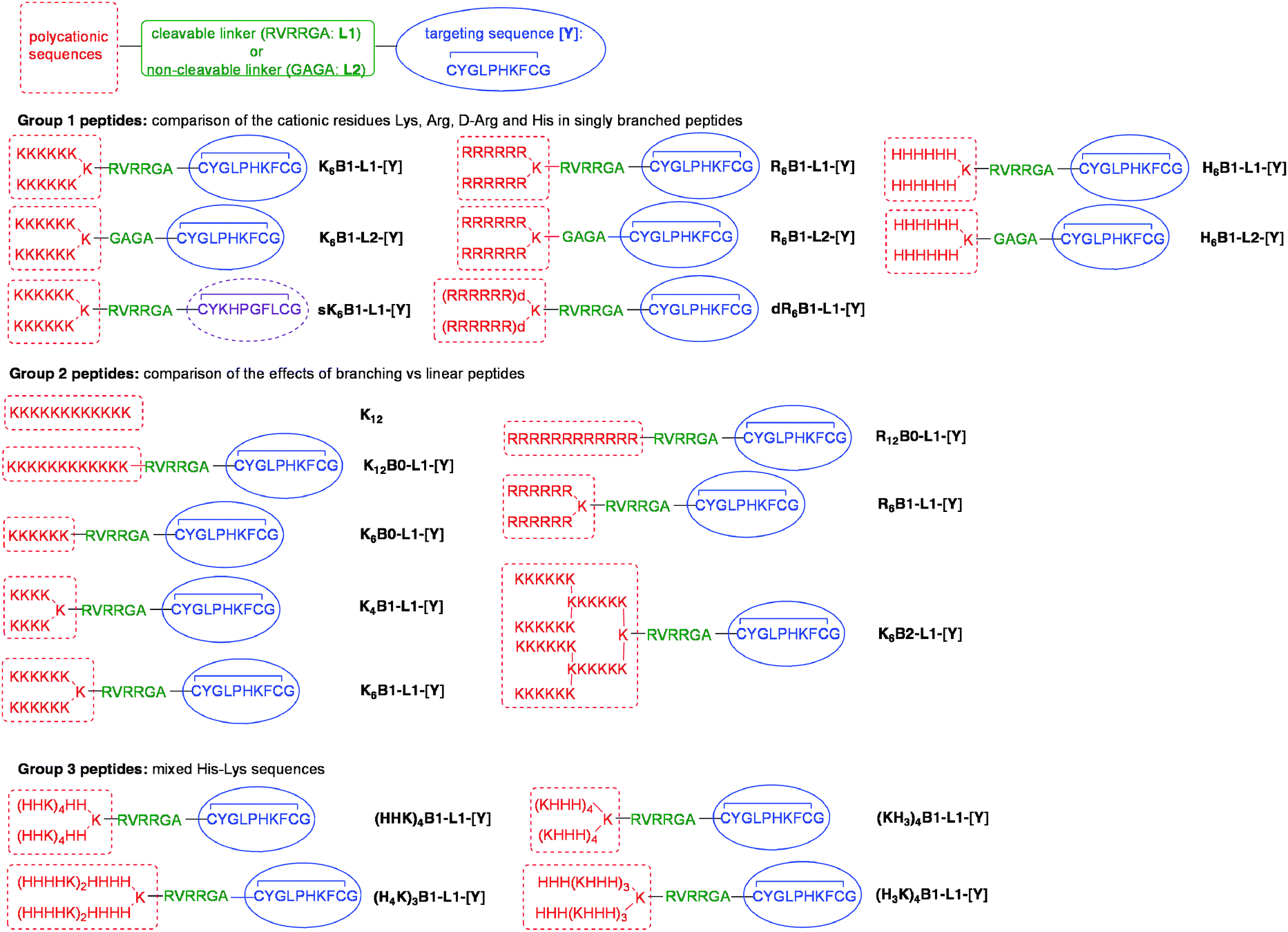 | ||
| Fig. 1 Structures of Group 1, Group 2 and Group 3 peptides. | ||
In vitro knockdown experiments
Knockdown experiments with LPRs, made using the various peptides at charge ratios of 0.5:6:1 or 0.5:12:1 and prepared either in water and diluted 1 in 4 in OptiMEM (water/OptiMEM) or OptiMEM diluted 1 in 4 with serum-containing growth medium (OptiMEM/Medium), were performed in a stably transfected A549-Luc cell line (Fig. 2). A comparable level of knockdown to the (water/OptiMEM) experiment was achieved when LPRs were prepared fully in OptiMem, and little knockdown was achieved when LPRs were prepared in water and diluted 1 in 4 with serum-containing growth medium (water/Medium) (Fig. S1, ESI†). For comparison, the knockdowns obtained with PRs when formulated fully in OptiMEM are given in Fig. S2, ESI.†
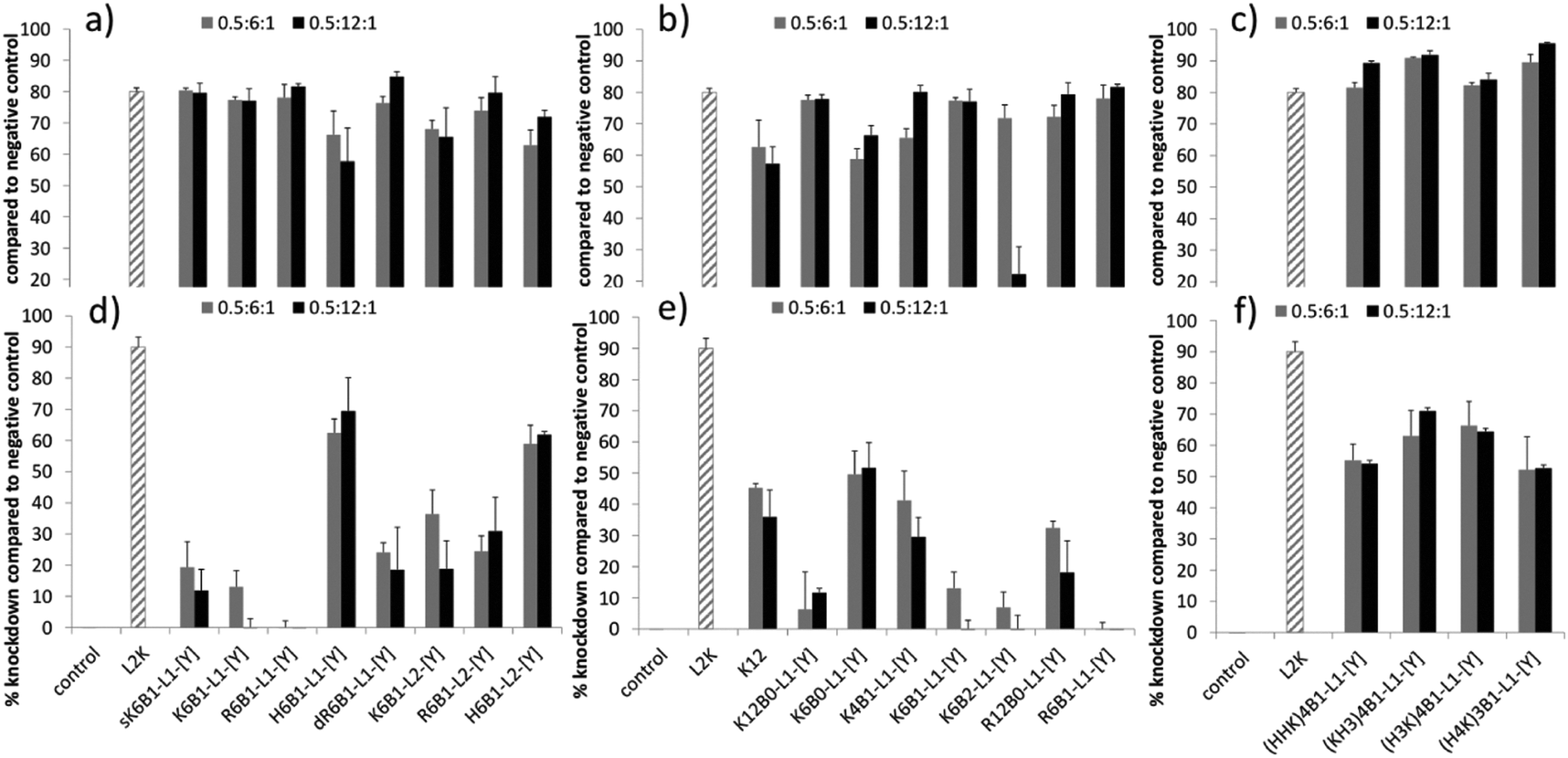 | ||
| Fig. 2 Percentage knockdown (calculated as % using luminescence produced by each complex when formulated with negative control siRNA and normalised according to the protein content) of LPRs at 0.5:6:1 (grey bars) or 0.5:12:1 (black bars) charge ratios, prepared using (a and d) Group 1, (b and e) Group 2, and (c and f) Group 3 peptides. LPRs were prepared either in (a–c) water then diluted in OptiMEM (water content = 12.5% v/v); or in (d–f) in OptiMEM then diluted in serum-containing growth media (OptiMEM content = 12.5% v/v). siRNA alone and L2K:siRNA at 5:1 weight ratio were used as controls. siRNA concentration was 50 nM per well. | ||
High levels of knockdown were achieved in the water/OptiMEM preparation (Fig. 2a–c), and when LPRs were prepared fully in OptiMEM (Fig. S1a–c, ESI†), for the majority of peptide sequences. Poor or negligible knockdown was obtained with the PRs prepared under the same conditions, clearly showing the benefit of formulating LPRs rather than PRs.
The greatest level of knockdown, in fact greater than that exhibited by the positive control, L2K, was observed with LPRs prepared using Group 3 peptides (Fig. 2c). These Group 3 peptides included the HHHK sequences identified by Mixson and co-workers13 as highly effective for siRNA delivery (peptides (KH3)4B1-L1-[Y] and (H3K)4B1-L1-[Y]) as well as two other alternating HK sequence peptides. Furthermore, with the exception of doubly branched K6B2-L1-[Y] peptide (and K6B1-L1-[Y] and K12B0-L1-Y when prepared fully in OptiMEM) increasing the charge ratio of the peptide in the LPR from 0.5:6:1 to 0.5:12:1 in the LPR formulations prepared in OptiMEM or water/OptiMEM did not significantly influence knockdown efficiency. This result is in contrast to LPD formulations, where DNA transfection increased upon increasing charge ratio of the peptide from 0.5:6:1 to 0.5:12:1.29
LPRs formulated in water/OptiMEM using the Group 1 peptides did not show major differences in knockdown efficiency (Fig. 2a). Furthermore, no difference in knockdown efficiency was seen when comparing LPRs prepared using peptides containing the enzymatically cleavable RVRRGA (L1) linker to those possessing the non-cleavable GAGA (L2) linker. It is also notable that the control peptide, sK6B1-L1-[Y], with a scrambled version of the [Y] sequence, gave similarly good knockdown results, suggesting that under these conditions receptor binding and uptake of the LPR complexes is also not the crucial step for knockdown. Finally, whilst the His-containing peptides H6B1-L1-[Y] and H6B1-L2-[Y] should have a lower charge at pH 7 when compared to the other sequences, this also does not have any influence on knockdown efficiencies.
LPRs containing the singly branched peptides in Group 2, K4B1-L1-[Y] and K6B1-L1-[Y], were slightly more effective knockdown agents than the LPRs with the linear K6B0-L1-[Y] in water/OptiMEM. However, the extent of peptide branching did not play a significant role in knockdown as further branching of the peptide (K6B2-L1-[Y]) did not improve knockdown efficiency (Fig. 2b). A further control peptide, K16, lacking any targeting sequence, also gave equally good knockdown results.
The effect of the presence of serum on the knockdown efficiency of the LPRs was also examined. It is of particular note that LPRs prepared in water and diluted in media containing 10% FBS showed very poor knockdown efficiencies, as did indeed the positive control when prepared under the same conditions (Fig. S1, ESI†).
This poor knockdown is attributed to the presence of negatively charged serum proteins, present in the growth media, neutralizing the cationic charge of the LPRs, most probably leading to their aggregation and thereby reducing their interaction with the negatively charged cell. In contrast, when the LPRs were prepared in OptiMEM and diluted in growth media (Fig. 2d–f), the knockdown activity of a number of the complexes, including those containing L2K was, at least partially, maintained.
The lower knockdown efficiencies seen in the presence of serum revealed differences in the extent of knockdown achieved using the various peptides, making certain interesting trends clear. Firstly, as with the LPRs prepared in water and then OptiMEM, and indeed fully in OptiMEM, no difference was seen in increasing the charge ratio of the peptide in the LPR from 0.5:6:1 to 0.5:12:1. Secondly, in the presence of serum the greatest knockdown was achieved using peptides containing histidine – in particular the results obtained using the Group 3 peptides which achieved knockdowns of up to ∼70%, with LPRs containing (KH3)4B1-L1-[Y] and (H3K)4B1-L1-[Y] being most effective. Similarly, within the Group 1 peptides (Fig. 2d), the complexes prepared using the His-containing peptides, H6B1-L1-[Y] and H6B1-L2-[Y] produced the greatest knockdown while LPRs prepared using K6B1-L1-[Y] and R6B1-L1-[Y] showed no activity whatsoever.
Again, with the exception of LPRs containing the peptides, H6B1-L1-[Y] and H6B1-L2-[Y], LPRs prepared (OptiMEM/Medium) using peptides containing the non-cleavable GAGA (L2) linker showed significantly better activity in the presence of serum than those containing the cationic Arg-rich RVRRGA (L1) linker. LPRs containing dR6B1-L1-[Y] were more effective than those containing R6B1-L1-[Y] (Fig. 2d). In addition, as can be seen from a consideration of the results obtained using the Group 2 peptides (Fig. 2e), increasing the degree of branching and number of cationic peptide residues in the Lys and Arg was detrimental to knockdown efficiency for LPRs. For example, the best knockdown was produced by LPRs containing the linear peptide, K6B0-L1-[Y], followed by the Lys containing singly branched peptide K4B1-L1-[Y], next K6B1-L1-[Y] and finally the doubly branched K6B2-L1-[Y]. Finally, complexes prepared using the linear Lys containing peptide, K12B0-L1-[Y], were less effective than those using K6B0-L1-[Y], or a K12 peptide devoid of targeting or linker sequences.
The toxicity exerted by the LPRs was measured indirectly using the protein assay (Fig. S3, ESI†). Most of the LPRs tested, regardless of their method of preparation, showed over 80% cell viability compared to untreated controls. In contrast however, the L2K containing lipoplexes exerted the lowest cell viability at ∼70%. Interestingly, LPRs prepared in the presence of serum-containing media showed higher viability than those prepared fully in OptiMEM or water/OptiMEM.
Dynamic light scattering and zeta potential
The apparent hydrodynamic size and zeta potential of the LPRs and PRs are shown in the ESI† (Fig. S4 and S5, respectively). The LPRs were small, being typically in the size range 45–75 nm and were positively charged with zeta potentials of between 12–50 mV, with the majority of LPRs exhibiting a zeta potential of greater than 30 mV. No correlation was observed between LPR size, zeta potential and the peptide ratio and structure of the peptide used to prepare the LPR, although there was a clear tendency for the LPRs containing His or His-rich peptides to exhibit lower zeta potentials.In contrast, the apparent hydrodynamic diameters of the PRs were much larger at up to 1500 nm and more polydisperse suggesting the presence of much more highly aggregated particles. Although the zeta potentials of the PRs were positive, typically in the range 5–38 mV (with the exception of PRs containing (H4K)3B1-L1-[Y] which were negative, probably due to the lower charge on the peptide at neutral pH) they were noticeably lower than the corresponding LPRs. This suggests that the cationic lipid plays a significant role in the structure of the complexes both in terms of complex size and charge.
Transmission electron microscopy
In agreement with dynamic light scattering measurements, transmission electron microscopy (TEM) imaging of selected LPR and PR formulations showed that the LPRs were small, homogeneous electron dense, spherical particles with no internal structure visible (Fig. 3a–e) while the PRs were much more heterogeneous, demonstrating extensive aggregation (Fig. S6 ESI†). By way of comparison, LR complexes prepared from DOTMA:DOPE:siRNA at 0.5:1 charge ratio showed particles containing a lamellar structure (indicated by the arrow in Fig. 3f) typically seen in DNA lipoplexes.26 This lamellar structure was not observed in any of the LPR formulations, suggesting that the structure of LPRs is indeed very different from that of binary complexes composed of either PR or LR components alone.
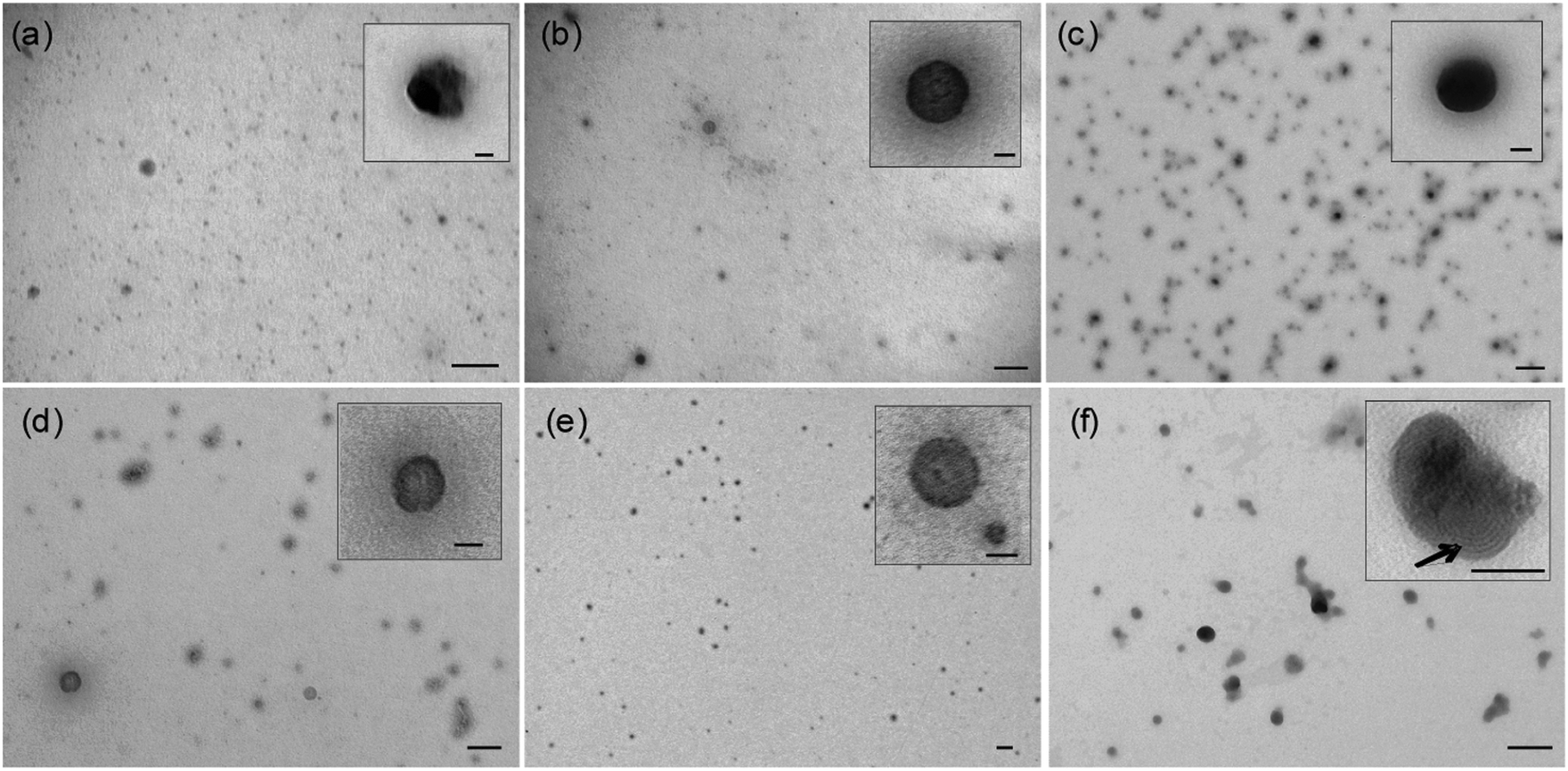 | ||
| Fig. 3 Transmission electron microscopy of LPRs prepared using DOTMA:DOPE with the peptides (a) K6B1-L1-[Y], (b) R6B1-L1-[Y], (c) H6B1-L1-[Y], (d) K12B0-L1-[Y] and (e) K6B2-L1-[Y] at a 0.5:6:1 LPR charge ratio and (f) DOTMA:DOPE:siRNA LR complexes at a 0.5:1 charge ratio. The siRNA concentration in all samples was 0.05 mg mL−1. Size bar represents 200 nm on original images and 50 nm on inserts. | ||
Gel retardation assays and PicoGreen fluorescence studies in the absence of serum
The gel electrophoresis studies indicated that the peptides, regardless of whether formulated as a LPR or PR, complexed siRNA to a significant degree at both of the charges ratios studied (Fig. 4, Lane 1). The only exceptions were the complexes containing K12B0-L1-[Y], R12B0-L1-[Y] and the Group 3 peptides (HHK)4B1-L1-[Y], (KH3)4B1-L1-[Y] and (H3K)4B1-L1-[Y], where the siRNA was more completely complexed at the higher peptide charge ratio of 0.5:12:1 for LPRs and 12:1 for PRs. In addition, incomplete siRNA complexation was observed with the histidine containing peptides, H6B1-L1-[Y], H6B1-L2-[Y] and (H4K)3B1-L1-[Y] at both peptide charge ratios studied. Treatment of the LPRs and PRs with pAsp resulted in the expected dissociation of siRNA from the complexes (Fig. 4, Lane 2). Significantly LPRs and PRs treated with RNAse A followed by RNAse inhibitor and pAsp (Fig. 4, Lane 3) in order to assess the stability of the complexed siRNA to enzymatic degradation, suggested that all the complexes significantly protected siRNA from RNAse (Fig. 4, Lane 3), while naked siRNA was completely degraded by RNAse A under the same circumstances (Fig. 4, Lane B).
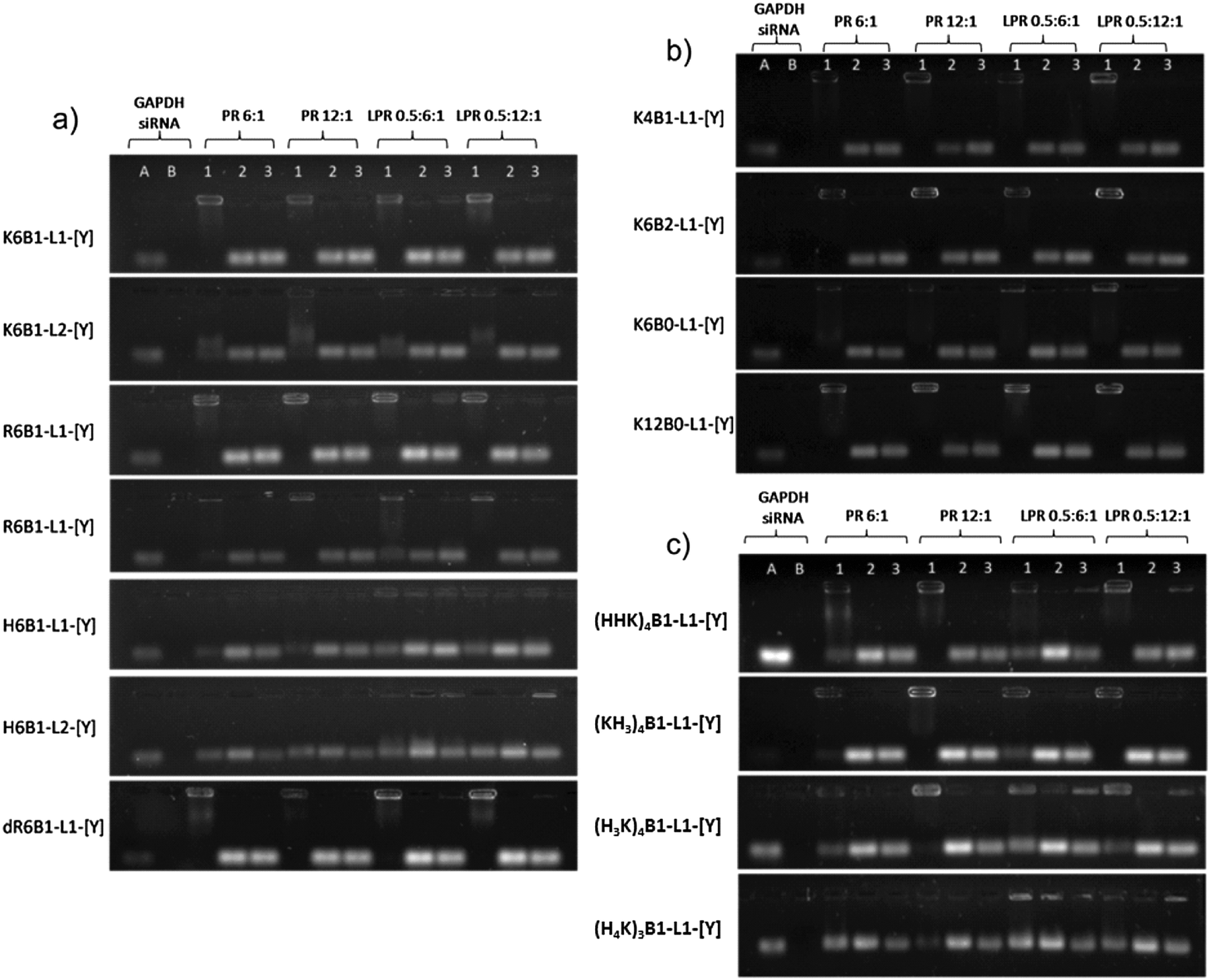 | ||
| Fig. 4 Gel electrophoresis of LPR and PR complexes containing (a) Group 1, (b) Group 2 and (c) Group 3 peptides. LPR complexes were prepared with a charge ratio of lipid:peptide:siRNA of 0.5:6:1 and 0.5:12:1 while PR complexes were prepared with a charge ratio of peptide:siRNA of 6:1 and 12:1. The effect of the peptide component on siRNA complexation, protection from degradation by RNAse A and on siRNA release is labelled by Lanes 1, 2, and 3 respectively. Lane 1: untreated complexes. Lane 2: complexes treated with RNAse A followed by pAsp. Lane 3: complexes treated with pAsp only. Lane A: siRNA control. Lane B: siRNA treated with RNAse A. | ||
PicoGreen fluorescence studies were performed to further study the effectiveness of the packaging of siRNA within the LPR complexes (Fig. 5). Accessibility of PicoGreen to siRNA results in a reduction of its fluorescence intensity. In agreement with the gel retardation studies, a drastic reduction in fluorescence intensity was seen upon increasing the peptide content in the LPR complexes, reaching a plateau (suggesting that maximum complexation had been reached for that complex) at LPR charge ratios of between 0.5:2:1 and 0.5:18:1, the exact ratio depending upon the peptide used. For most complexes the plateau (or maximum complexation) was reached at LPR charge ratios of 0.5:4:1, the exception being the complexes prepared using the Group 3 peptides, and in particular (H3K)4B1-L1-[Y], where a plateau was only reached at LPR charge ratios of 0.5:12:1. In addition, the extent of complexation, indicated by the fluorescence intensity of the plateau region, was strongly influenced by the nature of the peptide used to prepare the complex. For example, only a small reduction in fluorescence intensity of 10 and 40% of the intensity of free siRNA was observed in LPRs formulated with the His-only containing peptides H6B1-L1-[Y] and H6B1-L2-[Y], respectively (Fig. 5a). This contrasted with a ∼70% reduction in fluorescence seen with complexes containing Group 3 peptides comprising both His and Lys residues. The maximum reduction in fluorescence intensity of 95% was achieved with complexes prepared using the Arg and Lys rich peptides, namely R6B1-L1-[Y], R6B1-L2-[Y], dR6B1-L1-[Y] and K12B0-L1-[Y] (Fig. 5a and c).
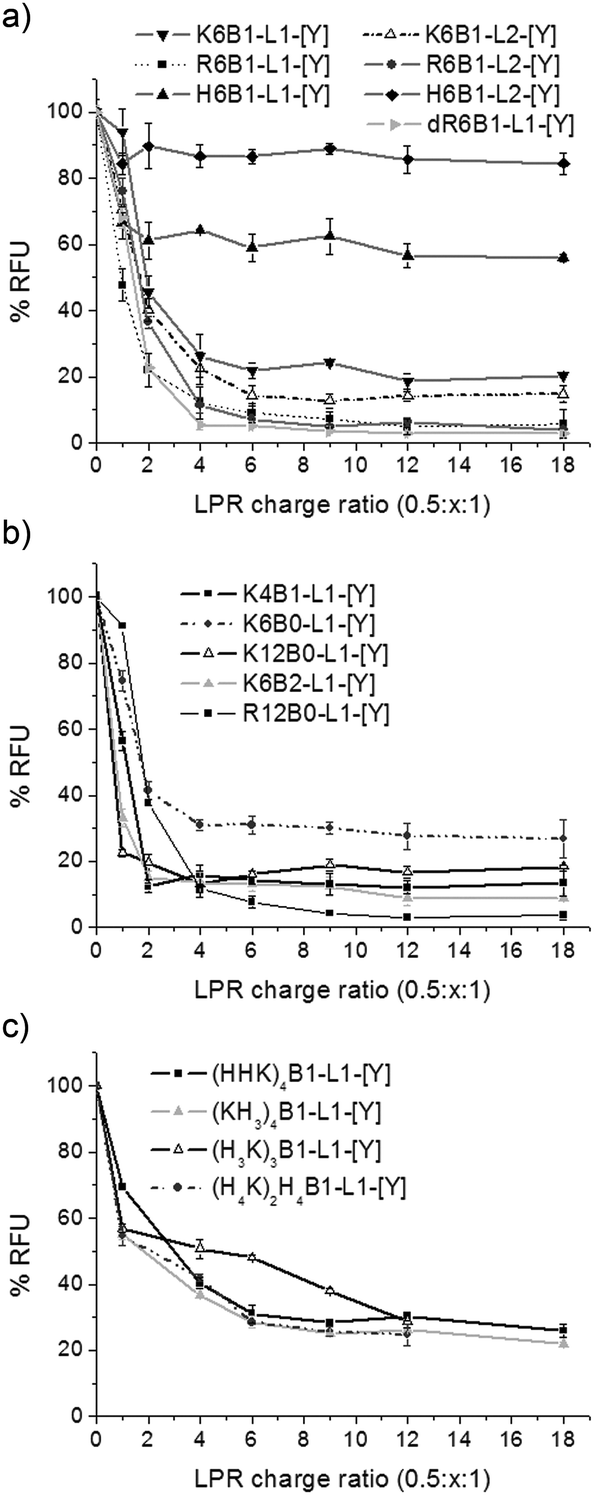 | ||
| Fig. 5 Picogreen fluorescence binding assay showing siRNA packaging within LPR complexes using (a) Group 1, (b) Group 2 and (c) Group 3 peptides. LPR complexes were prepared with constant charge ratios of lipid and siRNA (0.5:1) and varying peptide charge ratios (x = 1–18). Relative fluorescence units (RFU) of the LPR complexes were calculated as percentage fluorescence from free siRNA. | ||
Gel retardation assays in the presence of serum
Further gel electrophoresis studies were performed (Fig. 6) to study the complexation and stability under various conditions of LPRs prepared using 6 selected peptides, namely K6B1-L1-[Y], R6B1-L1-[Y], H6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y]. These peptides were selected for further study as they represented a cross selection of the peptides studied, with some facilitating a good degree of knockdown in the presence of serum while others appeared to be ineffective. The LPRs were therefore prepared in a range of aqueous solvents, namely fully in water, or 12.5% v/v water diluted in 10% FBS solution, OptiMEM or serum-containing growth medium or alternately fully prepared in OptiMEM or fully in serum-containing media. It should be noted that the degradation of naked siRNA observed when dissolved in 10% FBS in water was undoubtedly due to the RNAses naturally present in serum.30 In contrast no degradation was observed when the siRNA was dissolved in water before addition to serum-containing media (which also contains 10% FBS) or (FBS-free) OptiMEM (Fig. 6, Lanes C and D). Furthermore, partial degradation of the siRNA was observed when it was prepared directly in serum-containing media. These results suggest that the action of RNAses present in serum might to some extent be inhibited by components present in media. It is not clear what component in the growth medium exerts this effect especially since RNAse free media was not used in the study. When serum containing media (no siRNA or LPR present) was examined by electrophoresis a new high molecular weight band was seen on the gel, which was thought to be due to fluorescence produced by the interaction of serum protein with GelRed nucleic acid stain. The presence of this band is not thought to interfere with the bands observed in the presence of siRNA or LPR.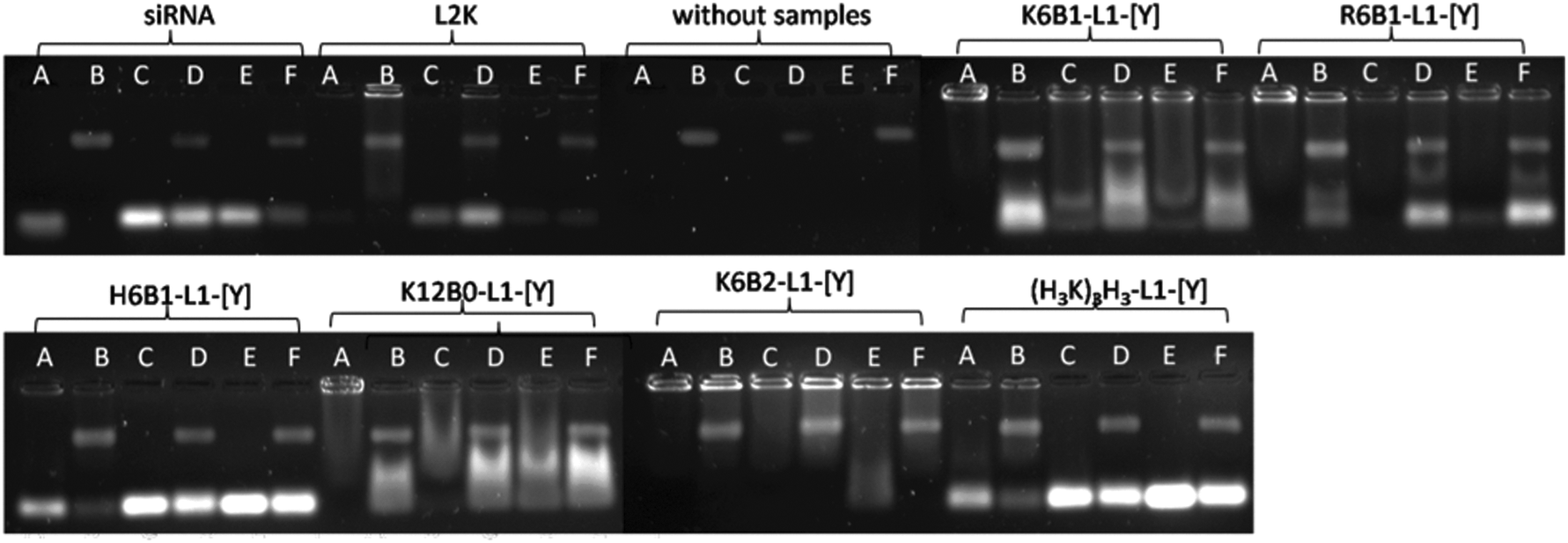 | ||
| Fig. 6 Gel electrophoresis of LPR complexes containing peptides K6B1-L1-[Y], R6B1-L1-[Y], H6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y] at a 0.5:6:1 charge ratio. Free siRNA and L2K:siRNA at 5:1 weight ratio were used as controls. Samples were prepared in (A) fully in water, (B) 12.5% v/v water diluted in 10% FBS, (C) 12.5% v/v water diluted in OptiMEM, (D) 12.5% v/v water diluted in media, (E) fully in OptiMEM and (F) fully in media and were incubated at 37 °C for 60 minutes prior to the gel electrophoresis experiment being performed. | ||
For the purposes of comparison, the corresponding results obtained using LRs prepared using L2K are shown. Although L2K is shown to complex siRNA efficiently in water (Fig. 6, Lane A), some siRNA release (and possible degradation) is detected in the other conditions tested. Interestingly, LPRs were shown to complex and protect siRNA to different extents in the different conditions. When comparing the complexation achieved using the Lys, Arg and His only-containing peptides – K6B1-L1-[Y], R6B1-L1-[Y] and H6B1-L1-[Y] – it is clear that the LPR containing R6B1-L1-[Y] exhibited the highest degree of siRNA complexation when prepared in water and in OptiMEM with all the electrolytes it contains (Fig. 6, Lanes A, C and E) but only in the absence of serum (Fig. 6, Lanes B, D and F). In the case of K6B1-L1-[Y], although well complexed in water, some dissociation of siRNA was observed in OptiMEM (Fig. 6, Lanes C, E) as well as in the serum containing aqueous solvents. As for H6B1-L1-[Y], inefficient complexation was evident under all the conditions tested. Furthermore, some siRNA degradation was evident in the presence of 10% FBS as evidenced by the fainter siRNA (lower) band seen in Lane B. The Group 3 peptide, (H3K)4B1-L1-[Y], behaved in a very similar manner to H6B1-L1-[Y].
When comparing the effect of peptide branching, it was clear that although the linear peptide, K12B0-L1-[Y] behaved in a very similar way to its singly branched counterpart, K6B1-L1-[Y], the double branched K6B2-L1-[Y] exhibited a significantly greater stability towards siRNA dissociation in the presence of both OptiMEM and serum, demonstrating an even higher degree of complexation than L2K. The reduced knockdown efficiency of the doubly branched peptide, K6B2-L1-[Y], could therefore be a consequence of excessive binding that the peptide exerts on siRNA, possibly preventing the dissociation of siRNA within the cell.
Circular dichroism studies
Circular dichroism (CD) spectra of siRNA, 6 selected peptides (namely, H6B1-L1-[Y], K6B1-L1-[Y], R6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y]) and their respective PR, LPR and lipid:siRNA (LR) complexes at different charge ratios using DOTMA:DOPE vesicles were measured (Fig. 7 and Fig. S7, ESI†). It should be noted that despite the relatively large size measured for some of the complexes, particularly the PRs, all preparations appeared clear, and showed very little evidence of light scattering, as assessed by simultaneous measurement of the UV spectroscopy of the samples (data not shown).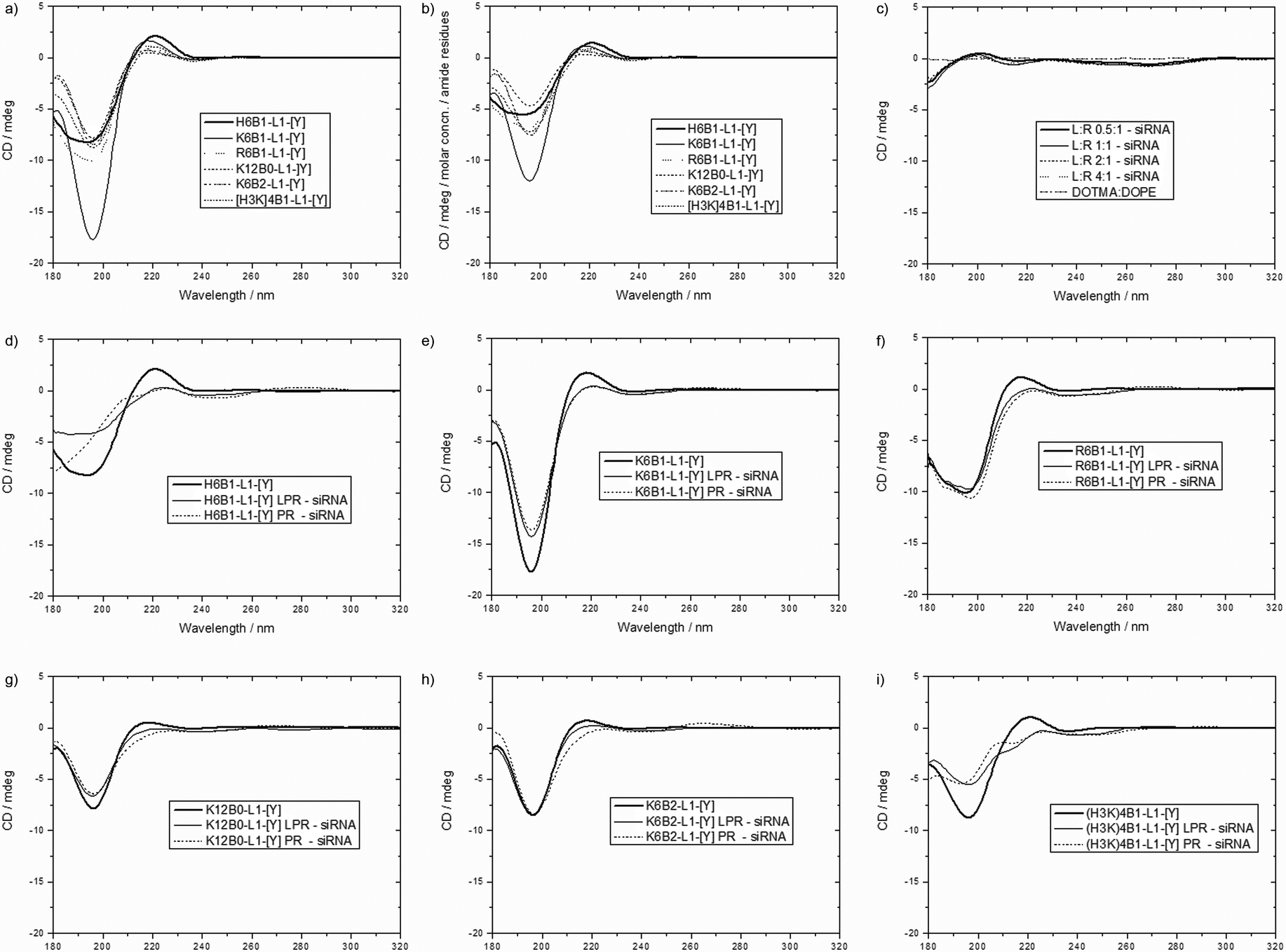 | ||
| Fig. 7 CD spectra of the peptides H6B1-L1-[Y], K6B1-L1-[Y], R6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y] (a) at concentrations equivalent to those used in PR and LPR complexes and (b) normalised according to amide residues per peptide, (c) CD spectra of DOTMA:DOPE vesicles alone (black line) compared to LR complexes at various charge ratios minus the spectrum of siRNA at 4 μM, (d-i) CD spectra of the above peptides alone (black line) at concentrations equivalent to those used in PR and LPR complexes, compared to the spectra of PR (light grey) and LPR complexes (dark grey) at 6:1 or 0.5:6:1 charge ratios respectively, from which the spectrum of free siRNA at 4 μM is subtracted to show differences in siRNA or peptide conformation. | ||
The CD spectra of individual LPR components were measured initially. Peptides alone dissolved in D2O exhibited a positive peak at ∼220 nm and a large negative signal at ∼200 nm, indicative of the peptides adopting, in solution a polyproline II (PII) conformation characterized by a left-handed helical turn31 (Fig. 7a and b). Normalising the CD spectra according to the number of amide residues per mole of peptide (Fig. 7b) revealed that K6B1-L1-[Y] exhibits the strongest PII conformation, while the rest of the peptides exist in equilibrium between PII and disordered state. siRNA alone was seen to adopt the characteristic A-form conformation with high positive and negative peaks at 260 nm and 210 nm respectively (Fig. S7, ESI†). Any change in the intensity of the peaks at 260 nm and 210 nm respectively, indicates changes in base stacking32 and/or the siRNA backbone structure,33 respectively. DOTMA:DOPE vesicles in D2O did not exhibit a CD spectrum at the range of concentrations tested.
To understand the nature of the ternary complexes, the binary complexes were first studied. Subsequently, the CD spectra of LRs were then measured to determine the siRNA conformation when complexed with DOTMA:DOPE at LR charge ratios of 0.5:1–4:1, the lowest lipid ratio being similar to that used in LPR complexes. Spectra of LRs showed a reduction in the intensity of the positive peak at ∼260 nm and a deepening of the trough at 210 nm at all ratios tested when compared to the spectrum of free siRNA (Fig. S7e ESI†). By subtracting the CD spectrum of free siRNA from that of the LR complexes, (and since DOTMA:DOPE itself does not exhibit a CD spectrum), any deviation in the shape of the subtracted CD spectra from baseline indicates a change in siRNA conformation (Fig. 7c).
The interaction of DOTMA:DOPE with siRNA is therefore thought to results in a change in base stacking of siRNA, as well as a change of siRNA backbone conformation. Interestingly, the reduction in band intensity at 260 nm in particular was not progressive upon increasing the lipid content, as previously observed in the case of lipid:DNA complexes,29 suggesting that a change in siRNA conformation was achieved at low (0.5:1) lipid:siRNA charge ratios, and the addition of further lipid did not cause any further conformational change in siRNA.
When examining the formation of PRs, the addition of peptide to siRNA did not cause significant changes in siRNA conformation, since the intensity of the positive band at 260 nm was preserved (Fig. S7b–f, ESI†). When studying the ternary, LPR complexes, the band at 260 nm was similarly preserved, despite containing the same lipid content as that of an LR at 0.5:1 charge ratio, suggesting that the binding of the cationic peptides with siRNA takes precedence over the interaction of the lipid with siRNA.
In order to determine any changes in peptide conformation within a PR or an LPR complex, the CD spectrum of free siRNA was subtracted from those obtained for PR and LPR complexes and compared to the spectrum of their respective peptides in solution (Fig. 7d–i). Any difference suggests a conformational change in either the siRNA or the peptide in the complex. However, since the peptides do not exhibit any CD at wavelengths greater than 230 nm, and very little change in the conformation of siRNA is observed in that region in both PR and LPR complexes, any changes in spectra below 230 nm were attributed to changes in peptide rather than siRNA conformation. Differences in peptide conformation are shown through a reduction in the intensity of the peaks and troughs at 220 nm and 200 nm respectively (Fig. 7d–i). Interestingly the most prominent differences are observed in peptides H6B1-L1-[Y], K6B1-L1-[Y] and (H3K)4B1-L1-[Y] (Fig. 7d, e and i), which have also shown the least siRNA binding capacity in gel electrophoresis and PicoGreen fluorescence studies. On the other hand, peptides R6B1-L1-[Y] and K6B2-L1-[Y], which were shown to bind siRNA the strongest, had preserved band intensities at 200 nm. It is unclear, however, whether the preserved conformation of peptides R6B1-L1-[Y] and K6B2-L1-[Y] in the complexes enhances their ability to complex siRNA more effectively, thereby hindering siRNA dissociation within the cell. In this context it is worth noting that gel electrophoresis (Fig. 6) indicates that the dissociation of siRNA in the presence of electrolytes contained within OptiMEM and serum-containing media was lowest in these two peptides.
Small angle neutron scattering
The small angle neutron scattering (SANS) profiles were recorded for DOTMA:DOPE vesicles (1:1 molar ratio, at a final DOTMA concentration of 1 mg mL−1 in D2O) and for LPR complexes formed from selected peptides at a charge ratio 0.5:6:1, namely (b) K12, (c) (H3K)4B1-L1-[Y] and (d) H6B1-L1-[Y] (Fig. 8). The corresponding data for LPRs containing peptides R6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y], K6B1-L1-[Y] and K6B0-L1-[Y] are given in Fig. S8, ESI.† Note that SANS experiments were not performed on the PRs due to their large size and high polydispersity coupled with the lack of any discernible internal structure in the complexes from TEM.
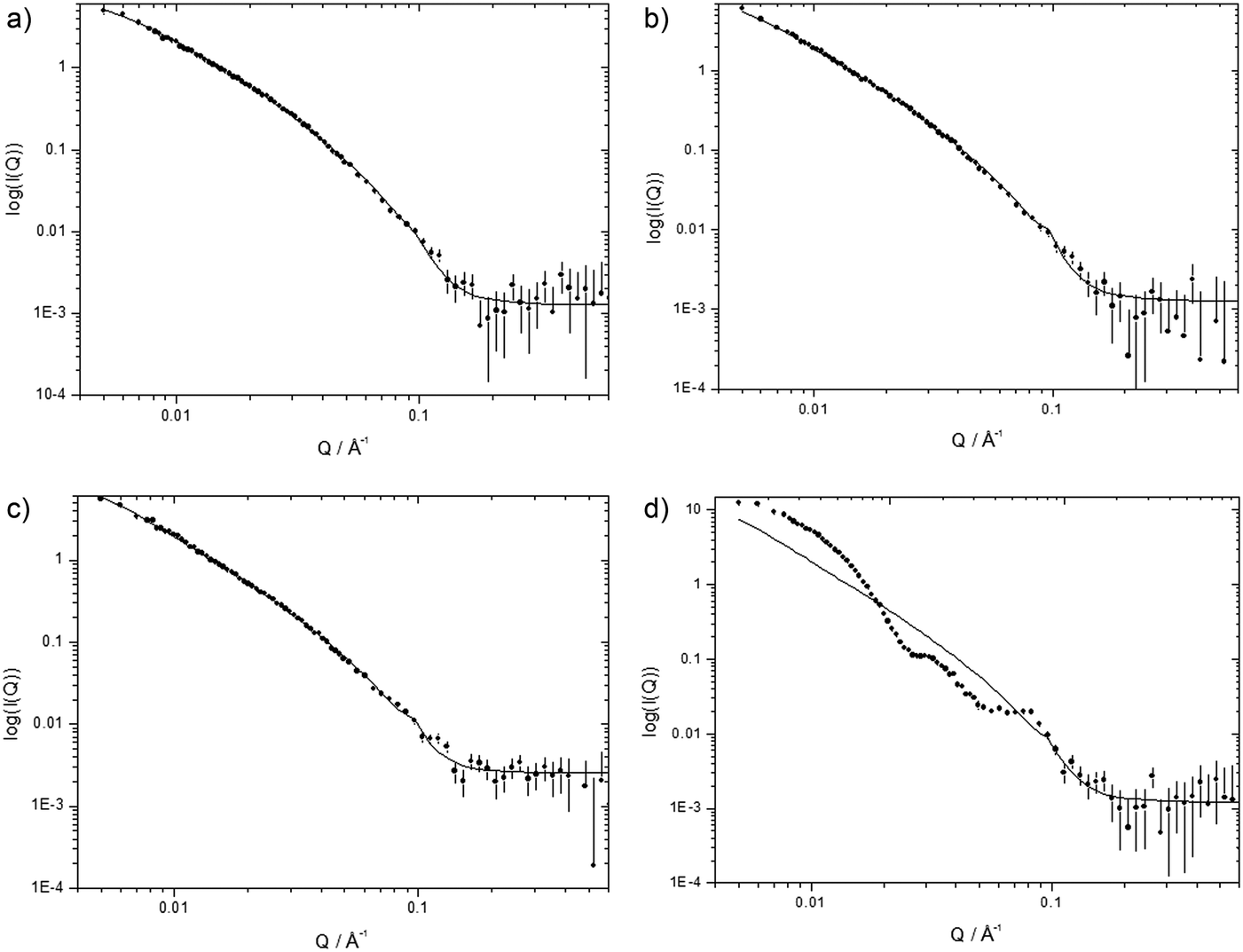 | ||
| Fig. 8 Small angle neutron scattering data (dots) at 298 K and the best fit to the data (solid line) obtained using the mixed sheet and stack model for (a) cationic vesicles prepared from a 1:1 molar ratio of DOTMA:DOPE diluted to give the same DOTMA concentration as is present in the LPRs and LPRs containing (b) K12, (c) (H3K)4B1-L1-[Y] and (d) H6B1-L1-[Y] at a 0.5:6:1 charge ratio. | ||
The variation in the intensity of the SANS as a function of Q for cationic vesicles prepared using a 1:1 molar ratio of DOTMA and DOPE at a DOTMA concentration of 1 mg mL−1 in D2O (Fig. 8a) is in agreement with that obtained in our previous studies.29 Due to the relatively large average size of the cationic vesicles, the SANS data was modelled assuming either single flat sheets (i.e., unilamellar vesicles) or a combination of sheets and stacks (i.e., multilamellar vesicles). The modelling suggested the presence of vesicles that were predominantly unilamellar in nature, and that only a few multilamellar vesicles were present with about 3–4% of the lipid being in the form of multilamellar vesicles. This result correlates well with the relatively small vesicle size measured by dynamic light scattering, which was indicative of the presence of very few multilamellar vesicles. The thickness and repeat distance (of D spacing) of 41.5 Å and 65 Å reported in this study agree well with values of 44.7 Å and 65 Å, previously reported for DOTMA:DOPE vesicles (1:1 molar ratio)27 and with 39 Å and 65 Å reported for related DOTAP:DOPE vesicles (1:1 mola ratio).34,35 Furthermore, the use of a Rσ of 250 Å to fit the data agrees well with that used in previous analysis of similar systems.27–29 Significantly, the DOTMA:DOPE vesicles examined using SANS were those used to prepare the LPR complexes.
SANS data for LPR complexes are shown in Fig. 8b–d and in Fig. S8, ESI.† With the exception of LPRs prepared using two peptides, namely H6B1-L1-[Y] and K6B0-L1-[Y] the scattering patterns recorded for the others LPRs are both qualitatively very similar to one another and to their parent DOTMA:DOPE vesicles. These scattering patterns suggest that the LPRs contain one or, in a small number of instances, more lipid bilayers. Furthermore, it was possible to fit the data obtained from all other LPRs, with the exception of those containing K6B2-L1-[Y] and K6B1-L1-[Y] using the same parameters as those used to fit the DOTMA:DOPE vesicles, suggesting that the bilayer present in the LPRs is by and large the same as in the vesicles. In order to fit the SANS data obtained for the LPRs containing peptides, K6B2-L1-[Y] and K6B1-L1-[Y] with comparable parameters to those obtained for the vesicle bilayers, it was necessary to use a higher Rσ of 380 and 520 Å respectively, suggesting that the lipid bilayer in these LPRs is in a more rigid state. Unfortunately, due to the limited data sets available, it is not possible to unambiguously state that the bilayer in these LPRs is more rigid as, for example, it is also possible to these data sets using different parameters, such as a bilayer of different thickness. However, from a knowledge of the physico-chemical properties of the systems under consideration and the known rigidity of siRNA, it would be reasonable to assume that it is most likely that the rigidity of the lipid bilayer was increased.
The fact that it was possible to fit the data obtained from the LPRs using the same parameters as for the parent vesicles was in line with similar observations made on LPDs prepared using the same peptides29 and from cationic lipids containing unsaturated C14 chains.27 In these LPDs it was assumed that the peptide and DNA complexed with each other and comprised the core of the lipid bilayer. There is no evidence from the SANS of the formation of multilayer structures produced from the interaction of cationic vesicles and siRNA. Furthermore, from inspection of the TEM data there is no evidence of the large, unstructured aggregates common for PDs. Hence, while it is not possible to unambiguously state if all the vesicles contain peptide/siRNA complexes, there is no evidence to suggest the presence of many, if any, empty vesicles.
Attempts to model the scattering patterns obtained from H6B1-L1-[Y] and K6B0-L1-[Y] were unsuccessful as it appears that the preparation consists of mixtures of more than one type of aggregate. Unfortunately, there was insufficient experimental data to unambiguously determine the nature of the mixed aggregates. Indeed close inspection of the TEM data (Fig. 3c, LPRs prepared using H6B1-L1-[Y]) suggested the presence of only one type of aggregate. This observation was unexpected and is currently under investigation.
In order to establish whether there was any internal structure inside the complexes, LPRs were prepared in 8.2 vol% D2O in H2O. This dispersion medium was specifically selected because the lipid was contrast matched to the medium and so therefore “invisible” to neutrons, and in this way it should be possible to determine both the size of any DNA and peptide “core” and to establish whether the peptide/DNA in the core possessed any regular structure. However, the concentration of the LPR complex used in the present study was too low to enable determination of either the size or the presence of any internal structure.
Confocal fluorescence microscopy
In order to assess the cellular uptake of the LPRs, confocal fluorescence microscopy studies were performed on A549-Luc cells transfected with LPRs formulated using six peptides, namely K6B1-L1-[Y], R6B1-L1-[Y], H6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y]. LPRs were prepared using Rhodamine-siRNA (red) and BODIPY-lipid (green) and formulated in water then diluted with OptiMEM, and incubated with the cells for 4 hours. No significant differences in fluorescence distribution (Fig. 9a–f) were observed. In all the samples, the green fluorescence due to the lipid appears very punctate, suggesting it is incorporated within endosomal compartments. Although co-localisation of (green) lipid with (red) siRNA (seen as yellow) is seen in some cells in a number of the images, red siRNA is observed in the majority of cells, suggesting that the siRNA has predominantly dissociated from the lipid and possibly escaped from the endosomal compartments. Indeed it is of note that there is very little nuclear localisation of siRNA or lipid.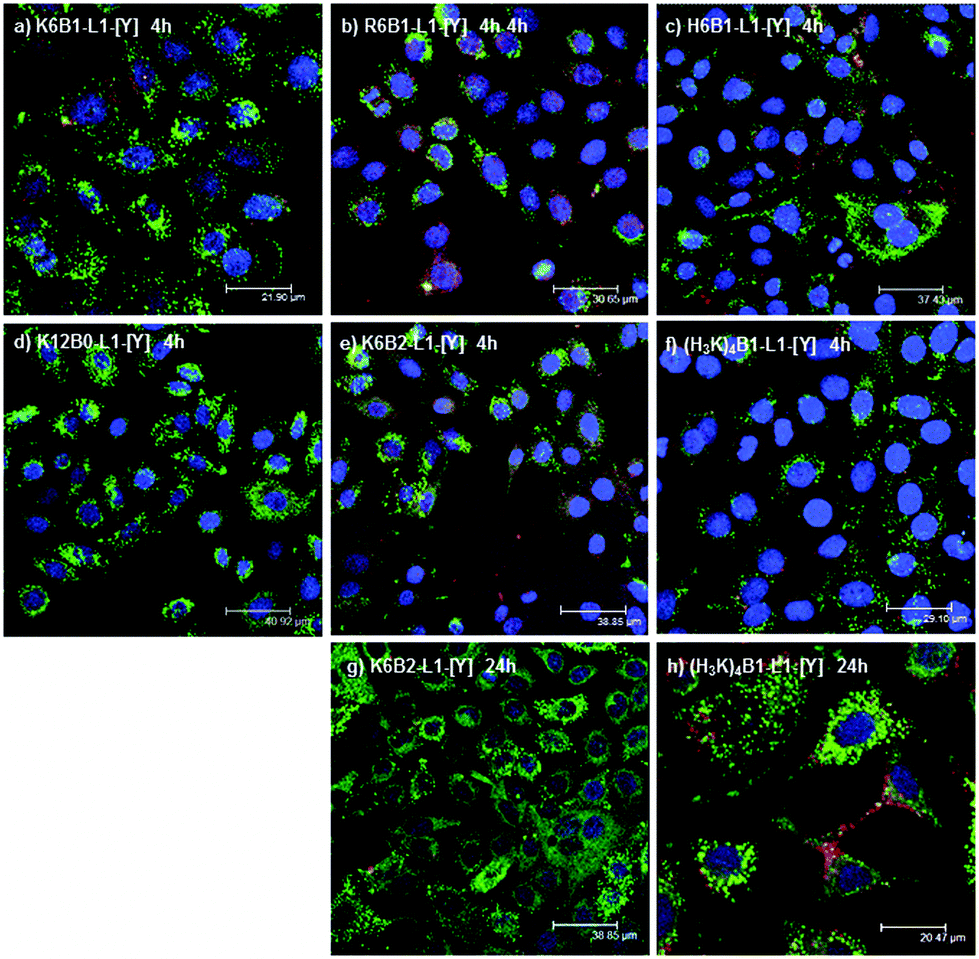 | ||
| Fig. 9 Confocal microscopy of A549-luc cells transfected with LPR complexes prepared in 12.5% water then diluted in OptiMEM containing peptides (a) K6B1-L1-[Y], (b) R6B1-L1-[Y], (c) H6B1-L1-[Y], (d) K12B0-L1-[Y], (e) K6B2-L1-[Y] and (f) (H3K)4B1-L1-[Y] after 4 hours of incubation or (g) K6B2-L1-[Y] and (h) (H3K)4B1-L1-[Y] after 24 hours of incubation. LPR complexes were prepared with BODIPY-HPC incorporated into DOTMA/DOPE lipid (green) and rhodamine labelled Silencer® Negative Control siRNA (red) at a lipid:peptide:siRNA charge ratio of 0.5:6:1. The cell nucleus was stained with DAPI (blue). All peptides were unlabelled. | ||
Furthermore, two peptides K6B2-L1-[Y] and (H3K)4B1-L1-[Y] were selected for detailed study as LPRs containing K6B2-L1-[Y] and (H3K)4B1-L1-[Y] exhibited very low and high knockdown activities, respectively. The LPRs were prepared either in water and diluted 1 in 4 in OptiMEM (Fig. 9e–h) or fully in OptiMEM (Fig. S9, ESI†). These showed no major differences in the distribution of either lipid (labelled green) or siRNA (labelled red) after 4 and 24 hours of incubation.
LPRs containing the peptides K6B2-L1-[Y] and (H3K)4B1-L1-[Y] were also observed after 24 hours of incubation with the cells (Fig. 9g and h). Interestingly, although the level of lipid due to green fluoresence was similar in both cases, less siRNA was detected within the cells in the case of K6B2-L1-[Y] than in the case of (H3K)4B1-L1-[Y], which could account for the difference in knockdown efficiency (separate green and red channel figures are shown in Fig. S10, ESI†). More red fluorescence due to siRNA was also observed in LPRs prepared using (H3K)4B1-L1-[Y] when prepared fully in OptiMEM (Fig. S9c and d, ESI†).
In order to determine the effectiveness of release of siRNA from the two peptides selected for further study, LPR complexes were formulated using unlabelled lipids, either FITC-K6B2-L1-[Y] or FITC-(H3K)4B1-L1-[Y] (labelled green), and siRNA (labelled red). The LPRs were prepared both in water and diluted in OptiMEM, or fully in OptiMEM. Again, after 24 hours of incubation, a clear difference between the amount of siRNA released from the complex with the two peptides could be detected (Fig. 10). With the LPRs formulated from the peptide which showed the lowest knockdown activity, K6B2-L1-[Y], after 24 hours the peptide and siRNA are still significantly co-localised (Fig. 10a and c). In contrast, with the LPRs formulated from the peptide with the highest knockdown activity, (H3K)4B1-L1-[Y], significant amounts of siRNA persist in the cytoplasm and are no longer co-localised with the peptide (Fig. 10b and d).
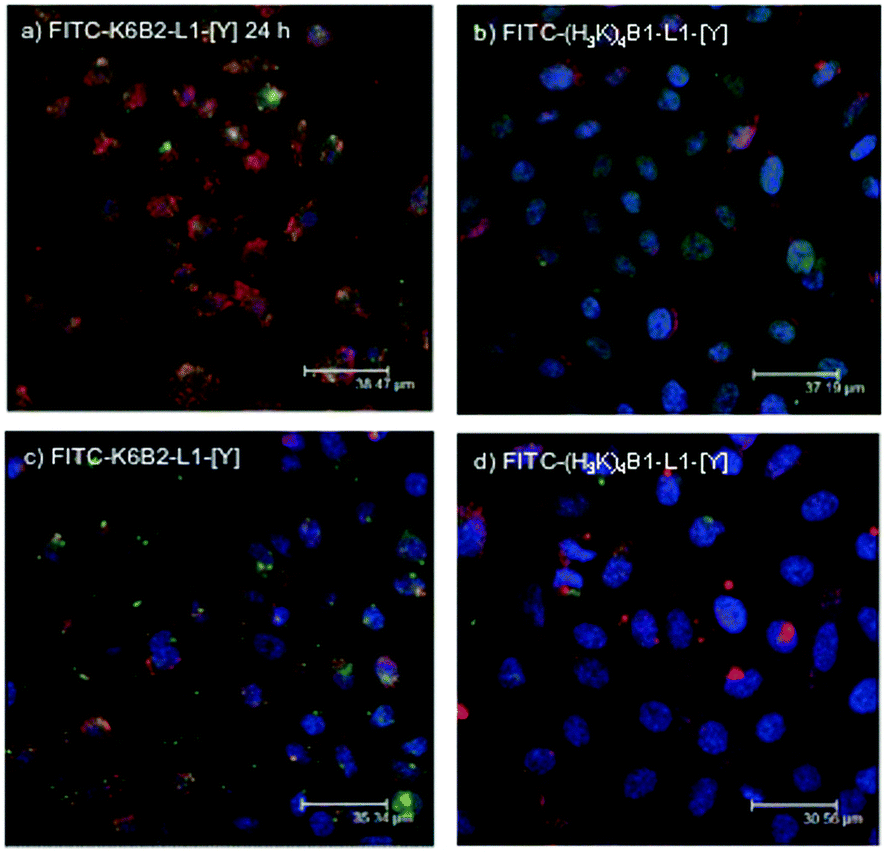 | ||
| Fig. 10 Confocal microscopy of A549-luc cells transfected with LPR complexes prepared in 12.5% water then diluted in OptiMEM containing peptides (a) FITC-K6B2-L1-[Y] and (b) FITC-(H3K)4B1-L1-[Y] after 24 hours of incubation, or A549-luc cells transfected with LPR complexes prepared fully in OptiMEM containing peptides (c) FITC-K6B2-L1-[Y] and (d) FITC-(H3K)4B1-L1-[Y] after 24 hours of incubation. LPR complexes were prepared with FITC-labelled peptides (green, 100%) and rhodamine labelled Silencer® Negative Control siRNA (red) at a lipid:peptide:siRNA charge ratio of 0.5:6:1. The cell nucleus was stained with DAPI (blue) and the lipids were unlabelled. | ||
Discussion
The use of cationic peptide sequences in PR and LPR formulations for the delivery of siRNA has previously been studied by a number of groups.6–17 However, each previous study has tended to focus on one specific type of cationic residues, whereas in this study we aimed to compare the effects on complex structure and knockdown efficiency of complexes formed from Lys, Arg, His, D-Arg, and mixed Lys/His sequences.Using the cell line studied in this paper, virtually all peptides gave impressive knockdown results when formulated into LPR complexes in water and diluted in OptiMEM (Fig. 2(a–c)). Under these conditions, even peptides lacking a targeting sequence, or with the cell targeting sequence scrambled, performed well. However, under physiologically relevant conditions, in which the LPR formulations were diluted in serum-containing growth media, major differences in knockdown efficiency resulted when both peptide sequence and formulation conditions were varied (Fig. 2(d–f) and Fig. S1, ESI†). Firstly, LPR formulations prepared in water and diluted in growth media showed uniformly poor knockdown efficiencies, whereas certain LPR formulations prepared in OptiMEM and diluted in growth media retained good knockdown activities. As the main difference between the composition of OptiMEM and growth media is the presence of serum, this observation suggests that, although there was no difference in the knockdown achieved in the absence of serum, that the ions present in OptiMEM influenced the structure of the LPRs in a beneficial way with respect to their interaction with serum.
The formulations which retained the greatest knockdown activity when prepared in OptiMEM and diluted in growth media were those prepared using His-rich peptides. Thus those LPRs containing the Group 1 peptides, H6B1-L1-[Y] and H6B1-L2-[Y] produced the greatest levels of knockdown, while those containing peptides possessing His-rich sequences from the Group 3 peptides gave good to excellent knockdown. The sequences (KH3)4B1-L1-[Y] and (H3K)4B1-L1-[Y], which were identified by Mixson13 as being most effective for siRNA delivery, also performed the best in our study. However, interestingly there was no major difference between LPRs formulated from these peptides, and LPRs formulated from the other His-rich sequences which were previously identified as being suboptimal for siRNA delivery.13 These observations suggest that the charge on the peptide plays an important role in the knockdown efficiency of the complexes in the presence of serum and that the partially charged histidine peptides were less affected by its presence than the more highly charged peptides. This point is further reinforced by the fact that LPRs prepared from the more highly charged peptides based on Lys or Arg sequences showed significantly better activities when the non-charged linker, GAGA, was used instead of RVRRGA (Fig. 2e). In comparison, in our previous work studying the effectiveness of these peptides in delivering DNA in LPD complexes, the His-rich sequences showed very poor transfection efficiencies and the RVRRGA linker was also critical for good transfection.27 The present results suggest that the cleavage of the peptide by furin within the endosome is not the crucial step for knockdown, but rather release of the siRNA from the complex in the cytoplasm is. This confirms the observations of other groups that the best delivery vehicles for DNA may not be optimal for siRNA delivery.1–4 It should also be noted that, whilst the best knockdown efficiencies in growth media were obtained with LR complexes prepared using L2K (Fig. 2d–f) these complexes also exhibited the lowest cell viability (Fig. S3, ESI†), whereas the LPRs studied had over 80% viability compared to controls.
In order to understand more fully the reasons for these observations, we carried out a series of biophysical studies comparing the LPR complexes. PicoGreen fluorescence studies, and gel shift assays in both the absence and presence of serum, indicated that the majority of the peptides packaged siRNA in tightly bound complexes, and that the siRNA packaging efficiency of the complexes was not improved by using more highly branched peptides. However, the LPR complexes which showed the best knockdown in the presence of serum – the His-rich Group 3 peptides and the Group 1 peptides H6B1-L1-[Y] and H6B1-L2-[Y] – showed reduced siRNA complexation and protection from siRNA degradation. As siRNA must be released into the cytoplasm in order to exert its knockdown effect, this in turn suggests that incomplete complexation of the siRNA and therefore more effective release from the nanocomplex is key to the greater knockdown efficiencies of these LPR complexes.
We next compared the effects of LPR complex formulation with the different peptides on the structural properties of these nanocomplexes. Whilst the sizes of the LPR complexes were all very similar, regardless of the peptide used, LPRs prepared with the His- or His-rich peptides had a lower surface charge (Fig. S5, ESI†), probably due to the lower charges on these peptides at neutral pH. TEM imaging of the LPR complexes showed them to be small, homogeneous and electron dense with no internal structure visible. By comparison, some LPD nanocomplexes formulated with these peptides had ordered core structures29 again emphasising the key differences between DNA and siRNA carriers. CD studies indicated that in contrast to LPDs, where the conformation of DNA in the complex was generally found to be different to that free in solution,29 the conformation of siRNA was unaltered when in the form of an LPR. Furthermore, CD studies showed that the His- and His-rich peptides, H6B1-L1-[Y] and (H3K)4B1-L1-[Y] exhibited the greatest change in their spectra when in the form of an LPR while the peptides, R6B1-L1-[Y] and K6B2-L1-[Y], which were shown by PicoGreen fluorescence studies and gel shift assays to bind siRNA to the greatest extent, preserved their spectra. This suggests that in order to complex the siRNA, the His and His-rich peptides have to undergo an amount of structural change, possibly explaining the greater tendency of the complexes containing these peptides to dissociate.
For the majority of peptides, including the His-rich peptide, (H3K)4B1-L1-[Y], SANS studies indicated that the LPRs contained a lipid bilayer that possessed virtually identical physico-chemical properties to the bilayer of the parent vesicles from which they were formed. Similar results were previously found for LPDs prepared from the same peptides as studied here.29 Interestingly however, the bilayer present in the LPRs containing K6B2-L1-[Y] and K6B1-L1-[Y] although of a similar thickness is thought to be slightly more rigid than in the parent vesicle. It is not known what effect the slightly more rigid bilayer may have on the transfection ability of the LPRs. Furthermore, for reasons as yet unknown, it was not possible to model the SANS data obtained for LPRs containing H6B1-L1-[Y] and K6B0-L1-[Y] suggesting the presence of a mixture of aggregates.
What effects do these difference in complexation efficiency, surface charge and internal structure have on the fate of the components once the LPR particles have entered the cell? Confocal studies on LPR particles formulated either in water/diluted in OptiMEM, or formulated in OptiMEM/diluted in OptiMEM, were carried out with the lipid, siRNA and peptide components variously labelled. When using LPRs prepared with labelled lipid and siRNA, it was clear that in all cases the lipid had largely dissociated from the LPR complex after 4 hours, regardless of the peptide sequence, and appeared to be distributed within endosomal compartments. Similarly, the siRNA is seen to have dissociated from the lipid and is distributed throughout the cells, rather than remaining in the endosomal compartment or being trafficked to the nucleus. We have previously shown for LPD complexes that endosomal release of the components is most likely mediated by the helper lipid DOPE, with the “proton sponge” effect of the cationic peptides in the formulations having only a negligible effect.27 However, the most effective LPR formulations, using either (H3K)4B1-L1-[Y] or H6B1-L1-[Y], showed larger amounts of siRNA within the cells and that this higher level of siRNA persisted, at least up to 24 h after incubation (Fig. 9h and Fig. S8d, ESI†). Further confocal experiments using labelled H6B1-L1-[Y] supported the observation that siRNA after 24 hours incubation was in the cytoplasm of the cell and showed that the siRNA was no longer co-localised with the peptide (Fig. 10b and d). By contrast, using labelled lipid and siRNA indicated that little siRNA was observed in cells treated with LPRs formulated using the peptide with the lowest knockdown activity but the tightest siRNA binding, K6B1-L1-[Y]. Furthermore, when studying LPRs formulated using labelled K6B2-L1-[Y] and siRNA showed that the labelled peptide was significantly co-localised with siRNA after 24 hours (Fig. 10a and c).
Conclusions
We have carried out a comprehensive comparative study of the siRNA delivery and gene knockdown properties of nanoparticles formulated from different cationic peptide sequences, a 1:1 mixture of the lipids DOTMA and DOPE, and siRNA. We have compared polyplex (PR) and lipopolyplex (LPR) formulations and have demonstrated that the LPR formulations have significantly better gene knockdown under a range of conditions. In the cell line studied, and in these ternary complexes, branched peptides containing His or mixed His-Lys sequences are the most effective, particularly when knockdown experiments are carried out in physiologically relevant growth media. Furthermore, these lipopolyplex formulations were much less toxic than those prepared using the current “standard” reagent, L2K.
In order to understand the molecular basis for these findings, we have studied the biophysical properties of these nanocomplexes in detail. We have shown that the looser binding of the mixed His/Lys peptides to their siRNA cargo is key to their higher knockdown efficiencies, and that this is an effect arising from both lower peptide charge at pH7 and a different geometry of interaction between peptide and siRNA. These in turn lead to nanoparticles in which the siRNA is more efficiently delivered and persists for longer in the cells. Importantly, we have also shown that the conditions under which the LPR complexes are formulated are critical to the knockdown efficiencies in serum, with nanocomplexes formulated in OptiMEM and subsequently diluted in serum being most effective. Whilst OptiMEM would not be regarded as a suitable formulation solvent for clinical studies, where well defined, simple electrolyte solvents are preferred, these results suggest that formulating LPR complexes under conditions of higher salt would be beneficial for delivery of siRNA under physiologically relevant conditions. Further studies in this area are currently in progress and will be reported in due course.
Experimental procedures
Materials
The synthesis of the bifunctional peptides used in the present study has been previously described.29 Characterisation data for the control (scrambled) peptide sequence, sK6B1-L1-[Y] is given in the ESI.† The methods used for the FITC labelling of selected peptides (namely K6B2-L1-[Y] and (H3K)4B1-L1-[Y]) are detailed in the ESI.† Silencer® Firefly Luciferase (GL2 + GL3) (Luc-siRNA) and Silencer® Negative Control siRNA #1 were purchased from Ambion (USA). A custom-made siRNA sequence, CUU ACG CUG AGU ACU UCG dTdT, termed here Sigma siRNA was purchased from Sigma-Aldrich (UK) and used for samples requiring high amounts of siRNA, namely small angle neutron scattering (SANS), dynamic light scattering, zeta potential and circular dichroism (CD) measurements. Uranyl acetate, agarose, RNAse A, RNAse inhibitor, poly-L-aspartic acid (pAsp) and all cell culture growth media and other reagents including 1,4-diazabicyclo[2.2.2]octane (DABCO) were purchased from from Sigma (UK), unless otherwise stated. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) and Pierce BCA Protein Assay kit were was purchased from Thermo Scientific (UK). OptiMEM, Lipofectamine 2000 (L2K), PicoGreen reagent and 2-(4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (β-BODIPY® 500/510 C12-HPC) (BODIPY-HPC) were purchased from Invitrogen Molecular probes, UK. Label-IT® TM-Rhodamine siRNA Tracker Intracellular Localization Kit was supplied by Mirus (USA). GelRed nucleic acid stain was obtained from Cambridge Bioscience (UK). N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA) was obtained from TCI Europe, Belgium, and dioleylphosphatidylethanolamine (DOPE) was obtained from Avanti Polar lipids, Alabama, Alabaster, USA. GelRed was purchased from Gencompare, Belgiun and Luciferase assay kit was purchased from Promega (UK). Human alveolar A549-Luc cells, stably transfected with an episomal S/MAR luciferase vector as described by Argyros et al.36 were a gift from Dr M. T. Thanou (Institute of Pharmaceutical Science, King's College London, UK). All reagents were of the highest grade possible and were used without further purification. Double distilled, filtered water was used throughout. D2O (>99% purity) was obtained from Aldrich (UK).Preparation of liposomes and stock solutions
Cationic lipid vesicles composed of DOTMA:DOPE at 1:1 molar ratio were prepared as previously described.29 Briefly a thin film of lipid, obtained by evaporating chloroform from a lipidic chloroform solution, was hydrated with sufficient filtered, double distilled water to produce a DOTMA concentration of 1 mg mL−1. The resulting crude lipid suspension was then probe sonicated (using a Lucas Dawes probe sonicator operating at 50% output for 10 minutes at room temperature) to produce small unilamellar vesicles of ∼60 nm in diameter.
Peptide solution (1 mg mL−1), siRNA solution and vesicle suspensions were diluted, as required, using either double distilled, filtered water (for dynamic light scattering, zeta potential, gel shift assays and TEM) or D2O (for CD and SANS), or in the case of knockdown studies, water or OptiMEM to give the required siRNA and peptide stock solutions, and the lipid stock suspensions for the preparation of PR and LPR complexes.
Formation of PR and LPR
Polyplexes (complexes of Peptide and siRNA, PR complexes) were formulated by adding equal volumes of the required siRNA stock solution to a peptide solution containing sufficient peptide to produce PRs at either a 6:1 or 12:1 (nominal) charge ratio, i.e. assuming all peptides are fully ionised under the conditions of use. The concentrations used depending on the nature of the experiment to be performed.
Lipopolyplexes (complexes of Lipid, Peptide and siRNA, LPR complexes) were prepared at LPR (nominal) charge ratios of 0.5:6:1 and 0.5:12:1 (i.e. assuming all peptides are fully ionised under the conditions of use) by first mixing equal volumes of cationic vesicles suspension and peptide solution and then adding with thorough mixing an equal volume of siRNA solution. For example, for the in vitro cell studies, 75 μL of a peptide solution (at the required concentration) was added to 75 μL DOTMA:DOPE vesicle suspension (containing 5.7 μg mL−1 of DOTMA). To this lipid:peptide mixture, an equal volume of siRNA solution (i.e. 150 μL of a 2.7 μg mL−1 siRNA solution) was added and the LPDs prepared by the gentle shaking of the resulting suspension.
Dynamic light scattering and zeta potential measurements
The apparent hydrodynamic size and the zeta potential of the LPR and PR complexes were measured by dynamic light scattering at 25 ± 0.1 °C using a Zetasizer Nano ZS (Malvern Instruments, UK). LPR and PR complexes were prepared in double distilled, filtered water at L:P:R charge ratios of 0.5:6:1 and 0.5:12:1 and P:R charge ratios of 6:1 and 12:1 using Sigma siRNA (final siRNA concentration of 26 μg mL−1 for the DLS measurements and 2.9 μg mL−1 for the zeta potential measurements). Three repeat measurements were made for each complex. In all cases the complexes exhibited polydispersities of less than 0.3.
Circular dichroism spectroscopy
The circular dichroism (CD) and ultra violet (UV) spectra of siRNA, LPRs and PRs, dispersed in D2O, were measured at 20 ± 2 °C using a Chirascan Plus spectrometer (Applied Photophysics, UK) operated with a scan speed of 30 nm min−1, a bandwidth of 1 nm and a time per point of 2 s. Spectra were recorded between 320 and 180 nm using a 1 mm path length cell. CD and UV absorbance spectra were acquired simultaneously. All spectra were corrected for a D2O background, which was measured periodically throughout the experiment. The CD and UV spectra of siRNA were measured at a concentration 4 μM (0.050 mg mL−1). The spectra of the LPR and PR complexes were measured at charge ratios of 0.5:6:1 and 6:1, and a siRNA concentration of 0.050 mg mL−1.
Transmission electron microscopy
LPR and PR complexes, prepared at charge ratios of 0.5:6:1 and 6:1, respectively and a final siRNA concentration of 0.05 mg mL−1, were examined using transmission electron microscopy (TEM) by negative staining with an aqueous solution of 4% w/v uranyl acetate. Briefly, a drop of sample was placed on a formvar 200 mesh copper grid for one minute before the excess liquid was removed using a wick of filter paper. A drop of uranyl acetate was then added to the grid for approximately five minutes after which time the grid was washed with an aqueous solution of 50% v/v ethanol. After drying the samples were visualized using an FEI TecnaiTM transmission electron microscope (USA).
Small angle neutron scattering
The small angle neutron scattering (SANS) of LPRs and DOTMA:DOPE vesicles was measured on the SANS2D and LoQ beamlines at the ISIS pulsed neutron source (ISIS Facility, the Rutherford-Appleton Laboratories, Didcot, UK). Freshly prepared DOTMA:DOPE vesicles dispersed in D2O were measured at a DOTMA concentration of 1 mg mL−1. LPR complexes were similarly freshly prepared in either 8.2 vol% D2O in H2O or D2O at a Sigma siRNA concentration of 0.1 mg mL−1 and a L:P:R charge ratio of 0.5:6:1. The SANS of the LPRs and vesicles and the relevant solvents were measured in 2 mm path length circular silica cells at 25 ± 0.1 °C. Where possible, the SANS of the parent vesicles and the LPRs were measured on more than one occasion to ensure the reproducibility of the data. Dynamic light scattering measurements of the vesicles and LPRs prepared in D2O confirmed that their preparation did not alter the size of the resulting nanoparticles.
The SANS intensity, I(Q), of the LPDs and vesicles as a function of the scattering vector, Q = (4π/λ)sin(θ/2), where θ/2 is the scattering angle, was determined by normalising the scattering to the appropriate sample transmission after subtraction of the scattering from the relevant solvent also normalised to its corresponding transmission. The fitting of the SANS data always included flat background corrections to allow for any mismatch in the incoherent and inelastic scattering between the sample and the solvent, with the levels of the fitted background being checked to ensure that they were physically reasonable. The SANS data for the DOTMA:DOPE vesicles and LPR complexes dispersed in D2O were routinely modelled either assuming a mixture of (isolated/single) infinite planar (lamellar) sheets with or without one-dimensional paracrystals (stacks) to account for the presence in the sample of any multilamellar vesicles. When modelling the vesicles and LPR complexes dispersed in D2O as (single) lamellar sheets, the fits to the SANS data were obtained by the least-squares refinement of three parameters, namely L, Rσ, and the absolute scale factor (together with the background, as described above), where Rσ is the Lorentz correction factor which provides information about the extent of rigidity/curvature of the lamellar sheets. Unless otherwise stated a Rσ of 250 Å was used. In this study the polydispersity on the thickness of the bilayer (σ(L)/L) was fixed at 0.1. When stacks were added to the (single) lamellar sheet model, the fit to the SANS data was obtained by least-squares refinement of seven parameters, namely Rσ, the mean bilayer thickness (L), the number of bilayers in the stack (M), their mean separation or d-spacing (D), the width of the Gaussian distribution in the plane, (σ(D)/D), and the absolute scale factors for the unilamellar and multilamellar vesicles. In the present study, σ(L)/L was again fixed as 0.1 and σ(D)/D at 0.05, with a Rσ of 250 Å was used. In addition, when modeling the SANS data using a mixed population of sheets and stacks, L, σ(L)/L, and Rσ were constrained to be the same for the isolated/single and stacked lamellae, a not unreasonable assumption. If no Bragg peak was seen in the SANS data, it was fitted using a stack with a maximum of 2 bilayers. In such cases the data was fitted using a higher number of bilayers comprising the stacks to ensure that it did not improve the quality of the fit obtained. For all models, the least-squares refinements were performed using the model-fitting routines provided in the FISH software.
Gel band shift assay
Gel electrophoresis experiments were performed to determine (a) the extent of siRNA complexation, (b) the dissociation of siRNA in the presence of pAsp and (c) the extent of siRNA protection from enzymatic degradation by RNAse A, when the siRNA was formulated as either an LPR or PR complex – either at a charge ratio of 0.5:6:1 and 0.5:12:1 for the LPRs and a charge ratios of 6:1 and 12:1 for the PRs. Complexes were prepared in a volume of 10 μL and at a siRNA concentration of 0.01 μg μL−1 (i.e. 0.1 μg siRNA per well). To determine the extent of siRNA complexation, LPR or PR complexes were used without further treatment. For siRNA release studies, 1.25 μL of a 1 mg mL−1 aqueous pAsp solution was added to the complexes. Experiments to determine the level of siRNA protection afforded by the complexes from enzymatic degradation were prepared by adding an aqueous solution of RNAse A (0.4 μL, 0.1 mg mL−1), incubating the resultant mixture for 30 min at 37 °C, then adding an aqueous solution of RNAse inhibitor (0.4 μL, 30–50000 U mL−1) to stop the enzymatic reaction. The mixture was further incubated at room temperature for 10 min after which time an aqueous pAsp solution (1.25 μL, 1 mg mL−1) was added to release any protected siRNA. To all prepared samples, 2 μL of gel loading buffer (0.25% w/v bromphenol blue and 40% w/v sucrose) was added prior to loading the samples onto a 2% w/v agarose gel in Tris/Borate/EDTA (TBE) buffer containing 3 μL of GelRed nucleic acid stain at 80 V for 45 min (Fisher Brand, Model HU12 electrophoreses chamber) and visualized under UV light using an Alphalmage EP MultiImage Light Cabinet (Alpha Innotech, South Africa).
Effect of media on lipopolyplex complexation and stability
Gel electrophoresis experiments were also used to determine the extent of siRNA complexation and stability in LPR complexes when prepared in different conditions. LPRs were prepared in a total volume of 10 μL at charge ratios of 0.5:6:1 and a siRNA concentration of 0.01 mg mL−1 using the peptides (K6B1-L1-[Y], R6B1-L1-[Y], H6B1-L1-[Y], K12B0-L1-[Y], K6B2-L1-[Y] and (H3K)4B1-L1-[Y]), either fully in water, OptiMEM, or in serum-containing growth media. LPR complexes were also prepared initially in water (in 12.5% of the total volume) then diluted with OptiMEM, serum-containing growth media or 10% fetal bovine serum (FBS) in water. Free siRNA and complexes of L2K:siRNA prepared at a 5:1 weight ratio in different media conditions and 10 μL of the media alone in the absence of samples were used as controls. After incubation of all the samples at 37 °C for 60 minutes, 2 μL of loading buffer was added to each sample before being loaded onto the gel and visualised as described above.
PicoGreen fluorescence experiments
PicoGreen assays were performed in black 96-well plates. For each well, 50 μL of PicoGreen reagent (1:150 v/v diluted with Tris-EDTA (TE) buffer, pH 7.4) was mixed with 50 μL of 4 μg mL−1 of negative control siRNA in TE buffer (giving a final siRNA content of 0.2 μg per well). LPR complexes were then formulated by adding 100 μL of the siRNA:PicoGreen mixture to a mixture of equal volumes (25 μL each) of the appropriate concentration of lipid and peptide. The final LPR ratios were prepared such that the lipid:siRNA ratio remained constant at 0.5:1, but the peptide:siRNA charge ratio was increased (to 18:1). Fluorescence was measured at λex/λem = 480/520 nm using a Varian Cary Eclipse fluorescence plate spectrophotometer. In each experiment, naked negative control siRNA mixed with PicoGreen was regarded as 100% fluorescence and was used to normalize the PicoGreen signal detected from the complexes.
Cell culture
A549-Luc cells were maintained in RPMI-1640 growth medium supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v of 100× strength non-essential amino acids (NEAA), 1% v/v penicillin/streptomycin solution (10000 U mL−1/10 mg mL−1) and 1% v/v L-glutamine 200 mM. All cells were maintained at 37 °C, 5% CO2 and 90% relative humidity. The cells were passaged every three to four days when around 70% confluent using 0.25% w/v trypsin–EDTA solution.
In vitro knockdown experiments
L2K was used in all knockdown experiments as a positive control when mixed with siRNA at a L2K:siRNA weight ratio of 5:1. Free Luc-siRNA was used as a negative control. A549-Luc cells were seeded onto 96-well plates at 1.2 × 104 cells per well and incubated for 24 h at 37 °C in a 5% CO2 and 90% relative humidity.
The growth medium (100 μL) in each well was then replaced with 50 μL of either OptiMEM or fresh (serum-containing) growth medium followed by the addition of 50 μL of the appropriate LPR complex at a charge ratio of either 0.5:6:1 or 0.5:12:1 or a PR complex at a charge ratio of 6:1. The effect of the ionic strength of the solvent used to prepare the complexes and the presence of serum on the knockdown efficiency was tested. For these studies, complexes were either formulated in water and then diluted 1 volume in 4 in OptiMEM or serum-containing growth medium, or prepared in OptiMEM and diluted 1 volume in 4 in OptiMEM or serum-containing growth media before adding to the cells, which brought the final water or OptiMEM content to 12.5% v/v. All complexes were tested at least in quadruplicates but generally sextuplicates. The final concentration of siRNA in the 100 μL in each well was 50 nM. Preliminary experiments using LRs prepared using L2K and LPRs made with K4B1-L1-[Y] were performed at siRNA concentrations of 10, 30, 50 and 100 nM in 100 μL per well to determine the optimal siRNA concentration (Fig. S11, ESI†). In the case of L2K:siRNA complexes prepared at a lipid:siRA weight ratio of 5:1, knockdown efficiency increased with increasing siRNA concentration up to 50 nM whereafter an increase in concentration resulted in a levelling off of knockdown, while cell toxicity (indirectly measured using a protein assay) increased with siRNA concentration, being greatest at 100 nM siRNA. When studying LPRs prepared using K4B1-L1-[Y] at a charge ratio of 0.5:12:1, a progressive increase in knockdown efficiency was observed upon increasing siRNA dose, however some toxicity was also observed at 100 nM siRNA. For these reasons, 50 nM of siRNA in 100 μL per well was used in all subsequent knockdown experiments.
Knockdown experiments were carried out at LPR ratios of 0.5:6:1 and 0.5:12:1, in line with previous studies.29,37 Peptide:siRNA (PR) complexes were also tested for selected peptides at 6:1 charge ratio (Fig. S2, ESI†).
Complexes were added to the cells and incubated for 24 h at 37 °C. The supernatant was removed and replaced by growth medium and incubated for a further 24 h. Each complex was formulated and tested using Luc-siRNA as well as the negative control siRNA and all knockdown results were expressed as % knockdown efficiency compared to the luciferase expression produced by the same formulation prepared using the negative control siRNA. A linear relationship between luminescence and protein cell content was measured at protein concentrations of up to ∼0.018 mg of protein per well (Fig. S12, ESI†). The protein content in all subsequent knockdown experiments was therefore monitored to ensure that it fell within the linear region of the luminescence graph.
Luciferase activity, analyzed 48 h after addition of the complexes, was determined in cell lysates using a commercial luciferase assay according to the manufacturer’s protocol. Briefly, cells were rinsed with 50 μL phosphate buffered saline (PBS) then lysed by the incubation of 50 μL of 1 x Reporter Lysis Buffer (provided in the luciferase assay kit) for 1 h at 25 °C. After freezing the cells for 30 min at −80 °C, the lysed cells were thawed and 30 μL of the lysate transferred to a white 96-well plate. The luciferase activity was then measured over 10 s using a MLX Microtitre® Plate Luminometer (Dynex Technologies, USA) after delivering 100 μL of the reconstituted Luciferase assay reagent into each well through an automated feeding system.
The amount of protein in each cell lysate was determined using a Pierce BCA Protein Assay kit according to manufacture’s instructions. Briefly, the cell lysate (20 μL) and bovine serum albumin (BSA) protein standards (20 μL) were transferred into a transparent 96 well plate to which mix reagent A/B (200 μL, provided in the kit) was added. The plate was incubated with slight shaking at 37 °C for 30 min after which time the absorbance was measured at 562 nm using a SpectraMax 190 plate reader (Molecular Device, USA). All luciferase measurements were normalised against the protein content of each well. Knockdown experiments were repeated on at least two different occasions to confirm that the results were reproducible (results of only one experiment reported).
Confocal fluorescence internalisation studies
A549-luc cells (1.2 × 105 cells per well) were incubated with LPR complexes in 24-well plates fitted with sterilized 13 mm circular cover slips (Agar Scientific, UK) as described above with the exception of using fluorescently labelled lipid (10% of lipid replaced with BODIPY-HPC (green)) and siRNA labelled with TM-Rhodamine (red) using a Label-IT® TM-Rhodamine siRNA Tracker Intracellular Localization Kit according to the manufacturer’s protocol. In addition LPRs were also made using FITC labelled peptides, either FITC-K6B2-L1-[Y] or FTIC-(H3K)4B1-L1-[Y] (green, 100%) and TM-Rhodamine (red) labelled siRNA and (unlabelled) lipid. The total volume in each well was 400 μL, with a siRNA concentration of 50 nM.After incubation for 4 h or 24 h, the complexes were removed and the cells washed once with PBS before fixation of the cells with 4% w/v paraformaldehyde in PBS for 20 min, and permeabilization using 0.2% v/v Triton X-100 solution for 20 min followed by treatment with 1 mg mL−1 of sodium borohydride in PBS for 5 min. The cell nucleus was stained blue by the addition of an aqueous DAPI solution (1 μg mL−1) for 1 min. The cover slips were washed with PBS several times and mounted onto microscope slides using Mowiol 4-88 mounting media containing 2.5% w/v DABCO. Confocal fluorescence images were acquired on a Leica DMIRE2 confocal fluorescence laser-scanning microscope (Leica Microsystems, Germany).
Acknowledgements
The authors would like to thank Dr O. Argyros and Prof R. P Harbottle for the episomal S/MAR luciferase vector and Dr M. T. Thanou for providing the stably transfected A549-Luc cell line. EPSRC are thanked for the award of a Nanotechnology Grand Challenge Grant (EP/G061521/1) (to L. K., K. W. and F. C.) and for a DTG studentship to A. M. The MRC are thanked for a studentship to N. D. (G0900207). We also gratefully acknowledge ISIS for the award of neutron beam time and Dr Ann Terry (Instrument Scientist, ISIS) for assistance with the SANS experiments. The expert help of Dr Alex Drake and Dr Tam Bui (Biomolecular Spectroscopy Centre, King's College London) is gratefully acknowledged for the circular dichroism/UV measurements, and the help of Dr Alice Warley (Centre for Ultrastructural Imaging, King's College London) with the TEM measurements is acknowledged.Notes and references
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- H. M. Aliabadi, B. Landry, C. Sun, T. Tang and H. Uludag, Biomaterials, 2012, 33, 2546–2569 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- D. L. Gary, N. Puri and Y. Y. Won, J. Controlled Release, 2007, 121, 64–73 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122–142 CrossRef CAS PubMed.
- T. Endoh and T. Ohtsuki, Adv. Drug Delivery Rev., 2009, 61, 704–709 CrossRef CAS PubMed.
- Y.-H. Wang, Y.-W. Hou and H.-J. Lee, J. Biochem. Biophys. Methods, 2007, 70, 579–586 CrossRef CAS PubMed.
- S. W. Kim, N. Y. Kim, Y. Bin Choi, S. H. Park, J. M. Yang and S. Shin, J. Controlled Release, 2010, 143, 335–343 CrossRef CAS PubMed.
- P. Kumar, H. Wu, J. L. McBride, K.-E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–43 CrossRef CAS PubMed.
- S.-S. Kim, C. Ye, P. Kumar, I. Chiu, S. Subramanya, H. Wu, P. Shankar and N. Manjunath, Mol. Ther., 2010, 18, 993–1001 CrossRef CAS PubMed.
- P. Kumar, H.-S. Ban, S.-S. Kim, H. Wu, T. Pearson, D. L. Greiner, A. Laouar, J. Yao, V. Haridas, K. Habiro, Y.-G. Yang, J.-H. Jeong, K. Y. Lee, Y.-H. Kim, S.-W. Kim, M. Peipp, G. H. Fey, N. Manjunath, L. D. Shultz, S.-K. Lee and P. Shankar, Cell, 2008, 134, 577–586 CrossRef CAS PubMed.
- P. Midoux, C. Pichon, J.-J. Yaouanc and P.-A. Jaffr”s, Br. J. Pharmacol., 2009, 157, 166–178 CrossRef CAS PubMed.
- Q. Leng, P. Scaria, J. Zhu, N. Ambulos, P. Campbell and A. J. Mixson, J. Gene Med., 2005, 7, 977–986 CrossRef CAS PubMed.
- Q. Leng, P. Scaria, P. Lu, M. C. Woodle and A. J. Mixson, Cancer Gene Ther., 2008, 15, 485–495 CrossRef CAS PubMed.
- S.-T. Chou, Q. Leng, P. Scaria, M. Woodle and A. J. Mixson, Cancer Gene Ther., 2011, 18, 707–716 CrossRef CAS PubMed.
- S.-T. Chou, Q. Leng, P. Scaria, J. D. Kahn, L. D. Tricoli, M. Woodle and A. J. Mixson, Biomacromolecules, 2013, 14, 752–760 CrossRef CAS PubMed.
- B. Langlet-Bertin, C. Leborgne, D. Scherman, B. Bechinger, A. J. Mason and A. Kichler, Pharm. Res., 2010, 27, 1426–1436 CrossRef CAS PubMed.
- D. L. McKenzie, E. Smiley, K. Y. Kwok and K. G. Rice, Bioconjugate Chem., 2000, 11, 901–909 CrossRef CAS PubMed.
- M. L. Read, S. Singh, Z. Ahmed, M. Stevenson, S. S. Briggs, D. Oupicky, L. B. Barrett, R. Spice, M. Kendall, M. Berry, J. A. Preece, A. Logan and L. W. Seymour, Nucleic Acids Res., 2005, 33, e86 CrossRef PubMed.
- M. F. Mohd Mustapa, P. C. Bell, C. A. Hurley, A. Nicol, E. Guénin, S. Sarkar, M. J. Writer, S. E. Barker, J. B. Wong, M. A. Pilkington-Miksa, B. Papahadjopoulos-Sternberg, P. A. Shamlou, H. C. Hailes, S. L. Hart, D. Zicha and A. B. Tabor, Biochemistry, 2007, 46, 12930–12944 CrossRef PubMed.
- A. D. Miller, Angew. Chem., Int. Ed., 1998, 37, 1768–1785 CrossRef.
- M. J. Writer, B. Marshall, M. A. Pilkington-Miksa, S. E. Barker, M. Jacobsen, A. Kritz, P. C. Bell, D. H. Lester, A. B. Tabor, H. C. Hailes, N. Klein and S. L. Hart, J. Drug Targeting, 2004, 12, 185–194 CrossRef CAS PubMed.
- J. B. Wong, S. M. Grosse, A. B. Tabor, S. L. Hart and H. C. Hailes, Mol. BioSyst., 2008, 4, 532–541 RSC.
- C. A. Hurley, J. B. Wong, J. Ho, M. Writer, S. A. Irvine, M. J. Lawrence, S. L. Hart, A. B. Tabor and H. C. Hailes, Org. Biomol. Chem., 2008, 6, 2554–2559 CAS.
- M. F. Mohd Mustapa, S. M. Grosse, L. Kudsiova, M. Elbs, E.-A. Raiber, J. B. Wong, A. P. R. Brain, H. E. J. Armer, A. Warley, M. Keppler, T. Ng, M. J. Lawrence, S. L. Hart, H. C. Hailes and A. B. Tabor, Bioconjugate Chem., 2009, 20, 518–532 CrossRef PubMed.
- S. M. Grosse, A. D. Tagalakis, M. F. Mohd Mustapa, M. Elbs, Q.-H. Meng, A. Mohammadi, A. B. Tabor, H. C. Hailes and S. L. Hart, FASEB J., 2010, 24, 2301–2313 CrossRef CAS PubMed.
- L. Kudsiova, B. Fridrich, J. Ho, M. F. Mohd Mustapa, F. Campbell, K. Welser, M. Keppler, T. Ng, D. J. Barlow, A. B. Tabor, H. C. Hailes and M. J. Lawrence, Mol. Pharmaceutics, 2011, 8, 1831–1847 CrossRef CAS PubMed.
- L. Kudsiova, J. Ho, B. Fridrich, R. Harvey, M. Keppler, T. Ng, S. L. Hart, A. B. Tabor, H. C. Hailes and M. J. Lawrence, Mol. BioSyst., 2011, 7, 422–436 RSC.
- K. Welser, F. Campbell, L. Kudsiova, A. Mohammadi, N. Dawson, S. L. Hart, D. J. Barlow, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Pharmaceutics, 2013, 10, 127–141 CrossRef CAS PubMed.
- J. J. Turner, S. W. Jones, S. A. Moschos, M. A. Lindsay and M. J. Gait, Mol. BioSyst., 2007, 3, 43–50 RSC.
- G. Siligardi and A. F. Drake, Biopolymers, 1995, 37, 281–292 CrossRef CAS PubMed.
- D. Gray, Circular Dichroism of Protein–Nucleic Acid Interactions, Circular Dichroism and the Conformational Analysis of Biomolecules, (The Language of Science), ed. G. Fasman, Plenum Press, New York, 1996, pp. 469–500 Search PubMed.
- R. W. Woody, Methods Enzymol., 1995, 246, 34–71 CAS.
- J. O. Radler, I. Koltover, T. Salditt and C. R. Safinya, Science, 1997, 275, 810–814 CrossRef CAS PubMed.
- B. Pitard, O. Aguerre, M. Airiau, A.-M. Lachages, T. Boukhinkachvili, G. Byk, C. Dubertret, C. Herviou, D. Scherman, J.-F. Mayaux and J. Crouzet, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14412–14417 CrossRef CAS.
- O. Argyros, S. P. Wong, K. Gowers and R. P. Harbottle, PLoS One, 2012, 7, e47920 CAS.
- A. D. Tagalakis, L. He, L. Saraiva, K. T. Gustafsson and S. L. Hart, Biomaterials, 2011, 32, 6302–6315 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00754b |
| ‡ Present address: Sandoz GmbH, Biochemiestraße 10, A-6250 Kundl, Austria. |
| § Present address: Leiden Institute of Chemistry, Gorlaeus Laboratories, Einsteinweg 55, 2333 CC Leiden, Netherlands. |
| This journal is © The Royal Society of Chemistry 2016 |