
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysyl oxidase promotes epithelial-to-mesenchymal transition during paraquat-induced pulmonary fibrosis
Jinfeng
Wang
,
Yong
Zhu
,
Jiuting
Tan
,
Xiaoxiao
Meng
,
Hui
Xie
and
Ruilan
Wang
*
Department of Intensive Care Unit, Shanghai General Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai, 201620, China. E-mail: wangyusun@hotmail.com; Fax: +86-21-37798527; Tel: +86-21-37798528
First published on 1st December 2015
Abstract
Lysyl oxidase (LOX) is a copper-dependent amine oxidase that plays a critical role in pulmonary fibrosis. Our previous study demonstrated that epithelial-to-mesenchymal transition (EMT) was strongly associated with paraquat (PQ) induced pulmonary fibrosis. This present study was aimed to evaluate the potential involvement of LOX on EMT in the process of pulmonary fibrosis induced by PQ. We established an in vivo rat model and an in vitro cell model induced by PQ treatment and found that LOX protein expression was significantly up-regulated and collagen deposition was enhanced in rats. The EMT process was strongly found in A549 and RLE-6TN cells after PQ exposure. After inactivating LOX with an inhibitor, pulmonary fibrosis was significantly reduced and EMT was also suppressed. Additionally, small interfering RNA (siRNA) targeting LOX was used to silence LOX expression to observe EMT in A549 cells. As a result, LOX could promote the progress of EMT, and inactivating LOX alleviated the EMT process in PQ-induced pulmonary fibrosis and mesenchymal-to-epithelial transition (MET) occurred after inactivating LOX in vitro and in vivo. In conclusion, LOX could promote the progress of EMT and inactivating LOX alleviated EMT in PQ-induced pulmonary fibrosis. Therefore, LOX could potentially be a new candidate therapeutic target for pulmonary fibrosis induced by PQ by regulating the balance between EMT and MET.
1. Introduction
Paraquat (PQ) is an effective and widely used herbicide and deaths due to acute PQ poisoning are frequently reported over the last few decades. Lung is the main target organ for the pathological effects of PQ where the PQ concentration can be 6 to 10 times higher than that in plasma, and PQ is retained in the lungs even when blood levels start to decrease.1 PQ rapidly accumulates in lungs after administration, causing acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) and extensive pulmonary fibrosis, resulting in dyspnea and eventually death from respiratory failure.2 No specific antidote has been currently recommended, leading to high mortality of PQ poisoning. Pulmonary fibrosis is a major manifestation and a leading cause of death due to PQ poisoning.3Pulmonary fibrosis is characterized by the loss of alveolar structures and functions following the apoptosis of epithelial and endothelial cells, proliferation of fibroblasts and excessive deposition of the extracellular matrix (ECM). Myofibroblasts are responsible for the deposition of collagen,4 which come from the differentiation of resident fibroblasts, bone marrow-derived precursors, and EMT.5 EMT is characterized by the decreased expression of an epithelial marker (e.g. E-cadherin, ZO-1), the loss of cell–cell adhesion and cell polarity, as well as the increased expression of mesenchymal markers (e.g. vimentin, N-cadherin and α-SMA), the reorganization of cytoskeleton and the gain of cell motility. Recently, increasing research efforts have been devoted to the role of EMT in fibrosis. According to several studies, over 30% of the myofibroblasts are derived from epithelial cells by means of EMT.6 Alveolar epithelial cells (AECs) are the sources of EMT in injured lungs, which undergo rapid proliferation and differentiation into mature myofibroblasts, secrete collagen and eventually lead to fibrosis. A large amount of evidence suggests that EMT has been implicated in the pathogenesis of pulmonary fibrosis.7–9
As fibrosis develops, collagen fibrils become permanently cross-linked as a result of the action of lysyl oxidase (LOX), which is responsible for the oxidation of specific lysine residues in collagen and elastin,10 leading to the formation of molecular crosslink essential for the stabilization of the ECM. This conversion plays an important role in growth, stabilization, remodeling and repairing of the organism.11,12 Although initially identified by their ability to crosslink collagen and elastin fibers, LOX has been shown to carry out intracellular functions beyond extracellular cross-linkage. It is reported that HIF-1 up-regulated the expression of LOX in hypoxic tubular epithelial cells to enable EMT, migrate toward the interstitium and promote fibrosis.13 In addition, LOX is also involved in eye and myocardial fibrosis.14 However the relationship between LOX and EMT is still unknown in pulmonary fibrosis. This study was conducted to investigate the role of LOX in the process of EMT in PQ-induced pulmonary fibrosis.
2. Methods and materials
2.1 Material and reagents
Standardized PQ, BAPN (Sigma, St. Louis, MO, USA). F-12K, RPMI-1640 media (Beijing Genosys Tech-Trading Co., Ltd, China). Bovine fetal serum, Trypsin (Gibco, Co., USA). RIPA, SDS-PAGE, BCA kits, Cell Counting Kit-8 (CCK-8), ECL reagents (Beyotime Institute of Biotechnology). 20% PQ solution (Syngenta Nantong Crop. Protection Co., Ltd, China). Antibodies: LOX, ZO-1, vimentin (Santa Cruz, USA); GAPDH, E-cadherin, α-SMA (Abcam, Cambridge, MA, USA); GSK-3β, Snail (Cell Signaling Technology, Danvers, MA, USA); β-Tubulin (Abmart, Shanghai, China), Secondary antibody (Biomart, China). Histostain-Plus Kit, Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA); siRNA (Gene Pharma, Shanghai, China).2.2 Animal experiment
Forty healthy male Spragne-Dawley (SD) rats (weight of 250 ± 30 g) were obtained from Shanghai Laboratory Animal Center (Animal permit number: SYXK (Hu) 2009-0086) and housed individually in a standard animal room. The rats were then randomly divided into four groups: the control (CTL) group, the β-aminopropionitrile (BAPN) treating group, the paraquat (PQ) treating group and the BAPN + PQ (BPQ) treating group (Fig. 4A). BAPN is the irreversible inhibitor of LOX. The mechanism of inhibition is postulated, which involves the formation of a covalent bond between an enzyme nucleophile and a ketenimine formed from BAPN by enzyme-assisted beta-proton abstraction.15 Rats in the CTL group underwent gavage with 1 mL of normal saline (NS) after prior 24 h intraperitoneal injections with 100 mL of NS. The BAPN group underwent gavage with 1 mL of NS and the BPQ group received an intragastric infusion of 20% PQ solution (50 mg kg−1) after prior 24 h intraperitoneal injections with 100 mL of BAPN (100 mg kg−1). The PQ group received an intragastric infusion of 20% PQ solution (50 mg kg−1) after intraperitoneal injections with 100 mL of NS for 24 h. BAPN and NS intraperitoneal injections were performed daily from the beginning until the end of the experiment. The rats were sacrificed by intraperitoneal injection of pentobarbital after PQ poisoning for 72 h. The upper lobe of the right lung was extracted and frozen in liquid nitrogen for subsequent assays. The left lung was soaked in formalin solution and processed for embedding in paraffin. However, some rats in PQ and BPQ groups could not survive till the predetermined time points, therefore only six rats in PQ and BPQ groups were used for the analysis. All the experiments were performed under the guidelines for the use of experimental animals and with the permission of the animal ethical committee of Shanghai Jiao Tong University.2.3 Cell culture
RLE-6TN cells (rat lung epithelial cell line) and A549 cells (human lung adenocarcinoma epithelial cell line) were purchased from ATCC. RLE-6TN cells were cultured in RPMI-1640 media containing 10% fetal bovine serum (FBS). A549 cells were cultured in F-12K media containing 10% FBS. The tissue culture flasks were put into a 37 °C incubator with 5% CO2 and the media were changed every other day.2.4 Cytotoxicity assay
The CCK-8 assay was utilized to determine the survival rate of the RLE-6TN and A549 cells. Cells were cultured in the 96-well plates with a concentration of 1 × 104 cells per well. After 24 h, the A549 cells were treated with different concentrations of PQ (100, 200, 300, 400, 500, 600, 700, 800, 900 and 1000 μmol L−1) and RLE-6TN (20, 40, 60, 80, 100, 120, 140, 160, 180 and 200 μmol L−1) for 24 h (our previous study has developed that cells underwent EMT, under review). Then 10 μL of the CCK-8 agent was added to each pole to react for 3 h. Finally, the absorbance (A) was measured at 450 nm using a microplate reader. The cell survival rate (%) = ((the mean of Aexperiment![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) group − the mean of Ablankgroup)/(the mean of Acontrolgroup − the mean of Ablankgroup)) × 100%. There were 3 parallel experiments for every group and the average value was selected.
group − the mean of Ablankgroup)/(the mean of Acontrolgroup − the mean of Ablankgroup)) × 100%. There were 3 parallel experiments for every group and the average value was selected.
2.5 Inhibitor intervention
The RLE-6TN cells were cultured in 60 mm petri dish at a concentration of 1.5 × 105 L−1. Cells were divided into four groups: CTL, BAPN, PQ, BPQ and subsequently treated with different concentrations of BAPN (0, 200, 0, 200 μmol L−1) for 2 h,16 then 80 μmol L−1 of PQ was added to PQ and BPQ groups for 24 h. The A549 cells were cultured in 60 mm petri dish with a concentration of 7 × 105 L−1. Cells were divided into six groups: CTL, BA300, PQ, BA100 + PQ, BA200 + PQ and BA300 + PQ. Subsequently, the cells were treated with different concentrations of BAPN (0, 300, 0, 100, 200, 300 μmol L−1) for 2 h, then 800 μmol L−1 PQ was added to PQ, BA100 + PQ, BA200 + PQ and BA300 + PQ groups. The morphology of cells was observed using a microscope. Three parallel experiments were set for every group.2.6 siRNA transfection
The A549 cells were cultured in 60 mm petri dish with a concentration of 5 × 105 L−1 and divided into 6 groups, including the control group (CTL), the positive control group (P), the negative control group (N), the transfection group (SI), the PQ-treated group (PQ) and the transfection + PQ group (SIPQ). A549 cells were transfected with 200 pmol LOX siRNA using 10 μL of lipofectamin2000 according to the instructions of the manufacturer. All siRNA experiments included positive control (GAPDH, to ensure transfection, RNA extraction and detection method is reliable, sense 5′-UGA CCU CAA CUA CAU GGU UTT-3′), a nonspecific negative control group (N) (specific control and non-specific control siRNAs, non- homologous with target genes, sense 5′-UUC UCC GAA CGU GUC ACG UTT-3′). Then cells in the SIPQ group were treated with 800 μmol L−1 of PQ after 48 h. After 24 h of incubation, the effect of LOX silencing was determined and the change in the expression of other proteins was determined by western blot.2.7 Immunofluorescence
The A549 cells were washed three times with phosphate-buffered saline (PBS) and fixed in 4% (w/v) formaldehyde for 10 min at room temperature. They were then permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 10% FBS for 1 h. Subsequently, the cells were incubated with an anti-primary antibody (ZO-1, 1:10; α-SMA, 1:50; LOX, 1:10) at 4 °C overnight and then with FITC-conjugated (ZO-1, LOX) or TRITC-conjugated (α-SMA) secondary antibodies for 1 h in the dark. Nuclei were counterstained with DAPI for 7 min. Finally, the cells were observed using a Leica confocal microscope (Leica TCS SP5, Germany) and five visions were randomly selected for each group.
2.8 Migration assays
RLE-6TN (5 × 104) and A549 (1 × 104) cells after the corresponding treatment were placed into the upper wells of a 24-well transwell chamber (8 μm pore size, Costar, USA) in serum-free medium. The cells were allowed to invade through the Matrigel for 10 h at a 37 °C incubator with 5% CO2, using medium containing 10% FBS in the lower chamber as a chemoattractant. Following migration, cells on the upper membrane surface were removed by scraping with a cotton swab. Cells migrating to the pores of the underlying membrane were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The number of cells was counted under a fluorescence microscope (×100) with three random fields for each well. Data presented are representative of four individual wells.2.9 Western blot
Samples from the cells and rat lungs were lysed with RIPA. After centrifugation, the protein concentration was measured using BCA protein assay kits with bovine serum albumin (BSA) as the standard. A total of 60 μg of protein from each sample were separated by 8% SDS-PAGE and transferred onto the polyvinylidene fluoride (PVDF) membrane. Then the membranes were blocked with 5% dehydrated milk in TBST for 2 h at room temperature, followed by incubation with primary antibodies (LOX, 1:100; E-cadherin, 1:300; ZO-1, 1:200; α-SMA, 1:120; vimentin, 1:200; GSK-3β, 1:200; Snail, 1:200; GAPDH, 1:500; β-Tubulin, 1:1000) at 4 °C overnight and an appropriate HRP-conjugated secondary antibody for 1 h. The immune complexes were detected using ECL reagents. All experiments were repeated at least three times. The protein expression levels were also quantified using Image J software for windows.
2.10 Immunohistochemistry, H&E and Masson's trichrome
Lung samples were fixed with 10% formalin, embedded in paraffin and sectioned for H&E staining, immunohistochemical staining and Masson's trichrome staining. The slides were examined using a light microscope and photographed. Immunohistochemical staining was performed using the Histostain-Plus Kit followed by DAB development. The sections were incubated with primary antibodies of anti-LOX (1:10) and fluorescence-conjugated secondary antibodies and stained with DAPI. Random 4 fields were chosen and photographed, and LOX-positive cells were counted.
2.11 Statistical analysis
All data were expressed as means ± SD. Comparisons between different groups were conducted using 2-tailed student's t test. Values of P < 0.05 were considered to be significantly different.3. Results
3.1 LOX expression elevated and EMT increased after PQ-induced pulmonary fibrosis in rats
In our previous study, we found that PQ exposure resulted in an increased expression of α-SMA and collagen deposition at an early stage (2 h) and in a significant decrease of E-cadherin at 48 h, which indicated that EMT participated in the development of lung fibrosis induced by PQ at an early stage.17 Then we set 72 h as the point time of PQ exposure to investigate the relationship between LOX and EMT. According to the results, the expression level of LOX was notably increased after PQ treatment (Fig. 1A and B). Lungs of rats in CTL showed a normal lung structure and no lesions. The acute widespread lesions of capillary endothelial cells and alveolar epithelial cells were observed with the infiltration of inflammatory cells around the interstitial lungs of rats in the PQ group (upper of Fig. 1C). Besides, an amount of collagen in the blue accumulation was observed in lungs of rats in the PQ group and the lung structure was obviously damaged (lower of Fig. 1C) as compared with the CTL group. In addition, the expression of EMT transcriptional factors Snail, mesenchymal markers α-SMA and vimentin was increased while epithelial markers E-cadherin and ZO-1 (Fig. 1D) were significantly decreased after PQ exposure (P < 0.05). Our results demonstrated that LOX was up-regulated and EMT was enhanced in the PQ-induced pulmonary fibrosis model.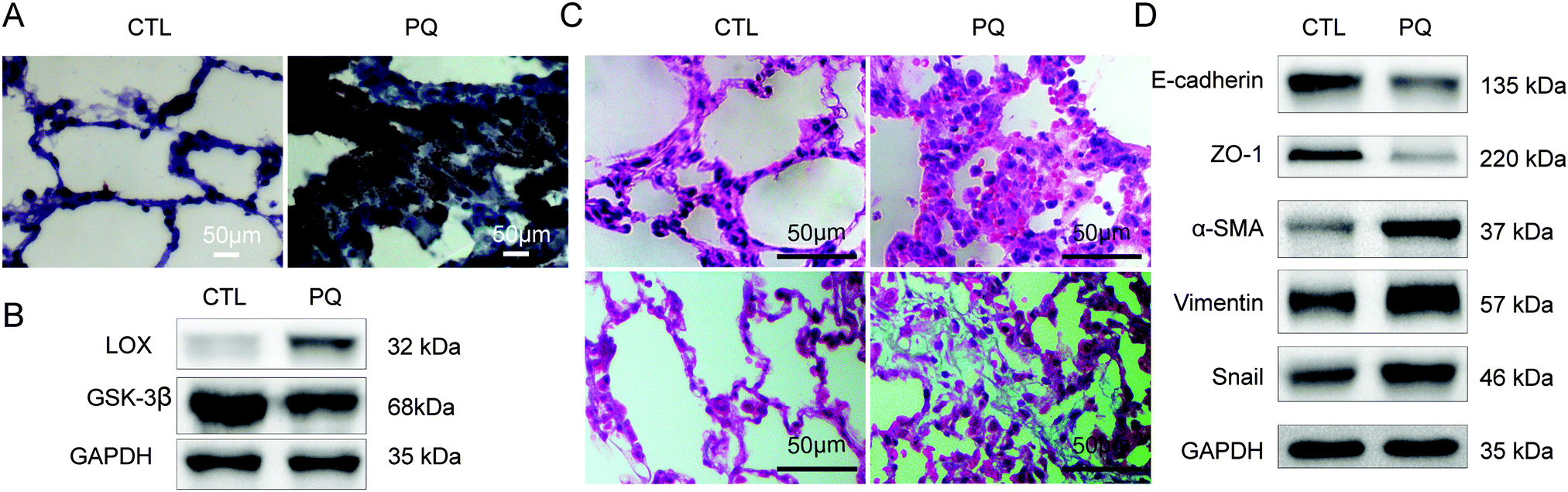 | ||
| Fig. 1 LOX expression was elevated during PQ-induced pulmonary fibrosis in rats. (A) Histological comparison of the expression of LOX in the control group (CTL) and PQ after treatment for 72 h (PQ). (B) Western blot for LOX, GSK-3β, expression. (C) Lung H&E staining (up); collagen deposition with Masson's trichrome staining (down) of rats (×400). (D) Western blot for EMT markers ZO-1, E-cadherin, α-SMA, vimentin and Snail. | ||
3.2 Overexpressed LOX and the enhanced EMT process were found after PQ poisoning in vitro
RLE-6TN and A549 cells were used to study the relationship between LOX and EMT in vitro after PQ treatment. The survival rate of the RLE-6TN cells and A549 cells was decreased after incubation in PQ solutions for 24 h in a concentration-dependent manner. The PQ concentration of 80 μmol L−1 with the RLE-6TN cell survival rate of 47.88% (Fig. 2A) and 800 μmol L−1 with the A549 cells survival rate of 54.96% (Fig. 2B) was selected as the intervention point for RLE-6TN cells and A549 cells, respectively.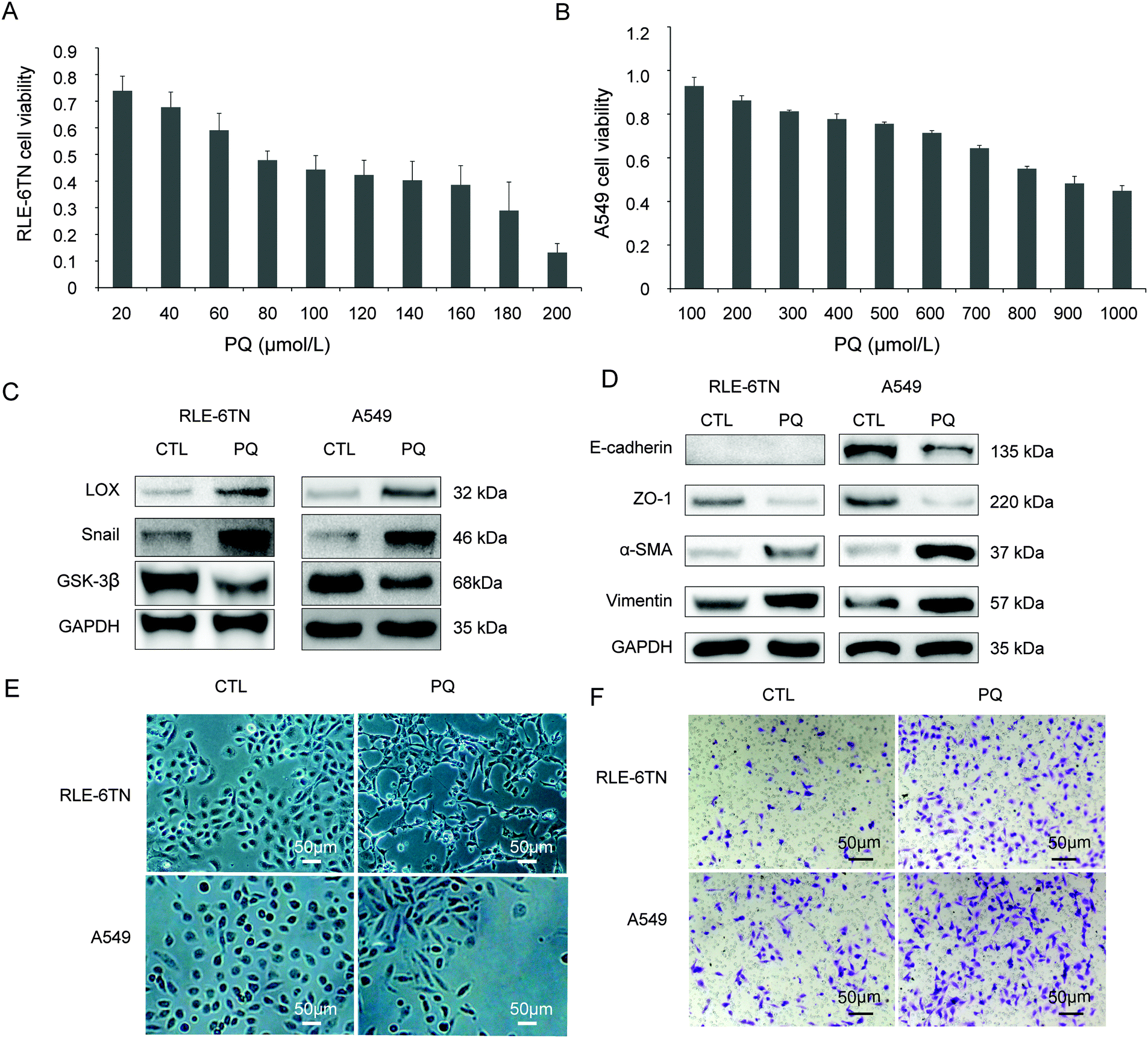 | ||
| Fig. 2 PQ induced EMT in RLE-6TN and A549 cells. (A) The survival rate of the A549 cells decreased after incubation in PQ solutions for 24 h as the concentration increased. (B) The survival rate of the RLE-6TN cells. (C) RLE-6TN and A549 were exposed to PQ for 24 h followed by analysis of LOX, Snail, GSK-3β protein levels by western blot. (D) RLE-6TN and A549 were exposed to PQ for 24 h followed by analysis of ZO-1, E-cadherin, α-SMA, vimentin protein levels by western blot. (E) Morphology characteristics of RLE-6TN and A549 cells between the control group (CTL) and the group to PQ (×100). (F) Cell migration in a transwell chamber between CTL and PQ groups. | ||
In the CTL group, RLE-6TN and A549 cells showed an epithelial-specific round or ellipse morphology. After PQ treatment for 24 h, the epithelial monolayer was disrupted and cells acquired a spindle-shaped morphology (Fig. 2E). In the PQ group, the protein levels of LOX, Snail (Fig. 2C), α-SMA and vimentin (Fig. 2D) were enhanced, while the expression of epithelial markers E-cadherin and ZO-1 (Fig. 2D) and GSK-3β (Fig. 2C) was down-regulated. The loss of epithelial phenotypes and the acquisition of mesenchymal traits through the process of EMT have been observed. Additionally, the migration rates of RLE-6TN cells and A549 cells were remarkably enhanced after PQ poisoning (Fig. 2F).
3.3 Inactivating LOX alleviated EMT induced by PQ in vitro
After inactivating LOX by BAPN, the expression of LOX and Snail was significantly reduced by in RLE-6TN cells (Fig. 3A, P < 0.05) and A549 cells (Fig. 3B, P < 0.05). The epithelial monolayer was changed into a spindle-shaped morphology after PQ poisoning for 24 h in RLE-6TN (Fig. 3C) and A549 cells (Fig. 3D). No obvious change of cell morphology was found in the group treated with 200 μmol L−1 of BAPN or 300 μmol L−1 of BAPN (Fig. 3C and D) as compared with the CTL group. However, the degree of the changes from epithelial monolayer to spindle-shaped morphology was decreased in the BPQ group (Fig. 3C and D). Particularly, A549 cells partially uniformed spindle-shaped morphology and the degree of changes was decreased with the increasing concentration of BAPN (Fig. 3D). | ||
| Fig. 3 Inactivating LOX by BAPN alleviated PQ-induced EMT in RLE-6TN and A549 cells. (A) Western blot for LOX, Snail, GSK-3β protein levels in RLE-6TN cells in different groups. CTL: control group, BAPN: treated with 200 μmol L−1 BAPN, PQ: exposed to 80 μmol L−1 PQ for 24 h, BPQ: pretreated with 200 μmol L−1 BAPN + exposed to 80 μmol L−1 PQ. (B) Western blot for LOX, Snail, GSK-3β protein levels in A549 cells in different groups. (C) Morphology characteristics of RLE-6TN cells between different groups (×100). (D) Morphology characteristics of A549 cells between different groups (×100). The control group (CTL), treated with 300 μmol L−1 BAPN (BA300), exposed to 800 μmol L−1 PQ (PQ) for 24 h, pretreated with 100 μmol L−1 BAPN + exposed to 800 μmol L−1 PQ (BA100 + PQ), 200 μmol L−1 BAPN + 800 μmol L−1 PQ (BA200 + PQ), 300 μmol L−1 BAPN + 800 μmol L−1 PQ (BA300 + PQ). (E) Western blot for EMT markers E-cadherin, ZO-1, α-SMA, vimentin in RLE-6TN cells in different groups. (F) Western blot for EMT markers ZO-1, E-cadherin, α-SMA, vimentin in A549 cells in different groups. | ||
The expression of α-SMA and vimentin (Fig. 3E and F, P < 0.05) was significantly increased and ZO-1, E-cadherin and GSK-3β (Fig. 3A and B, P < 0.05) were significantly decreased after PQ exposure. However, these changes were weakened after BAPN treatment in a concentration-depended manner (Fig. 3E and F). These results demonstrated that LOX is a key regulator of EMT induced by PQ in RLE-6TN and A549 cells.
3.4 Inactivating LOX by BAPN alleviated PQ-induced ALI and pulmonary fibrosis in rats
In the process of the experiment, we observed that the death rate of rats was 40% (n = 10) both in BPQ and PQ groups within 72 h after PQ treatment. It suggested that BAPN is not really effective in reducing the mortality after acute PQ exposure. LOX and Snail were reduced after inactivating LOX by BAPN (Fig. 4B and C). α-SMA and vimentin were increased and E-cadherin, ZO-1 (Fig. 4D) and GSK-3β (Fig. 4B) were decreased after PQ treatment. However, the degree of changes was decreased in the BPQ group (Fig. 4B–D, P < 0.05). | ||
| Fig. 4 Inactivating LOX by BAPN alleviated ALI, EMT and pulmonary fibrosis in rats. (A) Treatment of different group mice. CTL: control group, BAPN: treated with 100 mg kg−1 per day BAPN, PQ: exposed to 50 mg Kg−1 20% PQ for 72 h, BPQ: pretreated with 100 mg kg−1 per day BAPN + exposed to PQ, NS: normal saline. (B) Western blot for LOX, Snail, GSK-3β protein levels in rats in different groups. (C) Histological comparison of rat lung expression of LOX in different groups. (D) Western blot for EMT markers ZO-1, E-cadherin, α-SMA, vimentin in different groups. (E) H&E staining of rat lung (up) and collagen deposition with Masson's trichrome staining (down) in different groups (×400). | ||
The destruction of the alveolar architecture, infiltration of inflammatory cells and thickening of the alveolar septum were observed in the PQ group by H&E staining (upper in Fig. 4E). These situations were obviously improved and the collagen accumulation was also decreased in the BPQ group compared with those in the PQ group (lower in Fig. 4E). The manifestation of lung injury and lung fibrosis was reduced by inactivating LOX with BAPN.
3.5 Silencing of LOX alleviated PQ-induced EMT
Compared with the CTL group, the positive transfection group showed a significant decrease of GAPDH (P < 0.05), suggesting a successful silence (Fig. 5A and B). The LOX expression was obviously lower after transfected by siRNA than that in the CTL group (Fig. 5A and C). LOX was down-regulated in the SI group in A549 cells (Fig. 5D and E) without influencing the cell morphology compared with the CTL group (Fig. 5F). ZO-1 (Fig. 5G and H), E-cadherin (Fig. 5H) and GSK-3β (Fig. 5D) were down-regulated, while Snail (Fig. 5D), α-SMA (Fig. 5G and H) and vimentin (Fig. 5H) were up-regulated after treatment with PQ. Furthermore, these changes were significantly attenuated in the SIPQ group.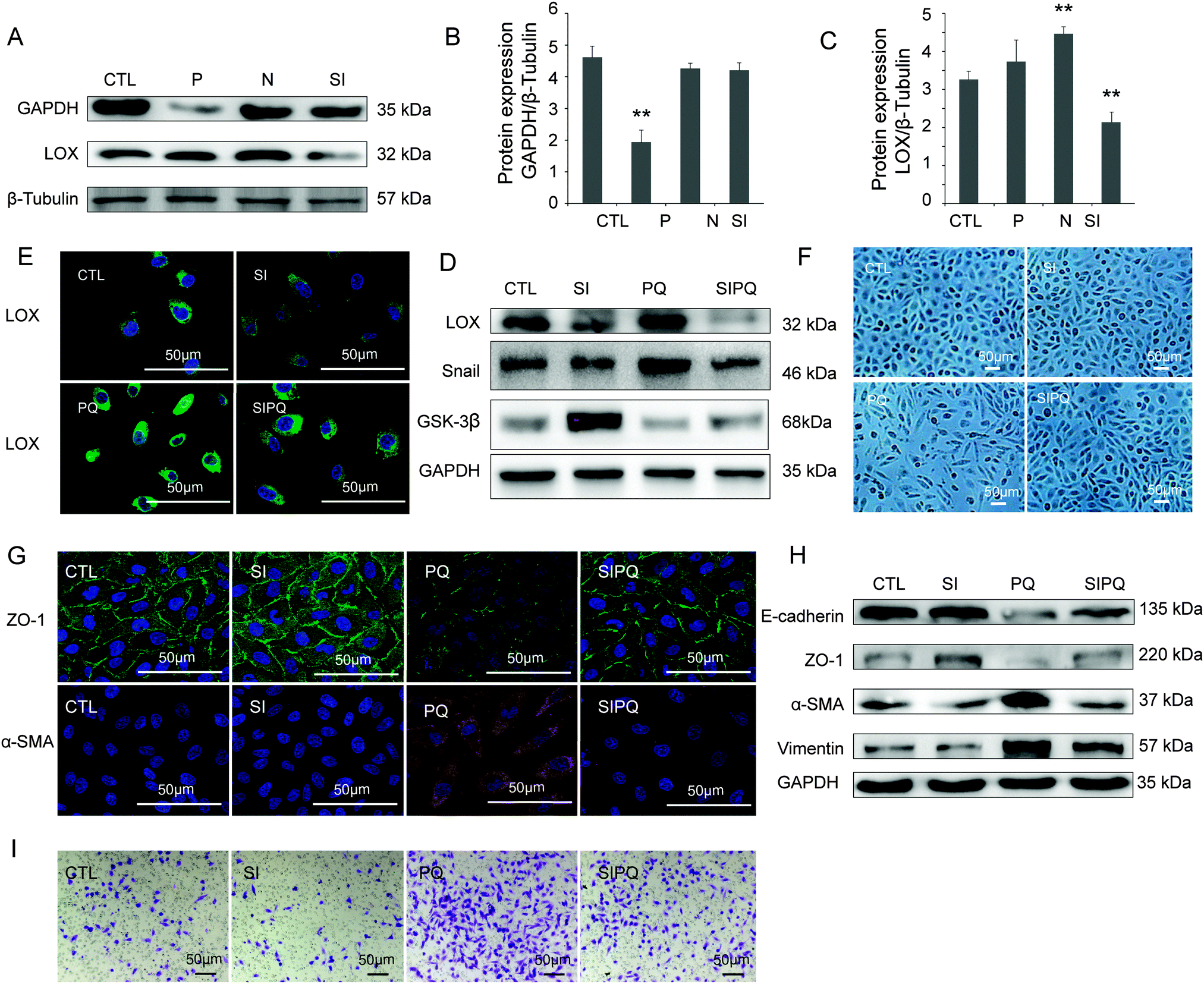 | ||
| Fig. 5 Knockdown of LOX by specific small interfering RNA (siRNA) attenuated PQ-induced EMT and migration of A549 cells after PQ poisoning. (A) Representative western blot showing the LOX, GAPDH protein expression of A549 cells in different groups. CTL: control group, P: positive control group (transfected GAPDH for 72 h), N: negative control group, SI: transfection group (transfected LOX 72 h), PQ: PQ-treated group (72 h), SIPQ: PQ treating 24 h after transfected LOX for 48 h. (B and C): Quantification of LOX, GAPDH protein expression related to β-Tubulin in different groups. β-Tubulin served as a loading control. (D) After silencing of LOX, representative western blot showing the LOX, Snail, GSK-3β protein levels in A549 cells in different groups. (E) Immunostaining images of LOX in different groups (×400). (F) Morphological characteristics of A549 cells in different groups (×100). (G) Immunostaining images of ZO-1, α-SMA in different groups (×400). (H) ZO-1, E-cadherin, α-SMA, vimentin protein expression of A549 cells with or without PQ treatment for 24 h. (I) A549 cells migration in a transwell chamber in different groups. | ||
3.6 Cell migration was reduced after silencing LOX
The transwell migration assay, together with the LOX siRNA transfection method, was performed to study the effect of LOX on the migration capability of A549 cells. Compared with the CTL group, the migration capability of A549 cells was increased after PQ exposure. However, the increase of migration capability of A549 cells was significantly weakened in the SIPQ group (Fig. 5I, P < 0.05), which suggested that LOX is involved in the regulation of the migration capability of A549 cells.4. Discussion
PQ rapidly accumulates in the lungs after poisoning, causing acute injury and pulmonary fibrosis. Kevin K9 found that AECs underwent EMT both in vitro and in vivo which can contribute significantly to pulmonary fibrosis after lung injury. Our previous studies demonstrated that EMT occurred during pulmonary fibrosis induced by PQ in the rat model.17 EMT is characterized by the loss of epithelial cell polarity, the loss of cell–cell contacts, and the acquisition of mesenchymal markers and increased cell motility. In the present experiment, we also found that EMT was developed in PQ-induced pulmonary fibrosis in vivo and in vitro.LOX is copper-dependent enzymes with extracellular activities that catalyze the oxidation of lysine residues in collagen and elastin. It also had been shown to carry out intracellular functions beyond extracellular cross-linkage, including cell migration, cell polarity and EMT.18 Jansen MK demonstrated that the expression of intracellular LOX was up-regulated after the induction of EMT in MDCK II cells.19 Both the Cu(II) cofactor and the lysyl-tyrosylquinone cofactor are required for the oxidase activity.20,21 Hence, the inhibitor BAPN could irreversibly inactivate LOX as it can trap Cu. The mechanism of inhibition is postulated which involves the formation of a covalent bond between an enzyme nucleophile and a ketenimine formed from BAPN by enzyme-assisted beta-proton abstraction.15 A lot of evidence showed that LOX played an important role in the process of EMT.13,16,22–24 We also found that LOX was up-regulated accompanied with EMT in PQ poisoning. We used BAPN to inactivate LOX to study the relationship between LOX and PQ-induced EMT and found that the inactivation of LOX in RLE-6TN, A549 cells and rats gradually attenuated the process of EMT as the concentration of the inhibitor increased. However, LOX-like proteins (LOXL1, LOXL2, LOXL3 and LOXL4) with varying degrees of similarities to LOX have been described. We used silencing of LOX to further confirm the role of LOX in EMT as BAPN could inhibit all the LOX family members. We found that silencing of LOX could suppress EMT and migration in A549 cells induced by PQ. This finding further indicated that the occurrence of EMT in pulmonary fibrosis induced by PQ relied on the expression of LOX.
Peinado and colleagues reported that LOX exerted an effect on the stability of the Snail protein25,26 at the post-transcriptional level.27 Snail, the zinc finger transcription factor, has been described as a direct repressor of E-cadherin expression in epithelial cells; the expression of Snail induces EMT and increases cell migration. We found that the Snail protein expression could be reduced by LOX silencing and LOX inactivation, which indicated that LOX could promote EMT by stabilizing Snail protein expression during PQ-induced pulmonary fibrosis. In addition, Snail protein stability is finely controlled by GSK3β-dependent phosphorylation and ubiquitination.18 We also found AEC PQ-exposure could decrease the level of GSK-3β, while the level of GSK-3β was increased after silencing LOX and inactivating LOX. We guessed that PQ-induced LOX could indirectly affect Snail by decreasing the expression of GSK-3β; however, the mechanism is unclear.
Interestingly, the RLE-6TN and A549 cells simply treated with the inhibitor BAPN showed no obvious change in the cell shape, but the mesenchymal markers α-SMA and vimentin of EMT expression levels were decreased while epithelial markers ZO-1, E-cadherin expression levels were increased which were evidence of that MET has occurred. MET is the inverse process of EMT, which has been shown to occur during the development of fibrotic disorders and cancer. Roy-Akira Saito28 found that TTF-1 might partially restore the epithelial properties. Ramos29 found that FGF-1 can revert EMT induced by TGF-β through the MAPK/ERK kinase pathway. Huang HY showed that LOX silencing induces a MET-like process.30 However, which signals induced MET is unknown. Since less is known about the reversibility of lung fibrosis, it is difficult to reverse it when pulmonary fibrosis is formed.31 MET may be a key point to prevent early pulmonary fibrosis by inactivating LOX. LOX expression is known to be regulated by HIF-1, TGF-β, the tumour necrosis factor α, the platelet derived growth factor and the fibroblast growth factor.32 Future studies will aim to investigate precisely the roles of LOX in epithelial cells and EMT during pulmonary fibrosis induced by PQ.
In summary, the findings of this study reveal that LOX could promote the progress of EMT and inactivating LOX may alleviate EMT in PQ-induced pulmonary fibrosis. The relationship between LOX and EMT could potentially be a new identified candidate therapeutic target for pulmonary fibrosis induced by PQ.
Competing interests
No conflicts of interest, financial or otherwise, are declared by the authors.Acknowledgements
This project was supported by National Natural Science Foundation of China (81272071, 81471891).References
- R. Dinis-Oliveira, J. Duarte, A. Sanchez-Navarro, F. Remiao, M. Bastos and F. Carvalho, CRC Crit. Rev. Toxicol., 2008, 38, 13–71 CrossRef CAS PubMed.
- F. Candan and H. Alagözlü, Hum. Exp. Toxicol., 2001, 20, 637–641 CAS.
- I. B. Gawarammana and N. A. Buckley, Br. J. Clin. Pharmacol., 2011, 72, 745–757 CrossRef CAS PubMed.
- N. A. Hosper, P. P. van den Berg, S. de Rond, E. R. Popa, M. J. Wilmer, R. Masereeuw and R. A. Bank, Exp. Cell Res., 2013, 319, 3000–3009 CrossRef CAS PubMed.
- D. J. Schneider, M. Wu, T. T. Le, S.-H. Cho, M. B. Brenner, M. R. Blackburn and S. K. Agarwal, FASEB J., 2012, 26, 503–512 CrossRef CAS PubMed.
- S. N. Flier, H. Tanjore, E. G. Kokkotou, H. Sugimoto, M. Zeisberg and R. Kalluri, J. Biol. Chem., 2010, 285, 20202–20212 CrossRef CAS PubMed.
- M. Guarino, A. Tosoni and M. Nebuloni, Hum. Pathol., 2009, 40, 1365–1376 CrossRef CAS PubMed.
- H. Kage and Z. Borok, Curr. Opin. Pulm. Med., 2012, 18, 517 CAS.
- K. K. Kim, M. C. Kugler, P. J. Wolters, L. Robillard, M. G. Galvez, A. N. Brumwell, D. Sheppard and H. A. Chapman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13180–13185 CrossRef CAS PubMed.
- M. R. Chinoy, S. E. Zgleszewski, R. E. Cilley and T. M. Krummel, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 279, L312–L318 CAS.
- S. Liu, H. Parameswaran, S. M. Young and B. M. Varisco, Respir. Res., 2014, 15, 34–45 CrossRef PubMed.
- L. I. Smith-Mungo and H. M. Kagan, Matrix Biol., 1998, 16, 387–398 CrossRef CAS PubMed.
- J. Chi, Z. Wang, D. S. Nuyten, E. H. Rodriguez, M. E. Schaner, A. Salim, Y. Wang, G. B. Kristensen, A. Helland and A. Borresen-Dale, PLoS Med., 2006, 3, e47 Search PubMed.
- A. Sethi, R. J. Wordinger and A. F. Clark, Exp. Eye Res., 2012, 104, 97–98 CrossRef CAS PubMed.
- S. S. Tang, P. C. Trackman and H. M. Kagan, J. Biol. Chem., 1983, 258, 4331–4338 CAS.
- D. F. Higgins, K. Kimura, W. M. Bernhardt, N. Shrimanker, Y. Akai, B. Hohenstein, Y. Saito, R. S. Johnson, M. Kretzler and C. D. Cohen, J. Clin. Invest., 2007, 117, 3810–3820 CAS.
- H. Xie, J.-t. Tan, R.-l. Wang, X.-X. Meng, X. Tang and S. Gao, Exp. Biol. Med., 2013, 238, 1062–1068 CrossRef PubMed.
- Q. Xiao and G. Ge, Cancer Microenviron., 2012, 5, 261–273 CrossRef CAS PubMed.
- M. K. Jansen and K. Csiszar, Matrix Biol., 2007, 26, 136–139 CrossRef CAS PubMed.
- H. Lucero and H. Kagan, Cell. Mol. Life Sci., 2006, 63, 2304–2316 CrossRef CAS PubMed.
- R. B. Rucker, T. Kosonen, M. S. Clegg, A. E. Mitchell, B. R. Rucker, J. Y. Uriu-Hare and C. L. Keen, Am. J. Clin. Nutr., 1998, 67, 996S–1002S CAS.
- C. Sahlgren, M. V. Gustafsson, S. Jin, L. Poellinger and U. Lendahl, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6392–6397 CrossRef CAS PubMed.
- R. Schietke, C. Warnecke, I. Wacker, J. Schödel, D. R. Mole, V. Campean, K. Amann, M. Goppelt-Struebe, J. Behrens and K.-U. Eckardt, J. Biol. Chem., 2010, 285, 6658–6669 CrossRef CAS PubMed.
- X. Yang, S. Li, W. Li, J. Chen, X. Xiao, Y. Wang, G. Yan and L. Chen, Oncol. Rep., 2013, 29, 541–548 CAS.
- H. Peinado, D. Olmeda, K. Csiszar, K. S. Fong, S. Vega, M. A. Nieto, A. Cano and F. Portillo, EMBO J., 2005, 24, 3446–3458 CrossRef CAS PubMed.
- H. Peinado, F. Portillo and A. Cano, Cell Cycle, 2005, 4, 1749–1752 CrossRef CAS PubMed.
- L. Wei, X.-R. Song, J.-J. Sun, X.-W. Wang, L. Xie and L.-Y. Lv, Cancer Biother. Radiopharm., 2012, 27, 672–677 CrossRef CAS PubMed.
- R.-A. Saito, T. Watabe, K. Horiguchi, T. Kohyama, M. Saitoh, T. Nagase and K. Miyazono, Cancer Res., 2009, 69, 2783–2791 CrossRef CAS PubMed.
- C. Ramos, C. Becerril, M. Montaño, C. García-De-Alba, R. Ramírez, M. Checa, A. Pardo and M. Selman, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2010, 299, L222–L231 CrossRef CAS PubMed.
- H.-Y. Huang, S.-Z. Chen, W.-T. Zhang, S.-S. Wang, Y. Liu, X. Li, X. Sun, Y.-M. Li, B. Wen and Q.-Y. Lei, Stem Cell Res., 2013, 10, 278–287 CrossRef CAS PubMed.
- T. Kisseleva and D. A. Brenner, Exp. Biol. Med., 2008, 233, 109–122 CrossRef CAS PubMed.
- M. Damjanac, A. R. Bilan, M. Paccalin, R. Pontcharraud, B. Fauconneau, J. Hugon and G. Page, Neurobiol. Dis., 2008, 29, 354–367 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |