
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolomic analysis of riboswitch containing E. coli recombinant expression system†
Howbeer
Muhamadali
a,
Yun
Xu
a,
Rosa
Morra
b,
Drupad K.
Trivedi
a,
Nicholas J. W.
Rattray
a,
Neil
Dixon
*b and
Royston
Goodacre
*a
aSchool of Chemistry, Manchester Institute of Biotechnology, University of Manchester, Manchester, UK. E-mail: roy.goodacre@manchester.ac.uk; Tel: +44 (0)161 306-4480
bFaculty of Life Sciences, Manchester Institute of Biotechnology, University of Manchester, Manchester, UK. E-mail: neil.dixon@manchester.ac.uk; Tel: +44 (0)161 306-4537
First published on 23rd November 2015
Abstract
In this study we have employed metabolomics approaches to understand the metabolic effects of producing enhanced green fluorescent protein (eGFP) as a recombinant protein in Escherichia coli cells. This metabolic burden analysis was performed against a number of recombinant expression systems and control strains and included: (i) standard transcriptional recombinant expression control system BL21(DE3) with the expression plasmid pET-eGFP, (ii) the recently developed dual transcriptional–translational recombinant expression control strain BL21(IL3), with pET-eGFP, (iii) BL21(DE3) with an empty expression plasmid pET, (iv) BL21(IL3) with an empty expression plasmid, and (v) BL21(DE3) without an expression plasmid; all strains were cultured under various induction conditions. The growth profiles of all strains together with the results gathered by the analysis of the Fourier transform infrared (FT-IR) spectroscopy data, identified IPTG-dependent induction as the dominant factor hampering cellular growth and metabolism, which was in general agreement with the findings of GC-MS analysis of cell extracts and media samples. In addition, the exposure of host cells to the synthetic inducer ligand, pyrimido[4,5-d] pyrimidine-2,4-diamine (PPDA), of the orthogonal riboswitch containing expression system (BL21(IL3)) did not display any detrimental effects, and its detected levels in all the samples were at similar levels, emphasising the inability of the cells to metabolise PPDA. The overall results obtained in this study suggested that although the BL21(DE3)-EGFP and BL21(IL3)-EGFP strains produced comparable levels of recombinant eGFP, the presence of the orthogonal riboswitch seemed to be moderating the metabolic burden of eGFP production in the cells enabling higher biomass yield, whilst providing a greater level of control over protein expression.
Introduction
Since the large-scale production of the first human recombinant protein (Humulin) in the early 1980s,1 the global biopharmaceutical industry has transformed in to a thriving market. Currently, biopharmaceutical pipelines contain a range of therapeutics such as recombinant proteins, peptides, monoclonal antibodies and antibody fragments, vaccines, nucleic acid based products and therapeutic enzymes.2 Recombinant proteins represented the biggest biopharmaceutical class of products in 2009, accounting for more than 65% of the total global sales.3 Such products not only benefit the biopharmaceutical industry but have also proven useful in other sectors including the agricultural industry.4Despite extensive research and strategies employed towards the optimisation of recombinant systems, the metabolic effects of introducing foreign DNA into the host organism, known as metabolic load or burden,5 is inevitable. This effect can be explained by the required energy, such as GTP or ATP, for maintenance of the plasmid or via depletion of the cellular resources such as amino acid, aminoacyl–tRNA pools and free ribosomal subunits due to their consumption and redirection towards the production of recombinant products.6–8 The imposed metabolic load may not only affect the metabolic and physiological characteristics of the cell but it can also limit the yield and quality of the final product through activation of different stress response mechanisms resulting in increased protease activity, leading to lower product yield.9 Therefore, a deeper understanding of the physiological and global changes in the metabolome of the host cell may provide further insight into understanding this metabolic burden, and aid metabolic engineering strategies towards improvement and re-design of superior host organisms.
The metabolome has been described as the collection of all the metabolites found in a biological system that are participating towards the normal function, maintenance and growth of the cell.10 Metabolites can be viewed as downstream products of gene expression and protein function, thus studying the metabolome may provide a clearer image of the biochemical state of a system whilst it can also reflect on gene function.11–13
Even though a substantial amount of progress has been made in the field of metabolomics, due to the highly complex nature of the metabolome, achieving comprehensive detection and identification of all the metabolites in a biological systems is not yet possible. Thus the heterogeneity of the metabolome has been the driving force behind the development and optimisation of different protocols,14 analytical platforms15,16 and metabolomics approaches,17–19 for a general review on metabolomics and its applications the reader is directed to the following reviews.15,19,20
The increasing demand and potential new applications of recombinant products has motivated the development of different protocols and approaches for optimising recombinant protein production processes. Several strategies have been successfully applied to improve and optimise the yield and quality of recombinant products in E. coli, including: strain engineering,21 vector design,22–24 re-engineering the translational regulatory regions25 and optimisation of cultivation conditions26,27 to name a few. In addition, during the past decade riboswitches have also attracted a lot of attention as regulatory mechanisms for gene expression control, for a detailed review on riboswitches the reader is directed to ref. 28 and 29.
The specificity and structural versatility of riboswitches have led to them being proposed for use in a number of potential applications, as their mechanism for controlling gene expression may prove to be a simpler strategy compared to other protein-based systems such as that of the lac operon.30
Recently Dixon and colleagues re-engineered the aptamer domain of the naturally occurring translation ON add-A riboswitch, producing an orthogonal riboswitch that no longer responds to the natural inducer, but was however specifically controlled via a synthetic inducer.31 Later, these authors also demonstrated the potential application of combining different synthetic riboswitches as a strategy for co-expression of multiple genes independently, controlled via the synthetic ligands in a dose-dependent manner.32 More recently, the orthogonal riboswitch has been incorporated into the genome of E. coli controlling the bacteriophage T7 RNA polymerase (T7 RNAP),33 with additional transcriptional IPTG inducible (lac promoter/operator) control over T7 RNAP affording a dual transcription-translation control expression strain BL21(IL3).
The aim of this study was to investigate the metabolic effects of recombinant eGFP production upon different E. coli systems, including (BL21(IL3)) (Table 1) under separate inducing conditions (Table 2). The BL21(IL3) strain was transformed with a pET-eGFP vector to create an expression system that combines both transcriptional and translation control of the T7 RNAP with transcriptional control over the gene of interest. Although the regulatory component reengineering31 and expression system performance33 of these systems have been studied, the metabolic effects of gene expression up on the host cell was not entirely understood. Thus, FT-IR spectroscopy was employed as a metabolic fingerprinting tool to investigate the overall effects of recombinant protein production on the biochemical composition of the cells. In addition, gas chromatography combined with mass spectrometry (GC-MS) was used for the analysis and identification of different metabolites of the cell extracts and spent media, as metabolic profiling and footprinting approaches respectively. Further GC-MS analysis was employed in a targeted approach to quantify any potential metabolism/catabolism of the synthetic orthogonal riboswitch inducer PPDA.34
| BL21 strains | Labels | pET15b | Riboswitch | T7 RNA polymerase | Green fluorescent protein (GFP) | Kanamycin resistant | Ampicillin resistant |
|---|---|---|---|---|---|---|---|
| DE3 | Wild | − | − | + | − | − | − |
| DE3 | PET | + | − | + | − | − | + |
| DE3 | EGFP | + | − | + | + | − | + |
| IL3 | iL3 PET | + | + | + | − | + | + |
| IL3 | iL3 EGFP | + | + | + | + | + | + |
| Inducer compound | Final concentration |
|---|---|
| 0.9% NaCl solution (no inducer) | — |
| IPTG (lac inducer) | 50 μM |
| PPDA (riboswitch inducer ligand) | 200 μM |
| IPTG + PPDA | 50 μM, 200 μM |
Materials and methods
Luria Bertani (LB) broth
LB broth was made from a preparatory mixture (tryptone 10 g L−1, NaCl 10 g L−1, yeast extract 5 g L−1) following the manufacturer's recommended protocol. The prepared solution was autoclaved and stored at 4 °C.Strains and culture conditions
The following E. coli strains were used in this study (Table 1). The BL21 DE3 strain not harbouring the pET15b plasmid was used as a control strain and referred to as wild type. All remaining strains carried the pET15b plasmid with the lac operator driving the expression of the plasmid downstream of a T7 RNA polymerase promoter. The pET and EGFP strains carried the T7 RNA polymerase gene in their genome under the control of the lac promoter/operator. The IL3-pET and IL3-EGFP strains also carried a genomic copy of the T7 RNA polymerase; however its expression is further regulated by an orthogonal riboswitch downstream of the lac promoter/operator region.33 Both EGFP and IL3-EGFP strains carried the eGFP coding gene on their recombinant plasmids. The riboswitch construct is described in more detail in the ESI.†All strains were streak plated three times on LB agar prior to every experiment to ensure the purity of the stocks. 100 mg L−1 ampicillin and/or 10 mg L−1 kanamycin were added to the LB broth/agar as selectable plasmid markers where necessary. Starting inocula were prepared by inoculating 25 mL of LB broth with a single colony of the appropriate strain followed by overnight incubation at 37 °C with 200 rpm shaking in a Multitron standard shaker incubator (INFORS-HT Bottmingen Switzerland). Different inducing conditions examined in this study are described in Table 2.
FT-IR analysis
FT-IR sample preparation
Overnight grown cells were used to inoculate 10 mL of LB broth to a final OD600nm = 0.1. At this point no antibiotics were used to avoid its metabolic effects on the cells. Samples were transferred as 200 μL aliquots (biological replicates, n = 9) to a 100 well Bioscreen plate under aseptic conditions. Growth of the strains was monitored using a Bioscreen spectrophotometer (Labsystems, Basingstoke, UK) at 600 nm wavelength. The Bioscreen was setup as follows: 10 min preheating, continuous medium shaking, OD600nm measurement with 10 min intervals, incubation at 37 °C. Upon reaching OD600nm = 0.5, different inducing conditions (Table 2) were applied to appropriate samples, followed by decreasing the incubation temperature to 20 °C for a total incubation time of 11 h. All other incubation parameters were kept unchanged.A Bruker 96-well silicon microtitre plate (Bruker Ltd, Coventry, UK) was washed three times with 5% sodium dodecyl sulfate (SDS) solution, twice with 70% ethanol and finally rinsed three times with deionised water and dried at 55 °C. 100 μL aliquots from each replicate (biological replicates, n = 9) was pooled together and centrifuged at 4 °C, 5000 × g for 10 min using a bench top Eppendorf Microcentrifuge 5424R with an FA-45-24-11 rotor (Eppendorf Ltd, Cambridge, UK). The supernatant were removed and cell pellets were washed twice using 2 mL of sterile 0.9% saline solution followed by 10 min centrifugation at 5000 × g at 4 °C. After the final washing step the supernatant was removed and the pellets were normalized to the same OD600nm using 0.9% saline solution. 20 μL of each sample was randomly spotted on to the FT-IR silicon plate and heated to dryness at 55 °C.
Instrument setup and data pre-processing
A Bruker Equinox 55 infrared spectrometer was used for the analysis of all samples. FT-IR spectra were collected in transmission mode35 in the 4000–600 cm−1 range at a resolution of 4 cm−1, with 64 spectra co-added and averaged to improve the signal-to-noise ratio. All spectra were scaled using extended multiplicative signal correction (EMSC) method36 to compensate for any sample size variations, followed by removal of CO2 vibrations (2400–2275 cm−1) which were replaced with a linear trend.GC-MS analysis
Quenching and extraction
50 mL of LB broth, three biological replicates per condition, was inoculated with the appropriate strains using the overnight grown cells to a final OD600nm = 0.1, followed by incubation at 37 °C with 200 rpm shaking for 3 h. Upon reaching the OD600nm = 0.5 the samples were exposed to one of the inducing conditions (Table 2), and the incubation temperature was decreased to 20 °C with 200 rpm for 8 h in shaking incubators, which sums up to a total of 11 h of incubation. 15 mL samples from each flask were quenched using 30 mL, 60% aqueous methanol (−48 °C) following procedures described in previous studies.14 Extraction protocol was also adapted from14 with the exception of centrifugation speed being set at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 871 × g. All extracts were normalized according to OD600nm followed by combining 100 μL from each of the samples in a new tube, to be used as the quality control (QC) sample. 100 μL internal standard solution (0.2 mg mL−1 succinic-d4 acid, 0.2 mg mL−1 glycine-d5, 0.2 mg mL−1 benzoic-d5 acid, and 0.2 mg mL−1 lysine-d4) was added to all the samples (including QCs) followed by an overnight drying step using a speed vacuum concentrator (Concentrator 5301, Eppendorf, Cambridge, UK).
871 × g. All extracts were normalized according to OD600nm followed by combining 100 μL from each of the samples in a new tube, to be used as the quality control (QC) sample. 100 μL internal standard solution (0.2 mg mL−1 succinic-d4 acid, 0.2 mg mL−1 glycine-d5, 0.2 mg mL−1 benzoic-d5 acid, and 0.2 mg mL−1 lysine-d4) was added to all the samples (including QCs) followed by an overnight drying step using a speed vacuum concentrator (Concentrator 5301, Eppendorf, Cambridge, UK).
Derivatization
Derivatization was carried out by oximation (using methoxyamine-hydrochloride in pyridine) followed by a silylation step (using N-methyl-N-(trimethylsilyl) trifluoroacetamide), as described by ref. 37 and 38.GC-MS setup and data pre-processing
A Gerstel MPS-2 autosampler (Gerstel, Baltimore, MD) used in conjunction with an Agilent 6890N GC oven (Wokingham, UK) coupled to a Leco Pegasus III mass spectrometer (St Joseph, USA) was used for the analysis of all samples following previously published methods.39,40 Collected data were deconvolved using Leco ChromaTOF software39 and initial identification (Table S2, ESI†) was carried out according to metabolomics standards initiative (MSI) guidelines,41 followed by removal of mass spectral features with high deviation within the QC samples.37 The chromatographic peaks corresponding to PPDA and IPTG were removed from the data before subjecting the data to CPCA-W, to eliminate any variation that might result from the presence of these compounds.PPDA quantification
A concentrated stock (20 mM) of PPDA solution was used for preparing the standard solutions (triplicates). The standard solutions were made in LB broth to account for any ion suppression that might result from the constituents within the complex media. Sample preparation, derivatization and GC-MS analysis followed the standard protocol as described above. PPDA specific fragments (305, 177 and 138 m/z) were used for identification of PPDA chromatographic peak (retention time = 896.08 s), followed by normalization of all chromatographic peaks area values to the area of the internal standard (succinic acid-d4) and using the normalized TICs for generating the standard curve and quantification of PPDA in the media and cell extract samples.Data analysis
MATLAB version 9 (The Mathworks Inc., Natwick, US) was used for analysis of all collected data. As there are multiple interacting factors within this study (5 different strains and 4 inducing conditions), the variation caused by each factor might not be effectively accounted for by using the classical PCA approach, therefore we have employed a form of multiblock PCA (MB-PCA)42,43 known as weighted consensus PCA (CPCA-W)44 for the analysis of both GC-MS and FT-IR data. In multiblock PCA, the data was re-arranged into blocks according to the experimental design and by doing so the effects of one factor would become baseline and no longer have its influence while that of the other factors would become a common-trend across different blocks. Without the inference from the “baseline” factor the effect of the “common trend” factor can be detected by the MB-PCA model more easily.42 In this study, the data were first arranged into five blocks based on the number of different strains to examine the effects of the inducing condition on each strain. Then the data were rearranged into four blocks based on the different inducing conditions to observe the effects of each condition on all the strains in each block. Of course it should be noted that despite the rearrangement of the data, CPCA-W can still be considered as an unsupervised method and the risk of over-fitting of the results is therefore considered low.45eGFP quantification
Cells were harvested and washed twice in PBS/0.2% tween. Relative fluorescence and OD600 were measured in a BioTek Synergy HT Microplate Reader (BioTek, Bedfordshire, UK). Relative fluorescence units over optical density (RFU/OD) were plotted to compare the effect of the various combinations of inducer concentrations.Results
In this study five different E. coli strains (Table 1) were grown in LB broth and examined under four separate inducing conditions (Table 2) to dissect the metabolic effects of plasmid maintenance, recombinant protein production, as well as to understand the direct effects of the small molecule inducers on the different strains.Growth profiles
All strains displayed similar growth patterns under un-induced condition (Fig. 1a), therefore this data set was used as a control to check the growth behaviour of all strains under no stress. As expected the growth rates for all strains reduced after dropping the incubation temperature from 37 °C to 20 °C. Furthermore, the wild strain (not carrying a pET expression plasmid) displayed almost unchanged growth behaviour under all culturing conditions (Fig. 1b), indicating a lack of inherent toxicity of either IPTG or PPDA inducers under the experimental conditions assessed here.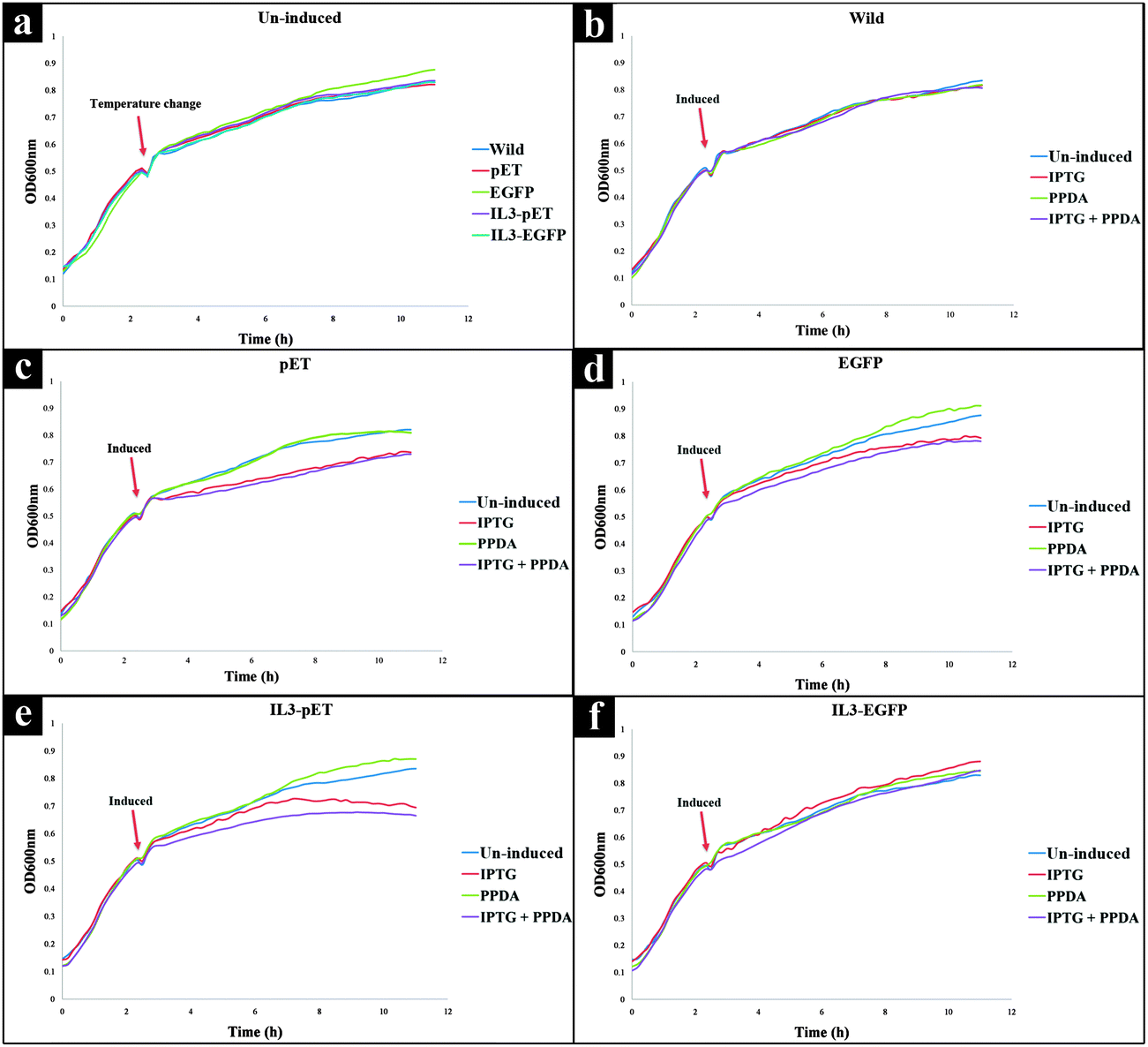 | ||
| Fig. 1 Growth curves of E. coli strains grown under different culturing condition: (a) all strains without the addition of any inducer compounds, (b) wild type, (c) pET strain, (d) EGFP strain, (e) IL3-pET strain, and (f) IL3-EGFP strain. All strains were grown at 37 °C, upon reaching mid exponential phase (OD600nm = ∼0.5) incubation temperature was dropped down to 20 °C, while the cells were exposed to different inducing conditions. Growth curves are average of 9 biological replicates. | ||
Upon addition of IPTG or IPTG + PPDA a decrease in growth rate for pET, EGFP and IL3-pET strains was observed (Fig. 1c–e). By contrast, the growth profile of IL3-EGFP strain displayed similar behaviour to the wild type, (Fig. 1f). However, the presence of PPDA alone did not display any detrimental effects on growth of any of the strains used in this study (Fig. 1a–f).
Metabolic fingerprinting using FT-IR spectroscopy
FT-IR spectroscopy is a high-throughput, non-invasive, low-cost and reproducible vibrational spectroscopy technique, which is based on the intermolecular bond vibrations of a sample caused by the incident and absorption of infrared light, allowing the acquisition of biochemical fingerprint of the investigated samples. In this study we have employed FT-IR as a metabolic fingerprinting approach for assessing the phenotypic changes in different strains upon exposure to different induction conditions.As there were interacting factors in the analysis – viz. (i) host strains and (ii) induction conditions – the FT-IR spectra were pre-processed and re-arranged according to these two main set of variables in this experiment (host or induction) and subjected to weighted consensus principal component analysis (CPCA-W) model.42,45 On the first approach all FT-IR data were arranged into five blocks based on strain types (strain blocking), to identify the effects of all inducing conditions on each strain separately. The second approach was carried out by re-arranging all data into four blocks according to inducing conditions (inducer blocking), to examine the effects of each of the inducing conditions on all the strains.
Scores plot of the wild type block displayed no significant effects under different inducing conditions, this is evident in Fig. 2a as the coloured circles, representing the scores of the data collected under different examined conditions relative to the principal components, do not show any specific clustering patterns according to the various induction conditions. This is perhaps not surprising, as the wild type does not carry the riboswitch nor the recombinant plasmid and thus does not produce the eGFP protein, making this strain an ideal candidate to be used as the control strain. Although this strain also carries the T7 RNAP gene which is expressed upon the addition of IPTG/IPTG + PPDA, yet the FT-IR CPCA-W results did not display any significant effects resulting from the production of T7 RNAP under IPTG and IPTG + PPDA conditions in this strain (Fig. 2a).
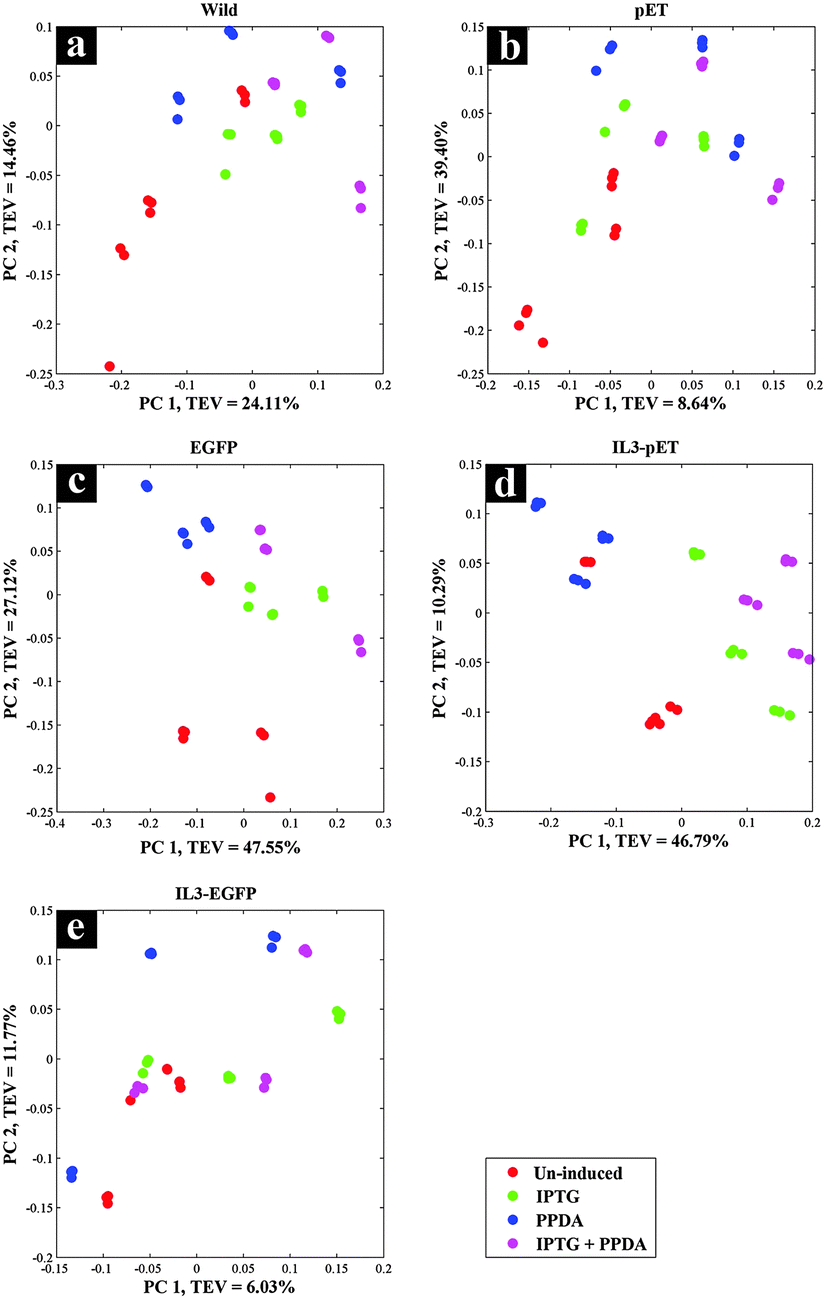 | ||
| Fig. 2 CPCA-W scores plots of FT-IR strain-blocked spectroscopy data. Different strain-blocked scores plots are presented on a–e plots, and the inducing conditions are presented by different coloured dots. | ||
There were also no significant effects on the metabolic fingerprints of pET and IL3-EGFP strains (Fig. 2b and e) under any of the inducing conditions. However, the IPTG and IPTG + PPDA seemed to be significantly affecting the EGFP and IL3-pET strains, as the data collected under these conditions are clustering on the positive side of PC1, whilst the data collected under un-induced and PPDA conditions are clustering on the negative side of PC1 (Fig. 2c and d).
The super scores plot (Fig. 3a) obtained from the five block model (strain-blocked), demonstrated a clear separation between different inducing conditions, with the first principal component (PC1) accounting for 26.68% of the total explained variance (TEV), where PPDA and un-induced conditions cluster together and further away from the IPTG and IPTG + PPDA conditions, illustrating the significant effects of IPTG and IPTG + PPDA on cellular metabolism as the dominant influencing factors.
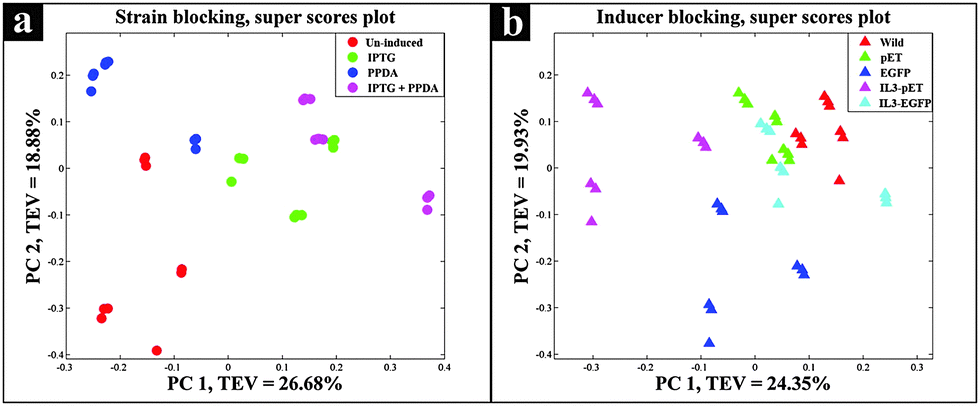 | ||
| Fig. 3 Super scores plots of the FT-IR strain blocking model. Different colours represent (a) various inducing conditions, (b) different strains. | ||
The second approach (inducer-blocked) aimed at assessing the E. coli strains with regard to each of the four inducing conditions (Table 2) as separate blocks. Scores plots of the un-induced and PPDA blocks (Fig. 4a and c) did not exhibit any specific trends, whilst IPTG and IPTG + PPDA blocks (Fig. 4b and d) once again displayed a clear separation, reflecting the dominant effects of these conditions on the fingerprints, which is in agreement with the strain-blocked findings (Fig. 2).
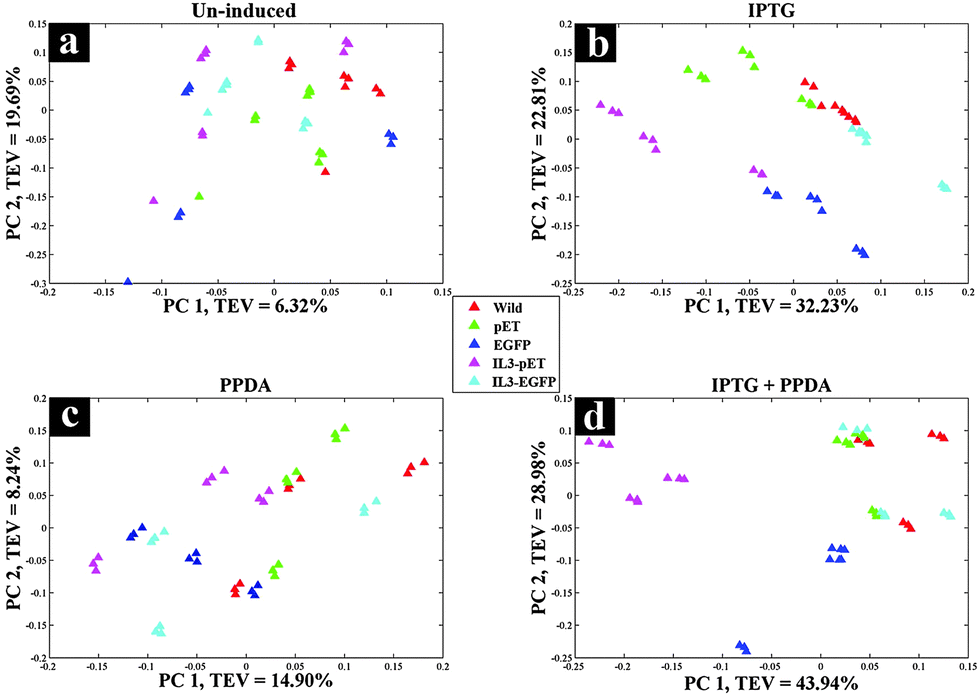 | ||
| Fig. 4 CPCA-W scores plots of FT-IR inducer-blocked data. Different strains are presented by different colours and a–d the various inducers applied: (a) un-induced; (b) IPTG; (c) PPDA; and (d) IPTG + PPDA. | ||
Scores plot of the IPTG block (Fig. 4b), according to PC1 and PC2 with TEV of 32.23% and 22.81% respectively, displayed the clustering of the IL3-pET and EGFP strains. IPTG + PPDA block also displayed similar trends (Fig. 4d). These findings were in agreement with the strain-blocked scores plots of IL3-pET and EGFP (Fig. 2c and d), suggesting that these strains are most affected under IPTG and IPTG + PPDA conditions. Loadings plot of the inducer-blocked data (Fig. S1, ESI†) further confirms these findings, as the IPTG (Fig. S1b and f, ESI†) displayed very similar loadings to IPTG + PPDA (Fig. S1d and h, ESI†), whilst PPDA (Fig. S1c and g, ESI†) and un-induced (Fig. S1a and e, ESI†) conditions were closely related.
Super scores plot of the inducer-blocked FT-IR data (Fig. 3b) also suggested that the two strains most affected under these conditions are IL3-pET and EGFP strains. However, although the IL3-pET strain is clustering away from the wild type, according to PC2 axis it is separated from the EGFP strain and closer to other strains, suggesting that its separation on PC1 is not due to recombinant eGFP production, and could be due to other underlying factors.
Metabolic profiling and footprinting by GC-MS
Although FT-IR spectroscopy has been applied in various fields of research due to its many advantages, when it comes to molecule-specific identification in complex samples it lacks the required chemical resolution. Thus, in this study GC-MS was also employed in parallel to FT-IR, due its high precision and sensitivity, as a metabolic profiling and footprinting approach. We have selected GC-MS combined with specific metabolite extraction protocol, aiming at detection and identification of the metabolites of the central carbon metabolism and amino acids biosynthetic pathways, whilst using CPCA-W for investigating the collected data to distinguish between the metabolic profiles and footprints of different strains under the examined conditions.The gathered GC-MS data were subjected to CPCA-W algorithm following very similar strategies as used for the FT-IR data analysis. The five block CPCA-W model of the GC-MS data collected from the cell extracts displayed no significant clustering pattern for the wild type strain (Fig. 5a) under different induction conditions, which is in agreement with our FT-IR findings (Fig. 2a). However, there appears to be a clear separation between different induction conditions for all other strains, with PPDA and un-induced conditions, once again clustering together and away from IPTG and IPTG + PPDA conditions (Fig. 5b–e).
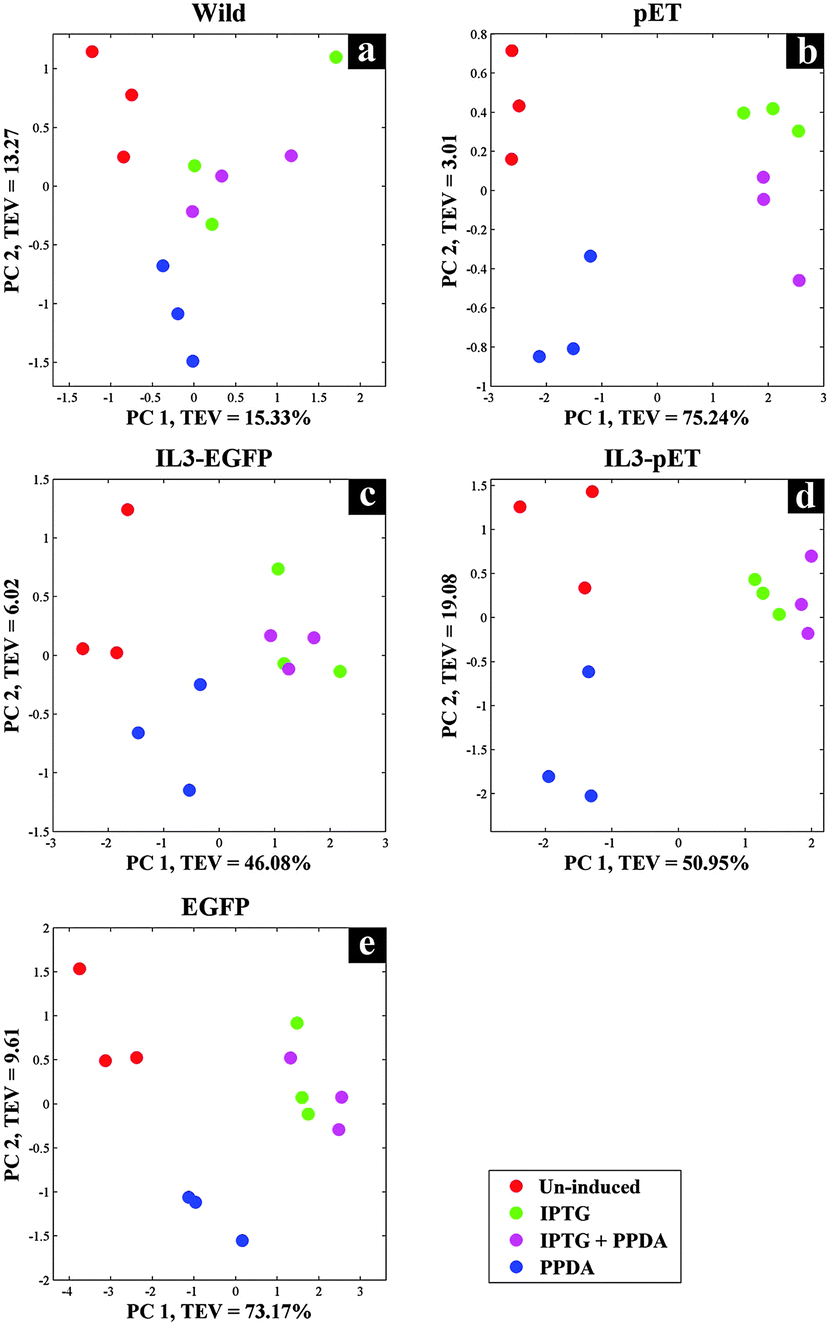 | ||
| Fig. 5 CPCA-W scores plots of GC-MS extracts, strain-blocked data. Different strain-blocked scores plots are presented on a–e plots, examined inducing conditions are presented by different colours. | ||
Protein expression analyses indicated that both EGFP and IL3-EGFP strains expressed comparable amounts of recombinant eGFP under fully induced conditions, IPTG or IPTG + PPDA respectively (Fig. 6). However, according to PC1 of IL3-EGFP (TEV = 46.08%) and EGFP (TEV = 73.17%) blocks (Fig. 5c and e), it appears that the metabolic burden of eGFP biosynthesis is affecting the EGFP strain to a greater extent in comparison to IL3-EGFP. Super scores plots of the five block model (Fig. 6a) also indicated the dominant effects of IPTG and IPTG + PPDA in comparison to PPDA and un-induced conditions, with PC1 accounting for 58.07% of the TEV. The inducer-blocked super scores plot (Fig. 6b) also displayed the separation of EGFP, IL3-EGFP and IL3PET from the wild type and the PET strain.
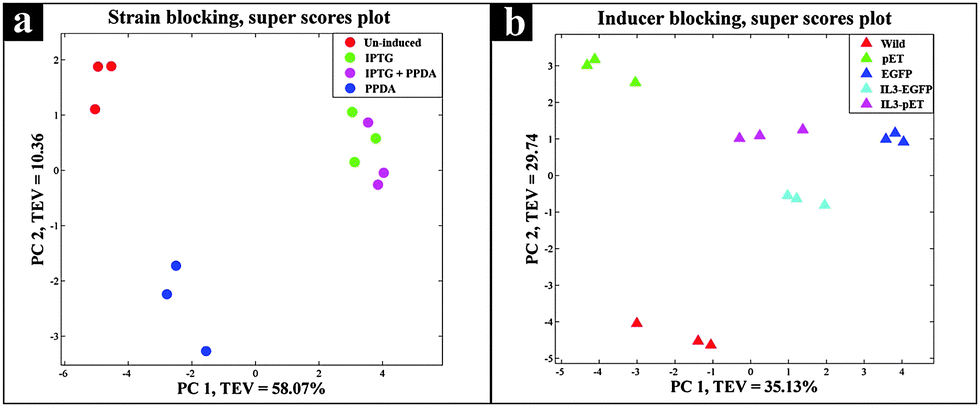 | ||
| Fig. 6 Super scores plot of the GC-MS extracts. Different colours represent (a) various inducing conditions, and (b) different strains. | ||
CPCA-W scores plot of the un-induced block displayed a clear separation between all strains (Fig. 8a). However the main separation is on PC1 (TEV = 47.40%), where EGFP is separated from all other strains. This could be a reflection of the metabolic effects of eGFP production on the EGFP strain, due to the established leaky nature of the engineered lac promoter used in the plasmids46–48 where even in the absence of the inducer compound (IPTG) the repression effect is incomplete, thus allowing a basal level of eGFP expression (Fig. 7).
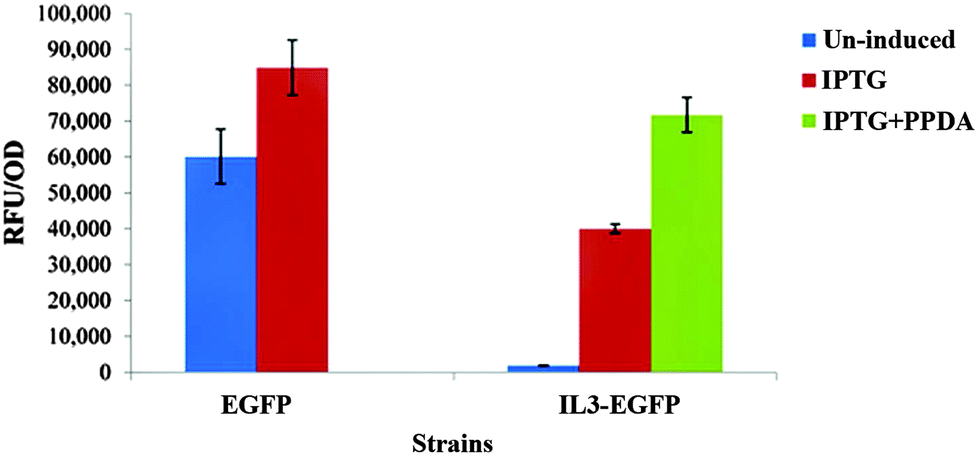 | ||
| Fig. 7 Comparison of recombinant eGFP production by the EGFP and IL3-EGFP strains under different inducing conditions. Each bar represents the mean of three biological replicates with error bars indicating the standard deviation. | ||
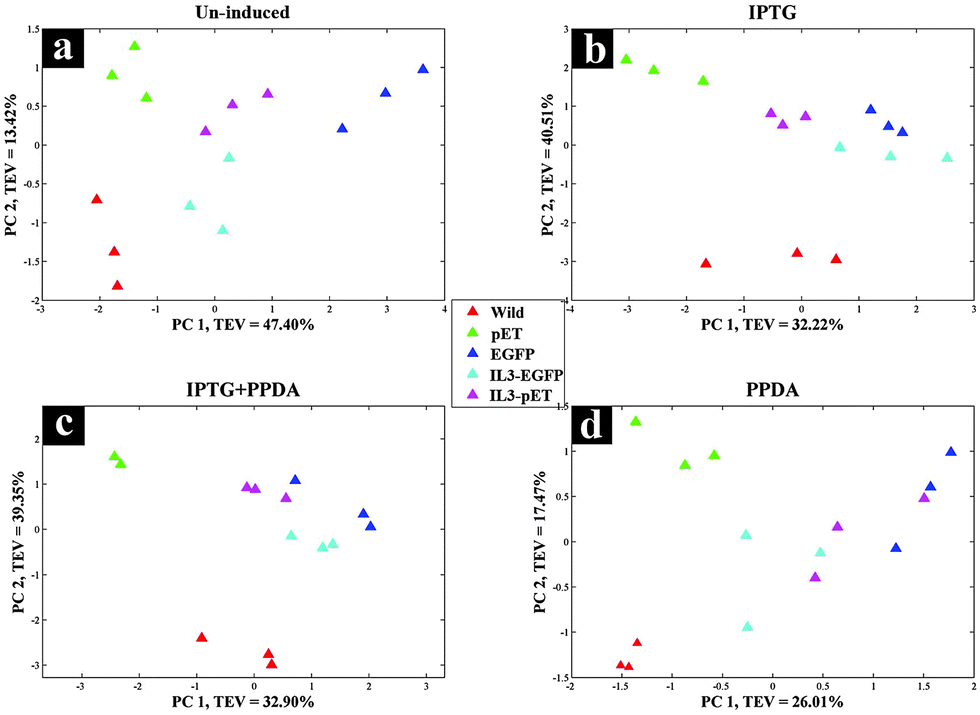 | ||
| Fig. 8 CPCA-W scores plots of GC-MS extracts, inducer-blocked data. Different strains are presented by different colours. a–d the various inducers applied: (a) un-induced; (b) IPTG; (c) IPTG + PPDA; and (d) PPDA. | ||
The PPDA block (Fig. 8d) also displayed similar trend to the un-induced block, whilst on the IPTG and IPTG + PPDA blocks (Fig. 8b and c) a clear separation between the wild type and all other strains was observed.
GC-MS footprint data were also subjected to CPCA-W following the same approach as for the GC-MS extracts. The results displayed clustering patterns very similar to those of the cell extracts, where strain-blocked data exhibited the dominant effect of IPTG and IPTG + PPDA in comparison to un-induced and PPDA conditions (Fig. S2 and S3a, ESI†), whilst the inducer-blocked scores plots confirmed these findings (Fig. S4 and S3b, ESI†).
Interpretation of GC-MS findings
Using the combined CPCA-W loadings plots, generated from the GC-MS data-blocks of the cell extracts and media, the significant metabolites were identified and their relative peak intensities were plotted for comparison purposes (Fig. 9). In addition, to investigate the effects of different induction conditions and eGFP production on the free amino acid pools in the media and cell extracts further, chromatographic peaks of 18 different amino acids were identified by GC-MS and their detected relative peak areas were compared in different strains under the examined four inducing conditions (Fig. 9).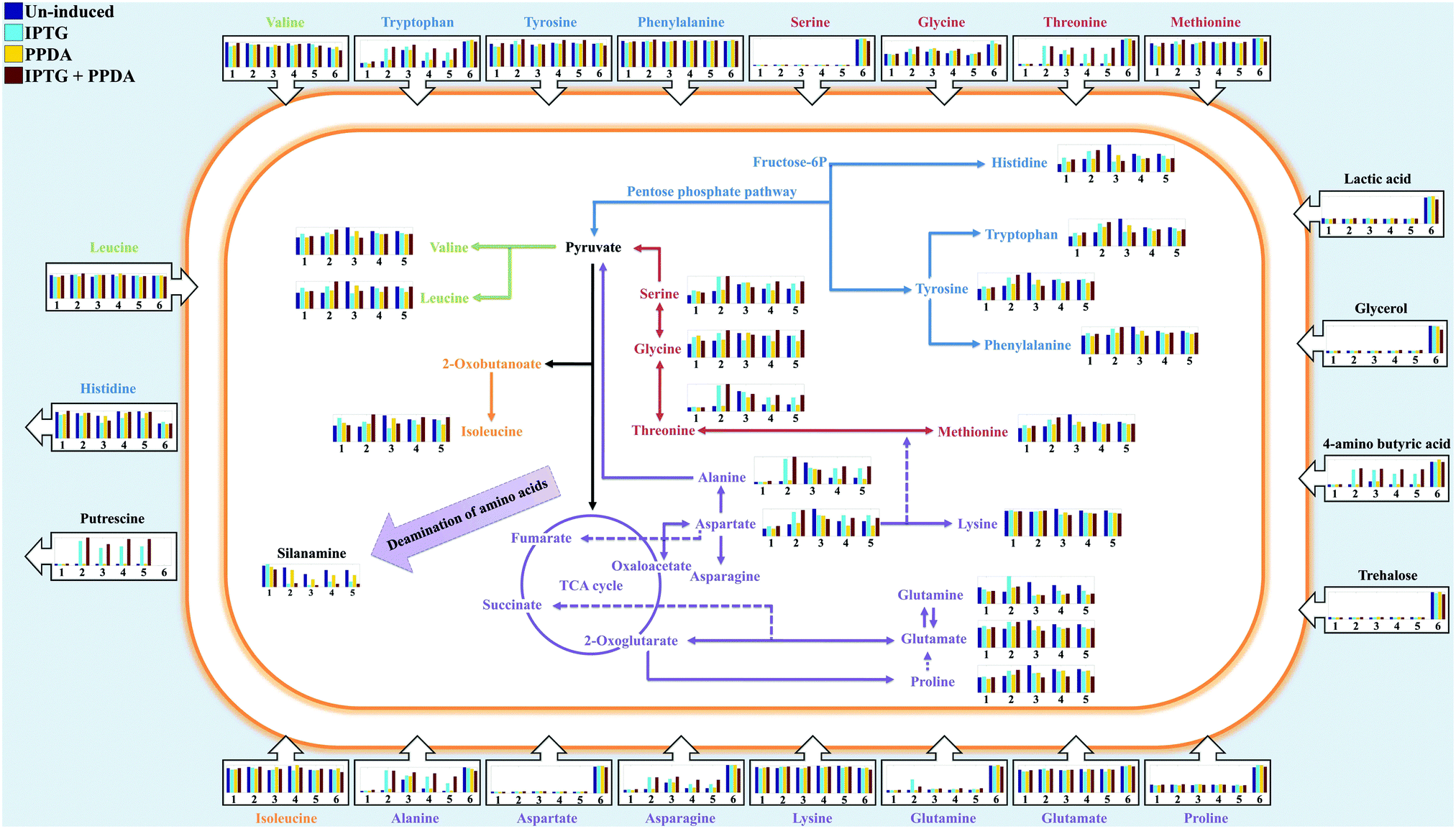 | ||
| Fig. 9 The relative peak intensities of metabolites (bar charts) in the cell extracts (inside the cell) and media (outside the cell) detected via GC-MS are plotted onto the amino acid biosynthetic and metabolic map of E. coli BL21. Different colours of the bar charts represent the examined inducing conditions (described on the top left of the figure), while numbers 1–5 represent different strains (wild, pET, EGFP, IL3-pET, IL3-EGFP respectively) used in this study. Number 6 represent LB media used as control for the footprint analysis. The arrows on the bar charts indicate uptake (inward arrows) or excretion (outward arrows) of the metabolites by E. coli cells. Detailed box plots of all the metabolites can be found in the ESI† (Fig. S6–S14). | ||
GC-MS data obtained from the media (metabolic footprints or the so called exometabolome) displayed a reduced level of aspartic acid, proline, serine, glycine, glutamine and methionine (Fig. 9). The detected levels of threonine, tryptophan, alanine and asparagine in the media displayed very similar response, as they showed a decrease of concentration in all strains (except for EGFP) cultured under the un-induced and PPDA conditions, whilst their concentrations remained almost unchanged under the IPTG and IPTG + PPDA conditions. Surprisingly, the EGFP strain displayed minor change in the detected levels of the above amino acids under the different induction conditions (Fig. 9).
The detected levels of the remaining amino acids in the media, including isoleucine, leucine, valine, lysine, phenylalanine and tyrosine did not display a significant change in comparison to their starting concentrations (Fig. 9). However, histidine is a special case in this study, as all the strains displayed an increase of histidine levels in the media under all culturing conditions. This may be attributed to the inability of E. coli in using histidine as carbon/nitrogen source due to the lack of histidine utilization genes,49–51 resulting in the excretion of excess histidine generated via the degradation of the imported peptides from the LB broth.
GC-MS data obtained from cell extracts (so called endometabolome) revealed a comparable trend for alanine, aspartic acid, serine and threonine (Fig. 9). The levels of these amino acids were at their lowest in the wild type with no significant effects from different inducing conditions (Fig. 9), whilst EGFP strain exhibited the highest levels of these amino acids. Furthermore, under un-induced and PPDA conditions both strains containing the empty plasmid (pET and IL3-pET) displayed similar levels of amino acids to the wild type, whilst exposure to IPTG and IPTG + PPDA resulted in an increasing trend (Fig. 9). Surprisingly, the amino acid profile of IL3-EGFP strain was very similar to the wild type for all conditions, except under IPTG + PPDA where higher levels of amino acids were detected (Fig. 9).
Silanamine, the derivatized (silylated) form of amine (formed from deamination of the amino acids), was also identified as a significant metabolite in the cell extracts. The silanamine levels displayed a complete opposite trend to those observed for the above amino acids, the highest levels were detected under un-induced and PPDA conditions, whilst IPTG and IPTG + PPDA exhibit the lowest levels (Fig. 9).
Trehalose, glycerol, lactic acid, and 4-aminobutyric acid were also amongst the significant metabolites in the media. An overall decrease in trehalose levels in the media was observed for all the strains (Fig. 9), which could be linked to the uptake of trehalose by the cells and its consumption as a carbon source or as an osmoprotectant.52 Furthermore, the levels of lactic acid and glycerol in the media were also decreased (Fig. 9). This is probably a result of the uptake and contribution of these metabolites towards pyruvate and glycerolipid metabolism, respectively. Adenosine and guanosine were also amongst the significant metabolites in the media, displaying a decreased trend (Fig. S16, ESI†) which is perhaps not surprising, considering their role as the building blocks of DNA and RNA during replication and transcription.
Putrescine was another significant metabolite detected in the culture media, exhibiting a rather interesting trend. It can be seen that putrescine is not initially present in LB media (Fig. 9), suggesting that it is excreted at a later stage by the cells during growth. Although the wild type strain displayed comparable levels of putrescine under all induction conditions, the plasmid bearing strains exhibited significantly higher levels of putrescine under IPTG and IPTG + PPDA conditions.
PPDA quantification
As the metabolic fate of PPDA inside E. coli cells has not been investigated by any other studies, we were concerned that this inducer could be used as a substrate (carbon and/or nitrogen source) and alter cellular metabolism through PPDA catabolism. Therefore we decided to quantify the level of PPDA that remained in the system. The PPDA calibration curve (R2 = 0.998) generated by targeted GC-MS (Fig. S5 and Table S1, ESI†) was used to quantitate PPDA in the cell extracts and footprint samples. The sum of concentrations detected in the footprints and cell extracts (Table 3) displayed comparable levels of PPDA for all strains under the examined conditions, suggesting that PPDA functions merely as an inducer compound and is not metabolized under the examined conditions. Moreover, the results from the quality control (QC) samples further supports the precision of the quantitation, as it displayed a final PPDA concentration of 102.92 ± 8.97 μM, which considering the starting concentration of 200 μM and the two fold dilution of PPDA due to the different conditions (un-inducer and IPTG), equates to a relevant concentration. This further confirms that the inducer was not being metabolized and any effects on cellular metabolism were a consequence of PPDA-dependent induction within the orthogonal riboswitch containing expression strain BL21 (IL3).| Strain | Condition | Footprint (μM) | Extract (μM) | Concentration ± SD (μM) |
|---|---|---|---|---|
| The data presented above represent average of three biological replicates. | ||||
| Wild | PPDA | 185.20 | 35.22 | 220.42 ± 10.14 |
| Wild | IPTG + PPDA | 168.35 | 41.27 | 209.62 ± 27.51 |
| PET | PPDA | 172.79 | 42.85 | 215.64 ± 18.42 |
| PET | IPTG + PPDA | 176.66 | 49.98 | 226.64 ± 30.37 |
| EGFP | PPDA | 166.99 | 43.15 | 210.14 ± 19.98 |
| EGFP | IPTG + PPDA | 143.59 | 34.76 | 178.35 ± 28.95 |
| IL3 | PPDA | 192.64 | 41.87 | 234.51 ± 24.09 |
| IL3 | IPTG + PPDA | 169.89 | 42.80 | 212.69 ± 4.77 |
| IL3EGFP | PPDA | 150.55 | 41.41 | 191.96 ± 27.61 |
| IL3EGFP | IPTG + PPDA | 153.36 | 35.24 | 188.60 ± 4.6 |
| LB | PPDA | 183.31 | 0.00 | 183.31 ± 10.86 |
| LB | IPTG + PPDA | 211.13 | 0.00 | 211.13 ± 20.92 |
| QC | All | 74.84 | 28.08 | 102.92 ± 8.97 |
Discussion
The use of E. coli as a bacterial host for heterologous protein production, remains an attractive option with almost 30% of the recombinant therapeutic proteins being produced using this organism.53 The well-characterized genome,54 proteome55 and metabolism of E. coli14 together with the availability of different expression systems and cloning tools, rapid growth and ability to grow to high cell density56 has made this microorganism the workhorse of the biotech industry. Extensive strategies and approaches57–59 have been employed towards improvement and optimisation of the yield and quality of the heterologous products in these important microorganisms.In this study we have employed metabolomics approaches to examine the metabolic burden of recombinant protein production in E. coli BL21(DE3) versus BL21(IL3) strains (Table 1) carrying an orthogonal riboswitch33 as additional gene control mechanism.
The growth profiles obtained for all the samples (Fig. 1), suggested that exposure to IPTG and IPTG + PPDA may trigger a stress response in IL3-pET, EGFP and pET strains, characterized by reduced growth, whilst the wild type and IL3-EGFP remained unaffected. Surprisingly, the IL3-EGFP displayed similar growth behaviour to the wild strain under all inducing conditions, suggesting that the presence of the riboswitch may have reduced the metabolic load of producing the recombinant eGFP, allowing for the normal growth behaviour in this strain (Fig. 1f).
Although PPDA belongs to a group of compounds known as pyrimido–pyrimidines, which are well known for their antimicrobial properties through inhibition of dihydrofolate reductase enzyme,60 the growth profiles obtained for all the strains did not point to any detrimental effects resulting from exposure to PPDA (Fig. 1). The CPCA-W super scores plot of the FT-IR strain blocked model (Fig. 3a), provided supporting evidence, as it displayed the clustering of the un-induced and PPDA induced conditions and their separation from IPTG and IPTG + PPDA conditions, suggesting that IPTG and IPTG + PPDA are the significant influencing conditions impairing normal cellular growth, whilst E. coli cells exposed to PPDA exhibited a metabolic fingerprint similar to that of the un-induced condition. Furthermore, the data obtained by GC-MS displayed comparable levels of PPDA in the media and cell extracts, suggesting that under the examined conditions PPDA is not metabolized/catabolized by E. coli cells, thus does not contribute towards cellular metabolism.
Although LB broth is one of the most popular and applied media in the fields of bacteriology and biotechnology due to its many advantages, these include: generally providing a high growth rate due to its nutrient rich content (yeast extract and tryptone), easy to prepare and supporting fast growth, it can be said that it also has its limitations. Some of the disadvantages of LB broth include limited availability of carbohydrates and utilizable sugars61 and also batch-to-batch variation due to the complex ingredients of the media (yeast extract and tryptone).
In 2007, Sezonov and colleagues62 examined the physiology and growth behaviour of E. coli in LB broth and detected the occurrence of a brief diauxic lag upon reaching the OD600nm of around 0.3 which was also reported by other studies.63 This behaviour was explained by the depletion of the limited available sources of sugars in LB, causing the cellular metabolism to switch to using the available catabolizable amino acids and oligopeptides as the carbon source. The authors went further and also suggested that the cells catabolize the available amino acids in a sequential manner, starting from serine, aspartate, tryptophan, glutamate, glycine, threonine, alanine and proline, as proposed by other studies,64,65 and later switch to using other amino acids such as arginine, glutamine, asparagine, cysteine and lysine.
Our metabolic footprint analysis of the free amino acids gathered by GC-MS was in agreement with the above statement as it displayed a decrease of aspartic acid, proline, serine, glycine, glutamine and methionine pools (Fig. 9), suggesting that these are the primary amino acids depleted from the media and utilized by E. coli cells (both as carbon and nitrogen source). This is not surprising considering that; aspartic acid can be used as a carbon source,66 through its direct contribution towards the biosynthesis of essential intermediates of the tricarboxylic acid cycle (TCA) such as fumarate and oxaloacetate, via the activity of aspartate ammonia-lyase (EC: 4.3.1.1) and aspartate oxidase (EC: 1.4.3.16) respectively. In addition, aspartic acid is linked to the biosynthesis of other important amino acids such as threonine, lysine, glycine and serine through various interconnecting pathways (Fig. 9). Glutamine and glutamic acid are also linked to energy production via their contribution towards several TCA intermediate metabolites (2-oxoglutarate and succinate) (Fig. 9).
Glycine and serine could also be consumed as nitrogen and/or carbon sources via different pathways such as; the glycine cleavage system, where glycine is cleaved to NH3, CO2 and single carbon units, or through the deamination of serine via the activity of serine ammonia-lyase (EC: 4.3.1.17) resulting in the production of NH3 and pyruvate. Gschaedler and Boudrant also reported the rapid utilization of serine as a carbon and energy source (even before glucose) in E. coli, as it is easily transformed to pyruvate, entering the central metabolism.65
According to the GC-MS results of cell extracts obtained for different amino acids and silanamine, it seems probable that two processes could be occurring upon exposure of plasmid-bearing E. coli cells which carry different promoters that are affecting metabolism due to the different induction conditions: (i), when the cells are not under stress (un-induced and PPDA conditions) the available amino acids that could be used as carbon and/or nitrogen source are generally consumed and catabolized via different reactions (e.g. deamination) and directed towards the central metabolism supporting cellular growth and resulting in decreased intracellular pools of amino acids and increased levels of free ammonia, which may contribute to higher silanamine levels under these conditions. However, (ii) when the recombinant eGFP production is initiated and the cells are under stress (IPTG and IPTG + PPDA conditions), at this point the transcription of T7 RNA polymerase along with the lac operator, regulating the gene of interest in the pET plasmid, is initiated resulting in the consumption of intracellular pools of amino acids and aminoacyl–tRNA towards the production of the recombinant protein (eGFP) constraining the amino acid pools and directing the intermediates of the central metabolism toward different amino acids biosynthetic pathways to keep the homeostasis of the intracellular metabolites, resulting in growth impairment and lower silanamine production.
The detection of higher putrescine levels in the plasmid carrying strains was also a significant finding. Putrescine belongs to the polyamines family which are biogenic polycations and can be found in many different organisms. In E. coli, putrescine has been linked with the regulation of a variety of stress response mechanisms including; oxidative stress,67 starvation as well as acid resistance.68,69
The wild type strain in this study displayed comparable levels of putrescine under all inducing conditions (Fig. 9), whilst all other strains exhibited similar putrescine levels only under the PPDA and un-induced conditions. By contrast, under IPTG and IPTG + PPDA conditions an increased level of putrescine was detected in these strains. The higher putrescine levels could be due to a range of influencing factors such as; the stress caused by the maintenance and replication of the foreign DNA (recombinant plasmid),70,71 or the starvation effect caused by the initiation of plasmid transcription and eGFP synthesis under these conditions, resulting in the depletion of cellular resources and stimulation of various stress response mechanisms.72
Although, polyamines are required for normal function of cells, at high concentration and specifically at high pH they may interfere with normal growth due to their uptake as membrane-permeant weak bases resulting in blockage of porins and reduced membrane permeability.73 Therefore, their intracellular concentrations are strictly regulated and any excess amounts could be excreted.
Perhaps, this could explain the presence of putrescine in the media, since the cells are grown in LB media where the main carbon source is the catabolizable amino acids and peptides, and the excess ammonia generated via the deamination of such amino acids are secreted into the media elevating the pH to the alkaline range (pH of 8–9), resulting in the excretion of putrescine into the media via specific transport proteins.74
Conclusion
In conclusion, the data collected in this study through employment of various metabolomics techniques and strategies displayed the metabolic burden of carrying a foreign DNA, producing a recombinant protein and different induction conditions, reflected by the effects upon cell growth and the metabolic fingerprints, profiles and footprints of the cells. Furthermore, it seems the orthogonal riboswitch containing BL21(IL3) strain, that permits dual transcriptional–translational expression control over T7 RNA polymerase gene, may contribute towards balancing growth under recombinant protein producing conditions, whilst in comparison to the BL21(DE3) strain, it also provided a higher level of control over recombinant protein expression. Finally, our data suggests that the synthetic inducer compound (PPDA) does not impose any negative effects on growth and cellular metabolism, is not metabolized by E. coli cells under the examined conditions and together with the orthogonal riboswitch containing strain BL21(IL3) remains an attractive system for recombinant protein production.Authors contributions
Howbeer Muhamadali: Experimental design, sample collection and preparation, FT-IR analysis, data interpretation, manuscript preparation. Yun Xu: GC-MS data analysis. Rosa Morra: GFP quantification. Drupad K. Trivedi and Nicholas J. W. Rattray: GC-MS analysis. Neil Dixon: experimental design, manuscript preparation. Royston Goodacre: Principal investigator, experimental design, manuscript preparation.Acknowledgements
HM would like to thank the UK BBSRC for funding his PhD. ND holds BBSRC David Phillips fellowship, and RM is also supported by the BBSRC (BB/K014773/1) as is RG (BBSRC: BB/I017186/1), who also is grateful to BBSRC for support of SYNBIOCHEM (BB/M017702/1).References
- I. S. Johnson, Science, 1983, 219, 632–637 CAS.
- G. Walsh, Nat. Biotechnol., 2014, 32, 992–1000 CrossRef CAS PubMed.
- G. Walsh, Nat. Biotechnol., 2010, 28, 917–924 CrossRef CAS PubMed.
- P. Ahmad, M. Ashraf, M. Younis, X. Hu, A. Kumar, N. A. Akram and F. Al-Qurainy, Biotechnol. Adv., 2012, 30, 524–540 CrossRef CAS PubMed.
- B. R. Glick, Biotechnol. Adv., 1995, 13, 247–261 CrossRef CAS PubMed.
- S. Birnbaum and J. E. Bailey, Biotechnol. Bioeng., 1991, 37, 736–745 CrossRef CAS PubMed.
- J. H. Seo and J. E. Bailey, Biotechnol. Bioeng., 1985, 27, 1668–1674 CrossRef CAS PubMed.
- J. Vind, M. A. Sørensen, M. D. Rasmussen and S. Pedersen, J. Mol. Biol., 1993, 231, 678–688 CrossRef CAS PubMed.
- S. W. Harcum and W. E. Bentley, Biotechnol. Bioeng., 1993, 42, 675–685 CrossRef CAS PubMed.
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155–171 CrossRef CAS PubMed.
- O. Fiehn, J. Kopka, P. Dormann, T. Altmann, R. N. Trethewey and L. Willmitzer, Nat. Biotechnol., 2000, 18, 1157–1161 CrossRef CAS PubMed.
- R. J. Bino, R. D. Hall, O. Fiehn, J. Kopka, K. Saito, J. Draper, B. J. Nikolau, P. Mendes, U. Roessner-Tunali, M. H. Beale, R. N. Trethewey, B. M. Lange, E. S. Wurtele and L. W. Sumner, Trends Plant Sci., 2004, 9, 418–425 CrossRef CAS PubMed.
- L. M. Raamsdonk, B. Teusink, D. Broadhurst, N. Zhang, A. Hayes, M. C. Walsh, J. A. Berden, K. M. Brindle, D. B. Kell, J. J. Rowland, H. V. Westerhoff, K. van Dam and S. G. Oliver, Nat. Biotechnol., 2001, 19, 45–50 CrossRef CAS PubMed.
- C. L. Winder, W. B. Dunn, S. Schuler, D. Broadhurst, R. Jarvis, G. M. Stephens and R. Goodacre, Anal. Chem., 2008, 80, 2939–2948 CrossRef CAS PubMed.
- W. B. Dunn and D. I. Ellis, TrAC, Trends Anal. Chem., 2005, 24, 285–294 CrossRef CAS.
- Z. Lei, D. V. Huhman and L. W. Sumner, J. Biol. Chem., 2011, 286, 25435–25442 CrossRef CAS PubMed.
- R. Goodacre, S. Vaidyanathan, W. B. Dunn, G. G. Harrigan and D. B. Kell, Trends Biotechnol., 2004, 22, 245–252 CrossRef CAS PubMed.
- R. A. van den Berg, H. C. J. Hoefsloot, J. A. Westerhuis, A. K. Smilde and M. J. van der Werf, BMC Genomics, 2006, 7, 142 CrossRef PubMed.
- D. B. Kell, Curr. Opin. Microbiol., 2004, 7, 296–307 CrossRef CAS PubMed.
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263–269 CrossRef CAS PubMed.
- M. S. Wong, S. Wu, T. B. Causey, G. N. Bennett and K. Y. San, Metab. Eng., 2008, 10, 97–108 CrossRef CAS PubMed.
- S. Tabor and C. C. Richardson, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1074–1078 CrossRef CAS.
- N. A. Valdez-Cruz, L. Caspeta, N. O. Perez, O. T. Ramirez and M. A. Trujillo-Roldan, Microb. Cell Fact., 2010, 9, 18 CrossRef PubMed.
- M. D. Thomas and A. Van Tilburg, Methods in enzymology, Academic Press, 2000, vol. 305, pp. 315–329 Search PubMed.
- V. Vimberg, A. Tats, M. Remm and T. Tenson, BMC Mol. Biol., 2007, 8, 1–13 CrossRef PubMed.
- B. Fahnert, H. Lilie and P. Neubauer, Adv. Biochem. Eng./Biotechnol., 2004, 89, 93–142 CrossRef CAS PubMed.
- C. H. Schein, Nat. Biotechnol., 1989, 7, 1141–1149 CrossRef CAS.
- A. Serganov and D. J. Patel, Annu. Rev. Biophys., 2012, 41, 343–370 CrossRef CAS PubMed.
- E. Nudler and A. S. Mironov, Trends Biochem. Sci., 2004, 29, 11–17 CrossRef CAS PubMed.
- W. C. Winkler and R. R. Breaker, Annu. Rev. Microbiol., 2005, 59, 487–517 CrossRef CAS PubMed.
- N. Dixon, J. N. Duncan, T. Geerlings, M. S. Dunstan, J. E. G. McCarthy, D. Leys and J. Micklefield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2830–2835 CrossRef CAS PubMed.
- N. Dixon, C. J. Robinson, T. Geerlings, J. N. Duncan, S. P. Drummond and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 3620–3624 CrossRef CAS PubMed.
- R. Morra, J. Shankar, C. J. Robinson, S. Halliwell, L. Butler, M. Upton, S. Hay, J. Micklefield and N. Dixon, Nucleic Acids Res., 2015 DOI:10.1093/nar/gkv912.
- C. J. Robinson, H. A. Vincent, M.-C. Wu, P. T. Lowe, M. S. Dunstan, D. Leys and J. Micklefield, J. Am. Chem. Soc., 2014, 136, 10615–10624 CrossRef CAS PubMed.
- C. L. Winder, S. V. Gordon, J. Dale, R. G. Hewinson and R. Goodacre, Microbiology, 2006, 152, 2757–2765 CrossRef CAS PubMed.
- H. Martens, J. P. Nielsen and S. B. Engelsen, Anal. Chem., 2003, 75, 394–404 CrossRef CAS PubMed.
- D. C. Wedge, J. W. Allwood, W. Dunn, A. A. Vaughan, K. Simpson, M. Brown, L. Priest, F. H. Blackhall, A. D. Whetton, C. Dive and R. Goodacre, Anal. Chem., 2011, 83, 6689–6697 CrossRef CAS PubMed.
- O. Fiehn, J. Kopka, R. N. Trethewey and L. Willmitzer, Anal. Chem., 2000, 72, 3573–3580 CrossRef CAS PubMed.
- P. Begley, S. Francis-McIntyre, W. B. Dunn, D. I. Broadhurst, A. Halsall, A. Tseng, J. Knowles, R. Goodacre and D. B. Kell, Anal. Chem., 2009, 81, 7038–7046 CrossRef CAS PubMed.
- W. B. Dunn, D. Broadhurst, P. Begley, E. Zelena, S. Francis-McIntyre, N. Anderson, M. Brown, J. D. Knowles, A. Halsall, J. N. Haselden, A. W. Nicholls, I. D. Wilson, D. B. Kell and R. Goodacre, Nat. Protoc., 2011, 6, 1060–1083 CrossRef CAS PubMed.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. M. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden and M. R. Viant, Metabolomics, 2007, 3, 211–221 CrossRef CAS PubMed.
- Y. Xu and R. Goodacre, Metabolomics, 2012, 8, 37–51 CrossRef CAS.
- J. A. Westerhuis, T. Kourti and J. F. MacGregor, J. Chemom., 1998, 12, 301–321 CrossRef CAS.
- A. K. Smilde, J. A. Westerhuis and S. de Jong, J. Chemom., 2003, 17, 323–337 CrossRef CAS.
- Y. Kassama, Y. Xu, W. B. Dunn, N. Geukens, J. Anne and R. Goodacre, Analyst, 2010, 135, 934–942 RSC.
- P. Penumetcha, K. Lau, X. Zhu, K. Davis, T. T. Eckdahl and A. M. Campbell, Bios, 2010, 81, 7–15 CrossRef CAS.
- Y. Setty, A. E. Mayo, M. G. Surette and U. Alon, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7702–7707 CrossRef CAS PubMed.
- Y. Kuang, I. Biran and D. R. Walt, Anal. Chem., 2004, 76, 6282–6286 CrossRef CAS PubMed.
- G. R. Smith and B. Magasanik, J. Biol. Chem., 1971, 246, 3330–3341 CAS.
- R. B. Goldberg, F. R. Bloom and B. Magasanik, J. Bacteriol., 1976, 127, 114–119 CAS.
- R. A. Bender, Microbiol. Mol. Biol. Rev., 2012, 76, 565–584 CrossRef CAS PubMed.
- J. C. Arguelles, Arch. Microbiol., 2000, 174, 217–224 CrossRef CAS PubMed.
- N. Ferrer-Miralles, J. Domingo-Espin, J. L. Corchero, E. Vazquez and A. Villaverde, Microb. Cell Fact., 2009, 8, 17 CrossRef PubMed.
- F. R. Blattner, G. Plunkett, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau and Y. Shao, Science, 1997, 277, 1453–1462 CrossRef CAS PubMed.
- M.-J. Han and S. Y. Lee, Microbiol. Mol. Biol. Rev., 2006, 70, 362–439 CrossRef CAS PubMed.
- J. H. Choi, K. C. Keum and S. Y. Lee, Chem. Eng. Sci., 2006, 61, 876–885 CrossRef CAS.
- C. J. Huang, H. Lin and X. Yang, J. Ind. Microbiol. Biotechnol., 2012, 39, 383–399 CrossRef CAS PubMed.
- S. Carneiro, E. C. Ferreira and I. Rocha, J. Biotechnol., 2013, 164, 396–408 CrossRef CAS PubMed.
- D. I. Ellis and R. Goodacre, Curr. Opin. Biotechnol., 2012, 23, 22–28 CrossRef CAS PubMed.
- M. Banjanac, I. Tatic, Z. Ivezic, S. Tomic and J. Dumic, Food Technol. Biotechnol., 2009, 47, 236–245 CAS.
- J. W. Payne and C. Gilvarg, J. Biol. Chem., 1968, 243, 6291–6299 CAS.
- G. Sezonov, D. Joseleau-Petit and R. D'Ari, J. Bacteriol., 2007, 189, 8746–8749 CrossRef CAS PubMed.
- C. H. Wang and A. L. Koch, J. Bacteriol., 1978, 136, 969–975 CAS.
- B. M. Pruss, J. M. Nelms, C. Park and A. J. Wolfe, J. Bacteriol., 1994, 176, 2143–2150 CAS.
- A. Gschaedler and J. Boudrant, J. Biotechnol., 1994, 37, 235–251 CrossRef CAS.
- W. W. Kay, J. Biol. Chem., 1971, 246, 7373–7382 CAS.
- A. Tkachenko, L. Nesterova and M. Pshenichnov, Arch. Microbiol., 2001, 176, 155–157 CrossRef CAS PubMed.
- I. L. Jung and I. G. Kim, J. Biol. Chem., 2003, 278, 22846–22852 CrossRef CAS PubMed.
- A. G. Tkachenko, M. S. Shumkov and A. V. Akhova, Biochemistry. Biokhimiia, 2006, 71, 185–193 CrossRef CAS PubMed.
- W. E. Bentley and D. S. Kompala, Ann. N. Y. Acad. Sci., 1990, 589, 121–138 CrossRef CAS PubMed.
- W. E. Bentley, N. Mirjalili, D. C. Andersen, R. H. Davis and D. S. Kompala, Biotechnol. Bioeng., 1990, 35, 668–681 CrossRef CAS PubMed.
- F. Silva, J. A. Queiroz and F. C. Domingues, Biotechnol. Adv., 2012, 30, 691–708 CrossRef CAS PubMed.
- E. Yohannes, A. E. Thurber, J. C. Wilks, D. P. Tate and J. L. Slonczewski, BMC Microbiol., 2005, 5, 59 CrossRef PubMed.
- K. Kashiwagi, S. Miyamoto, F. Suzuki, H. Kobayashi and K. Igarashi, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4529–4533 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00624d |
| This journal is © The Royal Society of Chemistry 2016 |