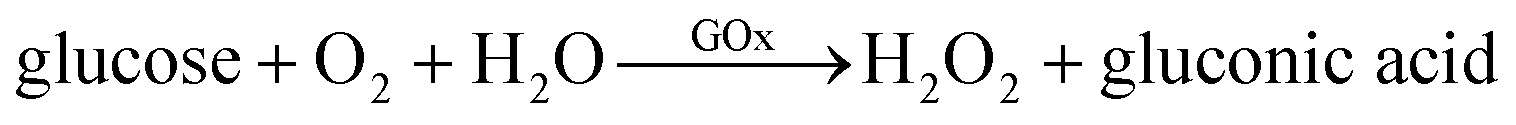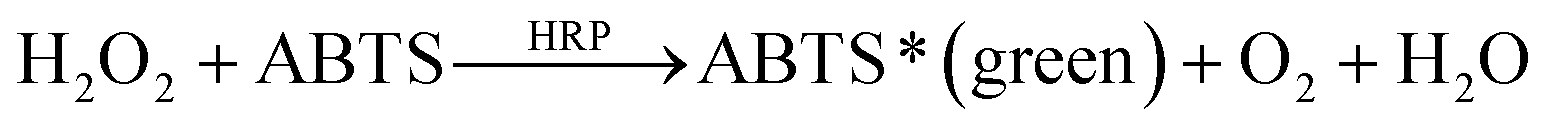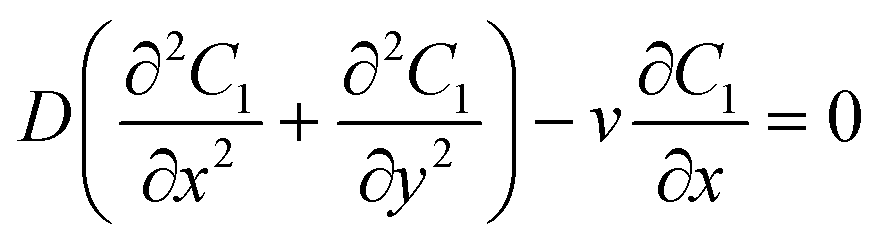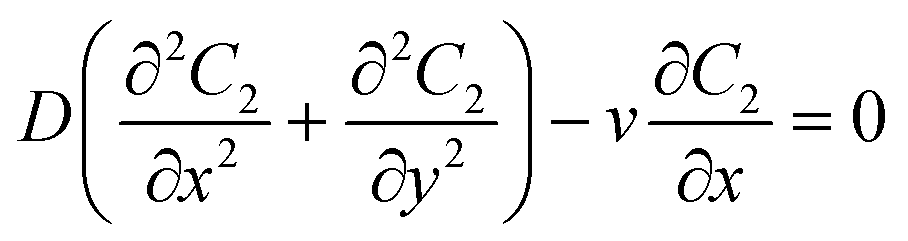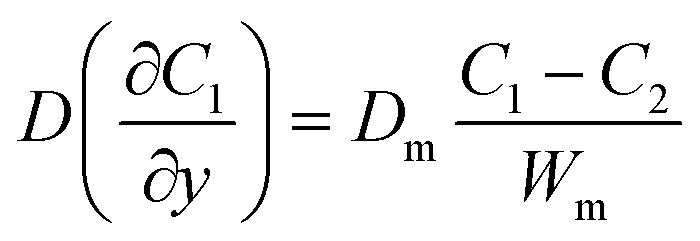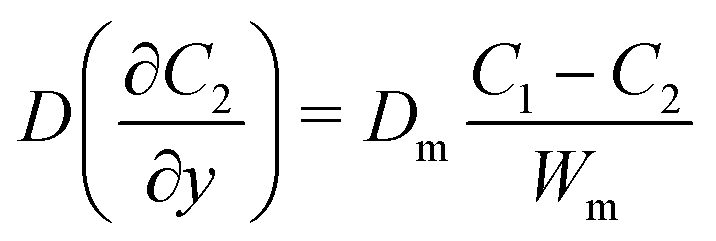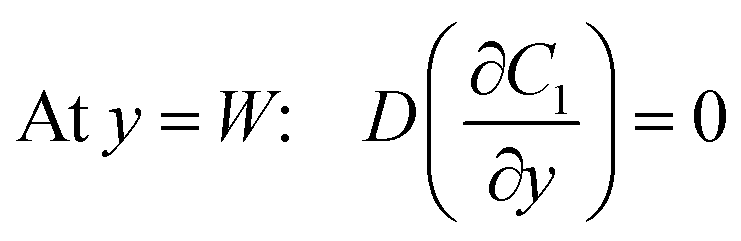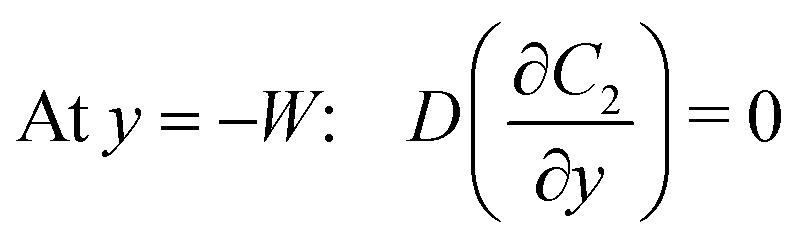DOI:
10.1039/C6LC01264G
(Paper)
Lab Chip, 2016,
16, 4725-4731
In situ formation of leak-free polyethylene glycol (PEG) membranes in microfluidic fuel cells
Received
12th October 2016
, Accepted 28th October 2016
First published on 28th October 2016
Abstract
Membraneless microfluidic fuel cells operated under two co-laminar flows often face serious fuel cross-over problems, especially when flow rates are close to zero. In this study, we show that polyethylene glycol (PEG) monomers can be cross-linked inside microfluidic channels to form leak-free PEG membranes, which prevent mixing of two incompatible electrolyte solutions while allowing diffusion of certain molecules (e.g. glucose) and ions. By using PEG monomers of different molecular weights and cross-linking conditions, we are able to tailor selectivity of the membrane to allow passage of glucose while blocking larger molecules such as trypan blue. As a proof of principle, a microfluidic fuel cell with a PEG membrane and two incompatible electrolytes (acid and base) is demonstrated. Thanks to the leak-free nature of the PEG membrane, these two electrolytes do not mix together even at very slow flow rates. This microfluidic fuel cell is able to generate a voltage up to ∼450 mV from 10 mM of glucose with a flow rate of 20 μL min−1. This microfluidic fuel cell is potentially useful as a miniature power source for many applications.
1. Introduction
A microfluidic fuel cell is a device in which fuels and oxidants are reacted and converted to electrical energy.1 Due to its small dimensions, a microfluidic fuel cell can be operated under a co-laminar regime in which fuels and oxidants flow as separated layers without any physical barriers between them.2 Due to the lack of physical barriers such as membranes, internal resistance is low and mass transfer is fast in the microfluidic fuel cell.3 Both factors make the microfluidic fuel cell perform much better than conventional fuel cells. However, fuel cross-over problem in the microfluidic fuel cell cannot be completely avoided. This problem becomes more serious when two electrolytes are incompatible (e.g. acid and base) or when the lateral diffusion is significant.4 As a result, voltage and electric current drop significantly when electrolyte solutions are neutralized or fuels are oxidized on both electrodes.5 In conventional fuel cells, a proton-exchange membrane (e.g. Nafion) is employed to separate fuels and oxidants. The membrane can be sandwiched between an anode and a cathode to separate fuels and oxidants. Meanwhile, the membrane also allows the passage of protons to complete the electric circuit.6 However, Nafion is very hydrophobic and cannot be synthesized in situ.7 In a microfluidic fuel cell, it is difficult to install a piece of pre-synthesized Nafion membrane inside a microchannel without leaking.
Because of the challenges mentioned above, poly(ethylene glycol) (PEG) hydrogel was considered as an alternative membrane to replace Nafion membrane. An advantage of the PEG membrane is that it can be cross-linked in situ in a microfluidic channel by using UV lithography. After cross-linking, PEG membrane can absorb water and become ion-conductive.8 Recently, PEG membrane draws much attention in biological and medical applications due to its water content and bio-compatibility.9 It also shows selectivity to certain biomolecules, and the selectivity is tunable when monomers of different molecular weight are used.10 For example, biological molecules including myoglobin, ovalbumin and albumin can be separated by using low-molecular-weight PEG membranes.11 Some studies showed that PEG membrane with a pore size of ∼1 nm is only permeable to small molecules (MW < 1000) and prevents the passage of large molecules (MW > 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000).12–14
000).12–14
In this study, microfluidic fuel cells with two parallel channels (side-by-side) were employed to study the diffusion of fuel molecules under different flow rates. A transport model was also proposed to simulate the diffusion process inside the channel. Secondly, we investigated how to prepare a leak-free PEG membrane inside microfluidic fuel cells to separate incompatible electrolytes and prevent diffusion of fuels through the membrane. Parameters which affect the conductivity, selectivity and permeability of the PEG membrane were studied in details.
2. Experimental
2.1 Materials
Polyethylene glycol diacrylate monomers (Mn250, Mn575, Mn750), 2,2-dimethoxy-2-phenyl-acetophenone (photo initiator), horseradish peroxidase (HRP), glucose oxidase (GOx), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and glucose were purchased from Sigma-Aldrich (Singapore). Trypan blue solution (0.4 wt%, 0.2 μm filtered) and ethanol were purchased from Thermo Scientific (Singapore). Sodium hydroxide and hydrochloric acid were purchased from Merck (Singapore). All chemicals were used as received without further purification.
2.2 Construction of microfluidic fuel cells
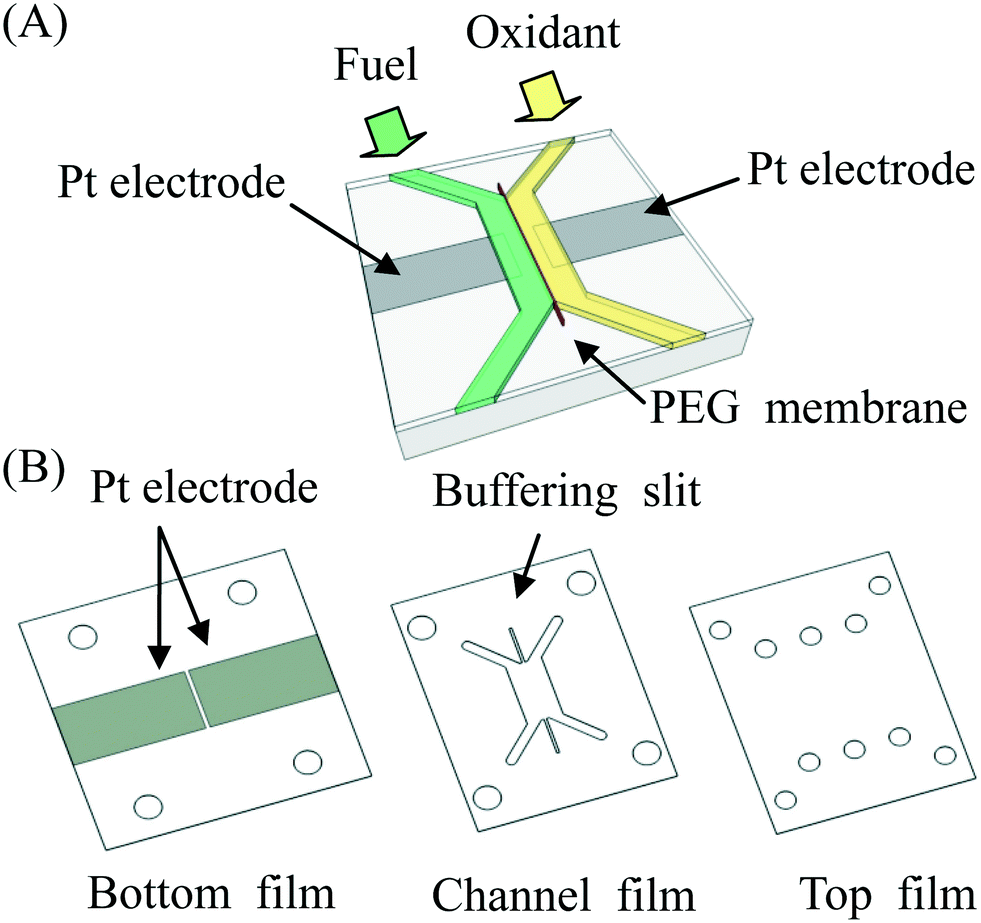
A schematic diagram of the microfluidic fuel cell was shown in Fig. 1. The main body of the fuel cell was made of three pieces (bottom, channel and top) of plastic films. The thickness of each film was approximately 1 mm. Patterns on the films were prepared by using a laser-cutting machine. Next, platinum electrodes (1 cm × 2 cm) were deposited on the bottom film for 6 min by using a sputtering machine (JEOL JFC-1300). The shape of the platinum electrodes was defined by using adhesive tapes to mask unwanted regions on the bottom film. After deposition of the electrodes, the tapes were removed, and the sample was cleaned by using ethanol and DI water. To assemble three pieces of plastic films together, a piece of double-sided tape was stuck on both sides of the channel film to glue them together to form a microfluidic fuel cell. In some cases, the plastic films were treated with oxygen plasma (Harrick Plasma PDC-32G) for 2 min to enhance adhesion of the films.
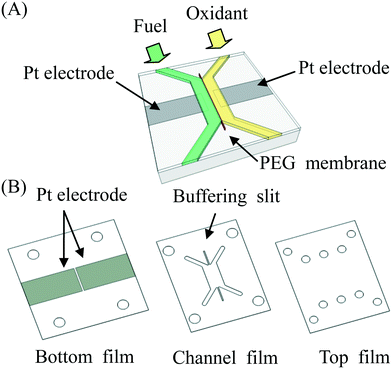 |
| | Fig. 1 (A) Schematic diagram of a microfluidic fuel cell with a PEG membrane to separate fuel and oxidant. (B) The microfluidic fuel cell was made from three pieces of plastic films (bottom, channel and top) bound together. Two platinum electrodes were deposited on the bottom film. | |
2.3
In situ formation of PEG hydrogel membranes
Two different PEG monomers (Mn250 and Mn575) were mixed together in 8:2 ratio to minimize swelling of PEG hydrogel. To cross-link PEG monomers, some photoinitiator (2.5% w/v) was mixed with the PEG monomer solution. After that, the PEG monomer solution was cured by using a UV lamp (365 nm, Spectroline ENF-260C) under a photomask with a desired pattern for 15 s. Unreacted PEG monomer solution was removed by using ethanol and DI water. The PEG membrane formed inside the microchannel was dried with nitrogen gas before use.
2.4 Operation of microfluidic fuel cells
Both electrolyte solutions were delivered into the microfluidic fuel cell with a syringe pump (PhD Ultra, Harvard, U.S.A). Inlets and outlets of the microfluidic fuel cell were connected to plastic tubing (EW-06406-60 FEP tubing from Cole-Parmer, U.S.A) through a chip-holder system (SIMTECH, Singapore). To avoid creating a significant pressure drop in microfluidic channels, one syringe pump was connected to the inlet and another syringe pump was connected to the outlet. The former was operated in the “infuse” mode whereas the latter was operated in the “withdraw” mode. The pressure inside the fuel cell was not altered when the same amount of solutions was infused and withdrawn at the same rate. Typical flow rates used in these studies were ranged from 0.5 μL min−1 to 20 μL min−1. In these studies, NaOH and HCl were used as electrolytes for anode and cathode, respectively. Fuels were either trypan blue or glucose in the NaOH solution.
2.5 Measurement of open circuit voltages (OCV) and loading voltage (LV)
To measure open circuit voltages of microfluidic fuel cells, copper wires were connected to the platinum electrodes by using conductive adhesion tapes. The open circuit voltage of the fuel cell was measured by using a high impedance voltmeter (eDAQ, U.S.A). The electrodes of the fuel cell were connected to the working and reference leads of the system. OCV at different times were recorded by using Chart and Scope software. To improve the accuracy of measurements, a low-pass filter was used to filter noises and high-frequency components of the signal. The loading voltage (LV) was measured by connecting a loading resistor (RL) to the fuel cell.
2.6 Detection of glucose
Glucose was detected by using two enzymes, HRP and GOx, following two enzymatic reactions shown below:15
An aqueous solution containing 1 μg mL−1 of HRP, 200 μg mL−1 of GOx and 5 mg mL−1 of substrate ABTS was injected into the microfluidic channel and incubated for 2 h. In the presence of glucose, the color of the solution will be changed to green due to the presence of ABTS*.
3. Results and discussion
3.1 Simulation of glucose diffusion through the membrane
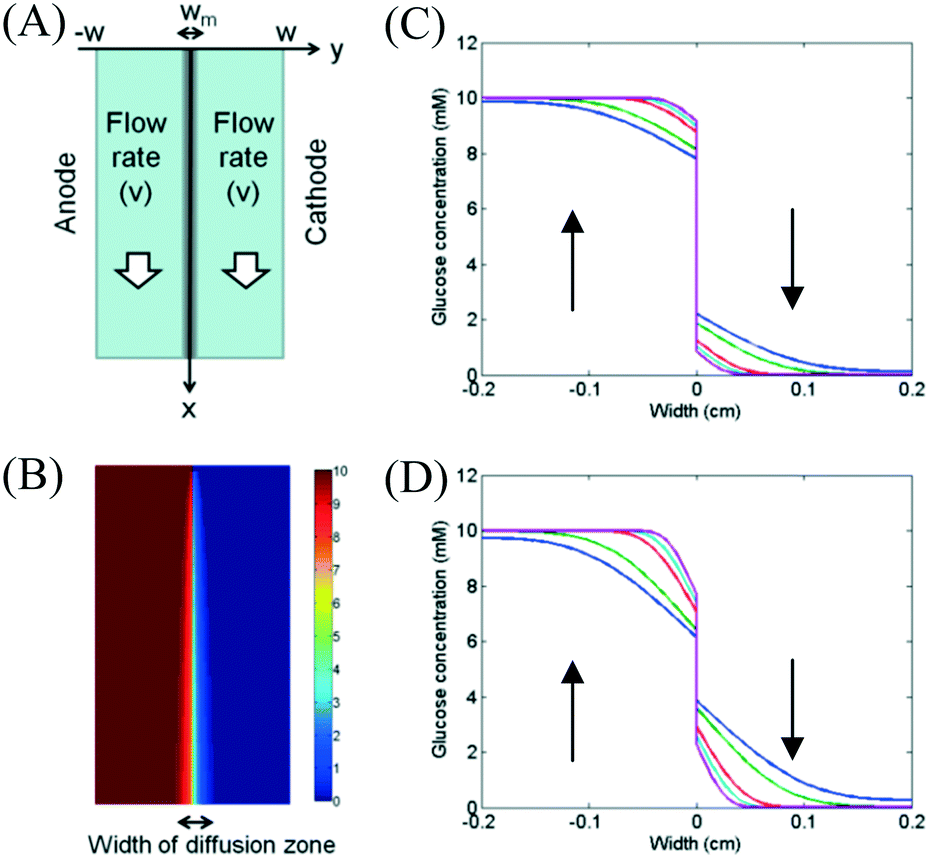
A schematic diagram of the microfluidic fuel cell was shown in Fig. 2A. The diffusions of glucose in two microfluidic channels are governed by the following equations:| | 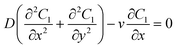 | (1) |
| | 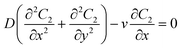 | (2) |
where C1 and C2 are the concentration of glucose in the anode and cathode compartments, respectively. D is the diffusion coefficient of glucose (6.7 × 10−6 cm2 s−1), v is the flow velocity. In the presence of a membrane, the following boundary conditions must be satisfied:| | 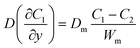 | (3) |
| | 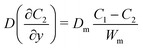 | (4) |
where Dm are the diffusion coefficient of glucose in the membrane, Wm is the thickness of the membrane.
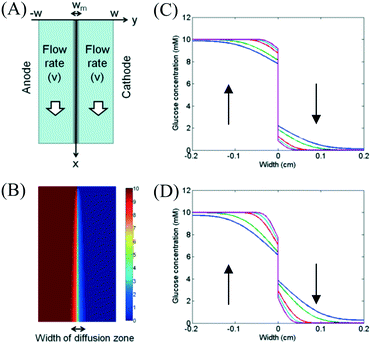 |
| | Fig. 2 (A) Schematic diagram of the fuel cell. (B) Concentration profile of glucose in microfluidic fuel cells when the flow rates were 20 μL min−1 in both channels. Effects of flow rates on the glucose concentration in a membraneless fuel cell for (C) Dm = 0.2Do and (D) Dm = 0.8Do under flow rates of 0.5 μL min−1 (blue), 1 μL min−1 (green), 5 μL min−1 (red), 10 μL min−1 (cyan) and 20 μL min−1 (magenta). | |
Under an open-circuit condition, there is no electric current. Thus, the following boundary conditions must be satisfied at y = W and y = −W,
| | 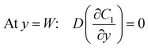 | (5) |
| | 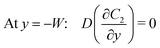 | (6) |
The model was simulated by using finite difference method. The widths of channels (W) and membrane (Wm) were 0.2 cm and 0.01 cm, respectively. For the boundary conditions at the inlets (x = 0), the values of C1 and C2 were set as 10 mM and 0 mM, respectively. The flow velocities were ranged from 0 cm s−1 to 0.1667 cm s−1 (equivalent to 20 μL min−1). The value of diffusion coefficient of glucose in membrane (Dm) was determined by the type of used membrane. Simulated glucose concentration profile was shown in Fig. 2B. Effects of flow rates on glucose concentration profiles at a location (x = 2 cm) downstream were shown in Fig. 2C and D. The diffusion coefficient of glucose in the membrane (Dm) was set to be 0.2Do or 0.8Do for different membranes. From the figures, we found that the width of the diffusion zone decreased when a higher flow rate was applied to both channels. For the membrane with a small permeability, glucose diffused to the cathode channel can be easily flushed away at high flow rate. The result shows that the membrane can significantly improve the fuel crossover problem.
3.2
In situ formation of PEG hydrogel membrane
To study behaviors of PEG hydrogel membrane, three PEG monomers of different molecular weights (Mn250, Mn575, and Mn700) were used to prepare three kinds of PEG membranes (PEG250, PEG575 and PEG700) for testing. For all monomer solutions, 2.5% (w/v) of photoinitiator was added to allow cross-linking of monomers with UV radiation. After exposure of PEG monomer solutions to UV for 15 s, PEG membranes were formed as shown in Fig. 3A. All PEG membranes were incubated in water for 1 day, and the degree of swelling was evaluated by measuring the length of each membrane. For PEG250 membrane, there was no change in its length, suggesting that PEG250 did not swell. This is because monomers of low molecular weights led to a very dense polymer matrix which did not adsorb water. Therefore, the conductivity of the PEG250 membrane was nearly zero, meaning that this type of membrane is not suitable for constructing fuel cells. In contrast, after incubation in water, the length of PEG575 and PEG700 membranes increased by 15% and 20%, respectively. Meanwhile, conductivities of both membranes increased to 0.071 μS and 0.076 μS, respectively. The results show that the degree of swelling and conductivity increased with increasing molecular weights of PEG monomers.10 This is probably because the PEG membrane formed by larger monomers is able to absorb more water and ions. The results also show that the conductivity can be tuned by using different monomers. Even though a high conductivity is desired, swelling posed a serious problem after the membrane was fixed inside the membrane and deformed after 2 h. The deformation caused leaking of fuels and electrolytes. To balance conductivity and degree of swelling, Mn250 and Mn575 monomers were mixed together in different ratios (6:4, 7:3 and 8:2) to tune the properties of PEG membranes. Among three membranes, the first two (6:4 and 7:3) still showed swelling and led to deformation of the membrane. The last sample (8:2) offered reasonable conductivity yet there was no obvious swelling problem as shown in Fig. 3D and E. Therefore, this mixing ratio (Mn250:Mn575 = 8:2) was chosen to prepare PEG membranes inside microfluidic fuel cells in the following study. To prevent buckling of PEG membrane when it swelled slightly, two buffering slits were designed at both ends to allow slight expansion of the PEG membrane as shown in Fig. 1B. They also provided mechanical support for the membrane.
 |
| | Fig. 3 Effect of monomer molecular weight on the swelling and deformation of PEG membranes. (A) PEG membranes prepared from three different monomer solutions (Mn250, Mn575 and Mn700). Deformation and buckling of PEG575 membrane (B) before and (C) after swelling. No deformation of PEG membrane (8:2 mixture) (D) before and (E) after incubation for 2 h. Color change of enzyme testing solution from (F) colorless to (G) green at lower channel due to the diffusion of glucose from upper channel. | |
3.3 Microfluidic fuel cell with PEG membrane
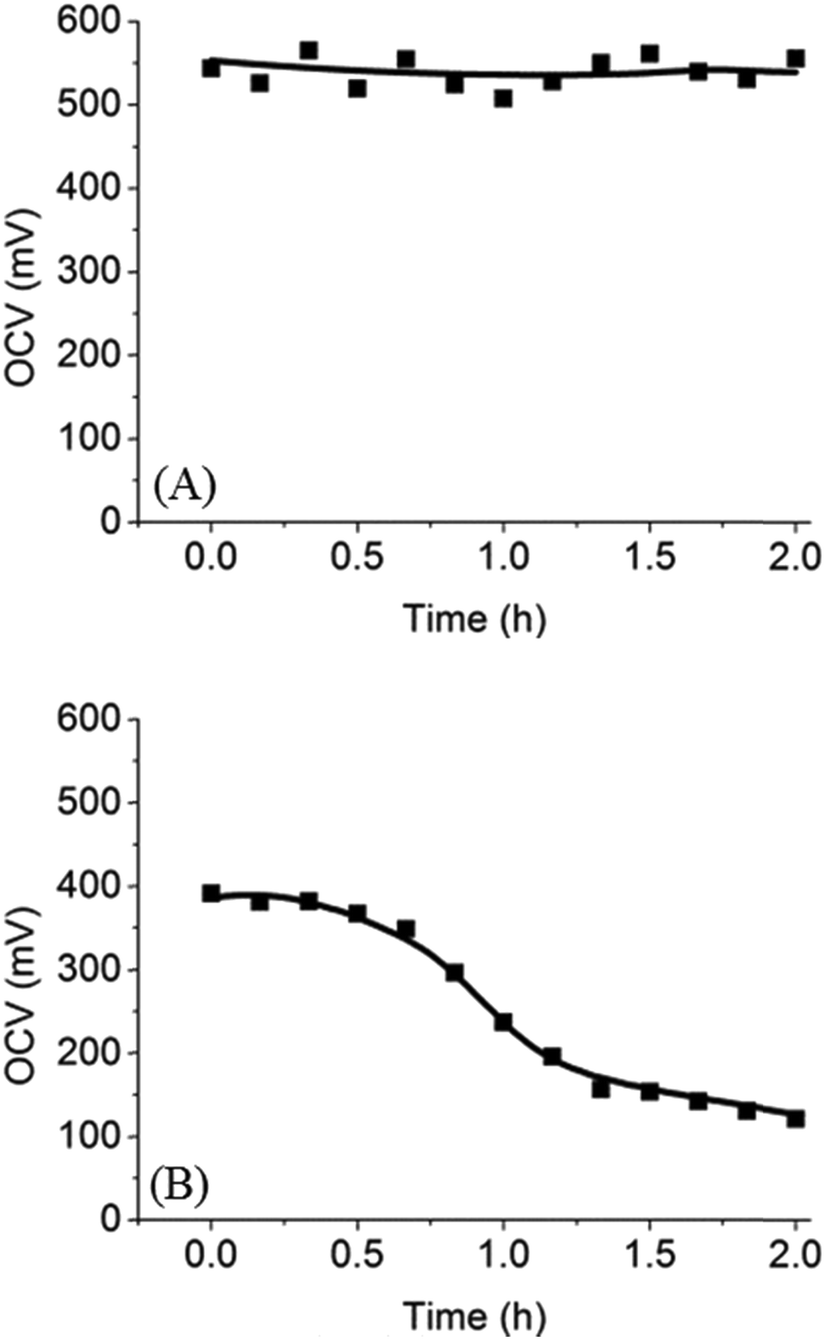
To test the performance of the microfluidic fuel cell with PEG membrane, we first chose trypan blue (MW = 872.88 g mol−1) as a fuel for three reasons. Firstly, trypan blue is an azo dye and has a strong blue color, which allowed us to track lateral diffusion of trypan blue across the membrane in real time. Secondly, trypan blue can be oxidized on a platinum electrode to release electrons. Finally, trypan blue is a neutral molecule such that it does not adsorb on a charged surface. Optical images and open circuit voltages (OCV) of the microfluidic fuel cell were used to evaluate the diffusion of trypan blue across the PEG membrane. As shown in Fig. 4A, the initial OCV was ∼550 mV, and there was no decrease in the OCV even after 2 h. Spatial distribution of the blue color indicated that trypan blue was confined to the anode side. Both results revealed that trypan blue could not diffuse through the PEG membrane to the cathode. Therefore, the fuel cross-over problem was successfully prevented by using the PEG membrane in the microfluidic fuel cell.
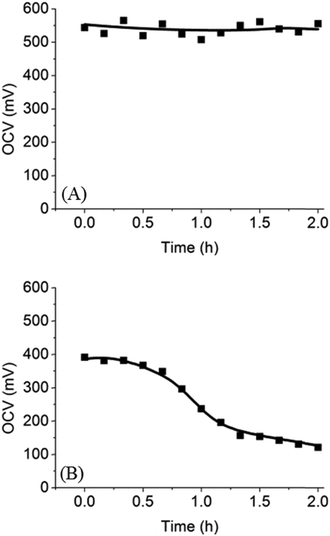 |
| | Fig. 4 Time-course OCV in microfluidic fuel cells with (A) trypan blue or (B) glucose as a fuel. Decreasing OCV was caused by the diffusion of glucose through the PEG membrane. | |
Next, trypan blue was replaced by glucose, a more commonly used fuel, for testing. In this case, since glucose is colorless, OCV was used to monitor the diffusion of glucose through the PEG membrane. The OCV in the glucose fuel cell as a function of time over 2 h was shown in Fig. 4B. It can be seen that the initial OCV was ∼390 mV, and then the OCV decreased slowly with time to ∼120 mV after 2 h. Based on the observation, we proposed that the decrease in OCV was due to slow diffusion of glucose through the PEG membrane into the cathode compartment. Because anode and cathodes were both platinum electrodes, presence of glucose in both compartments unavoidably decreased chemical potential of glucose and decreased OCV. To further confirm that glucose indeed diffused across the membrane into the cathode, a colorimetric assay was employed to detect glucose in the cathode compartment. Green color in Fig. 3F and G showed that glucose was present in the cathode compartment after 2 h of incubation. These results, when combined, strongly suggest that glucose was able to diffuse through the PEG membrane over a period of 2 h. In the literature, it has been shown that certain small molecules can pass through the PEG membrane by diffusion.16 In our case, the (8:2) PEG membrane used in the microfluidic fuel cell only allowed the diffusion of smaller glucose through the membrane while blocking larger molecules such as trypan blue. However, we noted that even though glucose could diffuse through the membrane, it only happened at a very slow diffusion rate. After 1 h, the OCV only dropped from ∼390 mV to ∼240 mV. In contrast, in the absence of a membrane, the OCV dropped to zero within a few minutes. These results suggest that the PEG membrane formed a good barrier to slow down the diffusion of glucose.
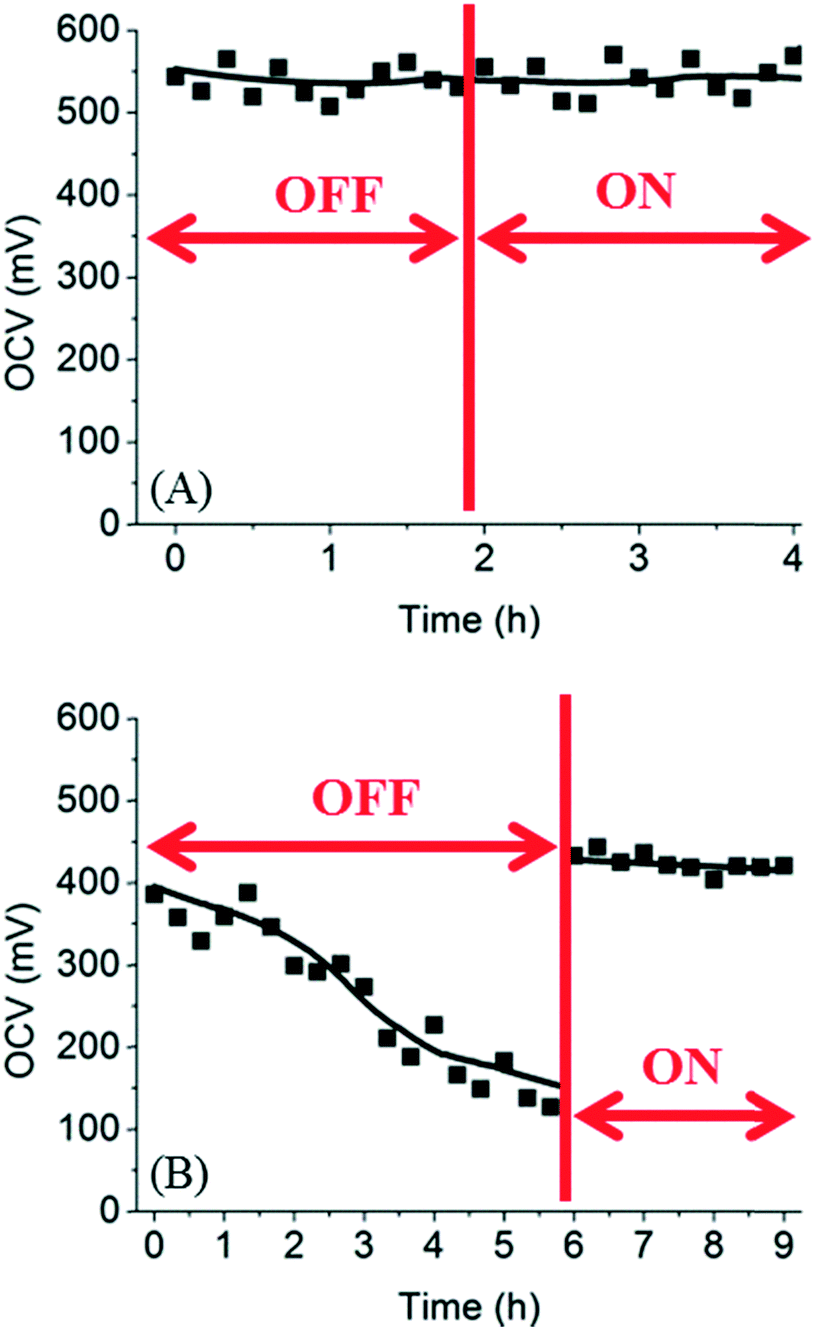
To completely prevent the decreasing OCV in the microfluidic fuel cell, a possible solution is to increase the flow rates of electrolyte solutions such that glucose diffused to the cathode compartment can be removed by the convective flow continuously. To test this hypothesis, the fuel cell was connected to a syringe pump for delivering electrolyte solutions at certain flow rates. The flow rates of fuel and oxidant were increased from 0.5 μL min−1 to 20 μL min−1 after a few hours. As shown in Fig. 5, for trypan blue, the OCV did not change before and after the flow was turned on. This result was expected because trypan blue did not diffuse to the cathode compartment such that flushing the cathode compartment had no effect on the OCV. In the case of glucose, the OCV dropped from 390 mV to 120 mV after 6 h when there was no flow in the cathode compartment. However, after the flow rate in the cathode was increased to 20 μL min−1, the OCV was restored to the original values within a few minutes. These results suggest that a flow rate of 20 μL min−1 was sufficient to remove all glucose in the cathode compartment.
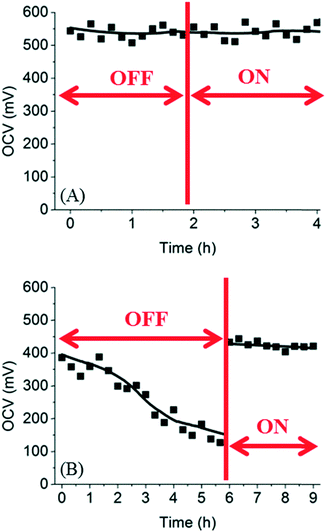 |
| | Fig. 5 Recovery of OCV in microfluidic fuel cells using (A) trypan blue and (B) glucose. The flow rate of the fuel was increased to 20 μL min−1 at the time indicated by the vertical line. | |
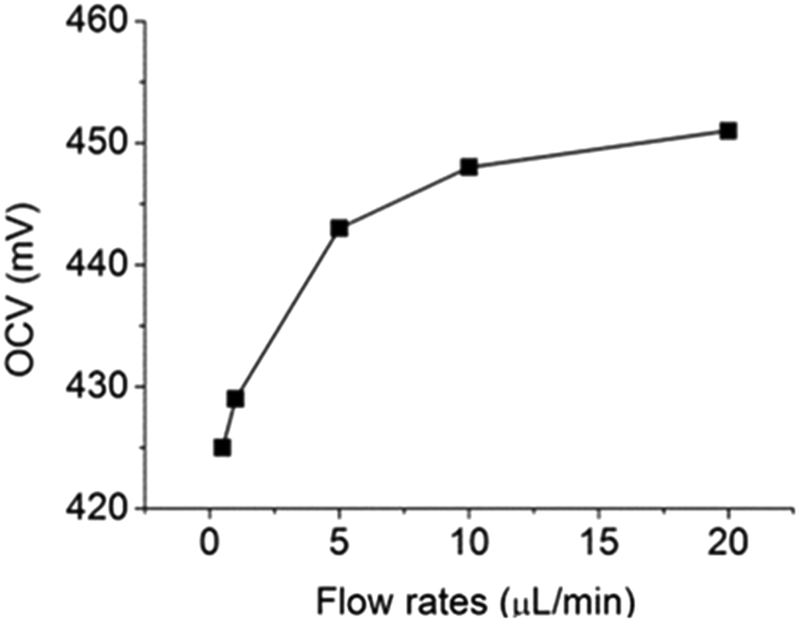
For further testing the effect of flow rate on OCV, different flow rates (between 0.5 μL min−1 and 20 μL min−1) were used to deliver electrolyte solutions and the results were shown in Fig. 6. When flow rate was lower than 10 μL min−1, the OCV was lower than 450 mV, suggesting that glucose diffused to the cathode compartment could not be completely removed by the convective flows. However, when the flow rates of both electrolyte solutions were increased to 20 μL min−1, the OCV was restored to 450 mV. Therefore, it can be concluded that 20 μL min−1 was the minimal flow rate required to flush out all glucose in the cathode compartment. Under this condition, the microfluidic fuel cell was able to generate an OCV of 450 mV from 10 mM of glucose. This result shows that the fuel cross-over problem in microfluidic fuel cells can be completely prevented by combining the use of the leak-free PEG membranes and convective flows at 20 μL min−1.
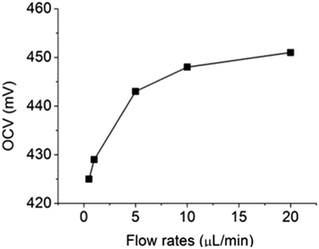 |
| | Fig. 6 Effect of flow rates on OCV in a microfluidic fuel cell. | |
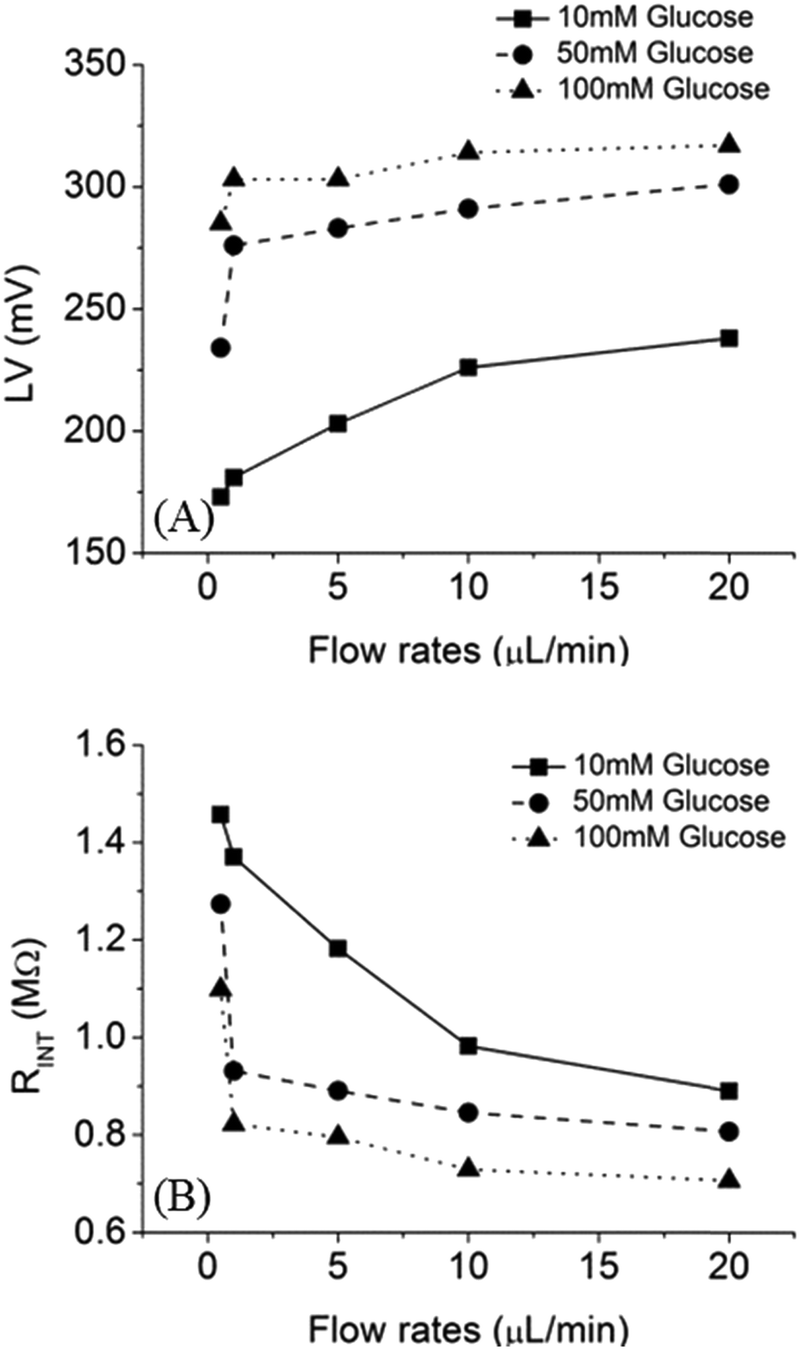
To characterize the performance of the fuel cell, loading voltage (LV) and internal resistance (RINT) of the fuel cell were measured as a function of flow rates, as shown in Fig. 7. With 10 mM of glucose and a flow rate of 20 μL min−1, LV was 240 mV and RINT was 0.9 MΩ. In contrast, when the flow rate was 0.5 μL min−1, the LV was 170 mV and RINT was 1.45 MΩ. Moreover, higher LV was observed with increasing glucose concentration. For example, 300 mV and 320 mV can be generated by using 50 mM and 100 mM of glucose fuel, respectively. Reduction of RINT was due to the increase of fuel concentration and mass transfer.
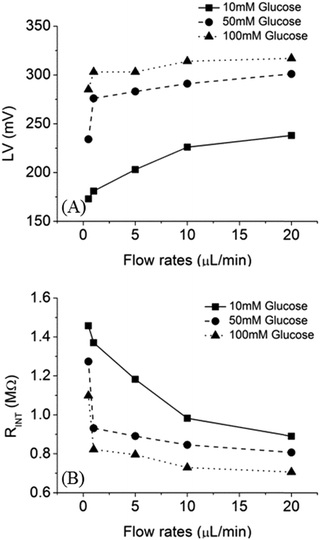 |
| | Fig. 7 Effect of flow rate on (A) LV (B) RINT in a microfluidic fuel cell with 10 mM (square), 50 mM (circle), 100 mM (triangle) of glucose as fuels. | |
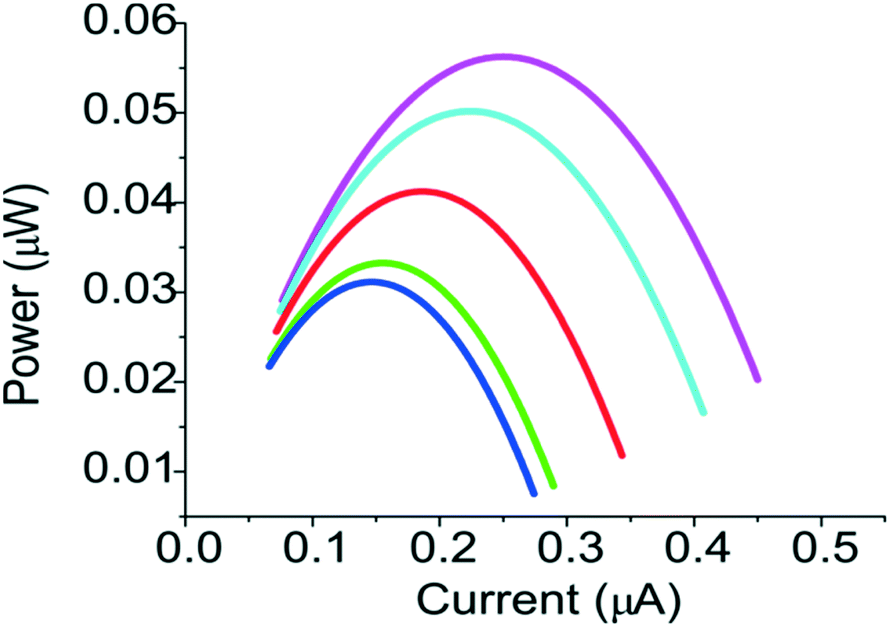
The power of the fuel cell as a function of the electric current was shown in Fig. 8. The power of the fuel cell depends on the flow rates. For example, the power increased from ∼0.03 μW to ∼0.06 μW when the flow rate was increased to 20 μL min−1. Since the internal resistance of the fuel cell in our system was limited by the conductivity of the PEG membrane, better performance could be achieved if a PEG membrane with higher conductivity and molecular selectivity can be used.17 Both properties can be tuned by using PEG monomers of different molecular weights. Moreover, the performance of the fuel cell can be greatly affected by the oxidation efficiency of the electrode for the glucose,18 better fuel cell can be achieved by choosing higher concentration of electrolytes and other electrode materials, such as nanoparticle supported electrode,19 and carbon-enzymatic electrodes.20
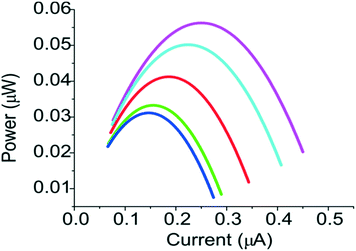 |
| | Fig. 8 Current–power curves for a membrane-type fuel cell under flow rates of 0.5 μL min−1 (blue), 1 μL min−1 (green), 5 μL min−1 (red), 10 μL min−1 (cyan) and 20 μL min−1 (magenta). | |
Conclusions
In situ formation of PEG hydrogel membranes for fuel separation in microfluidic fuel cells was proposed and demonstrated. By tuning the molecular weight of PEG monomers and mixing ratio, the swelling problem of PEG membrane was minimized. The best PEG membrane tested in this study completely blocked the diffusion of trypan blue and reduced the diffusion of glucose significantly. The fuel cross-over problem for glucose can be further improved by using a continuous flow of both electrolyte solutions at 20 μL min−1.
Acknowledgements
This project was funded by Agency for Science, Technology and Research (A*STAR) in Singapore under project no. 1421200079.
References
- E. Kjeang, N. Djilali and D. Sinton, J. Power Sources, 2009, 186, 353–369 CrossRef CAS.
- E. R. Choban, L. J. Markoski, A. Wieckowski and P. J. A. Kenis, J. Power Sources, 2004, 128, 54–60 CrossRef CAS.
- S. A. Mousavi Shaegh, N.-T. Nguyen and S. H. Chan, Int. J. Hydrogen Energy, 2011, 36, 5675–5694 CrossRef CAS.
- J. Atencia and D. J. Beebe, Nature, 2005, 437, 648–655 CrossRef CAS PubMed.
- B. H. Liu and S. Suda, J. Power Sources, 2007, 164, 100–104 CrossRef CAS.
- H. G. Haubold, T. Vad, H. Jungbluth and P. Hiller, Electrochim. Acta, 2001, 46, 1559–1563 CrossRef CAS.
- C. P. Andrieux, P. Audebert, B. Divisia-Blohorn, P. Aldebert and F. Michalak, J. Electroanal. Chem. Interfacial Electrochem., 1990, 296, 117–128 CrossRef CAS.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- B. Chakrabarty, A. K. Ghoshal and M. K. Purkait, J. Membr. Sci., 2008, 309, 209–221 CrossRef CAS.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287–1294 CrossRef CAS PubMed.
- A. D. Powers, B. Liu, A. G. Lee and S. P. Palecek, Analyst, 2012, 137, 4052–4061 RSC.
- R. J. Russell, A. C. Axel, K. L. Shields and M. V. Pishko, Polymer, 2001, 42, 4893–4901 CrossRef CAS.
- W. Zhan, G. H. Seong and R. M. Crooks, Anal. Chem., 2002, 74, 4647–4652 CrossRef CAS PubMed.
- M. Delvaux, A. Walcarius and S. Demoustier-Champagne, Biosens. Bioelectron., 2005, 20, 1587–1594 CrossRef CAS PubMed.
- Y. Wu, S. Joseph and N. R. Aluru, J. Phys. Chem. B, 2009, 113, 3512–3520 CrossRef CAS PubMed.
- A. ElMekawy, H. M. Hegab, X. Dominguez-Benetton and D. Pant, Bioresour. Technol., 2013, 142, 672–682 CrossRef CAS PubMed.
- M. R. Thorson, F. R. Brushett, C. J. Timberg and P. J. A. Kenis, J. Power Sources, 2012, 218, 28–33 CrossRef CAS.
- F. M. Cuevas-Muñiz, M. Guerra-Balcázar, F. Castaneda, J. Ledesma-García and L. G. Arriaga, J. Power Sources, 2011, 196, 5853–5857 CrossRef.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a