
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene-based microfluidics for serial crystallography†
Shuo
Sui
a,
Yuxi
Wang
a,
Kristopher W.
Kolewe
a,
Vukica
Srajer
b,
Robert
Henning
b,
Jessica D.
Schiffman
a,
Christos
Dimitrakopoulos
a and
Sarah L.
Perry
*a
aDepartment of Chemical Engineering, The University of Massachusetts Amherst, Amherst, MA 01003, USA. E-mail: perrys@engin.umass.edu
bBioCARS Center for Advanced Radiation Sources, The University of Chicago, Argonne, IL 60439, USA
First published on 24th May 2016
Abstract
Microfluidic strategies to enable the growth and subsequent serial crystallographic analysis of micro-crystals have the potential to facilitate both structural characterization and dynamic structural studies of protein targets that have been resistant to single-crystal strategies. However, adapting microfluidic crystallization platforms for micro-crystallography requires a dramatic decrease in the overall device thickness. We report a robust strategy for the straightforward incorporation of single-layer graphene into ultra-thin microfluidic devices. This architecture allows for a total material thickness of only ∼1 μm, facilitating on-chip X-ray diffraction analysis while creating a sample environment that is stable against significant water loss over several weeks. We demonstrate excellent signal-to-noise in our X-ray diffraction measurements using a 1.5 μs polychromatic X-ray exposure, and validate our approach via on-chip structure determination using hen egg white lysozyme (HEWL) as a model system. Although this work is focused on the use of graphene for protein crystallography, we anticipate that this technology should find utility in a wide range of both X-ray and other lab on a chip applications.
Introduction
The application of micro-crystallography in structural biology has accelerated in recent years due to technological advances that enable the use of smaller and ever more brilliant X-ray beams.1–4 One goal of this micro-focused technology is to enhance the diffraction signal from micro-crystals by matching the size of the beam to the size of the crystal. Here, the signal enhancement comes from a reduction in the level of background scatter associated with the solvent or mounting support surrounding the crystal. It has also been suggested that smaller crystals may have a lower probability of defects, and thus have the potential to yield higher quality diffraction data.3,5,6 Furthermore, micro-crystals are critical to enable aspects of time-resolved protein crystallography.7 However, illumination of a smaller crystal volume with a greater X-ray flux increases the level of radiation damage experienced by the sample, and can dramatically decrease the usable lifetime of the crystal. In the case of next-generation, ultra-brilliant X-ray free-electron lasers (XFELs), the X-ray beam is so intense that the crystal is destroyed after a single X-ray pulse.8,9To circumvent the issue of radiation damage and sample deterioration, ‘serial’ approaches to protein crystallography have become increasingly popular.9,10 These methods include the collection of data from a large number of independent volumes on a larger crystal,11–17 and have transitioned recently to the collection of as little as a single frame of data from many smaller crystals. Complete datasets can then be obtained by merging data from hundreds, thousands, or even tens of thousands of crystals.10,11,15,18–45 However, this data collection strategy suffers from the need to grow and efficiently manipulate a large number of fragile micro-crystals. Reported methods include the mounting of many crystals using traditional, loop-style holders,38,46 a variety of fixed-target architectures,4,6,11,47–55 and different micro-crystal injection strategies.10,52,56–63 However, nearly all of these strategies have focused on transferring a pre-prepared slurry of micro-crystals onto a mount or into an injector, rather than coupling the processes of crystallization and X-ray analysis.6,10,47,48,62 This requirement for physical handling introduces the need for additional experimental procedures, can potentially damage fragile or sensitive crystals, and particularly in the case of micro-crystal injection jets, can lead to inefficient sample utilization.6,10,47,48,62
The generation of a large number of high quality, isomorphous micro-crystals is an area where integrated microfluidic technologies excel. The small length-scales of microfluidic devices create an environment free of inertial or convective effects while providing exquisite control over local conditions and gradients. The reproducibility of the microfluidic environment allows for the formulation of identical crystallization conditions, without the uncontrolled variations in concentration that result from chaotic mixing in bulk crystallization strategies. The absence of these effects facilitates both the simultaneous growth of a large number of isomorphous crystals,17,30,31,33 and may provide additional benefits in crystal quality, as have been reported in other convection-free systems.64–71
The benefits of microfluidic crystallization strategies are best realized when coupled with on-chip diffraction analysis. By leaving crystals undisturbed in a sealed environment, on-chip analysis avoids both challenges associated with harvesting a large number of tiny crystals from a microfluidic device and avoids the potential for crystal damage due to physical handling and/or exposure to the ambient environment. This approach also facilitates high levels of sample utilization by avoiding losses due to sample transfer or low hit-rates associated with continuous sample injection strategies.18,19,52,57 Unfortunately, the presence of the microfluidic device inherently introduces additional material into the X-ray path, resulting in signal attenuation and adversely affecting subsequent diffraction analysis.
Traditional microfluidic devices have been constructed out of millimeter-thick layers of glass, plastic, and/or poly(dimethylsiloxane) (PDMS).72–76 However, the thickness of such materials does not permit the effective transmission of X-rays (Fig. 1).77 Recently, a variety of microfluidic design strategies have been reported to try and address the challenge of creating an X-ray compatible microfluidic device. Nearly all of these approaches have focused on decreasing the thickness of device materials along the X-ray path to ∼100 μm,6,29–33,78–91 a strategy that has been matched in more traditional well plate setups.2,28,92–96 These ∼100 μm-scale platforms have enabled the collection of X-ray diffraction data from a variety of both model and novel targets. Significantly, the quality of these data have been sufficient to enable detection of the small variations in signal necessary for both de novo structure determination via single-wavelength anomalous diffraction (SAD),30,80,81 and for capturing short-lived structural intermediates by Laue diffraction.32
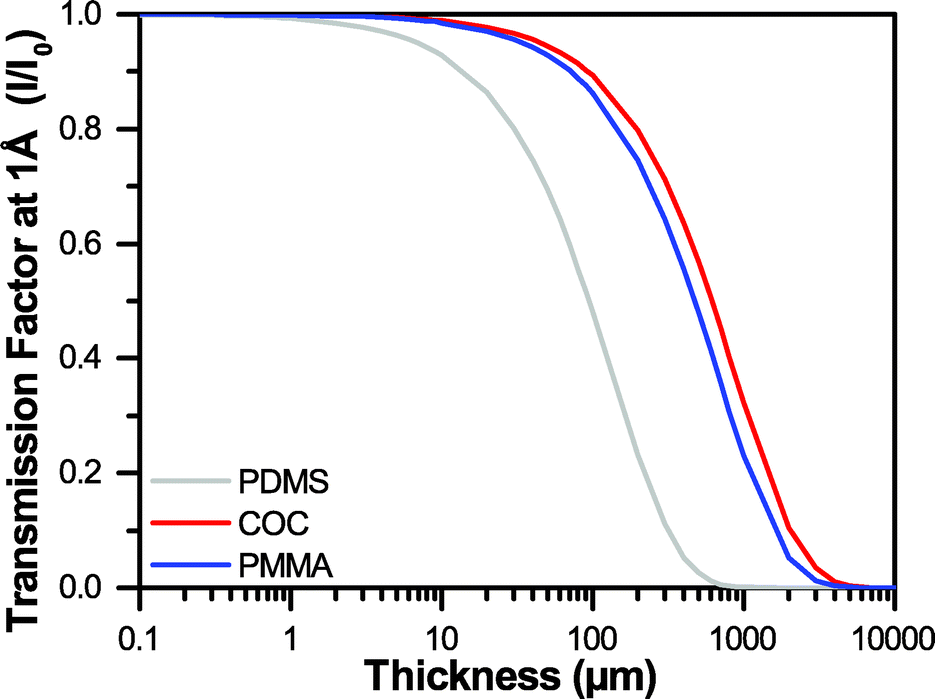 | ||
| Fig. 1 A comparison of the transmission factors I/I0 for varying thicknesses of PDMS, COC, and PMMA at an X-ray energy of 12.4 keV, or a wavelength of 1 Å. | ||
Despite these successes, microfluidic platforms for protein crystallography have typically been limited to data collection from relatively large crystals. This limitation is a consequence of needing to balance the strength of the diffraction signal from the crystal with losses associated with attenuation and background scattering from the device materials.31,32,48,97 For instance, in unrelated studies, relatively large crystals (50–200 μm) of hen egg white lysozyme (HEWL) were reported to diffract to a resolution limit of 1.55 Å in a 120 μm-thick device,33 while 10–20 μm micro-crystals of HEWL in a 300 μm-thick device only diffracted to 2.5 Å.6 While this comparison does not represent a well-controlled experiment, it is expected that resolution should suffer because of a decrease in signal-to-noise.97
Unfortunately, the development of even thinner devices has suffered from either intensive manufacturing requirements, or challenges related to sample stability and water loss. Many of the fixed-target approaches for serial crystallography rely on precision micro-manufacturing to fabricate device structures covered by an ultra-thin (50–500 nm-thick) silicon nitride membrane.47,51,55 These devices provide better stability against dehydration, compared to even thicker PDMS-based devices,6 and demonstrate the ability to collect high quality diffraction data from micro-crystals at a resolution equal to that observed for larger crystals.51 However, such devices cannot be manufactured with the same ease and low cost as soft lithographic and replica molding-based approaches, and are not amenable to the use of more easily-manufacturable ultra-thin polymer films because of challenges related to water loss and sample dehydration over time.
Here, we report a strategy for the straightforward incorporation of single-layer graphene into ultra-thin microfluidic devices to enable the in situ analysis of micro-crystals in a sample environment that is stable against evaporation for weeks. This work was inspired by two reports where graphene-wrapped protein crystals were shown to have good stability against dehydration and excellent signal-to-noise.98,99 To the best of our knowledge, this is the first report on the incorporation of large-sheet graphene into a microfluidic device to serve as a diffusion barrier against water loss.98–102 We establish the long-term stability of our devices by quantifying the rate of water loss through our graphene-based thin films. We further validate our approach and demonstrate the utility of graphene-based thin film microfluidics for on-chip X-ray crystallography by comparing the levels of signal-to-noise obtainable for diffraction signals as a function of device thickness. Although this work is focused on the use of graphene for protein crystallography, we anticipate that this technology should find utility in a wide range of lab on a chip applications.
Materials and methods
Graphene synthesis and film transfer
Monolayer graphene was grown on a copper substrate by chemical vapor deposition in a quartz tube furnace (Plana Tech).100,102–104 Solutions of 950PMMA A2 and 950PMMA A4 (poly(methylmethacrylate), Microchem) were spin coated (Specialty Coating Systems) onto the graphene film at 1000 rpm for 60 seconds, followed by curing at 120 °C for 10 minutes to form a PMMA film thickness of approximately 180 nm and 500 nm respectively, as measured by profilometry (Dektak 3). The PMMA/graphene film was released from the copper substrate by back-etching of the copper in an aqueous solution of copper etchant solution (Transene) for 3 hours, followed by three rinse cycles in MilliQ water (18.2 MΩ cm, Millipore Inc.) performed by floating the etched PMMA/graphene film on the surface of the water. The resultant film was then transferred directly from the surface of the water onto an adhesive-backed polyester film (McMaster Carr) for incorporation into the subsequent microfluidic device. Because of the way in which the graphene films are released from the copper substrate, it is most straightforward to transfer the PMMA/graphene film onto the backing support in such a way that the graphene film faces outward, rather than directly towards the channel. However, this assembly process can be performed in reverse, and no significant difference was observed in device performance comparing the two methods.Device architecture and assembly
The overall chip architecture consists of five layers, which allow for various different functional layouts (Fig. 2 and ESI† Fig. S8 and S9). The fluidic channels of the device were defined by a 100 μm thick cyclic olefin copolymer (COC, Topas, 6013) film. The channel structure was cut into this film using a cutting plotter (Graphtec CE6000). The channel was then sealed on one side with a PMMA/graphene film, supported by an adhesive-backed polyester film (McMaster Carr) with cut-out features to define fluidic inlets and/or window areas (Fig. 2 and 3 and ESI† Fig. S8 and S9). Here, the backing layer provides additional structural stability, while helping to define inlets, and providing a facile way to adhere the various layers together. Following assembly of the COC fluidic layer to the adhesive bottom layer containing a PMMA/graphene film, both the top and bottom halves of the device were coated with poly(dopamine) (PDA) to create a hydrophilic surface and facilitate channel wetting.105,106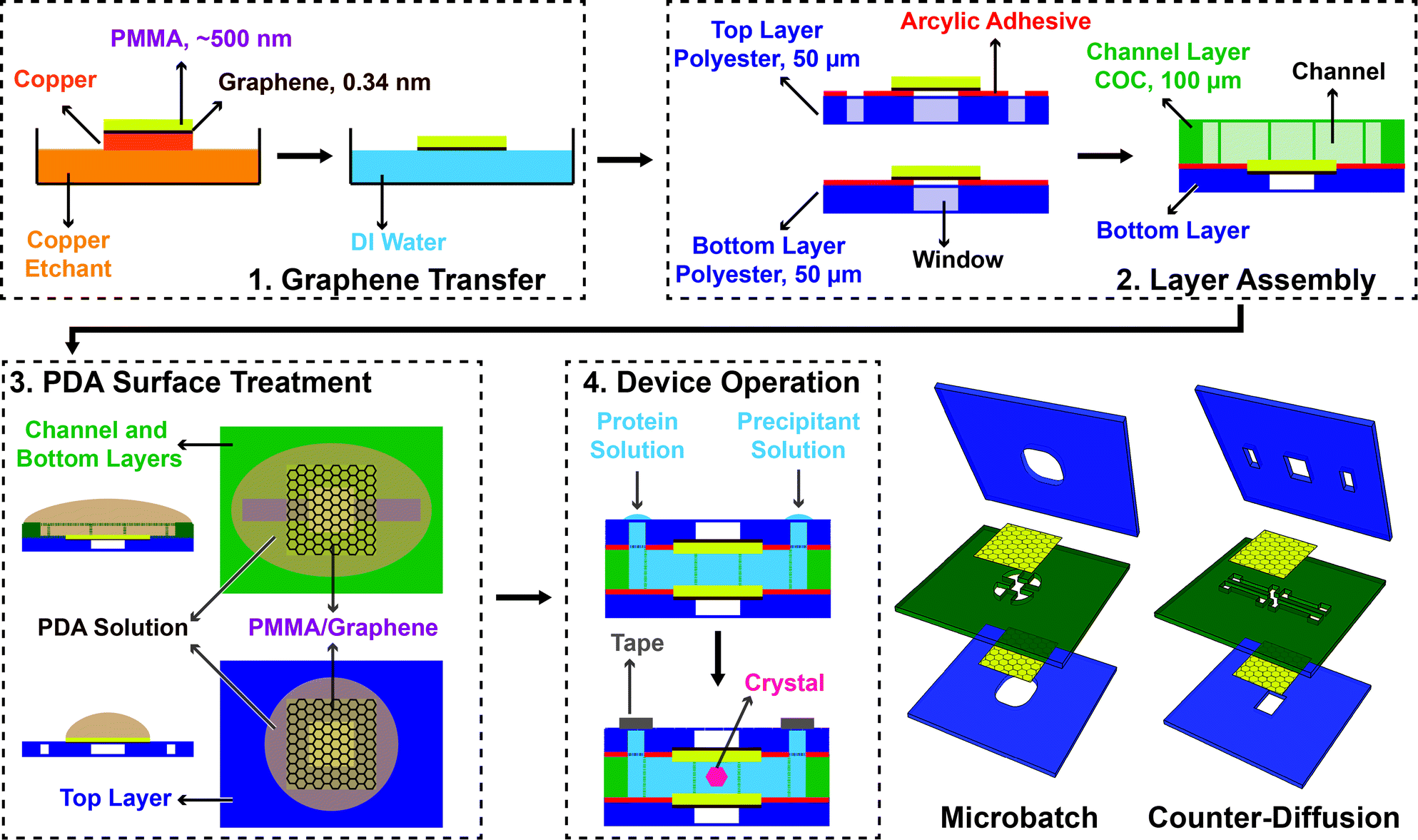 | ||
| Fig. 2 Schematic depiction of the fabrication scheme for thin-film graphene-based microfluidics, using a channel-based counter-diffusion as the example. (1) CVD-grown graphene on copper is first coated with a layer of PMMA, and then released from the copper substrate by etching. The subsequent film is floated on the surface of water for (2) transfer to an adhesive polyester support layer to define the window areas of the device. This layer is then adhered to a COC layer containing the cut-out pattern for the microfluidic channels. (3) A hydrophilic PDA surface treatment on both the top PMMA/graphene film and the bottom layer containing the microfluidic channel facilitates easy filling of the final device via capillary action. (4) The final, assembled device is held together by the adhesive layers defining the window structures, leaving the PMMA/graphene window areas free of excess material. Either microbatch or counter-diffusion crystallization trials can be set up by the addition of protein and precipitant solutions. The device inlets are sealed with Crystal Clear tape. | ||
 | ||
| Fig. 3 (a) A microbatch chip mounted on the 14-ID-B beamline at BioCARS, oriented at −30° towards the high resolution camera. (b) Photograph of a counter-diffusion chip that has been filled with blue and red food dye to simulate on-chip mixing. Optical micrographs under crossed-polarizers showing HEWL crystals grown in a (c) microbatch and (d) counter-diffusion chip. Crystals from 50–500 μm formed overnight. The inset shows a close-up view of selected crystals. | ||
Poly(dopamine) (PDA) surface treatment
A 2 mg mL−1 solution of dopamine hydrochloride (Sigma) in 10 mM Tris buffer pH 8.5105,106 (Fisher, Molecular Biology grade) was freshly prepared and then carefully dropped onto the PMMA/graphene films to cover the entire channel surface (Fig. 2 and ESI† Fig. S8 and S9). The surfaces were allowed to incubate for at least 5 hours at room temperature (23 °C) in a sealed Petri dish. Following treatment, the films were rinsed by dipping into MilliQ water three times and allowed to air dry before final device assembly and use.Raman spectroscopy
Raman spectroscopy was used to validate the composition and quality of the resulting graphene-based films. Films were placed onto a silicon wafer coated with a 200 nm layer of silicon dioxide. Analysis was performed using a DXR™2xi Raman imaging microscope system (Thermo Scientific) with an incident beam of light at 633 nm on PMMA/graphene/PDA and PMMA/graphene films. These data were compared to the spectra for graphene, PMMA, and PDA-only films coated directly onto the silicon wafer (Fig. 4).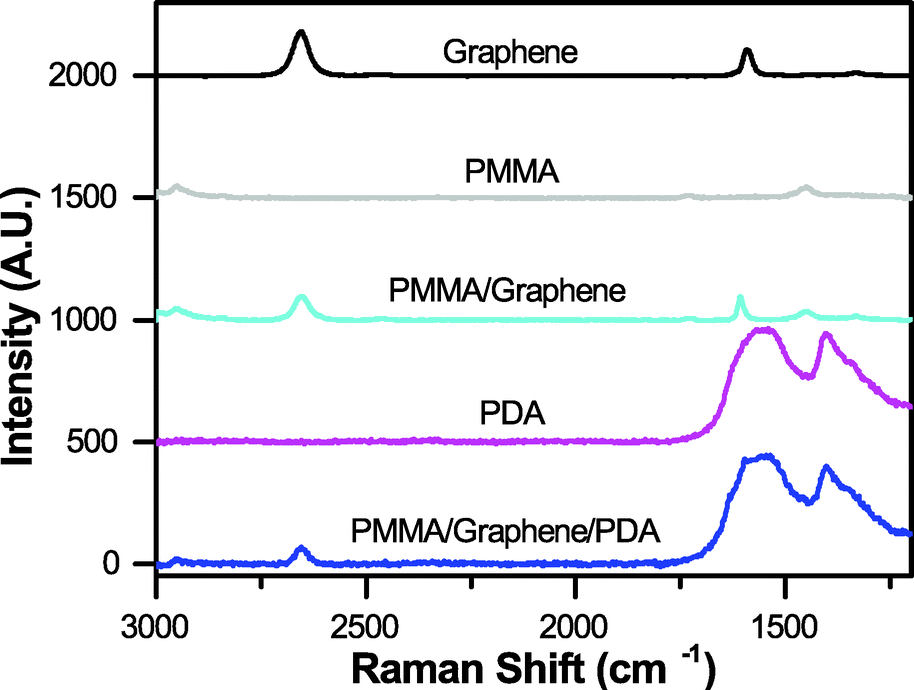 | ||
| Fig. 4 Raman spectra, demonstrating the expected signals for graphene, 500 nm PMMA, and PDA separately, and in our final composite films. | ||
Permeability measurements
The permeation of water through our various thin film materials was quantified as a function of time by measuring the change in absorbance of an aqueous solution of red food dye (Kroger). Films of A2 PMMA, A4 PMMA, A2 PMMA/graphene, and A4 PMMA/graphene were adhered to an adhesive-coated polyester layer, as in Fig. 2 and ESI† Fig. S8. These films, as well as a 100 μm COC were sealed with vacuum grease onto individual wells of a 96-well plate containing 300 μL of red food dye in water. The change in signal for these films was compared with wells that were left open to the air. All experiments were performed in triplicate. The solution absorbance was then monitored at 300 nm and 450 nm over time (Fig. 5a) using a plate reader (BioTek). Photographs of the well plate allowed for visual characterization of changes in the sample volume (Fig. 5b). In addition to direct measurements of water loss through films of the individual device materials, water loss from the both microbatch and counter-diffusion style devices was evaluated qualitatively using optical microscopy (ESI† Fig. S7).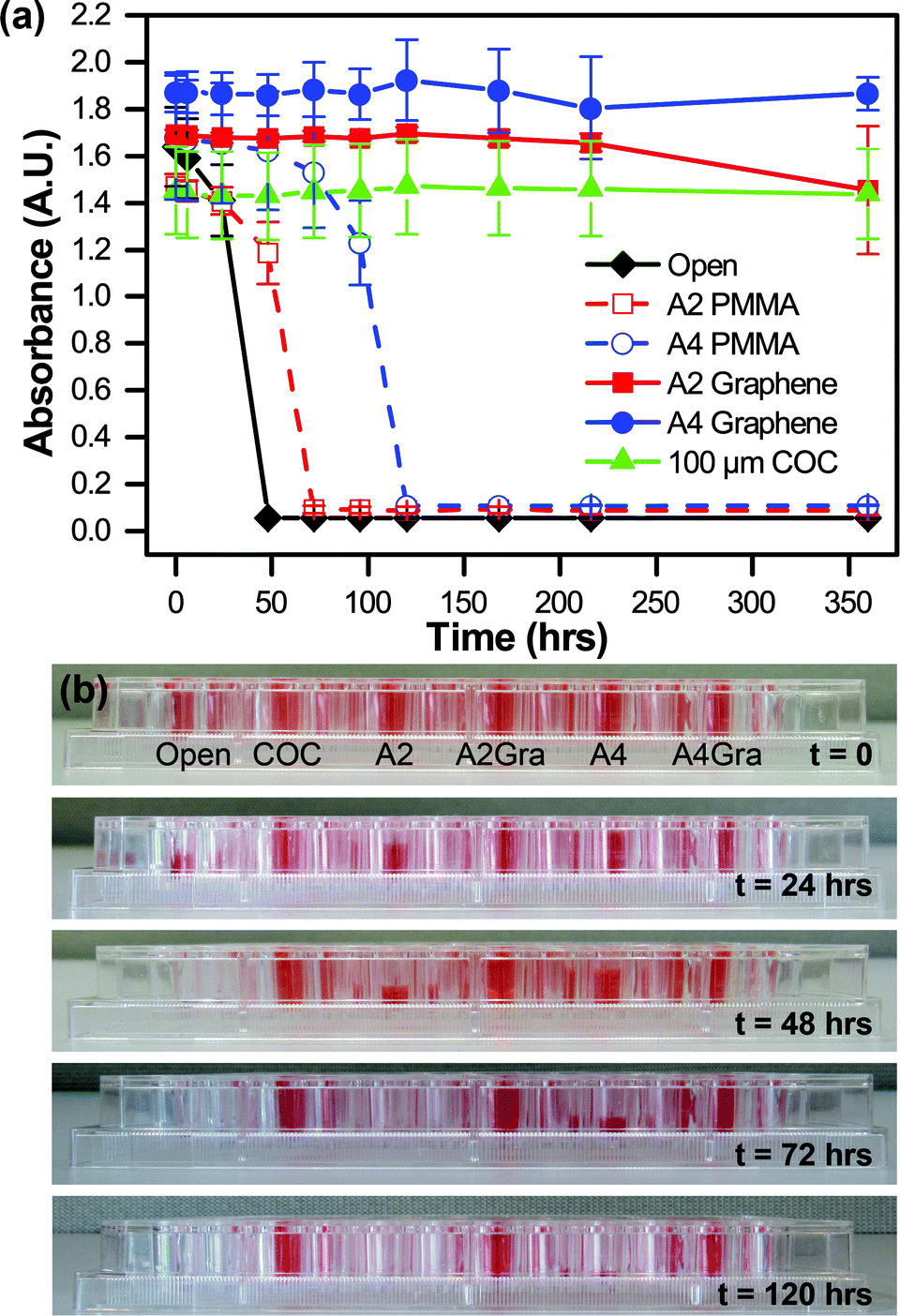 | ||
| Fig. 5 (a) Measurement of the absorbance of colored solutions as a function of time at 450 nm, demonstrating the water permeability of various PMMA, PMMA/graphene, and COC films. Both the thicker 100 μm COC film, and the two PMMA/graphene films show no significant decrease in signal over the course of more than two weeks. (b) Photographs of the experimental setup showing changes in the liquid levels as seen by a side-view. | ||
Contact angle measurements
A PMMA/graphene film was transferred from the aqueous rinse solution onto a silicon wafer (graphene-side down) and allowed to air dry 3 hours to facilitate strong binding between the film and the wafer surface. A PDA treatment was then applied to the PMMA surface. The contact angle of both a treated and an untreated PMMA/graphene film were measured using goniometry with MilliQ water (Ramé-Hart).AFM characterization
Atomic force microscopy (AFM) was used to characterize the surface roughness of all films. Surface scans of PMMA–graphene and PDA coated PMMA–graphene (ESI† Fig. S10) were acquired using a Cypher ES atomic force microscope (Asylum Research, Santa Barbara CA).107 Samples were imaged in AC mode with Tap-300G cantilevers (Budget Sensors).Protein crystallization
A solution of 80 mg mL−1 hen egg white lysozyme (HEWL, Hampton Research Inc.) was prepared in 50 mM sodium acetate buffer (Fisher Scientific, ACS grade), pH 4.8. A precipitant solution was prepared, containing 2 M sodium chloride (Sigma Aldrich, ACS reagent) in 50 mM sodium acetate, pH 4.8. For microbatch experiments (Fig. 3c), 12 μL protein solution and 4 μL precipitant solution were pre-mixed and pipetted into the chip before the layers were sealed by simply pressing the adhesive layers together by hand. For counter-diffusion experiments using a channel-based chip architecture (Fig. 3d), 12 μL protein solution and 4 μL precipitant solution were pipetted separately onto the two device inlets. Because of the hydrophilic PDA surface treatment, the solutions wetted the channels and flowed into the device. The chip can be filled either by the sequential or simultaneous addition of crystallization solutions. However, due to the impermeability of the PMMA/graphene film, simultaneous filling of crystallization and precipitant solutions can lead to trapping of air within the device. The filled chips were placed into a Petri dish alongside a microcentrifuge containing 200 μL of DI water. The Petri dish was sealed with Parafilm (Thermo Scientific), and stored at 4 °C. For both crystallization strategies, large lysozyme crystals (>100 μm) formed overnight. Crystals were visualized using a Zeiss V12 stereomicroscope with cross-polarizers (Carl Zeiss Microscopy LLC).On-chip X-ray diffraction
Data were collected in polychromatic mode at 12 keV (1.03 Å, 5% bandwidth) on the 14-ID-B beamline at the Advanced Photon Source at Argonne National Laboratory.108 The microfluidic chips were mounted directly on the φ spindle of the goniometer using a modified magnetic mount (Hampton Research; Fig. 3a). Positioning and alignment of the chips was performed using a high-resolution camera oriented at −30° with respect to the X-ray beam and a medium-resolution camera oriented at 60°, slightly modified from previous reports.31,32 This setup allowed for a range of motion of ±5 mm in x (focus, along the direction of the X-ray beam), ±6 mm in y (vertical with respect to the direction X-ray beam), and ±60 mm in z (horizontal with respect to the X-ray beam). Sample visualization and positioning were performed using an in-house graphical user interface at the 14-ID-B beamline. Sample positioning was achieved using a click- and -translate routine coupled to the high- and medium-resolution cameras. Sample centering along the path of the X-ray beam was achieved by visually focusing the sample, taking advantage of the very small depth of field of the high-resolution camera. The interface enabled identification and alignment of crystals on chip. BioCARS LaueCollect software was used for subsequent data acquisition.Data were collected from microfluidic chips oriented between −45° and 45° with respect to the X-ray beam. The large size (∼300 μm) and robustness of the HEWL crystals, combined with the small X-ray beam size (35 × 35 μm2 FWHM), permitted the collection of multiple frames (3 or 4, see Table 1 and ESI† Table S2) of data from a large number of fresh crystal volumes on each individual crystal at room temperature. Data were typically collected at 3° intervals over the range of 45° and 45°. This spacing was chosen to enable optimal coverage of reciprocal space given the available X-ray bandwidth. Complete data sets were obtained by merging data taken from multiple volumes of the same crystal. We compare four different crystals grown and/or analysed under different conditions (see Fig. 6, Table 1, and ESI† Table S2). A Rayonix MX340-HS detector was used with a sample-to-detector distance of 175 mm. Diffraction data were collected using a 1.5 μs exposure with the storage ring operating in 24-bunch mode (11 consecutive X-ray pulses of 100 ps duration).
| Parameter | Microbatch | Counter-diffusion PMMA/graphene | |
|---|---|---|---|
| PMMA/graphene | COC filma | ||
| a Values in parentheses are for the highest integrated resolution shell. | |||
| Data collection | |||
| Total # frames | 55 | 59 | 30 |
| # Frames/spot | 3 | 4 | 3 |
| Resolution (Å) | 50–1.40 | 50–1.41 | 50–1.46 |
| Space group | P43212 | P43212 | P43212 |
| Unit cell (Å) | a = b = 79.1, c = 37.7 | a = b = 79.1, c = 37.3 | a = b = 79.1, c = 37.8 |
| Single reflections | |||
| Total obs. | 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 678 678 |
143638 |
70996 |
| Unique obs. | 18294 |
17608 |
15629 |
| Redundancy | 8.2 | 8.2 | 4.5 |
| R merge on F2 | 0.054 | 0.077 | 0.047 |
| R merge on F | 0.036 | 0.049 | 0.031 |
| <F/σ(F)> | 63.9 (28.6) | 50.7 (19.9) | 60.4 (33.4) |
| Single and multiple reflections combined | |||
| Completeness (%) | 77.2 (25.1) | 76.6 (28.4) | 74.8 (27.1) |
| Structure refinement | |||
| R work | 0.149 | 0.156 | 0.145 |
| R free | 0.163 | 0.176 | 0.167 |
| Ramachandran statistics | |||
| Favored | 123 (96.9 %) | 122 (96.1 %) | 122 (96.1 %) |
| Allowed | 4 (3.1 %) | 5 (3.9 %) | 5 (3.9 %) |
| Disallowed | 0 (0.0 %) | 0 (0.0 %) | 0 (0.0 %) |
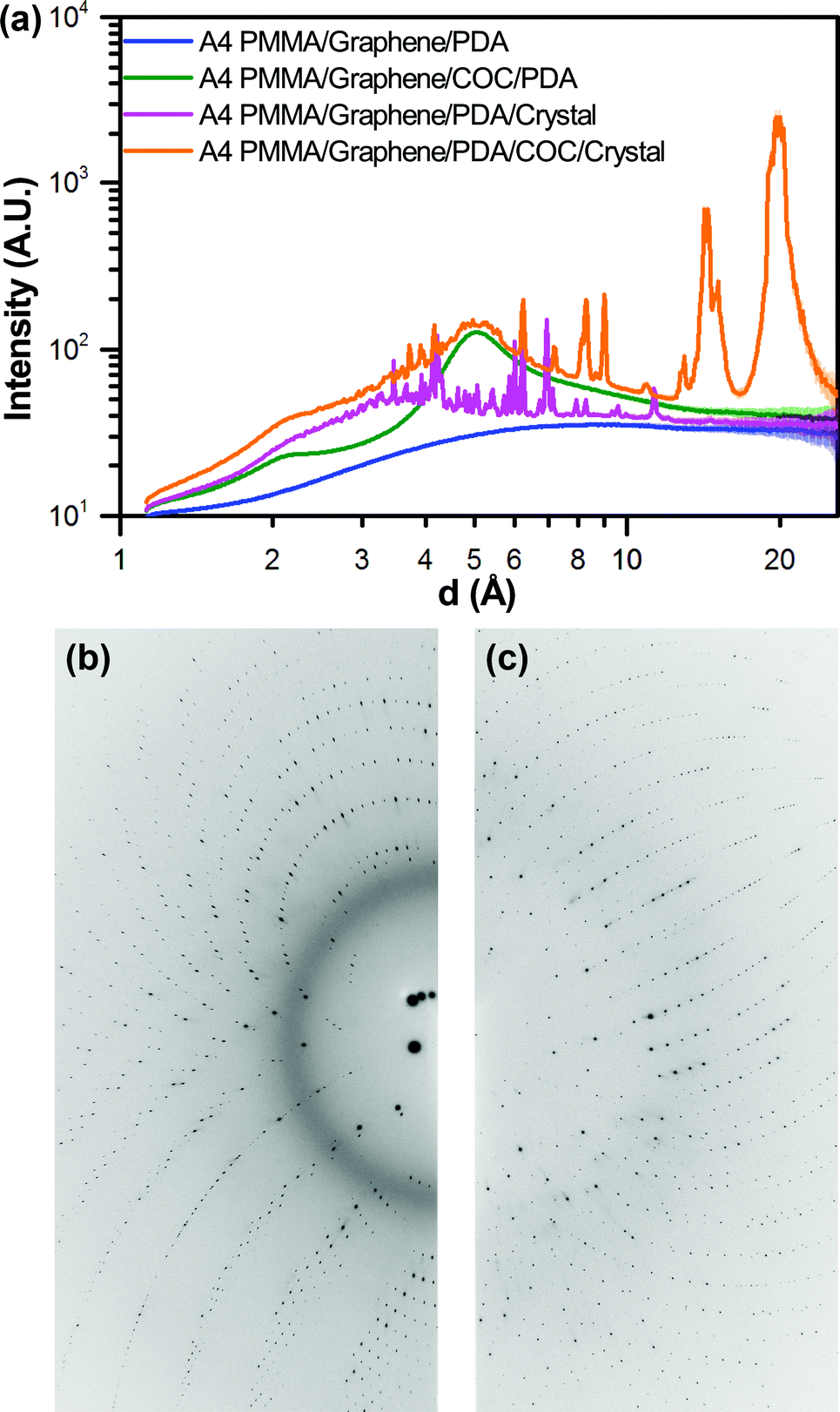 | ||
| Fig. 6 (a) One-dimensional integrated X-ray intensity profiles showing the relative strength of the observed diffraction signal from a HEWL crystal compared to the noise resulting from background scattering due to the presence of device materials as a function of resolution. The corresponding two-dimensional diffraction images for the (b) A4 PMMA/graphene/PDA/COC/crystal dataset (orange), and the (c) A4 PMMA/graphene/PDA/crystal dataset (magenta) shown in (a). | ||
Data analysis
Laue diffraction images were processed using the Precognition/Epinorm software (Renz Research; Table 1). Each crystal was processed separately. Microbatch data were integrated to 1.40 Å resolution, counter-diffusion data to 1.45 Å and ultimately merged to a final resolution that was chosen both to maintain a completeness in the highest resolution shell, of at least 25%, provided that I/σ(I) remains above 3. Subsequent processing of crystallographic data sets was carried out using both the CCP4 suite of programs109 and PHENIX.110 Structure refinement (Table 1) was carried out using PHENIX.refine starting from PDB model 193L70 in a fully automated fashion for 20 cycles, including optimization of atomic coordinates, real-space refinement, individual B-factors, TLS parameters and occupancies, while taking advantage of simulated annealing (Cartesian), automatically correcting N/Q/H errors, and updating waters.Analysis of the background signal-to-noise (Fig. 6) was done using the Nika software suite for 2D diffraction data reduction in Igor Pro (Wavemetrics Inc.).111 Integration of diffraction images in 2θ with log binning was performed using the calibrated beam center and sample-to-detector distance obtained from geometry refinement in Precognition.
Results and discussion
The goal of this work was the development of an ultra-thin X-ray compatible microfluidic platform to enable on-chip X-ray diffraction analysis of protein crystals with negligible contributions from the device materials. To this end, we developed a straightforward method for the incorporation of large-area, single-layer graphene films to serve as both X-ray compatible windows and as a diffusion barrier to prevent evaporative losses from the device. Our ultimate objective is to enable serial crystallography of micro-crystals (<10 μm in size) for both de novo structure determination and time-resolved crystallography, where the ability to detect small variations in the overall X-ray signal is critical. To achieve the low levels of background noise and high signal transmission necessary for these applications, we established design criteria for our X-ray transparent microfluidic device. We considered three main aspects of the interaction of our device materials with X-rays: (i) attenuation of the X-ray signal, (ii) background noise resulting from diffuse scattering, and (iii) the strength of the diffraction resulting from a crystal.Attenuation, or the change in intensity of the X-ray beam can be calculated for any material based on the atomic weight of the atoms present, the density of the material, and the wavelength of interest (see ESI† Table S1 and Fig. S1).77,112 Thus, we can minimize attenuation effects by decreasing the atomic weight of the atoms present in the material, decreasing the density, and/or decreasing the thickness. Based on these calculations, we show that at a wavelength of 1 Å (12.4 keV), achieving transmission levels of 99% would necessitate decreasing the thickness of our device to only 1 μm of PDMS, or approximately 10 μm of an organic polymer such as COC or PMMA. Achieving 99.9% transmission would require 200 nm-thick PDMS, or approximately 900 nm-thick COC or PMMA (Fig. 1 and ESI† Fig. S2). However, most sub-micron polymeric films suffer from relatively high rates of water permeation. We therefore developed a strategy for coupling sub-micron polymer films with large-area, single-layer sheets of graphene to create a diffusion barrier against water loss.
Traditional methods for graphene-transfer have taken advantage of thin-film PMMA as a support layer to aid during transfer of the graphene films from the copper substrate where they are grown to a functional device.104 Here, we adapted this procedure to facilitate incorporation of a sub-micron polymer film to serve as an impermeable X-ray window for our microfluidic protein crystallography platforms. Single-layer graphene was coated with a ∼500 nm-thick layer of PMMA and transferred to pre-cut adhesive polyester substrates containing the appropriate features to define a window and/or inlet structures for the top and bottom of the device (Fig. 2 and ESI† Fig. S8). The presence of the adhesive helped to secure both the PMMA/graphene films in place, as well as adhering these top and bottom layers to a spacer layer of 100 μm-thick COC, pre-cut to define the microfluidic features of the device. This strategy took advantage of commercially-available materials and avoided the need to apply a separate adhesive treatment that would potentially increase the overall thickness of the device. Furthermore, we anticipate that this strategy could be easily adapted to work with a variety of alternative strategies for defining microfluidic channels, including roll-to-roll manufacturing.
We utilized this manufacturing strategy to develop simple microfluidic geometries to enable microbatch and counter-diffusion crystallization experiments (Fig. 2 and 3). However, the various polymeric surfaces present in our device proved to be relatively hydrophobic, such that our crystallization solutions would not easily wet and flow into a microfluidic channel. We overcame this difficulty through the application of an ultra-thin, hydrophilic, PDA-based surface treatment.105,106 Here, a solution of dopamine at pH 8.5 was allowed to incubate on the various surfaces of our microfluidic channel (Fig. 2 and ESI† Fig. S8). Under basic conditions, dopamine has been shown to polymerize and create a ∼10 nm-thick hydrophilic surface coating.105,106,113 Contact angle measurements showed a change in wetting angle from 74° before treatment, to 36° after treatment (ESI† Fig. S11). We further confirmed the presence of both PDA and the integrity of our graphene film via Raman spectroscopy (Fig. 4). This level of wetting proved to be sufficient for aqueous solutions to easily wet and flow into our device (Fig. 3b).
Our main motivation for incorporating graphene into our microfluidic device architecture was to create a diffusion barrier to minimize the loss of water from our device. We tested the effectiveness of various polymeric films on preventing the evaporative loss of water in a well plate-style assay. Both visual inspection of the various wells (Fig. 5a and ESI† Fig. S6) and quantitative analysis via absorbance measurements at 300 nm (ESI† Fig. S6a) and 450 nm (Fig. 5b) demonstrated the efficacy of single layer of graphene as a diffusion barrier against water loss. Under our experimental conditions, complete evaporation of an open well occurred over the course of approximately 48 hours, while the presence of a 180 nm-thick A2 PMMA film only extended this lifetime to 72 hours. However, the addition of a single layer of graphene decreased the rate of water loss such that only minimal water loss was observed after 360 hours (15 days). Similarly, a 500 nm-thick A4 PMMA film showed complete evaporation after 120 hours, but the addition of graphene resulted in negligible water loss over the entire course of our experiment. These graphene-based, sub-micron films provided the same level of protection against water loss as a 100 μm-thick film of COC, a material known for its low water permeability.114 Furthermore, prior reports have demonstrated that the use of multiple layers of graphene can further improve the barrier properties of the film.100
We then extended our water permeation experiments from a simple film geometry, to a fully assembled microfluidic device architecture. Visual inspection of optical micrographs indicated no significant water loss over the course of 96 hours (4 days) for microbatch devices where the sample chamber was completely enclosed between two gas impermeable PMMA/graphene films (ESI† Fig. S7a). However, for the case of the counter-diffusion chip, if only the main sample chamber was protected by PMMA/graphene films, then significant water loss was observed from the unprotected areas of the device over the course of ∼1 day (ESI† Fig. S7b). While such evaporative losses can be mitigated by sample incubation in a controlled humidity environment, future device designs will incorporate complete isolation of the sample environment within a graphene film.
Having demonstrated the efficacy of our device materials to protect against dehydration and enable long-term sample incubation, we set up microbatch and counter-diffusion crystallization trials using HEWL. Large crystals (>100 μm, Fig. 3c and d) formed overnight for both of these methods, and were stable in the lab for days at 4 °C. As a precaution, devices were placed into a Petri dish along with a reservoir of water and sealed. These samples easily survived the rigors associated with overnight shipping to the synchrotron. This demonstration of stability significantly enhances the potential utility of these devices by allowing users to prepare crystallization trials at their home laboratory, incubate as needed, and transport their samples for analysis with the same relative ease as with more traditional samples.
We next investigated the levels of signal-to-noise achievable for on-chip X-ray diffraction measurements. While attenuation calculations (Fig. 1 and ESI† Fig. S2) describe the total signal lost via transmission through a material, this does not characterize the observed signal-to-noise. Here, we investigated the level of background scatter observed for our various device materials as a function of resolution. The introduction of a thin A2 PMMA film resulted in an approximately 10-fold increase in the observed background scattering, compared to air only (ESI† Fig. S3). No significant change in signal was observed with the subsequent addition of either graphene or a PDA treatment, and a minimal increase in scattering was observed when the PMMA film was increased from 180 nm-thick A2, to 500 nm-thick A4 PMMA. However, a further increase in scattering was observed with the addition of a 100 μm-thick film of COC. The signal for COC showed increases in the overall levels of background noise, with resolution-dependent scattering bands present at approximately 2.5 Å and 5 Å, characteristic of the internal structure of the polymer. It should be noted that Laue diffraction, due to the polychromatic nature of the X-ray beam, typically suffers from higher background and thus poorer signal-to-noise than monochromatic X-ray diffraction methods. Consequently, the experiments performed here are a powerful demonstration of the utility of our approach.
While basic characterization of the background scattering for the device materials was performed with the sample mounted perpendicular to the incident X-ray beam, protein structure determination can require sample rotation to enable data collection from a variety of different crystal orientations. Thus, we must consider not only the thickness of the device materials in terms of a minimal path length, but based on sample orientation. We therefore calculated the relative path length through a film as a function of rotation angle (ESI† Fig. S4). This path length varies as 1/cos(ϕ), and diverges as the angle ϕ approaches 90°. However, because of low signal attenuation, we are able to maintain a transmission of 99.9% of the incident signal through a rotation of +/− 86° from normal. This large range of rotation means that for low-symmetry space-group crystals, or for anomalous diffraction measurements, our devices are able to access 344° out of a possible 360° rotation at a better than 99.9% level. If we decrease our level of acceptable signal attenuation to 90% of the incident beam intensity, we can achieve +/− 89.5° of rotation from normal, meaning that only 2° of data would be out of reach. These high levels of transmission and the ability to access a wide range of sample orientations are a dramatic improvement over previously-reported PDMS/COC based devices which enable a maximum of only 77% of the incident X-ray intensity when normal to the incident beam.30–33,115–117
Having characterized the X-ray profile of our device materials, we next investigated the effect of this background scattering on the signal-to-noise associated with on-chip X-ray diffraction measurements. All images were well exposed, typically with several saturated diffraction spots (∼65000 counts). A comparison of both the 2D diffraction images and the corresponding 1D integrations shows the presence of strong diffraction peaks and an additional diffuse signal around 3 Å, which we attribute to the presence of solvent (Fig. 6 and ESI† Fig. S5).98 The observed strength of the diffraction signal decreases relative to the background noise at higher resolution, as expected. It should be noted that the data presented in Fig. 6 and ESI† Fig. S5 is the result of integration in 2θ across the entire image, rather than along a line. Thus, the presence of multiple diffraction peaks at nearly the same resolution could result in apparent broadening of the integrated signal.
The benefit of our ultra-thin device materials can be observed through a comparison of the observed signal-to-noise from a crystal analysed in one of our PMMA/graphene-based microbatch devices, compared with a second crystal from the same device that opportunistically grew underneath the COC spacer layer. Comparing the data from these two crystals, we can clearly observe a decrease in signal quality as a result of the higher background signal from the 100 μm-thick COC layer, both visually in the 2D diffraction images, and more quantitatively in the 1D integrations (Fig. 6 and ESI† Fig. S5). We have previously struggled to observe diffraction for relatively small crystals in a PDMS/COC device, and posit that the balance of signal-to-noise observed here explains our observations.
Finally, we collected complete datasets for different individual crystals using a micro-focused X-ray beam to enable the collection of small wedges of data from distinct locations across a crystal. We did not observe a significant variation in data obtained from different crystals within the same device (microbatch), or between crystallization techniques (microbatch vs. counter-diffusion; Table 1 and ESI† Table S2). However, in general, the counter-diffusion method would be expected to result in more reproducible crystal growth, due to the precise control over concentration gradients, diffusion, and mixing afforded by microfluidic devices.30,31,33
We also compared data from the two microbatch-grown crystals described in Fig. 6 and ESI† Fig. S5, examining the effect of an obscuring 100 μm-thick COC layer. Interestingly, we did not observed a significant decrease in resolution for the crystal present under the thicker COC film. This result may be due to the relatively strong diffraction signals expected from our large crystals. High quality crystallographic statistics were observed for all samples; however, the presence of the thick COC film did correspond with an increase in the values for Rmerge, and a decrease in signal-to-noise (i.e., <F/σ(F)>).
A closer analysis of <F/σ(F)> as a function of resolution (Fig. 7) demonstrated that the increased attenuation and background scatter resulting from the presence of a 100 μm-thick COC layer had a negative effect on the overall sensitivity of the measurement. This decrease in signal-to-noise was observed despite similar, levels of completeness and redundancy in the overall dataset, compared with data collected through PMMA/graphene windows (Table 1, ESI† Table S2, and Fig. S12). The effect of background material on signal-to-noise can be particularly highlighted with respect to a significant decrease in <F/σ(F)> at a resolution of ∼2.5 Å, corresponding to one of the scattering bands observed for COC (Fig. 6 and ESI† Fig. S5). We were unable to directly observe the impact of the larger scattering band observed at ∼5.0 Å, due to the range of resolutions over which the binned <F/σ(F)> data were provided by the processing software. However, we anticipate that contributions from this large scattering band are what cause the large difference in <F/σ(F)> at low resolution between the samples containing COC and those without.
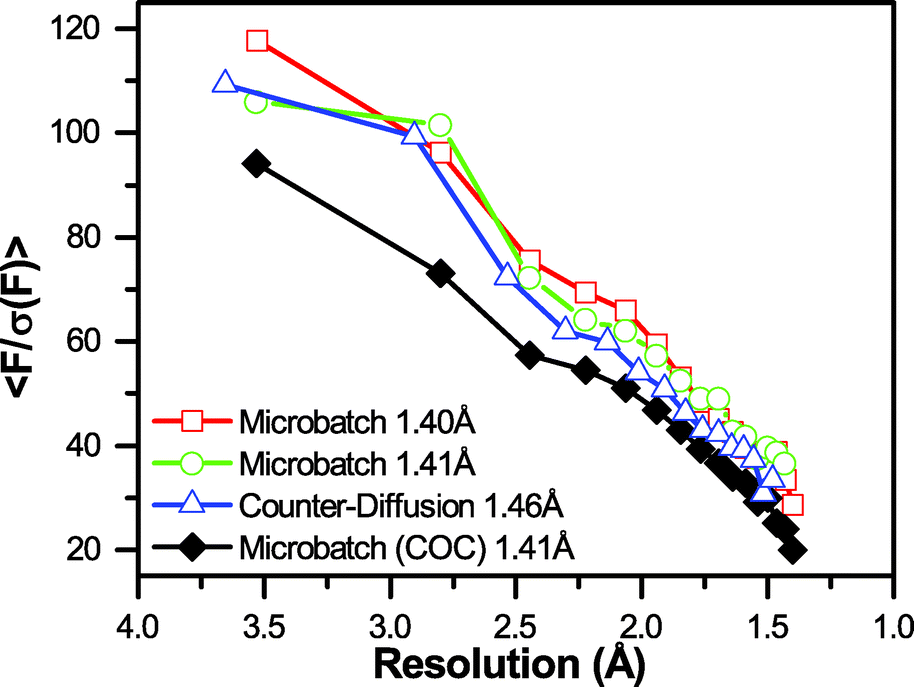 | ||
| Fig. 7 Graph of signal-to-noise <F/σ(F)> as a function of resolution for the four datasets shown in Table S2.† The final resolution of the dataset is indicated in the legend. A significant decrease in <F/σ(F)> is observed for the dataset collected in the presence of a 100 μm-thick COC film. | ||
Examination of the electron density maps generated from these various datasets show similar levels of structural detail, as would be expected from data extending to ∼1.40 Å (Fig. 8 and ESI† Fig. S13). The quality of the data allows for unambiguous interpretation of structural details, including side-chain conformations, the presence of aromatic groups, and the location of bound water molecules. Statistical measures associated with structure refinement (Rwork and Rfree) were very good, and showed no significant dependence on the details of data collection.
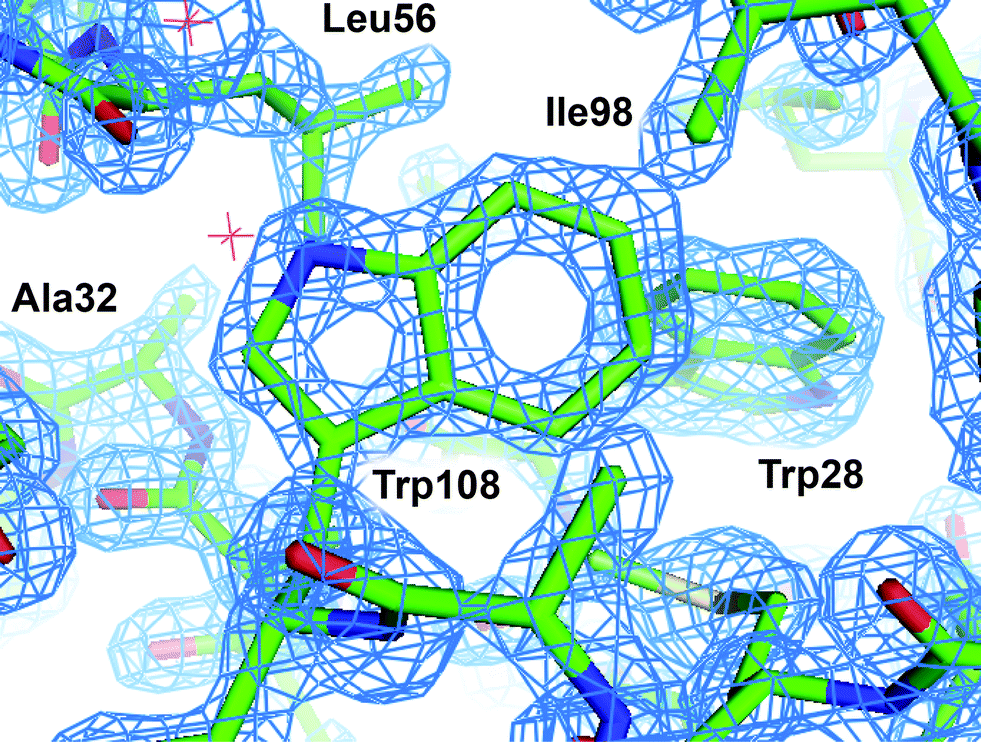 | ||
| Fig. 8 2Fo − Fc electron density map of HEWL grown in microbatch to 1.40 Å. The map was contoured at 2σ and superimposed over a licorice representation of the protein structure surrounding Trp108. | ||
It should be noted that the excellent levels of signal-to-noise obtained for all of these samples, as well as the informative differences between samples, and high levels of structural detail were observed based on the diffracted intensity produced by a 1.5 μs polychromatic X-ray exposure (11 consecutive X-ray pulses of 100 ps duration). The next phase of testing will focus on the serial analysis of micro-crystals. We will also investigate the potential for performing data collection using only a single ∼100 ps X-ray pulse. Such studies, particularly coupled with serial data collection strategies, have the potential to “out-run” the adverse effects of secondary radiation damage, such as radical generation and the breakage of chemical bonds to enhance the relevance of the resultant data.31,118
Conclusions
In summary, we have presented a straightforward method for the incorporation of single-layer graphene into ultra-thin, X-ray compatible microfluidic devices. We have demonstrated the utility of graphene as a diffusion barrier to mitigate the effects of evaporation over the course of several weeks. We further validated our approach via the on-chip structure determination of HEWL as a model system to facilitate quantification of the effects of device thickness on the signal-to-noise of the diffraction measurement and subsequent structure determination efforts. Looking forward, these graphene-enhanced, ultra-thin device fabrication strategies hold tremendous promise for enabling the on-chip crystallization and subsequent serial crystallographic analysis of micro-crystals at synchrotron sources, and could be further scaled to enable the analysis of nano-crystals at XFELs.The ability to collect data from micro-crystals has the potential to enable structure determination from targets that have proven resistant to the growth of larger crystals for more traditional analyses. This approach would take advantage of microfluidics to grow a large number of high-quality, isomorphous crystals while facilitating efficient, high-throughput serial diffraction analysis without the need for sample handling and mounting. Additionally, these types of serial methods can be used to potentially “out-run” radiation damage by, coupling the single-shot analysis of a large number of crystals with fast data collection.118 Strategies for implementing this type of approach include single X-ray pulse Laue diffraction at synchrotron sources, as well as more advanced, XFEL-based methods.
Ultimately, we see the intersection of microfluidics, micro-crystallography, and fast data collection as an opportunity to enable the study of protein structural dynamics. To this end, integrated microfluidic fluid-handling capabilities could be harnessed to enable chemical triggering (e.g., substrate addition or a pH jump) to investigate a wide range of biologically and medically relevant protein targets that have thus far been resistant to time-resolved studies. For these studies, micro-crystals are critical to enable efficient diffusion of the triggering species into the crystal at a timescale faster than the enzymatic reaction.7 Building on this idea, it would also be possible to extend these types of studies to examine multiple variables (e.g., structural changes due to ligand binding as a function of time, salt concentration, and pH) to expand the scope and context of the resultant dynamic structural information.119
Acknowledgements
We would like to acknowledge Ashley Kaiser for her assistance with graphene synthesis, Prof. Jungwoo Lee and Jun-Goo Kwak for help with the cutting plotter, Prof. Lili He for her assistance with Raman spectroscopy, Prof. Stephen S. Nonnenmann for the use of his atomic force microscope, Kerianne Dobosz from the Schiffman lab and Minchao Zhang from the McCarthy lab for assistance with contact angle measurements, Zoey Meng for assistance with profilometry, George Chang from the Emrick lab for suggestions regarding PDA treatments, and Rui Pereira for helpful discussions. K. W. K. was supported by National Research Service Award T32 GM008515 from the National Institutes of Health. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number R24GM111072. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- J. L. Smith, R. F. Fischetti and M. Yamamoto, Curr. Opin. Struct. Biol., 2012, 22, 602–612 CrossRef CAS PubMed.
- D. Axford, R. L. Owen, J. Aishima, J. Foadi, A. W. Morgan, J. I. Robinson, J. E. Nettleship, R. J. Owens, I. Moraes, E. E. Fry, J. M. Grimes, K. Harlos, A. Kotecha, J. Ren, G. Sutton, T. S. Walter, D. I. Stuart and G. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 592–600 CAS.
- S. Cusack, H. Belrhali, A. Bram, M. Burghammer, A. Perrakis and C. Riekel, Nat. Struct. Mol. Biol., 1998, 5, 634–637 CAS.
- C. Riekel, NanoWorld J., 2015, 1, 71–76 Search PubMed.
- C. Kupitz, I. Grotjohann, C. E. Conrad, S. Roy-Chowdhury, R. Fromme and P. Fromme, Philos. Trans. R. Soc., B, 2014, 369, 20130316 CrossRef PubMed.
- A. Y. Lyubimov, T. D. Murray, A. Koehl, I. E. Araci, M. Uervirojnangkoorn, O. B. Zeldin, A. E. Cohen, S. M. Soltis, E. L. Baxter, A. S. Brewster, N. K. Sauter, A. T. Brunger and J. M. Berger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 928–940 CAS.
- M. Schmidt, Adv. Condens. Matter Phys., 2013, 2013, 167276 Search PubMed.
- H. N. Chapman, C. Caleman and N. Timneanu, Philos. Trans. R. Soc., B, 2014, 369, 20130313 CrossRef PubMed.
- M. S. Hunter and P. Fromme, Methods, 2011, 55, 387–404 CrossRef CAS PubMed.
- H. N. Chapman, P. Fromme, A. Barty, T. A. White, R. A. Kirian, A. Aquila, M. S. Hunter, J. Schulz, D. P. DePonte, U. Weierstall, R. B. Doak, F. R. N. C. Maia, A. V. Martin, I. Schlichting, L. Lomb, N. Coppola, R. L. Shoeman, S. W. Epp, R. Hartmann, D. Rolles, A. Rudenko, L. Foucar, N. Kimmel, G. Weidenspointner, P. Holl, M. Liang, M. Barthelmess, C. Caleman, S. Boutet, M. J. Bogan, J. Krzywinski, C. Bostedt, S. Bajt, L. Gumprecht, B. Rudek, B. Erk, C. Schmidt, A. Hömke, C. Reich, D. Pietschner, L. Strüder, G. Hauser, H. Gorke, J. Ullrich, S. Herrmann, G. Schaller, F. Schopper, H. Soltau, K.-U. Kühnel, M. Messerschmidt, J. D. Bozek, S. P. Hau-Riege, M. Frank, C. Y. Hampton, R. G. Sierra, D. Starodub, G. J. Williams, J. Hajdu, N. Timneanu, M. M. Seibert, J. Andreasson, A. Rocker, O. Jönsson, M. Svenda, S. Stern, K. Nass, R. Andritschke, C.-D. Schröter, F. Krasniqi, M. Bott, K. E. Schmidt, X. Wang, I. Grotjohann, J. M. Holton, T. R. M. Barends, R. Neutze, S. Marchesini, R. Fromme, S. Schorb, D. Rupp, M. Adolph, T. Gorkhover, I. Andersson, H. Hirsemann, G. Potdevin, H. Graafsma, B. Nilsson and J. C. H. Spence, Nature, 2011, 469, 73–77 CrossRef PubMed.
- K. Hirata, K. Shinzawa-Itoh, N. Yano, S. Takemura, K. Kato, M. Hatanaka, K. Muramoto, T. Kawahara, T. Tsukihara, E. Yamashita, K. Tono, G. Ueno, T. Hikima, H. Murakami, Y. Inubushi, M. Yabashi, T. Ishikawa, M. Yamamoto, T. Ogura, H. Sugimoto, J.-R. Shen, S. Yoshikawa and H. Ago, Nat. Methods, 2014, 11, 734–736 CrossRef CAS PubMed.
- H. Ihee, S. Rajagopal, V. Srajer, R. Pahl, S. Anderson, M. Schmidt, F. Schotte, P. A. Anfinrud, M. Wulff and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7145–7150 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Srajer and H. Ihee, Nat. Chem., 2013, 5, 212–220 CrossRef CAS PubMed.
- Z. Ren, B. Perman, V. Srajer, T. Y. Teng, C. Pradervand, D. Bourgeois, F. Schotte, T. Ursby, R. Kort, M. Wulff and K. Moffat, Biochemistry, 2001, 40, 13788–13801 CrossRef CAS PubMed.
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthur, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. van Thor, J. C. H. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Science, 2014, 346, 1242–1246 CrossRef CAS PubMed.
- S. Tripathi, V. Srajer, N. Purwar, R. Henning and M. Schmidt, Biophys. J., 2012, 102, 325–332 CrossRef CAS PubMed.
- M. G. Bowler and M. W. Bowler, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 70, 127–132 CrossRef PubMed.
- S. Boutet, L. Lomb, G. J. Williams, T. R. M. Barends, A. Aquila, R. B. Doak, U. Weierstall, D. P. DePonte, J. Steinbrener, R. L. Shoeman, M. Messerschmidt, A. Barty, T. A. White, S. Kassemeyer, R. A. Kirian, M. M. Seibert, P. A. Montanez, C. Kenney, R. Herbst, P. Hart, J. Pines, G. Haller, S. M. Gruner, H. T. Philipp, M. W. Tate, M. Hromalik, L. J. Koerner, N. van Bakel, J. Morse, W. Ghonsalves, D. Arnlund, M. J. Bogan, C. Caleman, R. Fromme, C. Y. Hampton, M. S. Hunter, L. C. Johansson, G. Katona, C. Kupitz, M. Liang, A. V. Martin, K. Nass, L. Redecke, F. Stellato, N. Timneanu, D. Wang, N. A. Zatsepin, D. Schafer, J. Defever, R. Neutze, P. Fromme, J. C. H. Spence, H. N. Chapman and I. Schlichting, Science, 2012, 337, 362–364 CrossRef CAS PubMed.
- L. C. Johansson, D. Arnlund, T. A. White, G. Katona, D. P. DePonte, U. Weierstall, R. B. Doak, R. L. Shoeman, L. Lomb, E. Malmerberg, J. Davidsson, K. Nass, M. Liang, J. Andreasson, A. Aquila, S. Bajt, M. Barthelmess, A. Barty, M. J. Bogan, C. Bostedt, J. D. Bozek, C. Caleman, R. Coffee, N. Coppola, T. Ekeberg, S. W. Epp, B. Erk, H. Fleckenstein, L. Foucar, H. Graafsma, L. Gumprecht, J. Hajdu, C. Y. Hampton, R. Hartmann, A. Hartmann, G. Hauser, H. Hirsemann, P. Holl, M. S. Hunter, S. Kassemeyer, N. Kimmel, R. A. Kirian, F. R. N. C. Maia, S. Marchesini, A. V. Martin, C. Reich, D. Rolles, B. Rudek, A. Rudenko, I. Schlichting, J. Schulz, M. M. Seibert, R. G. Sierra, H. Soltau, D. Starodub, F. Stellato, S. Stern, L. Strüder, N. Timneanu, J. Ullrich, W. Y. Wahlgren, X. Wang, G. Weidenspointner, C. Wunderer, P. Fromme, H. N. Chapman, J. C. H. Spence and R. Neutze, Nat. Methods, 2012, 9, 263–265 CrossRef CAS PubMed.
- L. C. Johansson, D. Arnlund, G. Katona, T. A. White, A. Barty, D. P. DePonte, R. L. Shoeman, C. Wickstrand, A. Sharma, G. J. Williams, A. Aquila, M. J. Bogan, C. Caleman, J. Davidsson, R. B. Doak, M. Frank, R. Fromme, L. Galli, I. Grotjohann, M. S. Hunter, S. Kassemeyer, R. A. Kirian, C. Kupitz, M. Liang, L. Lomb, E. Malmerberg, A. V. Martin, M. Messerschmidt, K. Nass, L. Redecke, M. M. Seibert, J. Sjöhamn, J. Steinbrener, F. Stellato, D. Wang, W. Y. Wahlgren, U. Weierstall, S. Westenhoff, N. A. Zatsepin, S. E. B. Boutet, J. C. H. Spence, I. Schlichting, H. N. Chapman, P. Fromme and R. Neutze, Nat. Commun., 2013, 4, 2911 Search PubMed.
- T. R. M. Barends, L. Foucar, S. Botha, R. B. Doak, R. L. Shoeman, K. Nass, J. E. Koglin, G. J. Williams, S. Boutet, M. Messerschmidt and I. Schlichting, Nature, 2013, 505, 244–247 CrossRef PubMed.
- H. Demirci, R. G. Sierra, H. Laksmono, R. L. Shoeman, S. Botha, T. R. M. Barends, K. Nass, I. Schlichting, R. B. Doak, C. Gati, G. J. Williams, S. Boutet, M. Messerschmidt, G. Jogl, A. E. Dahlberg, S. T. Gregory and M. J. Bogan, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 69, 1066–1069 CrossRef CAS PubMed.
- J. Kern, R. Alonso-Mori, R. Tran, J. Hattne, R. J. Gildea, N. Echols, C. Glockner, J. Hellmich, H. Laksmono, R. G. Sierra, B. Lassalle-Kaiser, S. Koroidov, A. Lampe, G. Han, S. Gul, D. DiFiore, D. Milathianaki, A. R. Fry, A. Miahnahri, D. W. Schafer, M. Messerschmidt, M. M. Seibert, J. E. Koglin, D. Sokaras, T.-C. Weng, J. Sellberg, M. J. Latimer, R. W. Grosse-Kunstleve, P. H. Zwart, W. E. White, P. Glatzel, P. D. Adams, M. J. Bogan, G. J. Williams, S. Boutet, J. Messinger, A. Zouni, N. K. Sauter, V. K. Yachandra, U. Bergmann and J. Yano, Science, 2013, 340, 491–495 CrossRef CAS PubMed.
- J. Kern, R. Tran, R. Alonso-Mori, S. Koroidov, N. Echols, J. Hattne, M. Ibrahim, S. Gul, H. Laksmono, R. G. Sierra, R. J. Gildea, G. Han, J. Hellmich, B. Lassalle-Kaiser, R. Chatterjee, A. S. Brewster, C. A. Stan, C. G. O. ckner, A. Lampe, D. O. R. DiFiore, D. Milathianaki, A. R. Fry, M. M. Seibert, J. E. Koglin, E. Gallo, J. Uhlig, D. Sokaras, T.-C. Weng, P. H. Zwart, D. E. Skinner, M. J. Bogan, M. Messerschmidt, P. Glatzel, G. J. Williams, S. E. B. Boutet, P. D. Adams, A. Zouni, J. Messinger, N. K. Sauter, U. Bergmann, J. Yano and V. K. Yachandra, Nat. Commun., 2014, 5, 4371 CAS.
- J. Kern, J. Hattne, R. Tran, R. Alonso-Mori, H. Laksmono, S. Gul, R. G. Sierra, J. Rehanek, A. Erko, R. Mitzner, P. Wernet, U. Bergmann, N. K. Sauter, V. Yachandra and J. Yano, Philos. Trans. R. Soc., B, 2014, 369, 20130590 CrossRef PubMed.
- L. Redecke, K. Nass, D. P. DePonte, T. A. White, D. Rehders, A. Barty, F. Stellato, M. Liang, T. R. Barends, S. Boutet, G. J. Williams, M. Messerschmidt, M. M. Seibert, A. Aquila, D. Arnlund, S. Bajt, T. Barth, M. J. Bogan, C. Caleman, T.-C. Chao, R. B. Doak, H. Fleckenstein, M. Frank, R. Fromme, L. Galli, I. Grotjohann, M. S. Hunter, L. C. Johansson, S. Kassemeyer, G. Katona, R. A. Kirian, R. Koopmann, C. Kupitz, L. Lomb, A. V. Martin, S. Mogk, R. Neutze, R. L. Shoeman, J. Steinbrener, N. Timneanu, D. Wang, U. Weierstall, N. A. Zatsepin, J. C. H. Spence, P. Fromme, I. Schlichting, M. Duszenko, C. Betzel and H. N. Chapman, Science, 2013, 339, 227–230 CrossRef CAS PubMed.
- R. Koopmann, K. Cupelli, L. Redecke, K. Nass, D. P. DePonte, T. A. White, F. Stellato, D. Rehders, M. Liang, J. Andreasson, A. Aquila, S. Bajt, M. Barthelmess, A. Barty, M. J. Bogan, C. Bostedt, S. Boutet, J. D. Bozek, C. Caleman, N. Coppola, J. Davidsson, R. B. Doak, T. Ekeberg, S. W. Epp, B. Erk, H. Fleckenstein, L. Foucar, H. Graafsma, L. Gumprecht, J. Hajdu, C. Y. Hampton, A. Hartmann, R. Hartmann, G. Hauser, H. Hirsemann, P. Holl, M. S. Hunter, S. Kassemeyer, R. A. Kirian, L. Lomb, F. R. N. C. Maia, N. Kimmel, A. V. Martin, M. Messerschmidt, C. Reich, D. Rolles, B. Rudek, A. Rudenko, I. Schlichting, J. Schulz, M. M. Seibert, R. L. Shoeman, R. G. Sierra, H. Soltau, S. Stern, L. Strüder, N. Timneanu, J. Ullrich, X. Wang, G. Weidenspointner, U. Weierstall, G. J. Williams, C. B. Wunderer, P. Fromme, J. C. H. Spence, T. Stehle, H. N. Chapman, C. Betzel and M. Duszenko, Nat. Methods, 2012, 9, 259–262 CrossRef CAS PubMed.
- X. Wang, W. Peng, J. Ren, Z. Hu, J. Xu, Z. Lou, X. Li, W. Yin, X. Shen and C. Porta, Nat. Struct. Mol. Biol., 2012, 19, 424–429 CAS.
- M. Heymann, A. Opthalage, J. L. Wierman, S. Akella, D. M. Szebenyi, S. M. Gruner and S. Fraden, IUCrJ, 2014, 1, 349–360 CrossRef CAS PubMed.
- S. L. Perry, S. Guha, A. S. Pawate, A. Bhaskarla, V. Agarwal, S. K. Nair and P. J. A. Kenis, Lab Chip, 2013, 13, 3183–3187 RSC.
- S. L. Perry, S. Guha, A. S. Pawate, R. Henning, I. Kosheleva, V. Srajer, P. J. A. Kenis and Z. Ren, J. Appl. Crystallogr., 2014, 47, 1975–1982 CrossRef CAS PubMed.
- A. S. Pawate, V. Srajer, J. Schieferstein, S. Guha, R. Henning, I. Kosheleva, M. Schmidt, Z. Ren, P. J. A. Kenis and S. L. Perry, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 823–830 CrossRef CAS PubMed.
- S. Guha, S. L. Perry, A. S. Pawate and P. J. A. Kenis, Sens. Actuators, B, 2012, 174, 1–9 CrossRef CAS PubMed.
- F. Stellato, D. Oberthur, M. Liang, R. Bean, C. Gati, O. Yefanov, A. Barty, A. Burkhardt, P. Fischer, L. Galli, R. A. Kirian, J. Meyer, S. Panneerselvam, C. H. Yoon, F. Cherevinskii, E. Speller, T. A. White, C. Betzel, A. Meents and H. N. Chapman, IUCrJ, 2014, 1, 204–212 CAS.
- A. Yonath, J. Harms, H. Hansen, A. Bashan, F. Schlunzen, I. Levin, I. Koelln, A. Tocilj, I. Agmon and M. Peretz, Acta Crystallogr., Sect. A: Found. Crystallogr., 1998, 54, 945–955 CrossRef CAS.
- E. E. Fry, J. Grimes and D. I. Stuart, Mol. Biotechnol., 1999, 12, 13–23 CrossRef CAS PubMed.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis and B. K. Kobilka, Science, 2007, 318, 1258–1265 CrossRef CAS PubMed.
- S. Cornaby, D. M. Szebenyi, D. M. Smilgies, D. J. Schuller, R. Gillilan, Q. Hao and D. H. Bilderback, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 2–11 CAS.
- Q. Liu, Z. Zhang and W. A. Hendrickson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 67, 45–59 CrossRef PubMed.
- Q. Liu, Q. Liu and W. A. Hendrickson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1314–1332 CrossRef CAS PubMed.
- Z.-J. Liu, L. Chen, D. Wu, W. Ding, H. Zhang, W. Zhou, Z.-Q. Fu and B.-C. Wang, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 544–549 CrossRef CAS PubMed.
- Q. Liu, T. Dahmane, Z.-N. Zhang, Z. Assur, J. Brasch, L. Shapiro, F. Mancia and W. A. Hendrickson, Science, 2012, 336, 1033–1037 CrossRef CAS PubMed.
- M. Schmidt, K. Pande, S. Basu and J. Tenboer, Struct. Dyn., 2015, 2, 041708 CrossRef PubMed.
- C. Kupitz, S. Basu, I. Grotjohann, R. Fromme, N. A. Zatsepin, K. N. Rendek, M. S. Hunter, R. L. Shoeman, T. A. White, D. Wang, D. James, J.-H. Yang, D. E. Cobb, B. Reeder, R. G. Sierra, H. Liu, A. Barty, A. L. Aquila, D. Deponte, R. A. Kirian, S. Bari, J. J. Bergkamp, K. R. Beyerlein, M. J. Bogan, C. Caleman, T.-C. Chao, C. E. Conrad, K. M. Davis, H. Fleckenstein, L. Galli, S. P. Hau-Riege, S. Kassemeyer, H. Laksmono, M. Liang, L. Lomb, S. Marchesini, A. V. Martin, M. Messerschmidt, D. Milathianaki, K. Nass, A. Ros, S. Roy-Chowdhury, K. Schmidt, M. Seibert, J. Steinbrener, F. Stellato, L. Yan, C. Yoon, T. A. Moore, A. L. Moore, Y. Pushkar, G. J. Williams, S. Boutet, R. B. Doak, U. Weierstall, M. Frank, H. N. Chapman, J. C. H. Spence and P. Fromme, Nature, 2014, 513, 261–265 CrossRef CAS PubMed.
- A. Aquila, M. S. Hunter, R. B. Doak, R. A. Kirian, P. Fromme, T. A. White, J. Andreasson, D. Arnlund, S. Bajt, T. R. M. Barends, M. Barthelmess, M. J. Bogan, C. Bostedt, H. Bottin, J. D. Bozek, C. Caleman, N. Coppola, J. Davidsson, D. P. DePonte, V. Elser, S. W. Epp, B. Erk, H. Fleckenstein, L. Foucar, M. Frank, R. Fromme, H. Graafsma, I. Grotjohann, L. Gumprecht, J. Hajdu, C. Y. Hampton, A. Hartmann, R. Hartmann, S. Hau-Riege, G. Hauser, H. Hirsemann, P. Holl, J. M. Holton, A. Hömke, L. Johansson, N. Kimmel, S. Kassemeyer, F. Krasniqi, K.-U. Kühnel, M. Liang, L. Lomb, E. Malmerberg, S. Marchesini, A. V. Martin, F. R. N. C. Maia, M. Messerschmidt, K. Nass, C. Reich, R. Neutze, D. Rolles, B. Rudek, A. Rudenko, I. Schlichting, C. Schmidt, K. E. Schmidt, J. Schulz, M. M. Seibert, R. L. Shoeman, R. Sierra, H. Soltau, D. Starodub, F. Stellato, S. Stern, L. Strüder, N. Timneanu, J. Ullrich, X. Wang, G. J. Williams, G. Weidenspointner, U. Weierstall, C. Wunderer, A. Barty, J. C. H. Spence and H. N. Chapman, Opt. Express, 2012, 20, 2706–2716 CrossRef CAS PubMed.
- V. Cherezov, M. A. Hanson, M. T. Griffith, M. C. Hilgart, R. Sanishvili, V. Nagarajan, S. Stepanov, R. F. Fischetti, P. Kuhn and R. C. Stevens, J. R. Soc., Interface, 2009, 6, S587–S597 CrossRef CAS PubMed.
- M. S. Hunter, B. Segelke, M. Messerschmidt, G. J. Williams, N. A. Zatsepin, A. Barty, W. H. Benner, D. B. Carlson, M. Coleman, A. Graf, S. P. Hau-Riege, T. Pardini, M. M. Seibert, J. Evans, S. Boutet and M. Frank, Sci. Rep., 2014, 4, 6026 CAS.
- A. Zarrine-Afsar, T. R. Barends, C. Mueller, M. R. Fuchs, L. Lomb, I. Schlichting and R. D. Miller, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 321–323 CAS.
- E. L. Baxter, L. Aguila, R. Alonso-Mori, C. O. Barnes, C. A. Bonagura, W. Brehmer, A. T. Brunger, G. Calero, T. T. Caradoc-Davies, R. Chatterjee, W. F. DeGrado, J. S. Fraser, M. Ibrahim, J. Kern, B. K. Kobilka, A. C. Kruse, K. M. Larsson, H. T. Lemke, A. Y. Lyubimov, A. Manglik, S. E. McPhillips, E. Norgren, S. S. Pang, S. M. Soltis, J. Song, J. Thomaston, Y. Tsai, W. I. Weis, R. A. Woldeyes, V. Yachandra, J. Yano, A. Zouni and A. E. Cohen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2016, 72, 1–10 Search PubMed.
- A. E. Cohen, S. M. Soltis, A. González, L. Aguila, R. Alonso-Mori, C. O. Barnes, E. L. Baxter, W. Brehmer, A. S. Brewster, A. T. Brunger, G. Calero, J. F. Chang, M. Chollet, P. Ehrensberger, T. L. Eriksson, Y. Feng, J. Hattne, B. Hedman, M. Hollenbeck, J. M. Holton, S. Keable, B. K. Kobilka, E. G. Kovaleva, A. C. Kruse, H. T. Lemke, G. Lin, A. Y. Lyubimov, A. Manglik, I. I. Mathews, S. E. McPhillips, S. Nelson, J. W. Peters, N. K. Sauter, C. A. Smith, J. Song, H. P. Stevenson, Y. Tsai, M. Uervirojnangkoorn, V. Vinetsky, S. Wakatsuki, W. I. Weis, O. A. Zadvornyy, O. B. Zeldin, D. Zhu and K. O. Hodgson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17122–17127 CrossRef CAS PubMed.
- T. D. Murray, A. Y. Lyubimov, C. M. Ogata, H. Vo, M. Uervirojnangkoorn, A. T. Brunger and J. M. Berger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1987–1997 CAS.
- L. M. G. Chavas, L. Gumprecht and H. N. Chapman, Struct. Dyn., 2015, 2, 041709 CrossRef CAS PubMed.
- G. K. Feld, M. Heymann, W. H. Benner, T. Pardini, C.-J. Tsai, S. Boutet, M. A. Coleman, M. S. Hunter, X. Li, M. Messerschmidt, A. Opathalage, B. Pedrini, G. J. Williams, B. A. Krantz, S. Fraden, S. Hau-Riege, J. E. Evans, B. W. Segelke and M. Frank, J. Appl. Crystallogr., 2015, 48, 1072–1079 CrossRef CAS.
- P. Roedig, I. Vartiainen, R. Duman, S. Panneerselvam, N. Stübe, O. Lorbeer, M. Warmer, G. Sutton, D. I. Stuart, E. Weckert, C. David, A. Wagner and A. Meents, Sci. Rep., 2015, 5, 10451 CrossRef CAS PubMed.
- N. Coquelle, A. S. Brewster, U. Kapp, A. Shilova, B. Weinhausen, M. Burghammer and J.-P. Colletier, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1184–1196 CAS.
- United States Patent Office, US 2013/0308756, 2013, p. 34 Search PubMed.
- R. G. Sierra, C. Gati, H. Laksmono, E. H. Dao, S. Gul, F. Fuller, J. Kern, R. Chatterjee, M. Ibrahim, A. S. Brewster, I. D. Young, T. Michels-Clark, A. Aquila, M. Liang, M. S. Hunter, J. E. Koglin, S. Boutet, E. A. Junco, B. Hayes, M. J. Bogan, C. Y. Hampton, E. V. Puglisi, N. K. Sauter, C. A. Stan, A. Zouni, J. Yano, V. K. Yachandra, S. M. Soltis, J. D. Puglisi and H. Demirci, Nat. Methods, 2016, 13, 59–62 CAS.
- C. E. Conrad, S. Basu, D. James, D. Wang, A. Schaffer, S. Roy-Chowdhury, N. A. Zatsepin, A. Aquila, J. Coe, C. Gati, M. S. Hunter, J. E. Koglin, C. Kupitz, G. Nelson, G. Subramanian, T. A. White, Y. Zhao, J. Zook, S. Boutet, V. Cherezov, J. C. H. Spence, R. Fromme, U. Weierstall and P. Fromme, IUCrJ, 2015, 2, 421–430 CAS.
- R. M. Lawrence, C. E. Conrad, N. A. Zatsepin, T. D. Grant, H. Liu, D. James, G. Nelson, G. Subramanian, A. Aquila, M. S. Hunter, M. Liang, S. Boutet, J. Coe, J. C. H. Spence, U. Weierstall, W. Liu, P. Fromme, V. Cherezov and B. G. Hogue, Struct. Dyn., 2015, 2, 041720 CrossRef PubMed.
- W. Liu, D. Wacker, C. Wang, E. Abola and V. Cherezov, Philos. Trans. R. Soc., B, 2014, 369, 20130314 CrossRef PubMed.
- U. Weierstall, D. James, C. Wang, T. A. White, D. Wang, W. Liu, J. C. H. Spence, R. B. Doak, G. Nelson, P. Fromme, R. Fromme, I. Grotjohann, C. Kupitz, N. A. Zatsepin, H. Liu, S. Basu, D. Wacker, G. W. Han, V. Katritch, S. E. B. Boutet, M. Messerschmidt, G. J. Williams, J. E. Koglin, M. M. Seibert, M. Klinker, C. Gati, R. L. Shoeman, A. Barty, H. N. Chapman, R. A. Kirian, K. R. Beyerlein, R. C. Stevens, D. Li, S. T. A. Shah, N. Howe, M. Caffrey and V. Cherezov, Nat. Commun., 2014, 5, 3309 Search PubMed.
- W. Liu, D. Wacker, C. Gati, G. W. Han, D. James, D. Wang, G. Nelson, U. Weierstall, V. Katritch, A. Barty, N. A. Zatsepin, D. Li, M. Messerschmidt, S. Boutet, G. J. Williams, J. E. Koglin, M. M. Seibert, C. Wang, S. T. A. Shah, S. Basu, R. Fromme, C. Kupitz, K. N. Rendek, I. Grotjohann, P. Fromme, R. A. Kirian, K. R. Beyerlein, T. A. White, H. N. Chapman, M. Caffrey, J. C. H. Spence, R. C. Stevens and V. Cherezov, Science, 2013, 342, 1521–1524 CrossRef CAS PubMed.
- M. Caffrey, D. Li, N. Howe and S. T. A. Shah, Philos. Trans. R. Soc., B, 2014, 369, 20130621 CrossRef PubMed.
- A. Adawy, E. Rebuffet, S. Törnroth-Horsefield, W. J. de Grip, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2013, 13, 775–781 CAS.
- C. W. Carruthers Jr, C. Gerdts, M. D. Johnson and P. Webb, PLoS One, 2013, 8, e82298 Search PubMed.
- J. M. Garcıa-Ruiz, F. Otálora, M. L. Novella, J. A. Gavira, C. Sauter and O. Vidal, J. Cryst. Growth, 2001, 232, 149–155 CrossRef.
- J. R. Helliwell and N. E. Chayen, Nature, 2007, 448, 658–659 CrossRef CAS PubMed.
- C. E. Kundrot, R. A. Judge, M. L. Pusey and E. H. Snell, Cryst. Growth Des., 2001, 1, 87–99 CAS.
- C. Sauter, F. Otálora, J. A. Gavira, O. Vidal, R. Giegé and J. M. Garcia-Ruiz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1119–1126 CAS.
- M. C. Vaney, S. Maignan, M. Ries-Kautt and A. Ducruix, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 505–517 CrossRef CAS PubMed.
- H. Wada, N. Hirota, S. Matsumoto, H. Okada, M. Kiyohara, T. Ode, M. Tanokura, A. Nakamura, J. Ohtsuka, A. Kita, N. Numoto, T. Kashiwagi and E.-I. Suzuki, Phys. Procedia, 2012, 36, 953–957 CrossRef CAS.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- S. L. Perry, G. W. Roberts, J. D. Tice, R. B. Gennis and P. J. A. Kenis, Cryst. Growth Des., 2009, 9, 2566–2569 CAS.
- M. Madou, J. Zoval, G. Jia, H. Kido, J. Kim and N. Kim, Annu. Rev. Biomed. Eng., 2006, 8, 601–628 CrossRef CAS PubMed.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC.
- J.-U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. Jia, K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129, 8825–8835 CrossRef CAS PubMed.
- E. D. Greaves and A. Manz, Lab Chip, 2005, 5, 382–391 RSC.
- M. Maeki, A. S. Pawate, K. Yamashita, M. Kawamoto, M. Tokeshi, P. J. A. Kenis and M. Miyazaki, Anal. Chem., 2015, 87, 4194–4200 CrossRef CAS PubMed.
- M. Maeki, H. Yamaguchi, M. Tokeshi and M. Miyazaki, Anal. Sci., 2016, 32, 3–9 CrossRef PubMed.
- N. Y. Chirgadze, G. Kisselman, W. Qiu, V. Romanov, C. M. Thompson, R. Lam, K. P. Battaile and E. F. Pai, in Recent Advances in Crystallography, ed. J. B. Benedict, InTech, 2012, pp. 87–96 Search PubMed.
- G. Kisselman, W. Qiu, V. Romanov, C. M. Thompson, R. Lam, K. P. Battaile, E. F. Pai and N. Y. Chirgadze, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 533–539 CrossRef CAS PubMed.
- K. Dhouib, C. Khan Malek, W. Pfleging, B. Gauthier-Manuel, R. Duffait, G. Thuillier, R. Ferrigno, L. Jacquamet, J. Ohana, J.-L. Ferrer, A. Théobald-Dietrich, R. Giegé, B. Lorber and C. Sauter, Lab Chip, 2009, 9, 1412–1421 RSC.
- V. Stojanoff, J. Jakoncic, D. A. Oren, V. Nagarajan, J. C. Navarro Poulsen, M. A. Adams-Cioaba, T. Bergfors and M. O. Sommer, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 971–975 CrossRef CAS PubMed.
- S. Emamzadah, T. J. Petty, V. De Almeida, T. Nishimura, J. Joly, J.-L. Ferrer and T. D. Halazonetis, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 913–920 CrossRef CAS PubMed.
- J. D. Ng, P. J. Clark, R. C. Stevens and P. Kuhn, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 189–197 CrossRef CAS PubMed.
- C. J. Gerdts, M. Elliott, S. Lovell, M. B. Mixon, A. J. Napuli, B. L. Staker, P. Nollert and L. Stewart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 1116–1122 CrossRef CAS PubMed.
- C. P. Steinert, J. Mueller-Dieckmann, M. Weiss, M. Roessle, R. Zengerle and P. Koltay, IEEE, 2007, pp. 561–564 Search PubMed.
- C. Sauter, K. Dhouib and B. Lorber, Cryst. Growth Des., 2007, 7, 2247–2250 CAS.
- M. J. Anderson, B. DeLaBarre, A. Raghunathan, B. O. Palsson, A. T. Brunger and S. R. Quake, Biochemistry, 2007, 46, 5722–5731 CrossRef CAS PubMed.
- C. L. Hansen, S. Classen, J. M. Berger and S. R. Quake, J. Am. Chem. Soc., 2006, 128, 3142–3143 CrossRef CAS PubMed.
- M. J. Anderson, C. L. Hansen and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16746–16751 CrossRef CAS PubMed.
- D. Axford, J. Foadi, N.-J. Hu, H. G. Choudhury, S. Iwata, K. Beis, G. Evans and Y. Alguel, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1228–1237 CAS.
- A. S. Soliman, M. Warkentin, B. Apker and R. E. Thorne, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 646–656 CrossRef CAS PubMed.
- A. le Maire, M. Gelin, S. Pochet, F. Hoh, M. Pirocchi, J. F. Guichou, J. L. Ferrer and G. Labesse, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 747–755 CrossRef CAS PubMed.
- L. Jacquamet, J. Ohana, J. Joly, F. Borel, M. Pirocchi, P. Charrault, A. Bertoni, P. Israel-Gouy, P. Carpentier, F. Kozielski, D. Blot and J.-L. Ferrer, Structure, 2004, 12, 1219–1225 CrossRef CAS PubMed.
- N. Watanabe, H. Murai and I. Tanaka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1527–1530 CrossRef.
- J. M. Holton and K. A. Frankel, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 393–408 CAS.
- J. L. Wierman, J. S. Alden, C. U. Kim, P. L. McEuen and S. M. Gruner, J. Appl. Crystallogr., 2013, 46, 1501–1507 CrossRef CAS PubMed.
- A. J. Warren, A. D. Crawshaw, J. Trincao, P. Aller, S. Alcock, I. Nistea, P. S. Salgado and G. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2079–2088 CAS.
- C. Wirtz, N. C. Berner and G. S. Duesberg, Adv. Mater. Interfaces, 2015, 2, 1500082 Search PubMed.
- N. Mohanty, M. Fahrenholtz, A. Nagaraja, D. Boyle and V. Berry, Nano Lett., 2011, 11, 1270–1275 CrossRef CAS PubMed.
- H. W. Kim, H. W. Yoon, S.-M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J.-Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R. D. Piner, L. Colombo and R. S. Ruoff, Nano Lett., 2009, 9, 4359–4363 CrossRef CAS PubMed.
- C.-C. Chang, K. W. Kolewe, Y. Li, I. Kosif, B. D. Freeman, K. R. Carter, J. D. Schiffman and T. Emrick, Adv. Mater. Interfaces, 2016, 3, 1500521 Search PubMed.
- G. Pérez-Mitta, J. S. Tuninetti, W. Knoll, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, J. Am. Chem. Soc., 2015, 137, 6011–6017 CrossRef PubMed.
- D. E. Acevedo-Cartagena, J. Zhu, E. Trabanino, E. Pentzer, T. Emrick, S. S. Nonnenmann, A. L. Briseno and R. C. Hayward, ACS Macro Lett., 2015, 4, 483–487 CrossRef CAS.
- T. Graber, S. Anderson, H. Brewer, Y. S. Chen, H. S. Cho, N. Dashdorj, R. W. Henning, I. Kosheleva, G. Macha, M. Meron, R. Pahl, Z. Ren, S. Ruan, F. Schotte, V. Srajer, P. J. Viccaro, F. Westferro, P. Anfinrud and K. Moffat, J. Synchrotron Radiat., 2011, 18, 658–670 CrossRef CAS PubMed.
- S. Bailey, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral and R. W. Grosse-Kunstleve, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
- J. H. Hubbell and S. M. Seltzer, Tables of X-ray Mass Attenuation Coefficients and Mass Energy-Absorption Coefficients from 1 keV to 20 MeV for Elements Z=1 to 92 and 48 Additional Substances of Dosimetric Interest, National Institute of Standards and Technology, 1996 Search PubMed.
- D. Chai, Z. Xie, Y. Wang, L. Liu and Y.-J. Yum, ACS Appl. Mater. Interfaces, 2014, 6, 17974–17984 CAS.
- P. S. Nunes, P. D. Ohlsson, O. Ordeig and J. P. Kutter, Microfluid. Nanofluid., 2010, 9, 145–161 CrossRef CAS.
- D. S. Khvostichenko, E. Kondrashkina, S. L. Perry, A. S. Pawate, K. Brister and P. J. A. Kenis, Analyst, 2013, 138, 5384–5395 RSC.
- D. S. Khvostichenko, J. M. Schieferstein, A. S. Pawate, P. D. Laible and P. J. A. Kenis, Cryst. Growth Des., 2014, 14, 4886–4890 CAS.
- E. Kondrashkina, D. S. Khvostichenko, S. L. Perry, J. V. Osinski, P. J. A. Kenis and K. Brister, J. Phys.: Conf. Ser., 2013, 425, 012013 CrossRef PubMed.
- M. Warkentin, J. B. Hopkins, R. Badeau, A. M. Mulichak, L. J. Keefe and R. E. Thorne, J. Synchrotron Radiat., 2013, 20, 7–13 CAS.
- Z. Ren, P. W. Y. Chan, K. Moffat, E. F. Pai, W. E. Royer, V. Srajer and X. Yang, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 946–959 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of X-ray attenuation calculations, background scattering and signal-to-noise analysis, device fabrication, permeability tests, AFM characterization, contact angle measurements, and additional crystallographic analysis and electron density maps. See DOI: 10.1039/c6lc00451b |
| This journal is © The Royal Society of Chemistry 2016 |