
Chemogenomic analysis of neuronal differentiation with pathway changes in PC12 cells†
Jack Yu-Shih
Lin
ab,
Chien Liang
Wu
b,
Chia Nan
Liao
a,
Akon
Higuchi
ce and
Qing-Dong
Ling
*ad
aGraduate Institute of Systems Biology and Bioinformatics, National Central University, Chungli, Taiwan, Republic of China. E-mail: qdling@hotmail.com
bTaipei Medical University Municipal Wan-Fang Hospital, Taipei, Taiwan, Republic of China
cDepartment of Chemical & Materials Engineering, National Central University, Chungli, Taiwan, Republic of China
dCathay Medical Research Institute, Cathay General Hospital, No. 32, Ln 160, Jian-Cheng Road, Shi-Zhi, Taipei, Taiwan, Republic of China. E-mail: manto0719@gmail.com; Fax: +886-2-26907963; Tel: +886-2-26907954 ext. 2313
eDepartment of Botany and Microbiology, King Saud University, Riyadh, 11451, Saudi Arabia
First published on 5th November 2015
Abstract
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database creates networks from interrelations between molecular biology and underlying chemical elements. This allows for analysis of biologic networks, genomic information, and higher-order functional information at a system level. Through high throughput experiments and system biology analysis, we investigated the genes and pathways associated with NGF induced neuronal differentiation. We performed microarray experiments and used the KEGG database, system biology analysis, and annotation of pathway functions to study NGF-induced differentiation in PC12 cells. We identified 2020 NGF-induced genes with altered expressions over time. Cross-matching with the KEGG database revealed 830 genes; among which, 395 altered genes were found to have a 2-fold increase in gene expression over a two-hour period. We then identified 191 associated biologic pathways in the KEGG database; the top 15 pathways showed correlation with neural differentiation. These included the neurotrophin pathways, mitogen-activated protein kinase (MAPK) pathways, genes associated with axonal guidance and the Wnt pathways. The activation of these pathways synchronized with nerve growth factor (NGF)-induced differentiation in PC12 cells. In summary, we have established a model system that allows one to systematically characterize the functional pathway changes in a group of neuronal population after an external stimulus.
Introduction
The concept of systemic structures has been applied in areas such as information engineering and has recently been applied to the field of biology. Its value has become evident with the development of tools such as microarrays and Next Generation sequencing, which provide huge amounts of data related to basic molecules as well as biologic systems, resulting in the field of systems biology. In the past, science has utilized methods that transform complex things into simple elements to study their properties and interactions with other elements. However, this approach often falls short with respect to proven systemic characteristics. Systems biology identifies and develops networks via quantitative data analyses and integration of mathematical formulas.1 Many independent elements, such as tissues, cells, proteins, and genes, form complicated and nonlinear interacting networks, so-called biologic pathways.2 These pathways with various functions form a living system capable of responding to stimuli, reproducing, and maintaining a certain level of stability.The KEGG is part of the 15 year national research program funded by the Japanese government in 1995. It is a suite of databases and associated software for understanding and simulating higher-order functional behaviors of the cell or the organism from its genome information.3 The main focus of the KEGG was to understand how genes and gene products combine to represent a biologic system and make the system functional from analysis of previous accumulated sequencing and basic genomics databases.4 With advances in the study of cell functions, KEGG has become an exquisite information bank that analyzes gene functions from a systemic viewpoint.
NGF, a small secreted protein discovered 50 years ago, belongs to the neurotrophin family, and has been shown to promote peripheral nerve regeneration, stimulate morphological differentiation and regulate neuronal gene expression.5 In this study, we attempt to delineate neural differentiation pathway changes induced by NGF administration by studying the networks involved through quantitative data analysis and mathematical formula integration. In the process, we hope to obtain a deeper understanding on how NGF exerts its therapeutic effects in various neurodegenerative diseases. The aim of the present study was to understand the NGF interaction system by looking at the networks involved through quantitative data analysis and mathematical formula integration. We set up an external perturbation of NGF administration to the PC12 model system and analyzed the pathways involved through the analysis of the KEGG database. We found that the development of new pathways after NGF administration was synchronized with the time course of neuronal differentiation. The results may help us understand more the mechanism, through which NGF administration may offer a therapeutic role in the treatment of various neurological disorders. Furthermore, the methodology described in this study may also find clinical applications in setting up individualized dynamical models based on large scale databases, which can help design and test effective treatment tailored to each patient.
Materials and methods
PC12 cell culture
PC12 cells were obtained from the Bioresource Collection and Research Center (BCRC 60048).6 The cells were cultured on 100 mm dishes and 6-well poly-L-lysine coating plates and maintained in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% (v/v) heat-activated fetal bovine serum (FBS), 5% horse serum (HS), 100 IU ml−1 of penicillin and 100 μg ml−1 of streptomycin under a culture condition of 37 °C and 5% CO2. The culture medium was replaced every two or three days. Cells were subcultured when they reached 70% to 80% confluence. Subculture was done in the following manner. First, the culture medium was discarded and the cells were washed with PBS several times. After incubating with 3 ml of trypsin in a 37 °C oven for 5 minutes, the PC12 cells from the bottom of the plate fall off (trypsinization). Subsequently, 3 ml of culture solution was added to terminate the reaction. The supernatant was then collected into a centrifuge tube and centrifuged at 800 rpm for 5 min. The supernatant was then removed, culture medium was added, and the cells were again replated onto 10 cm culture dishes.Treatment with NGF
When the plated PC12 cells reached 60% to 70% confluence, the cells were then again washed with PBS to ensure the complete removal of the culture medium. Excessive shaking was avoided to prevent the falling off of the PC12 cells from the plate. Then 4 ml of Neurobasal medium (Invitrogen, Carlsbad, CA, USA) containing 50 ng ml−1 of NGF, 1% horse serum, 1% fetal bovine serum, 100 μg ml−1 of streptomycin, and 100 IU ml−1 of penicillin was added to the plate. The cells were then cultured at 37 °C and 5% CO2 for 1 hour and 3 hours as the experimental protocols dictate.RNA extraction
RNA extraction was conducted at 0 hour (control), 1 hour and 3 hours after the addition of the NGF. The extraction was performed by first removing the culture medium. Then 1 ml of the Trizol Reagent (Invitrogen) was added to the 10 cm culture dish. The collected sample was placed into the 1.5 ml centrifuge tube and incubated for 10 min. 200 μl of chloroform (Sigma) was then added. The tube was shaken for 10 s and incubated at room temperature for 15 min. Subsequently, the samples were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 15 minutes. The supernatant was removed, 500 μl of 100% isopropanol was added, and the samples were incubated at room temperature for 15 min. The centrifugation and isopropanol incubation steps were performed again. After the final centrifugation at 12000 rpm at 4 °C for 15 minutes, the supernatant was removed and 500 μl of ice-cold 70% ethanol was added. The samples were centrifuged at 12000 rpm for 5 min. The supernatant was removed and the resulting pellet was dissolved in RNase-free water and stored at −80 °C for future usage.
000 rpm at 4 °C for 15 minutes. The supernatant was removed, 500 μl of 100% isopropanol was added, and the samples were incubated at room temperature for 15 min. The centrifugation and isopropanol incubation steps were performed again. After the final centrifugation at 12000 rpm at 4 °C for 15 minutes, the supernatant was removed and 500 μl of ice-cold 70% ethanol was added. The samples were centrifuged at 12000 rpm for 5 min. The supernatant was removed and the resulting pellet was dissolved in RNase-free water and stored at −80 °C for future usage.
Gene expression profiling
After the RNA samples were purified, the samples were used as substrates for reverse transcription in the presence of fluor-derivatized nucleotides. The tagged mRNA samples were hybridized to the gene-specific cDNA detectors immobilized on the microarray. The resulting localized concentrations of fluorescent molecules were detected and quantitated. For this part of study, the Rat Exon 1.0 ST Array (Affymetrix) was the gene chip used to search for the genes involved after NGF stimulation. 2 gene chips were used for each of the time points (0 h, 1 h and 3 h).Data mining and clustering
Principal component analysis (PCA) was performed based on the default covariance matrix in the R statistical software to emphasize the variation and bring out patterns in the microarray dataset. The data were not pre-scaled. Data were then subsequently analyzed by two different methods. First, analysis of variance (ANOVA) was performed using the Partek Genomic Suite software (Partek Inc, St. Louis MO, USA). We first imported appropriately formatted chip information and defined the sample attribute according to the experimental protocols. Then we generated a sample histogram to evaluate the data distribution and performed the gene-level analysis and ANOVA. The purpose of ANOVA was to evaluate the changes in datasets when there are variations of different parameters. We used hypothesis testing methods to determine whether these parameters can indeed explain the changes in the datasets. The results of the analysis were screened with a p-value of less than 0.05.After the datasets were processed using ANOVA with R statistical software, we performed cluster analysis and generated heat maps for both the control and the experimental groups. The hierarchical clustering was done using the default complete linkage clustering methodology from the R software. The longest distance between 2 clusters was defined for cluster analysis. The formula is as follows:
| max{d(a,b): a ∈ A, b ∈ B}. |
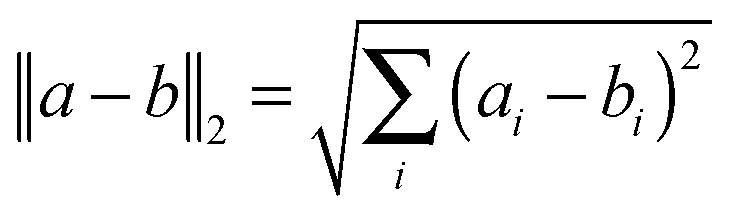
KEGG pathway analysis
The results from the ANOVA and cluster analysis were formatted using the Microsoft Excel to form an input database that is compatible with the KEGG software. Gene identity was denoted by the field (ORF, “Open Reading Frame”) in the KEGG database. In search of the possible pathways associated with the genes, we used the KEGG gene ID as the first preference, complemented with the gene IDs from other databases (i.e., NCBI Entrez, IPI, and Genbank). Fields X and Y denote the position of X and Y axes on the gene chip. The field “Ratio” represents the initial results from the analysis of the gene expression comparison between the control and the experimental groups.The converted dataset was an input into the KEGG using the KegArray tool. We also used the “Mapping to Pathway” function in the “Tools” section to map each gene to the associated functional pathway. If a dataset ID was not recognized by the KEGG as a gene ID, a conversion step was performed to transform the data to a KEGG gene ID.
For the comparison of gene information, we downloaded the Rattus norvegicus gene list from the KEGG database. This included gene names and KEGG gene IDs. We used the VLookup function of the Excel to locate each entire biologic pathway in the KEGG database and summarize it in the Excel.
To make a pathway table, the results of KegArray were categorized into several columns in the Excel. The Pathway ID and Pathway Name correspond to the KEGG database IDs and names. The number of matched genes corresponds to the fit gene number in the KegArray datasets. The total gene number represents the number of genes associated with the pathway. The Class field corresponds to the name of each KEGG pathway category.
Genes' associations with pathways
The Fisher exact test was used to test the null hypothesis that each gene was randomly associated with a given biologic pathway. We used R software to construct a 2 × 2 table (field A = fit gene number; field B = total gene number − fit gene number; field C = number of matched genes − fit gene number; field D = number of genes in the KEGG Rattus norvegicus database − number in field B). We used Fisher exact test R values, for calculating p values for each pathway. The table and formula are as follows:p < 0.05 (cutoff threshold for null hypothesis).| #Significance | #No significance | |
|---|---|---|
| #In category | A | B |
| #Not in the category | C | D |
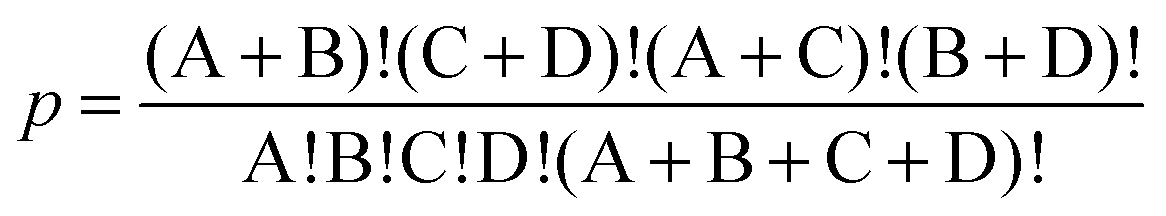
Results
Dataset screening
The consistency and stability of the gene chip experiments were evident as shown in the figure of frequency distribution of signal strengths (Fig. 1). After PCA was performed with the dataset of 2020 genes and 6 arrays, a biplot was generated using the function ggbiplot in R. The first two principal components accounted for 57.2% of the variance in the dataset. The PCA revealed a clear distinction among the 0 h, 1 h and 3 h experimental groups (Fig. 2). The datasets were then categorized into the control (0 h) and experimental (1 h and 3 h) groups before the ANOVA processing. Setting a significant level of p-value < 0.05, we identified 2020 NGF-induced PC12 genes after ANOVA. For each of the group sets in comparison, we listed the genes that were altered by the NGF treatment with significant changes of gene expressions over time (ESI,† Fig. S8–S10). We also performed heat map and cluster analysis with p-value < 0.05, which revealed that the experimental groups can be separated into 3 clusters based on the NGF treatment time (Fig. 3). We also found that in clustering, the distance between the control and 1 h group was less than the distance between the control and the 3 h group, which indicated that gene expression was more similar between the control and the 1 h group than the gene expression between the control and the 3 h group. We then cross-matched the 6035 genes from the KEGG database with the NGF induced PC12 genes found in our study, resulting in a total of 830 genes.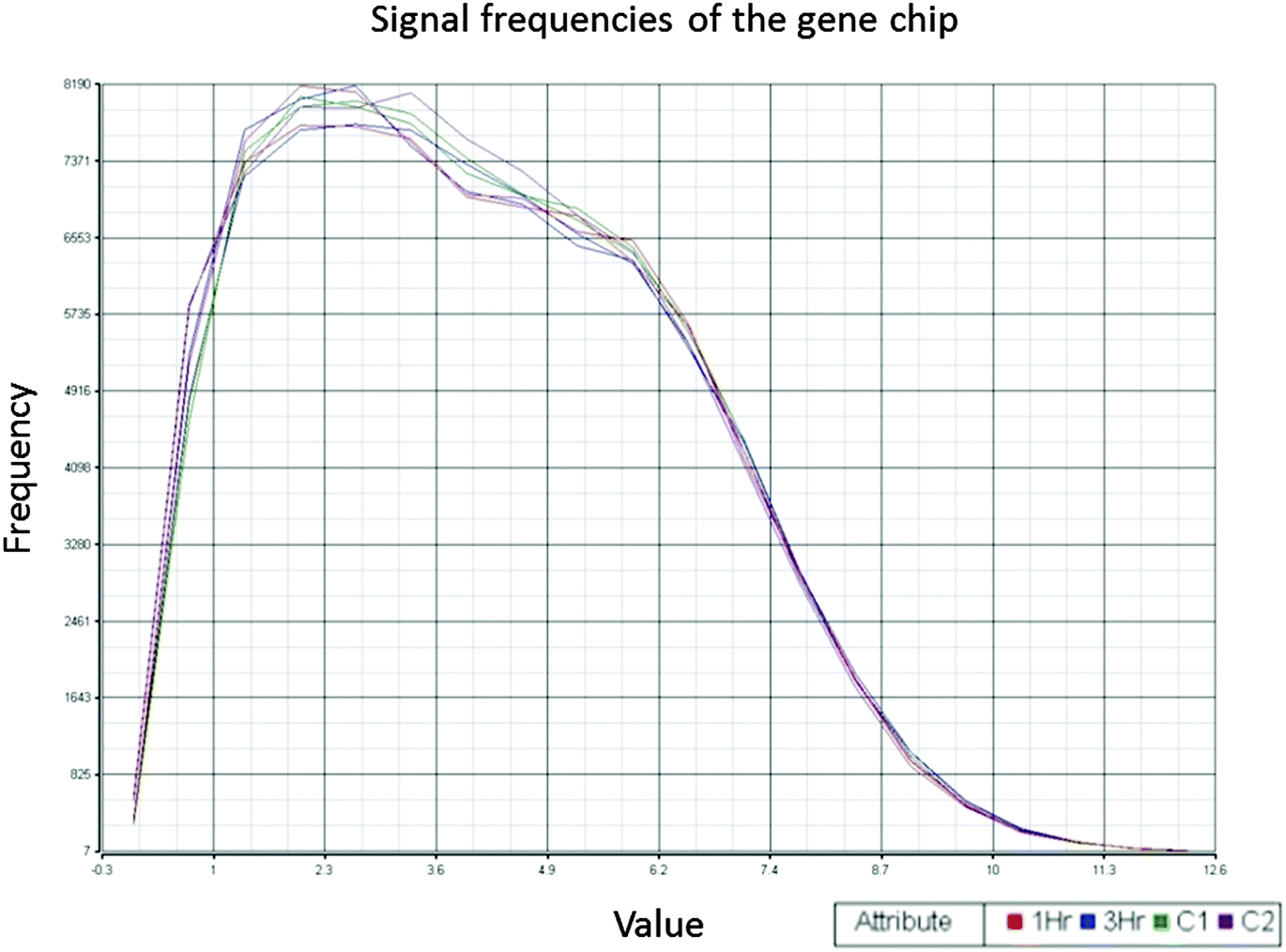 | ||
| Fig. 1 Quality control of gene chip experiments. Signal frequency distribution showed that the signal intensities among the six gene chips are consistent and stable. Two gene chips were done for every experimental group (1 h, 3 h, and control groups). | ||
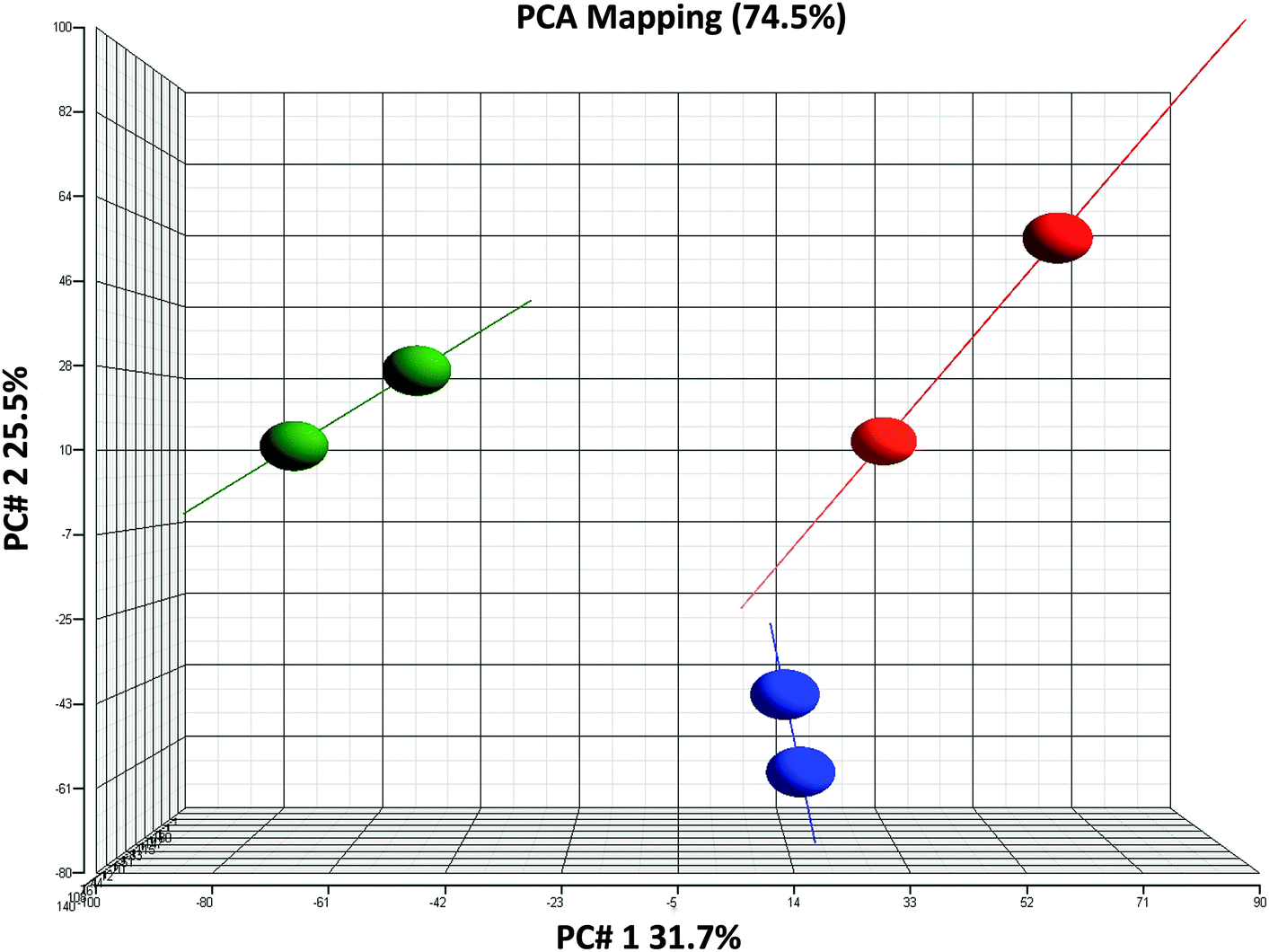 | ||
| Fig. 2 Quality control of gene chip experiments. PCA (principal component analysis) classification showed that the 0 h (red dots), 1 h (blue dots) and 3 h (green dots) groupings are distinct without overlapping. | ||
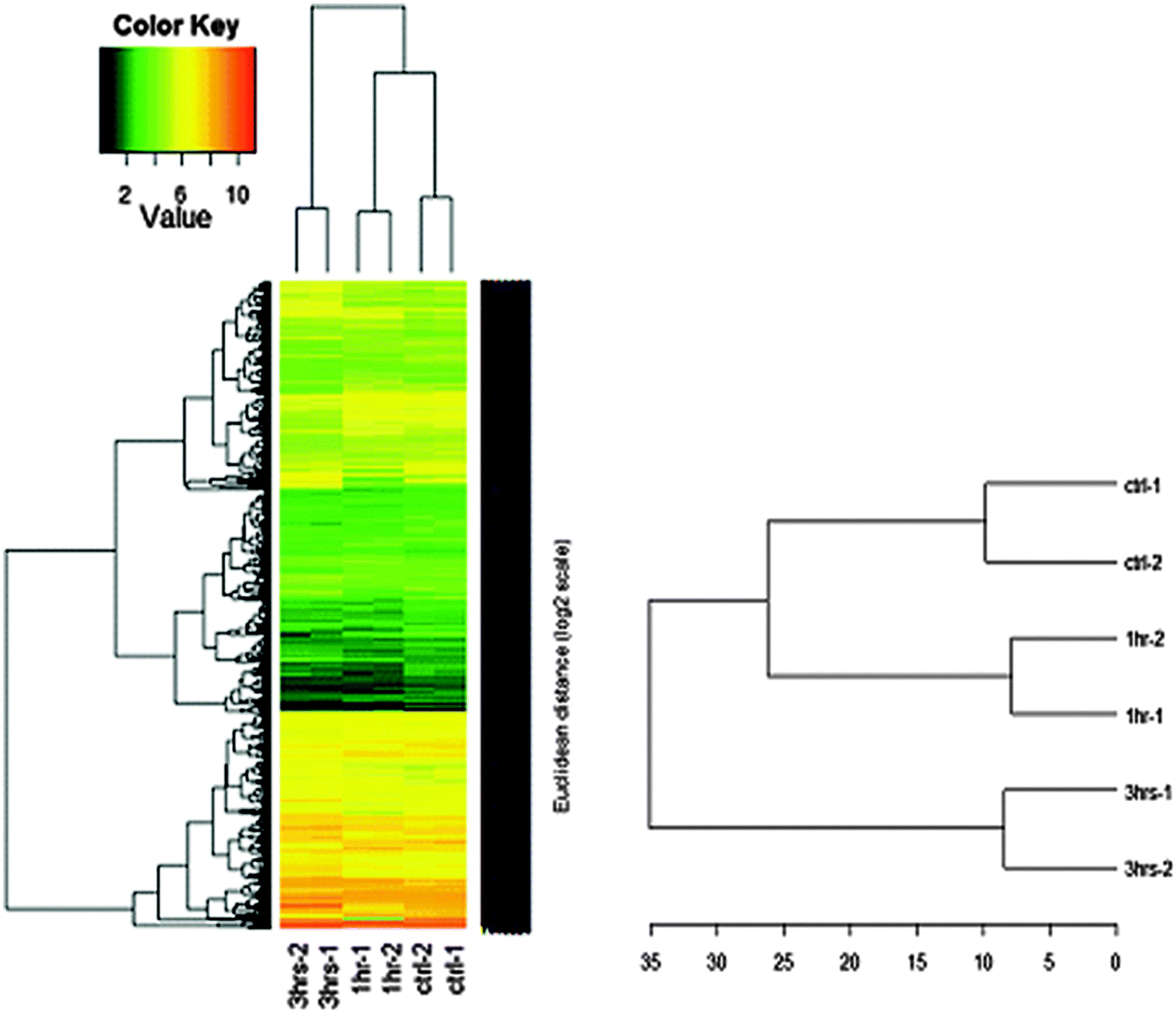 | ||
| Fig. 3 Heat maps and results of cluster analysis (p < 0.05). The results showed that the experimental groups could be broken down into 3 clusters based on the NGF treatment time. The distance between the control and 1 h NGF groups was less than that between the control and 3 h NGF groups, indicating that gene expression was more similar between the control and 1 h NGF groups than between the control and 3 h groups. | ||
Gene expression analysis
The KEGG database has a standardized threshold value for the change in the gene expression of 1.5-fold. We applied this threshold to our experiments. Among the 830 cross-matched genes, 395 showed altered expression according to this threshold. We performed clustering with the cross-matched 395 genes, and generated a heat map, which showed color enhancement over time (Fig. 4). This color enhancement indicates that this threshold of 1.5-fold can effectively screen out genes with significantly enhanced expression. Among the 395 altered genes, the number of genes with 1.5 fold gene expression changes in the 3 h NGF treated group was approximately twice that of the 1 h NGF treated group. The number of genes involved in the 3 h NGF treated group was 60.6% of the total number of genes involved, compared to 24% in the 1 h NGF treated group. In both the 1 h NGF and 3 h NGF treated groups, 15.4% of genes showed significant gene expression (Fig. 5). Based on the results of clustering of genes with changes in gene expression of 1.5-fold, we identified 4 gene sets with specific patterns of changes of gene expression over time, in response to NGF treatment (Fig. 6). Among the gene sets, gene set 4 has the most number of genes that was altered significantly by NGF treatment over the time courses (ESI,† Fig. S4). | ||
| Fig. 4 Heat map and clustering analysis after subjecting to the KEGG database. The results showed analysis for gene changes greater than 1.5 fold, which effectively selected out genes with significantly enhanced expression. | ||
 | ||
| Fig. 5 KEGG gene analysis. After subjecting initial data into the KEGG database, there are a total of 395 altered genes after NGF stimulation. The number of genes with 1.5 fold gene expression changes in the 3 h NGF treated group was approximately twice that of the 1 h NGF treated group. | ||
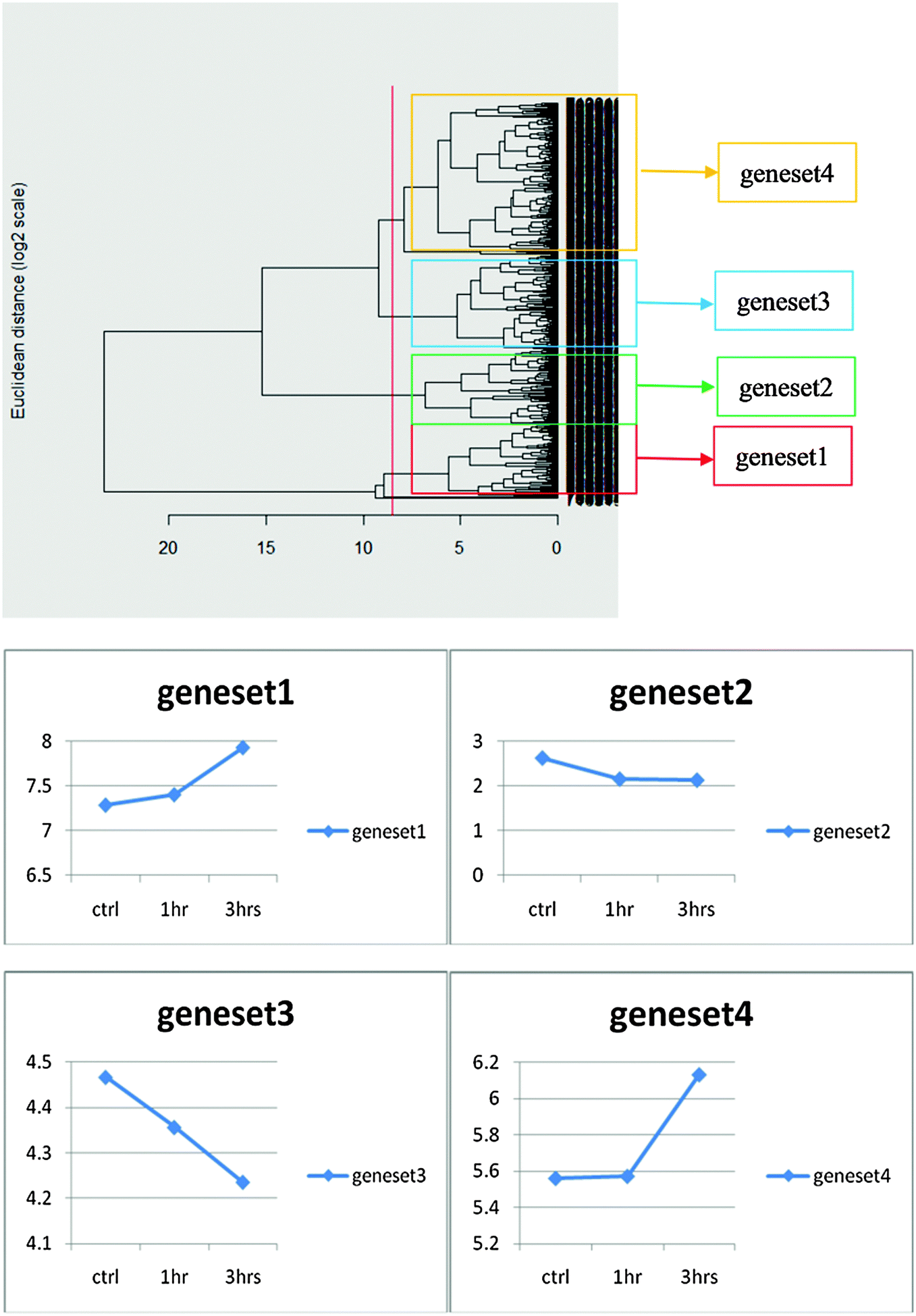 | ||
| Fig. 6 Time courses for changes in gene expression among different gene sets in response to NGF treatment. Before clustering, changes in gene expression occurred over time, with different gene sets showing the increase, decrease, or minimal changes with time in response to NGF treatment. The threshold for the change in gene expression was set at ±1.5-fold changes. | ||
KEGG pathway analysis
KEGG analysis revealed that there are 191 biologic pathways that could be affected by the observed alterations in gene expression. The top 15 pathways showed correlations with neuronal differentiation. Among these pathways, there are 4 pathways that showed a 2-fold increase in gene expression between the 3 h NGF group and the 1 h NGF group. They are the mitogen-activated protein kinase (MAPK) pathway (35 genes at 1 h and 54 genes at 3 h), genes associated with axonal guidance (12 genes at 1 h and 26 genes at 3 h), Wnt pathway (16 genes at 1 h, 25 genes at 3 h), and neurotrophin pathway (4 genes at 1 h and 14 genes at 3 h). A shortlist of genes that are significantly altered by NGF treatment over the time courses for each of the pathways is provided in ESI,† Fig. S7. In KegArray analysis, there is a two-fold change in the number of genes between the 1 h and 3 h NGF treated groups. The number of pathways associated with neuronal differentiation has also increased, indicating that the effect of NGF on PC-12 cell differentiation will increase over time. We performed the Fisher exact test for the pathway changes at different time points. The number of pathways involved at the 3 h time point was greater than that at the 1 h time point (Fig. 7). On the KEGG pathway map for the MAPK pathway, we observed that many nodes from the cell membrane to intracellular compartments were activated. We assessed the timing of the pathway changes and found synchrony with NGF-induced gene alterations. The nodes were mostly up-regulated from the 1 h time point to the 3 h time point, which indicated that the MAPK activity increased over time (Fig. 8). These results showed that the activation of pathways associated with neuronal differentiation was synchronized with changes in gene expression induced by NGF treatment. | ||
| Fig. 7 KEGG pathway analysis. The results showed that after NGF stimulation, the number of pathways involved at the 3 h time point was greater than that at the 1 h time point. New KEGG biologic pathways also developed between 1 h and 3 h after NGF treatment. These newly developed pathways such as axon guidance (red bracket) indicated changes in biologic behavior. | ||
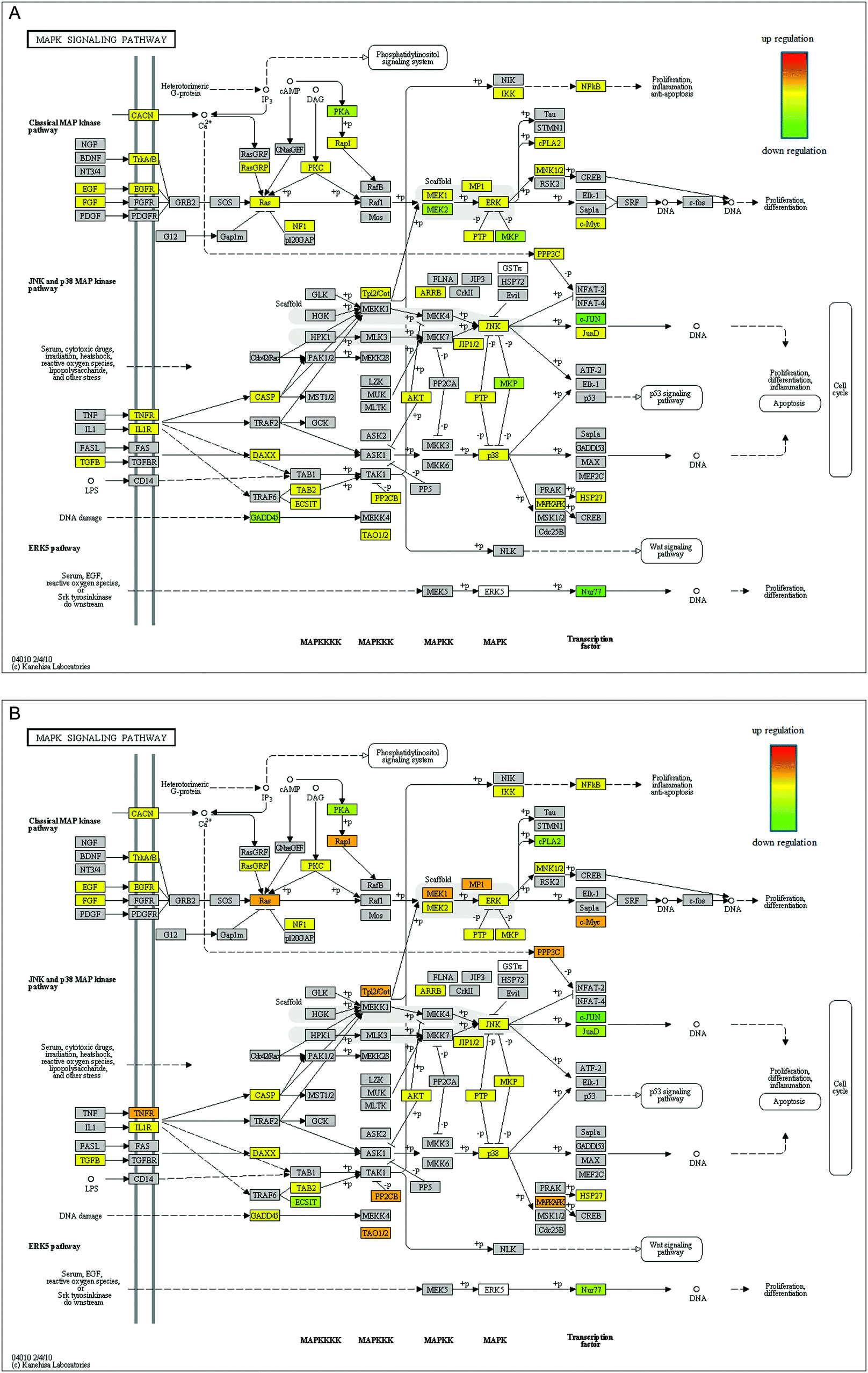 | ||
| Fig. 8 Changes in the mitogen-activated protein kinase (MAPK) signaling pathway 1 hour after NGF treatment (A) and 3 hours after NGF treatment (B). Pathway changes synchronized with NGF-induced gene alterations. Colored nodes represent genes that were activated differentially. The nodes were mostly upregulated after 3 hours. That is, if we looked at different points along the pathway, the internodal change was mostly upregulated. The results indicated that the MAPK activity increased over time. | ||
Discussion
In this study, we utilized the system biology methodology to investigate the changes in gene expression that occurred in PC12 cells after NGF administration. NGF is a polypeptide growth factor which functions to regulate the growth and survival of nerve cells, and has been shown to profoundly affect the development of both the young and the adult nervous systems.5 It prevents neuronal apoptosis in primary cultured neurons and reduces neuronal degeneration in animal models of neurodegenerative diseases. We identified 2020 NGF-induced PC12 genes in our arrays. The results showed changes in gene expression over time. We performed a gene ontology (GO) analysis (ESI,† Fig. S5 and S6), and then compared these genes with 6035 genes from the KEGG database. Cross-matching resulted in 830 genes. Among these, we identified 395 altered genes. Compatible with the GO analysis, most of these genes changes were transcription or development oriented as the cells were still in the early stages of differentiation. We then identified 191 associated biologic pathways in the KEGG database; the top 15 showed correlations with neuronal differentiation. Our data revealed that the modulation of the neurotrophin, Wnt/beta-catein, MAPK/ERK, and axonal guidance signaling pathways may contribute to cell regeneration and repair of nerve tissue. Thus, we identified changes in neuronal differentiation pathways using the KEGG database, which were synchronized with the time course of NGF-induced differentiation.The quantification of functionalities as a means to better understand biologic behavior is a key in contemporary biology.7 Although microarrays can generate huge amounts of data, interpreting such a large volume of data remains an obstacle. In 1998, the gene ontology (GO) project was initiated in an attempt to interpret microarray-derived data. GO was the first tool to utilize computer networks as a platform to assist in data analysis.8 However, there are certain limitations to the GO analytical tools. For example, in GO, certain databases can only be applied to certain genes, and there is also the lack of direct association with pathways. In contrast, the KEGG database standardizes biologic knowledge and serves as a link between genetic and high-level information. The KEGG database links genes, proteins, and interactive biologic pathways together. Presently, there are 16 major information banks in the KEGG databases; these can be roughly divided into categories of genomic (KEGG GENES), chemical (KEGG LIGAND), and system information (KEGG PATHWAYS and KEGG BRITE), and health information (KEGG DISEASE).9 Whereas the genomic and chemical information relate to the individual living beings, the system and health information relate to high-level aspects such as pathways and disease processes. The KEGG pathway database thus reflects integrated molecular networks, metabolism, and genetic information processing. To facilitate the analysis of a huge amount of data collected from multiple platforms, a Java application called KegArray was also developed to map these data into the KEGG databases individually or simultaneously. Tightly integrated with the KEGG database, KegArray is a visualization tool, which provides researchers an interactive environment with the KEGG databases.10,11 Although KegArray does not provide any statistical evaluations on the inputted data, it does give researchers the full flexibility in determining the significance level while inputting the data, as well as the corresponding color coding for the mapping.
The clustering process categorizes different subjects that share similar elements. Every subject in each group shares similar characteristics. In our time course graphs, these subjects are grouped together. There are many factors that affect grouping. Based on the heat map, values that are close together (e.g., 0.1 and 0.2, 0.2 and 0.3) are readily grouped together. From the KEGG data analysis, we know that genes activated in the pathway at a specific time point will be different. This is because each gene in the pathway plays a different role. From the viewpoint of systems biology, genes with different, complex time courses of expression make the pathway robust and complete. Explaining these differences in timing within the same pathway is difficult and requires additional calculations and time points.
PC12 cells have been widely used as models for studying nerve differentiation, neurotoxicity, neuropharmacology, neuronal protection mechanisms, and neuroendocrinology.12–17 Early studies with this cell line showed that NGF treatment induces neuronal differentiation.18 Under NGF stimulation, PC12 cells will slow down cell divisions and differentiation into sympathetic neuron-like cells with neurite outgrowth. This makes PC12 cells a useful model for studying the functions of neurotrophic factors and neuronal differentiation. During the first hour of NGF treatment, the cells are in the early stages of differentiation; therefore, most of the activated genes are transcription factor oriented.19 The KEGG pathways reflected the action of enzymes or regulating factors inside cellular compartments. While an increase in gene expression from 1 h to 3 h was evident, gene expression at 3 h represented approximately 60% of the total gene amount. This indicated that the majority of genes were activated after 1 h of NGF treatment. Clustering categorized the genes into different groups, and each gene was clustered based on the experimental group (i.e., 0, 1, or 3 h NGF treatment). Additional KEGG biologic pathways developed between 1 h and 3 h after NGF treatment. These pathways indicated changes in biologic behavior such as axon guidance, mitogenic signaling, and phosphorylation of phospholipase C-γ.20–22
The neurotrophin pathway is the main pathway whereby NGF enters the cell. The expression of increased TrkA expression after NGF coincides with the fact that NGF leads to significant changes in NGF receptor expression, leading to a 19 fold increase in the TrkA/p75 receptor ratio. In our study, there are 4 and 14 genes, associated with the neurotrophin pathway, which are activated at the 1 h and 3 h time points, respectively. It has been reported in the literature that Trk activity peaks from 1 h to 3 h after NGF treatment and decreases after 3 hours.23Via activation of the neurotrophin signaling pathway, NGF treatment is able to activate the MAPK pathway, which subsequently activates nuclear regulatory factors.24 Comparing the timing of activation on the pathway map, most nodes showed upregulation 1 h after NGF treatment, indicating that the neurotrophin pathway exerted its maximal effect 1 h after NGF treatment (ESI,† Fig. S1). It has been reported that Trk activity peaks from 1 h to 3 h after NGF treatment and decreases after 3 h.21 The activation of p38 and IkB/NFkB internodal expression in the Trk pathway decreases 3 h after NGF treatment.22 If we were to add more NGF or prolong the experiment time, the overall activity of the neurotrophin pathway would decrease.
Besides the activation of the neurotrophin pathway, there was also an increase in genes associated with MAPK and axonal guidance between 1 h and 3 h after the NGF administration. The MAPK signaling pathway is the central pathway for neuronal differentiation.25 Studies have shown that the activation of the MAPK pathway prevents apoptosis through various trophic factors like docosahexaenoic acid and exerts neuroprotective effects on axotomized retinal ganglion cells.25–27 On the KEGG pathway map, we observed many nodes localized on this pathway, from the cell membrane to intracellular compartments. We assessed the timing of the pathway changes and found synchrony with NGF-induced gene alterations. The nodes were mostly upregulated. That is, if we looked at different points along the pathway (i.e., from point A to point B or from point B to point C), the internodal change was mostly upregulated. Taking all of the changes into consideration, MAPK activity increased over time. While the activation of the MAPK pathway has been shown to be directly involved in neuronal differentiation, the genes involved in axonal guidance may also be required.28 This is shown in our data where there is a greater than 2 fold increase between 1 h and 3 h in the expression of the genes involved in axonal guidance (ESI,† Fig. S2).
Our study also identified an increase in Wnt pathway activity associated with NGF-induced neuronal differentiation. The Wnt family of signaling proteins participates in multiple developmental events during the embryogenesis and tissue homeostasis. Different Wnt proteins have specific and unique activity profiles. Purified Wnt-3a and Wnt1 proteins activate Rho-kinase and inhibit the NGF-dependent neurite outgrowth in PC12 cells.29 In contrast, a member of the Wnt family growth factors, Wnt 5a, is a key downstream effector in NGF-mediated axonal branching and growth in developing sympathetic neurons.30–31 We found a significant increase in genes associated with the Wnt pathway between 1 h and 3 h after NGF stimulation in PC-12 cells (ESI,† Fig. S3). Thus, we suggested Wnt proteins as key effectors in transducing extrinsic cell signals that specifically regulate neuron development in PC12 cells. The increase in Wnt pathway activity also synchronized with NGF-induced differentiation. As mutations of genes linked to the Wnt pathway often lead to human degenerative diseases and cancer, the activation of Wnt signaling may find a therapeutic application in diseases affecting neuron like Alzheimer's disease and Parkinson's disease.32
In summary, we have used PC12 cells to establish a model system that allows us to assess relationships between NGF-induced PC12 cell differentiation and pathway changes in the KEGG database. The results showed an increase in NGF-induced gene expression over time. Looking at KEGG biologic pathways, we noted that neuronal differentiation pathways changed as activated genes and their associated pathways were altered. The modulation of the Wnt/beta-catein, MAPK/ERK, and the Neurotrophin signal signaling pathways contributes significantly to the neuronal cell differentiation process, which is compatible with previous studies.24,29,35
Our study thus demonstrated that the KEGG database is useful in the elucidation of complicated biologic systems. In addition, choropleth map analysis like the heat map, showing the color enhancement and shifts over time, may also help to identify the activation and inactivation of specific pathways. The analysis of these color-shift maps at different time points may allow for categorization of these pathway changes. Given that we were able to follow KEGG pathway activation over time during neuronal differentiation of PC12 cells, it is entirely possible to use this system to look at specific disease models, for example, models of tumor growth or inflammation.10 We could target specific pathways, enhancing or inhibiting them, to determine if this might alter the time course of the disease or even cure it. This application of chemogenomic analysis and systems biology to address individual patient's disease process is important in the development of precision medicines.33 Likewise, the high-through put methodology combined with KEGG database analysis described in this study may be helpful in setting up a personalized dynamical model to analyze individual patient's disease and design specific targeted treatment. A KEGG editing program (KGML-ED), which combines semi-static and dynamic modes of pathway visualization and manipulation, is presently available.34 Utilization of this tool may further refine specific pathway details and timing of activation. In conclusion, our study has identified changes in neuronal differentiation pathways with the KEGG database, which were synchronized with NGF-induced differentiation; and also demonstrated that KEGG pathway map analysis is a valuable tool for biological interpretation of genome sequences and other high-throughput data, and may find application in the clinical setting.
Acknowledgements
The authors would like to thank Ms Yi-Lin Hung for her technical assistance in experiments and Mr Ji-Chian Tsai for his work in the bioinformatics. This study was fully supported by research grants from Cathay General Hospital research grants (CGH-MR-10103 and CGH-MR-10115) and the Deanship of Scientific Research at King Saud University project (No RG-1435-065).References
- H. Kitano, Systems biology: a brief overview, Science, 2002, 295, 1662–1664 CrossRef CAS PubMed.
- H. Li, Y. Sun and M. Zhan, Exploring pathways from gene co-expression to network dynamics, Methods Mol. Biol., 2009, 541, 249–267 CAS.
- M. Kanehisa, S. Goto, Y. Sato, M. Kawashima, M. Furumichi and M. Tanabe, Data, information, knowledge and principle: back to metabolism in KEGG, Nucleic Acids Res., 2014, 42, 199–205 CrossRef PubMed.
- H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono and M. Kanehisa, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res., 1999, 27, 29–34 CrossRef CAS PubMed.
- L. Lorigados, N. Pavón, T. Serrano and M. A. Robinson, Nerve growth factor and neurological diseases, Rev. Neurol., 1998, 26, 744–748 CrossRef CAS PubMed.
- A. Levi, J. D. Eldridge and B. M. Paterson, Molecular cloning of a gene sequence regulated by nerve growth factor, Science, 1985, 229(4711), 393–395 CAS.
- S. Baginsky, L. Hennig, P. Zimmerman and W. Gruissem, Gene expression analysis, proteomics, and network discovery, Plant Physiol., 2010, 152, 402–410 CrossRef CAS PubMed.
- P. Khatri and S. Dřghici, Ontological analysis of gene expression data: current tools, limitations, and open problems, Bioinformatics, 2005, 21, 3587–3595 CrossRef CAS PubMed.
- M. Kanehisa, S. Goto, M. Furumichi, M. Tanabe and M. Hirakawa, KEGG for representation and analysis of molecular networks involving diseases and drugs, Nucleic Acids Res., 2010, 38, D355–D360 CrossRef CAS PubMed.
- C. E. Wheelock, A. M. Wheelock, S. Kawashima, D. Diez, M. Kanehisa, M. van Erk, R. Kleemann, J. Z. Haeggström and S. Goto, Systems biology approaches and pathway tools for investigating cardiovascular disease, Mol. BioSyst., 2009, 5(6), 588–602 RSC.
- X. Y. Qin, Y. Kojima, K. Mizuno, K. Ueoka, K. Muroya, H. Zaha, H. Akanuma, Q. Zeng, T. Fukuda, J. Yoshinaga, J. Yonemoto, K. Kohri, Y. Hayashi, M. Fukami, T. Ogata and H. Sone, Identification of novel low-dose bisphnol a targets in human foreskin fibroblast cells derived from hypospadias patients, PLoS One, 2012, 7(5), e36711 CAS.
- B. B. Nankova, J. Chua, R. Mishra, C. D. Kobasiuk and E. F. La Gamma, Nicotinic induction of preproenkephalin and tyrosine hydroxylase gene expression in butyrate-differentiated rat PC12 cells: a model for adaptation to gut-derived environmental signals, Pediatr. Res., 2003, 53, 113–118 CrossRef CAS PubMed.
- E. Mazzio, J. Huber, S. Darling, N. Harris and K. F. Soliman, Effect of antioxidants on L-glutamate and N-methyl-4-phenylpyridinium ion induced-neurotoxicity in PC12 cells, Neurotoxicology, 2001, 22, 283–288 CrossRef CAS PubMed.
- P. Crisanti, A. Leon, D. M. Lim and B. Omri, Aspirin prevention of NMDA-induced neuronal death by direct protein kinase Czeta inhibition, J. Neurochem., 2005, 93, 1587–1593 CrossRef CAS PubMed.
- E. Lee, Z. Williams, C. B. Goodman, E. T. Oriaku, C. Harris, M. Thomas and K. F. Soliman, Effects of NMDA receptor inhibition by phencyclidine on the neuronal differentiation of PC12 cells, Neurotoxicology, 2006, 27, 558–566 CrossRef CAS PubMed.
- Y. Song, E. Q. Wei, W. P. Zhang, Q. F. Ge, J. R. Liu, M. L. Wang, X. J. Huang, X. Hu and Z. Chen, Minocycline protects PC12 cells against NMDA-induced injury via inhibiting 5-lipoxygenase activation, Brain Res., 2006, 1085, 57–67 CrossRef CAS PubMed.
- S. D. Santos, P. J. Verveer and P. I. Bastiaens, Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate, Nat. Cell Biol., 2007, 9, 324–330 CrossRef CAS PubMed.
- A. S. Tischler and L. A. Greene, Nerve growth factor-induced process formation by cultured rat pheochromocytoma cells, Nature, 1975, 258, 341–342 CrossRef CAS PubMed.
- M. E. Greenberg and E. B. Ziff, Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene, Nature, 1984, 311, 433–438 CrossRef CAS PubMed.
- D. P. Bartel, M. Sheng, L. F. Lau and M. E. Greenberg, Growth factors and membrane depolarization activate distinct programs of early response gene expression: dissociation of fos and jun induction, Genes Dev., 1989, 3, 304–313 CrossRef CAS PubMed.
- K. Asakura, A. Ueda, N. Kawamura, M. Ueda, T. Mihara and T. Mutoh, Clioquinol inhibits NGF-induced Trk autophosphorylation and neurite outgrowth in PC12 cells, Brain Res., 2009, 1301, 110–115 CrossRef CAS PubMed.
- F. Rezaee, S. L. Rellick, G. Piedimonte, S. M. Akers, H. A. O'Leary, K. Martin, M. D. Craig and L. F. Gibson, Neurotrophins regulate bone marrow stromal cell IL-6 expression through the MAPK pathway, PLoS One, 2010, 5(3), e9690 Search PubMed.
- E. Caminos, E. Becker, D. Martín-Zanca and E. Vecino, Neurotrophins and their receptors in the tench retina during optic nerveregeneration, J. Comp. Neurol., 1999, 404, 321–331 CrossRef CAS PubMed.
- D. Vaudry, P. J. Stork, P. Lazarovici and L. E. Eiden, Signaling pathways for PC12 cell differentiation: making the right connections, Science, 2002, 296, 1648–1649 CrossRef CAS PubMed.
- P. Sun, H. Watanabe, K. Takano, T. Yokoyama, J. Fujisawa and T. Endo, Sustained activation of M-Ras induced by nerve growth factor is essential for neuronal differentiation of PC12 cells, Genes Cells, 2006, 11, 1097–1113 CrossRef CAS PubMed.
- R. Roduit and D. F. Schorderet, MAP kinase pathways in UV-induced apoptosis of retinal pigment epithelium ARPE19 cells, Apoptosis, 2008, 13, 343–353 CrossRef CAS PubMed.
- J. C. Dreixler, F. C. Barone, A. R. Shaikh, E. Du and S. Roth, Mitogen-activated protein kinase p38alpha and retinal ischemic preconditioning, Exp. Eye Res., 2009, 89, 782–790 CrossRef CAS PubMed.
- S. Y. Jiang, Y. Y. Zou and J. T. Wang, p38 mitogen-activated protein kinase-induced nuclear factor kappa-light-chain-enhancer of activated B cell activity is required for neuroprotection in retinal ischemia/reperfusion injury, Mol. Vision, 2012, 18, 2096–2106 CAS.
- A. H. Chou and B. D. Howard, Inhibition by Wnt-1 or Wnt-3a of nerve growth factor-induced differentiation of PC12 cells is reversed by bisindolylmaleimide-I but not by several other PKC inhibitors, Oncogene, 2002, 21(41), 6348–6355 CrossRef CAS PubMed.
- D. Bodmer, S. Levine-Wilkinson, A. Richmond, S. Hirsh and R. Kuruvilla, Wnt5a mediates nerve growth factor-dependent axonal branching and growth in developing sympathetic neurons, J. Neurosci., 2009, 29(23), 7569–7581 CrossRef CAS PubMed.
- P. C. Salinas, Retrograde signaling at the synapse: a role for Wnt proteins, Biochem. Soc. Trans., 2005, 33, 1295–1298 CrossRef CAS PubMed.
- H. Clevers, Wnt/beta-catenin signaling in development and disease, Cell, 2006, 127(3), 469–480 CrossRef CAS PubMed.
- J. Hansen and R. Iyengar, Computation as the mechanistic bridge between precision medicine and systems therapeutics, Clin. Pharmacol. Ther., 2013, 93(1), 117–128 CrossRef CAS PubMed.
- C. Klukas and F. Schreibe, Dynamic exploration and editing of KEGG pathway diagrams, Bioinformatics, 2007, 23, 344–350 CrossRef CAS PubMed.
- J. C. Schwamborn, R. Fiore, D. Bagnard, J. KAppler, C. Kaltschmidt and A. W. Puschel, Semaphorin 3A stimulates neurite extension and regulates gene expression in PC12 cells, J. Biol. Chem., 2004, 279(30), 30923–30926 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00338e |
| This journal is © The Royal Society of Chemistry 2016 |