
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDroplet microfluidics for microbiology: techniques, applications and challenges
Tomasz S.
Kaminski†
a,
Ott
Scheler†
ab and
Piotr
Garstecki
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: garst@ichf.edu.pl
bInstitute of Molecular and Cell Biology, University of Tartu, Riia 23, 51010, Tartu, Estonia
First published on 6th May 2016
Abstract
Droplet microfluidics has rapidly emerged as one of the key technologies opening up new experimental possibilities in microbiology. The ability to generate, manipulate and monitor droplets carrying single cells or small populations of bacteria in a highly parallel and high throughput manner creates new approaches for solving problems in diagnostics and for research on bacterial evolution. This review presents applications of droplet microfluidics in various fields of microbiology: i) detection and identification of pathogens, ii) antibiotic susceptibility testing, iii) studies of microbial physiology and iv) biotechnological selection and improvement of strains. We also list the challenges in the dynamically developing field and new potential uses of droplets in microbiology.
Introduction
Recent introduction of droplet microfluidic technologies to studies in microbiology – on bacteria, yeasts, algae and bacteriophages – opened new possibilities in i) detection and identification of pathogens, ii) antibiotic susceptibility testing, iii) studies of microbial physiology and iv) biotechnological applications. Here we review the state of the art, from the classic works on encapsulation of microorganisms in droplets through the most interesting demonstrations of microfluidic technologies to the challenges and perspectives for the field.The use of droplets for studying microorganisms presents several advantages over classical methods that use bioreactors, flasks, Petri dishes and multi-well plates. Below we review the most outstanding features of cell encapsulation and handling them in droplets: (i) confinement to ultra-small volumes, (ii) ability to work with very large numbers of droplet reactors, and (iii) capability to incorporate complex liquid handling protocols in large numbers of droplets.
The first, most evident, advantage brought in by encapsulation of microorganisms in droplets is the stochastic confinement,1i.e. isolation of single cells from the bulk, each into its own tiny liquid compartment. The confinement of growth to a small volume of a droplet allows for the products of metabolism and molecules secreted by the cell or its progeny to accumulate faster than if the cell lived in a bulk culture. This sole feature opens up a number of possibilities – from early detection of cells and secreted molecules to the possibility of isolating and culturing rare individuals.
The second most important feature provided uniquely by droplet microfluidics is the ability of analysing massively large numbers (even millions) of individual droplets. This feature gives access to two new possibilities: i) examination of phenotypic and genetic variabilities at the level of cells or small populations and ii) high throughput screening, testing research hypotheses and selection over large pools of cells or populations (e.g. in biotechnology for selection of individuals with desired properties).
The third most pronounced advantage is the emerging possibility of executing iterative operations on droplets for more complex experimental protocols. Automated droplet chips are capable of controlled formation of droplets,2,3 merging them and mixing with additional reagents, splitting, sorting and incubation even over hundreds of generations and extended periods of time. This allows, for example, conducting multiple measurements on the same droplets4–6 or tracking the evolution of a population in controllably changing chemical environments.7
The combination of these features makes droplet microfluidics a uniquely attractive platform for many future technologies and for many applications in research. Although microorganisms have been cultured in droplets many years before the advent of droplet microfluidics, without the techniques developed in the recent years it was not possible to work with libraries of monodisperse liquid droplet-reactors and it was difficult to perform any operations (such as titration of an additional downstream reagent) on them. We start the review with a survey of the methods, from the first demonstrations of encapsulation of microorganisms in droplets through a short overview of the use of single phase microfluidics to the techniques of droplet microfluidics that are pivotal to the use of this technology in microbiology (Fig. 1).
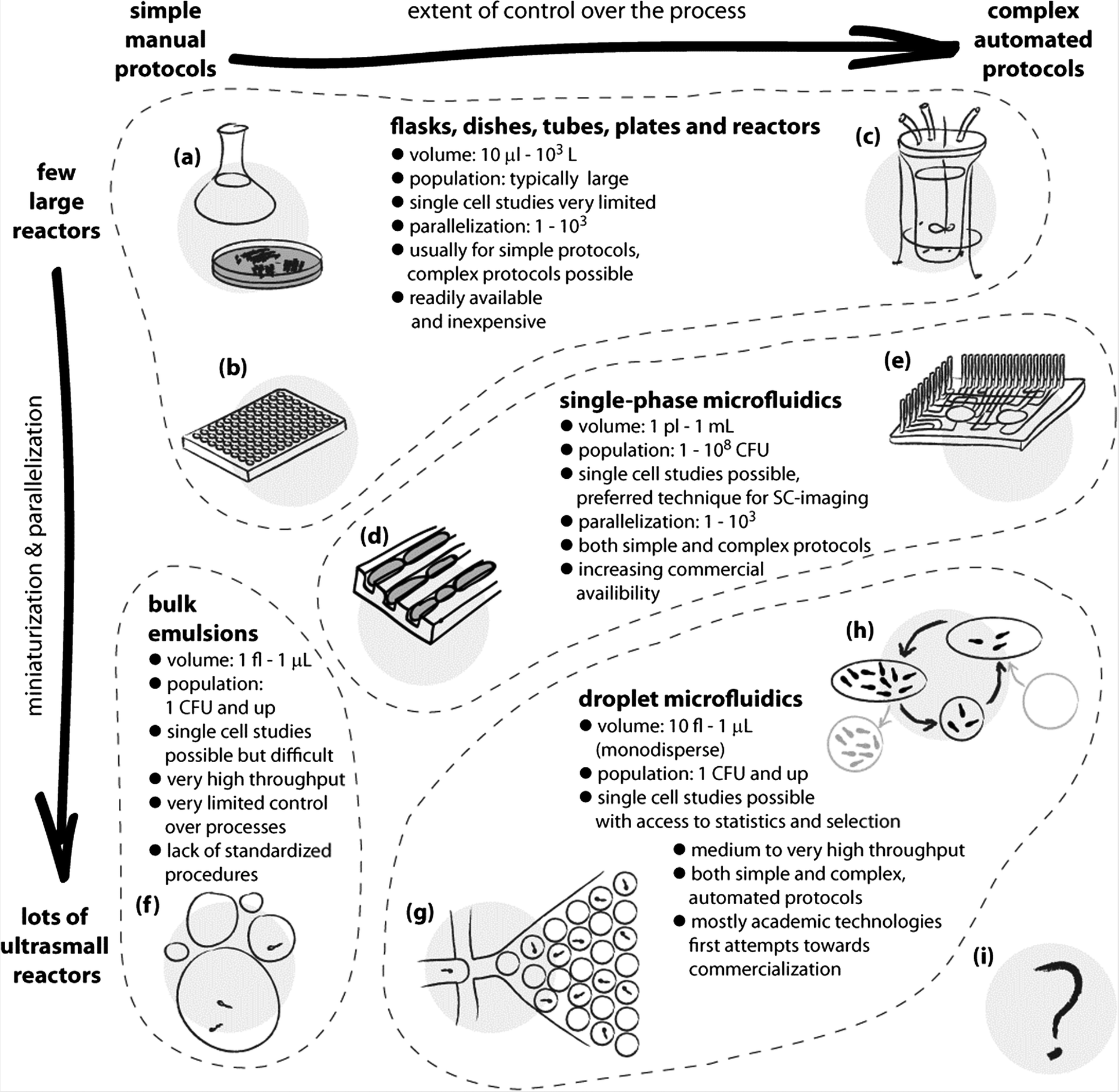 | ||
| Fig. 1 Schematic illustration of the use and applications of different technologies for culturing microorganisms. The horizontal axis spans the extent of control over the liquid handling processes and the chemical environment for growth. The vertical axis expresses the level of parallelization and miniaturization of the bioreactors. Classic methods span a wide range of techniques, from the oldest and simplest such as culture flasks and Petri dishes (a), and extend both towards miniaturization and parallelization, as in well plate cultures (b), and to complex growth protocols, as in automated bioreactors (c). The advent of microfluidics made it possible to control the chemical environments at the scale of individual cells and to observe their growth under chemical and mechanical stresses. This opened new possibilities in microbiology, such as studying the aging of bacterial cells in the ‘mother machine’ (d).8 Single phase microfluidics spans a broad range of applications, both for research and for diagnostics, and includes also highly automated systems (e) for culturing,9 yet it is not straightforward with these systems to incubate massively large numbers of single cells or small colonies in isolation. Already in the 1950s droplets have been proposed as isolated, ultra-small compartments for culturing.10 The bulk emulsion techniques (f), however, have no standardized protocols and use highly polydisperse droplets, which significantly limits their use. The advent of droplet microfluidics led to the development of technologies for controlling practically all the aspects of generation and handling of droplets. The ability to generate thousands or millions of almost ideally identical droplets allows one to controllably encapsulate individual cells (g), culture them in known and controlled environments, take full advantage of the confined volumes and concentration of products of metabolism, detect subtle differences between cells and their phenotypes and sort them. The droplet microfluidic systems span a wide range of sizes of droplet bioreactors. The techniques are also gradually developed towards automation of even highly complex protocols, such as exchange of media in droplets and long-term culturing (h).7 Further developments may expand these possibilities, for example, towards a fully automated system for directed evolution (i) supporting thousands of iterations of selection conducted on pools of billions of cultures each originating from a single encapsulated mutant. | ||
Methods for studying microbes in confinement
In this section, we briefly describe the three main approaches that enable studying microbial cells in parallel in microvolume bioreactors. The oldest method of growing microbes in bulk emulsions ensures a large number of tiny compartments that can be used for single cell studies. However, this approach does not allow for individual and continuous control of the conditions for each culture. The development of single-phase microfluidic systems and integrated valves opened the possibility of studying cells in a highly controlled manner, yet with limited throughput. Droplet microfluidics synergistically combines the advantages of the massively parallel execution of assay in emulsions with the robust control provided by microfluidic technologies.Bulk emulsions
Microdroplets were used as micro-bioreactors long before the development of droplet microfluidics. In 1954, Joshua Lederberg presented a method for the isolation and observation of single microbes in droplets deposited on a glass slide covered with a thin layer of mineral oil.10 Since then, the idea of encapsulation of single cells into droplets was adapted to multiple microbiological studies. Microorganisms were encapsulated in liquid droplets,11 agarose,12–16 polyacrylamide17 and alginate microgel droplets18 or in double emulsions.19 In order to increase the throughput of these experiments, droplets and hydrogel particles usually were not deposited on solid substrates but rather suspended in a continuous liquid medium. Agarose microgel particles brought the widest interest due to the possibility of interrogating and sorting them with flow cytometry,20,21 even if it required prior filtration of the suspension to select the appropriate size of the beads.17Before the advent of droplet microfluidics, several approaches were used to generate droplets and microgels: i) bulk homogenization12,13 ii) extrusion18 or iii) passing the liquid through a vibrating orifice.14,15 These straightforward methods were usually used in relatively simple applications, such as i) cell enumeration and metabolic activity monitoring,12 ii) antibiotic susceptibility testing,20,22 and iii) enrichment of slow-growing bacteria.23,24 Although simple, these methods continue to be interesting research tools. Bachmann et al. have recently demonstrated the use of encapsulation of Lactococcus lactis single cells in water-in-oil droplets for passive selection of the slower growing mutants with higher biomass yield – simply by isolating their nutrition from the faster-growing competitors.11 This work shows an interesting use of microdroplets and stochastic confinement in research on microbial ecology, including potential applications to studies of competition and co-operation in evolving populations or on the relationship between the environment and various metabolic strategies. In another interesting recent demonstration, Fitzsimons et al. used droplets to significantly improve a method of sequencing single bacteria.16 Prior to DNA isolation, they split a diverse microbial community into droplet-microgels, each containing a single cell. Then the cells were allowed to grow inside the microgels to obtain thousands of genetically identical cells as an input for more efficient whole genome amplification with a reduced bias.16
Despite the numerous applications of single cell encapsulation using bulk emulsification, these approaches suffer from important limitations and until recently could not be translated to other fields of microbiology. First of all, liquid and microgel droplets generated by bulk emulsification are highly polydisperse – and differences in the volume of microbioreactors may produce biases in the results of the assays. Moreover, the most commonly used analysis with FACS requires particles of a specific size, and polydisperse populations of droplets must be filtered20,21,23 prior to the introduction to a flow cytometer. Usually, the encapsulation of cells with bulk techniques requires separate instruments for each operation: formulation, incubation, detection and sorting. Manipulation over chemical composition in a single experiment is difficult. Also, some operations cannot be conducted, e.g. merging and splitting of droplets.
Single phase microfluidic systems
Single phase systems, i.e. ones that guide the aqueous solutions and suspensions of microorganisms directly in the channels without the addition of carrier oil, allow for a high degree of integration and automation of operations on the chip. Flow is usually controlled with syringe pumps and pneumatic microvalves25 or with the use of electrokinetic effects.26,27 The breakthrough momentum for continuous-flow microfluidics was the development of large-scale integration with elastomer microvalves that enabled manufacturing and handling simultaneously hundreds of individual micro-chambers on a single chip.28 Single phase systems have been successfully used for microbial analysis, including single cell assays. The most important examples include studies of bacterial antibiotic resistance29 and persistence,30,31 long-term microbial cultivation in a miniaturized chemostat,9 isolation and genetic analysis of single cells of uncultivated microbes,32,33 and other examples of single-microbe genomic studies.34 Single phase microfluidics cannot handle the massively large numbers of reactions available to droplet microfluidics; it is more difficult to work with truly tiny volumes in the pico- to nano-litre range and to provide for long-term experiments on small volumes due to the contact of the microbial colonies with solid surfaces. Still, continuous-flow microfluidics offers numerous unique advantages over droplet systems, including e.g. possibilities to execute assays involving complex microscopic imaging (e.g. on single cells) or experiments requiring the formation of specified and structured flows and gradients. The applications of continuous-flow microfluidics to the study of microbial physiology and ecology were presented in recent reviews35–37 and will not be discussed here further.Droplet microfluidics
Droplet microfluidics brings together the sample isolation and confinement offered by emulsion systems and the precise liquid handling delivered by single-phase microfluidics. Droplet microfluidic systems use two immiscible phases. One is typically organic liquid (oil) that preferentially wets the walls of the channels. The second, typically aqueous, immiscible phase is introduced into the system and is broken up into droplets. The droplets do not wet the wall of the channels and are carried along them pushed by the oil.The beginning of droplet microfluidic technology is usually associated with the first demonstrations of generation of aqueous droplets in microchannels using T-junction38 or flow-focusing geometry.39 These chip geometries bring together two immiscible liquids (e.g. oil and water) so that the aqueous phase is broken up by the biocompatible oil phase into uniformly sized droplets. In the following years the physics of droplet formation was described40–43 and modules for droplet handling were developed. The first systems demonstrated that droplets can be incubated,44–46 passively split47 and merged.48,49 Detailed reviews of the techniques for handling droplets have been extensively presented in multiple publications recently.50–53 From the point of view of usage in microbiology it is important to know that it is possible to manipulate both small (from a few microliters) and large (mL and bigger) volumes of aqueous suspensions of microorganisms. These can be split into droplets ranging in volume from pico- to microliters. The droplets can subsequently be manipulated, i.e. split, titrated with additional reagents, incubated, and analysed.
Current state of the art allows generation of droplets with frequencies of more than ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz and a coefficient of variation of droplet diameter of less than 2%.54 The interrogation and analysis of droplet fluorescence can be handled with speeds of up to ∼250000 droplets per second.55 One of the most important operations is the active sorting of droplets at high rates using electromagnetic,56 acoustic57 and mechanical forces.58 Monodisperse droplets generated in microfluidics devices can be also sorted with FACS provided that droplets are dispersed in an aqueous solution (they are in the form of double emulsions)59,60 or as microgels.61,62
000 Hz and a coefficient of variation of droplet diameter of less than 2%.54 The interrogation and analysis of droplet fluorescence can be handled with speeds of up to ∼250000 droplets per second.55 One of the most important operations is the active sorting of droplets at high rates using electromagnetic,56 acoustic57 and mechanical forces.58 Monodisperse droplets generated in microfluidics devices can be also sorted with FACS provided that droplets are dispersed in an aqueous solution (they are in the form of double emulsions)59,60 or as microgels.61,62
The possibility of single cell encapsulation and the following incubation and analysis is one of the main advantages of droplet microfluidics. In most of the systems the number of cells encapsulated in each droplet is dictated by Poisson statistics. This limits the ability to encapsulate a single cell in each droplet: typically, in order to obtain single-cell occupancy per droplet, the concentration needs to be low enough that only a small fraction of all the droplets is occupied. Interestingly, it is possible to enhance the efficiency of single-cell encapsulation in droplets.63
Using droplet microfluidics it is also possible to generate droplets with an arbitrarily predefined chemical composition. There are two main approaches for the generation of concentration gradients of chemical agents in the droplets: i) via active manipulation of flow velocities using pumps, valves and other external forces64 and ii) using passive techniques that are based on physical phenomena such as interfacial tension or gravity. A chemical gradient in droplets can be generated in active ways by varying the input flow rates of the constituent streams,65 by drawing liquid from a chromatography column66 or by formation of gradients in single-phase streams prior to droplet generation.67–69 Other methods are based on merging droplets of different volumes3 or using direct injection of portions of samples to already preformed droplets.70
In recent years, various techniques for passive operations on microdroplets have been developed, i.e. methods that do not require programmable syringe pumps or other automated instrumentation for controlling the flow of liquids on chips. The latest advances within this area include passive formation of monodisperse droplets71–73 and techniques for subsequent operations on droplets. For example, droplets can be passively merged in dedicated geometries such as pillar traps,48,74 static arrays75,76 or rails and anchors.77,78 Passive microfluidics can also be used for more complex operations such as formation of simple gradients in hydrodynamic traps.79,80 However, passive methods are of low throughput and typically produce individually addressed compositions of droplets at single Hz rates that are far too low for efficient screening, thus prompting for their implementation in autonomous devices dedicated to point-of-care testing (POCT).81
Droplet microfluidic techniques offer a very high degree of control over the generation and manipulation of emulsions and droplets.82,83 Easy access to a high number of experiments, in turn, provides for supreme statistics and better analytical results of the assays. Importantly, these features depend critically not only on the fluidic operations but also on chemistry. The droplet phase may not even partially wet the surfaces of the microchannels. Surfactants and the carrier liquid should be inert with respect to the microorganisms, often should allow for mass transport of gasses into and out of the droplets, and also should prevent transport of media, active substances and dyes between the droplets.84,85 These challenges prompted several breakthroughs in the chemical aspects of microdroplet technology – the most notable examples comprise the introduction of biologically inert fluorocarbons as a continuous phase86 and synthesis of biocompatible surfactants that provided long-term stability of emulsions.87
Importantly, if properly designed, surfactant-stabilized droplets may reduce the risk of cross-contamination between cultures – a common problem in classical methods (flasks, Petri dishes, multiwell plates) in which crucial operations are performed on cultures that are in contact with the external environment. In droplet microfluidic systems, an organic continuous phase prevents the transfer of cells between droplets and their adsorption on the walls of microfluidic channels. Lack of contact of the cultures with solid walls prevents formation of biofilms and eliminates additional steps of washing and sterilization of the devices.
In summary, droplet microfluidic systems present the following major unique properties and advantages in comparison to the use of bulk emulsification or continuous flow microfluidics: (i) ability to generate massively large numbers of highly monodisperse liquid compartments for culturing of microbes, from single cells to small populations, (ii) possibility to control and alter the chemical composition and to monitor and sort the droplet bioreactors, and (iii) capability of execution of multistep, iterative, automated long-term reaction protocols.
Below we review the applications of droplet microfluidics solely in microbiology, starting with various approaches to detection of cells and their metabolic activity in droplets, through studies on antibiotics and antibiotic resistance, on bacterial physiology and interactions and, finally, applications in biotechnology.
Applications of droplet microfluidics in microbiology
Detection and identification of microorganisms in droplets
Detection of bacterial cells and their metabolic activity is critical to most of the experimental protocols in microbiology. Different methods of detection contribute to the toolbox used in more complex studies and applications. Some of these methods have also been developed directly to serve an application in rapid identification of microbial pathogens.In 2003, Martin et al. published their pioneering work that demonstrated the detection of fluorescent proteins synthesized by bacteria that were encapsulated in 60 nL of aqueous droplets.88 They used a T-junction chip to encapsulate different bacterial strains and one yeast strain, with the droplets separated by an immiscible oil phase. Microbial growth was detected directly in a droplet by measuring the fluorescence of proteins expressed by Escherichia coli and, as a reference, off-chip by agar plate counting.88 Since then, detection of the fluorescence of expressed proteins has been widely popular and it has been used, for example, for the analysis of E. coli (Fig. 2A),89–93Salmonella typhimurium94 and Bacillus subtilis.95
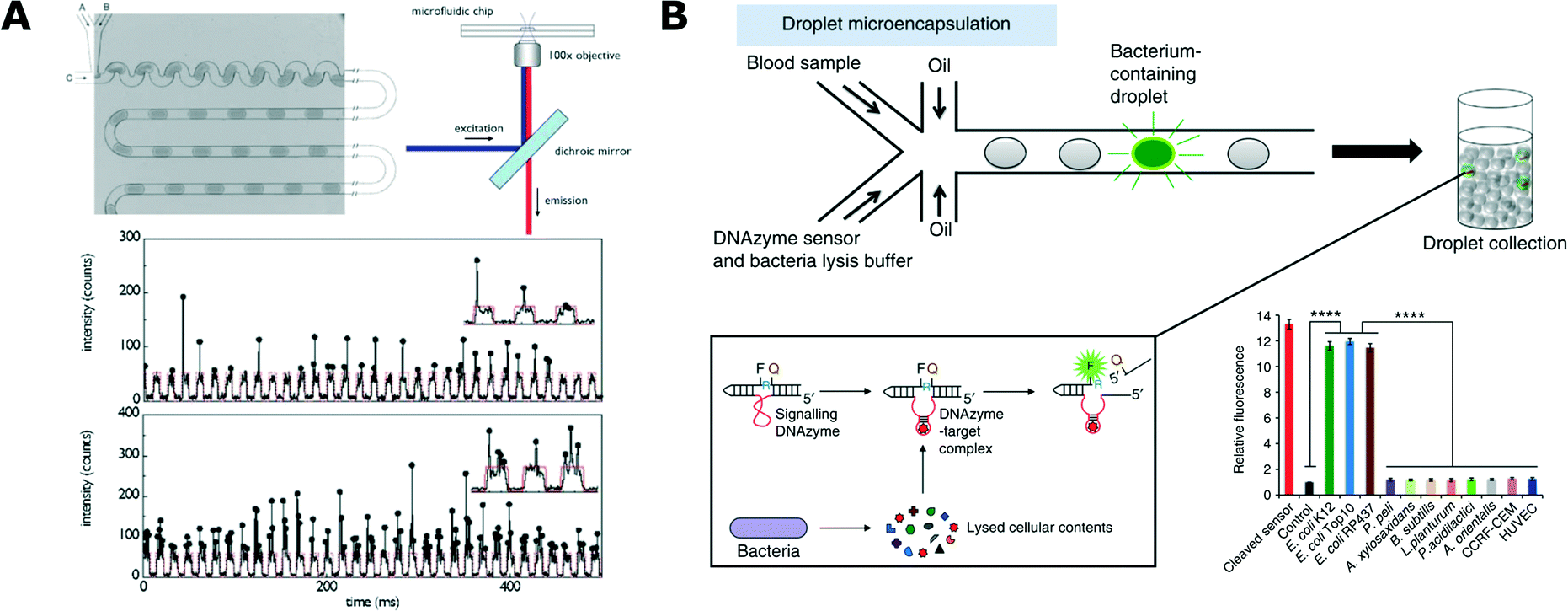 | ||
| Fig. 2 Detection and analysis of bacteria in droplets. A) Detection of bacteria expressing fluorescent protein. Microfluidic chip for generation of uniformly sized and spaced droplets (top left) together with a schematic of the fluorescence readout setup (top right). At the bottom, exemplary traces of the optical readout of the fluorescence signal. Each arch-shaped signal corresponds to a weakly fluorescent medium that forms the bulk volume of the droplet. Vertical spikes correspond to expressed fluorescence signal of bacteria. Different cell loading conditions are shown with low density on top and high density at the bottom (reproduced from ref. 89 with permission from The Royal Society of Chemistry).89 B) Specific detection of bacteria from blood using DNAzyme-based sensor. Blood samples and DNAzyme sensors are mixed prior to encapsulation in droplets (top). Bacteria produce target molecules that bind to DNAzyme, which then undergoes activation via a conformational change. Activated DNAzyme autocleaves the domain with a quencher molecule, thus resulting in the increase of fluorescence signal (box, bottom left). Specificity of the DNAzyme designed for E. coli detection against the panel of non-target organisms (bottom right) (figures adapted from ref. 108. Copyright 2014 Macmillan Publishers Limited).108 | ||
Droplets have also been demonstrated as a suitable platform for assessing the enzymatic activity of microbes. The first paper to demonstrate the monitoring of microbial enzymatic activity in droplets was presented by Huebner et al., who measured the activity of alkaline phosphatase expressed by bacteria.96 Other enzymes that have been also used as markers of microbial activity in droplet assays include β-galactosidase,46,56,97,98 β-glucuronidase,97,99 cellulase,100 glycosidase,101,102 SFP synthase.103 Droplet microfluidic assays have also been developed for estimating the alcohol production rate of microbes using enzymatic assays.104,105
Multiple fluorescent markers can be detected/monitored simultaneously in droplet cultures. Shim et al. presented a microfluidic device that was capable of trapping and storing picoliter droplets.106 The authors monitored simultaneously both the gene expression referenced by the red fluorescent protein and the enzymatic activity of alkaline phosphatase in bacteria over 20 h cultures starting from single cells.106 In other works, a similar dual parameter monitoring was carried out in droplet emulsion in order to estimate the level of dye leakage from the droplets.107,114
The presence and the activity of bacteria can also be measured indirectly by monitoring the changes in pH and oxygen level inside the droplets. Polymer micro-particles containing an immobilized pH-sensitive dye were used for determination of pH inside microdroplets. This technology allowed monitoring the metabolic activity of E. coli in 300 nL droplets over four days.109 A similar microfluidic setup was used to measure microbial growth using oxygen-sensitive fluorescent nanoparticles.110 Mahler et al. used oxygen-sensitive nanoparticles to analyse the oxygen availability and consumption in emulsions consisting of hundreds of thousands of droplets.111
Viability markers commonly used in traditional microbiology have found their way to droplet platforms as well. Boedicker et al. used resazurin to test the viability of Staphylococcus aureus in droplets.112 Resazurin is converted to fluorescent resorufin by metabolism of viable aerobic organisms. Their work demonstrated how confinement in small droplets shortens the detection time of bacteria comparing to macroscopic cultures.112 Since then, resazurin has been often used for the detection of viable bacteria.4,90,113,114 Kang et al. demonstrated the use of SYTO® 9 and propidium iodide (PI) dyes as live/dead markers in droplet microfluidics format.115 The membrane permeable SYTO® 9 stains both live and dead cells, while PI stains only dead cells with disrupted cell walls and membranes. This technology was demonstrated in droplets for measuring the fraction of dead and live E. coli cells.115
An interesting and important challenge in droplet microfluidics is to develop new schemes of detection with high specificity towards selected strains. Kang et al. proposed the use of DNAzymes for rapid detection of bacteria in clinical samples (Fig. 2B).108 DNAzymes are synthetic single-stranded DNA oligonucleotides with catalytic activities. These catalytic DNA molecules are activated when they recognize their targets (in a similar way to aptamer binding). DNAzymes are generated from a random library using in vitro evolution and their catalytic activity can be tied to the generation of a fluorescence signal when the DNAzyme meets its target. E. coli specific DNAzyme was used in a droplet assay combined with a custom developed ‘Integrated Comprehensive Droplet Digital Detection’ (IC 3D) device for specific detection and quantification of bacteria. This technology enabled absolute quantification of both stock and clinical isolates of E. coli in spiked blood within a broad range of concentrations, starting from a single cell per mL, in only 4 h.108
Lyu et al. combined droplet encapsulation with a BlaC-specific fluorogenic probe for quantitative detection of bacteria.116 BlaC is a beta-lactamase enzyme that is naturally expressed by Mycobacterium tuberculosis (Mtb) and can therefore be considered a suitable marker for specific detection of Mtb. The lowest detection limit of 10 colony forming units per milliliter was demonstrated with BlaC expressing E. coli. Quantification was shown with a dynamic range of up to 1 × 107 CFU mL−1 in million-fold higher background concentrations of other bacteria.116 Bacteria can also be detected using immunochemistry, as shown in the work of Sinn et al. who used magnetic beads coated with specific antibodies for detecting and monitoring the growth of E. coli.117 The detection was performed via measurements of the changes in magnetic bead rotation upon binding to a single bacterial cell.117
The droplet platform is also suitable for the direct analysis and detection of bacterial genes and genomes. For example, microbial cells can be sorted according to their gene sequences. Lim et al. used real-time PCR in droplets to amplify specific gene sequences from encapsulated bacteria.118 The fluorescence signal generated during PCR allowed sorting and further analysis of the droplets comprising bacteria with a desired DNA sequence.118 In another work, encapsulation in droplets enabled uniform amplification of genomes as demonstrated with single E. coli cells. This technique is highly useful as a preliminary step for the next-generation sequencing platforms.119
Stochastic confinement of microbes into small droplets allows for digital quantification (similarly to droplet digital PCR that has recently become available commercially).120–122 Assuming Poissonian distribution of bacteria,63 the fraction of positive droplets containing at least one bacteria can be used to calculate the initial microbial concentration in the sample. The positive droplets are usually detected by measuring the increase in fluorescence associated with the metabolic activity of viable bacteria.97,99 More specific detection and identification technologies (e.g. previously mentioned DNAzyme108- and BlaC116-assisted assays) can also be used for enumeration of their target cells. With these technologies the high detection sensitivities can be complemented by the ability to quantify targets over a wide dynamic range.
While the detection of fluorescence is the most common approach to monitoring and analysing the droplet content, many applications may greatly benefit from label-free schemes. The most straightforward option is to monitor the optical changes in the solution caused by the bacterial growth via bright-field microscopy. Actinobacteria detection and sorting was demonstrated using such an approach. Actinobacteria is a biotechnologically important phylum of microorganisms because many of the modern antimicrobial compounds are derived from them.123 The optical density or turbidity has also been used to monitor bacterial growth and adaptation to antibiotic stress in a micro-droplet continuous culture device.7 Recently, Liu et al. presented a system for high-throughput label-free detection of proliferating bacterial colonies in droplets using a system based on light scattering.124 This method also enabled simultaneous sorting of positive hits, e.g. mutants resistant to antibiotics. Another interesting method for detection that is unique to droplet microfluidics is the analysis of evolution of the droplet volume over time. The growth of microorganisms is usually associated with consumption of the nutrients and synthesizing new metabolites, thus changing the chemical composition of the droplet. As a result, the concentration of osmotically active solutes changes and induces mass transfer of water between droplets, resulting in respective shrinkage or swelling. The decrease in volume of a droplet can be correlated with the number of initially encapsulated cells and their ability to metabolize osmotically active components. This technology is suitable for label-free monitoring of cell metabolism at the single cell level for both yeast and bacteria125,126 and also can be used for droplet sorting based on their size.127
Antimicrobial susceptibility analysis in droplets
Boedicker et al. used droplet cultures to measure the antibiotic susceptibility of methicillin sensitive and resistant strains of S. aureus.112 In the study, different antibiotics with fixed concentration were encapsulated with both strains separately in order to test their susceptibility. Later, the minimum inhibitory concentrations (MICs) towards cefoxitin were also determined.112Antibiotic susceptibility tests are often conducted in millifluidic droplet systems that control droplets of volume ranging from ∼100 nL to a few microliters. These systems allow incubation of long sequences of droplets to monitor the growth of bacteria. Very dense gradients of antibiotic concentration in sequences of more than 1000 droplets can be generated for precise MIC determination of encapsulated bacteria (Fig. 3C).4,6,128 A similar microfluidic setup was used in another work presented by Cottinet et al., who investigated the phenotypic and genotypic changes in bacterial monocultures in starving conditions.129 After various time intervals of starvation in bulk the bacteria were encapsulated so that each droplet contained approximately a single cell. The growth of populations derived from these individual bacteria was then followed over time. Starvation resulted in different growth phenotypes and emergence of corresponding heritable genotypes that were subsequently characterized via sequencing.129
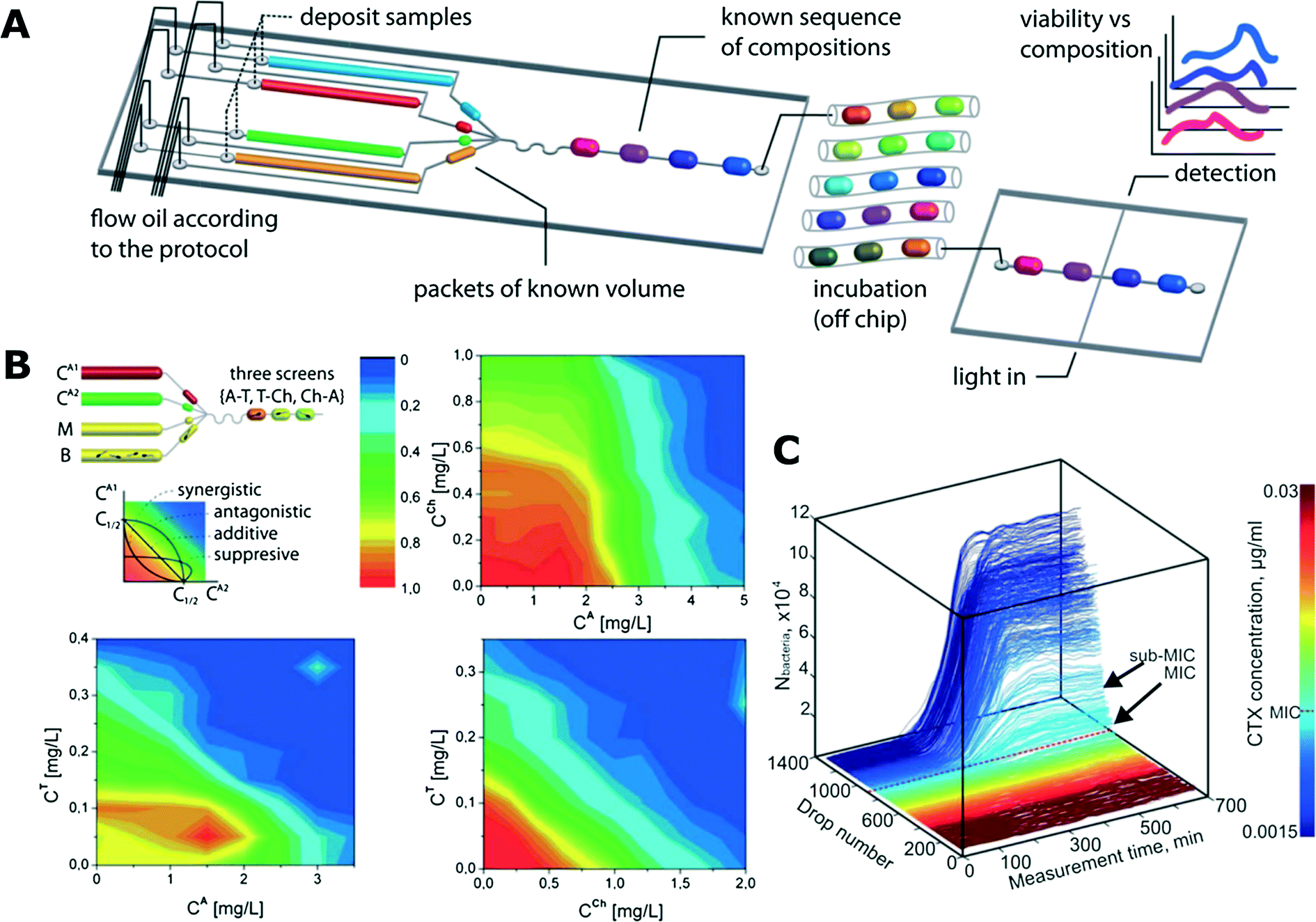 | ||
| Fig. 3 Antibiotic susceptibility studies using trains of droplets in microfluidic channels. A) A schematic of an exemplary microfluidic system for screening various antibiotic combinations. Microdroplets of controlled and predefined volume and composition (media, bacteria, reagents, fluorescence substrate resazurin) are generated and incubated in long microtubing as droplet trains. After the incubation, the fluorescence of droplets is measured (reproduced from ref. 113 with permission from The Royal Society of Chemistry).113 B) Interactions between antibiotic pairs measured in the droplet platform shown in (A). Antagonistic (top right), additive (bottom right) and suppressive (bottom left) interactions are demonstrated (reproduced from ref. 113 with permission from The Royal Society of Chemistry).113 C) Growth curves of bacterial cultures in the gradient of antibiotic concentration. The colour mapping on the right corresponds to droplet number that is linked to antibiotic concentration (reproduced from ref. 128 with permission from The Royal Society of Chemistry).128 | ||
Churski et al. used an automated microfluidic platform to screen the effect of both individual antibiotics and combinations of drugs (Fig. 3A and B).113 Automation allowed for rapid determination of MIC levels and pairwise antibiotic interaction mechanisms (synergy, antagonism and suppression).113
In a series of projects, the group of M. Köhler used automated droplet systems109 for studies on i) the toxicity of an industrial pollutant 2,4-dinitrophenol (DEP) as a single agent130 and in combination with silver and gold nanoparticles;131 ii) the antimicrobial function of a candidate peptide,130 also in combinations with nanoparticles;132 iii) the impact of caffeine and a hypertension drug to antibiotic co-administration133 and iv) the combinatorial effect of metal ions and media composition on heavy metal-tolerant bacteria.110,134,135 Other similar examples of studies that used droplet microfluidic platforms to generate antibiotic gradients to assess the antimicrobial susceptibility of bacteria can be found in ref. 115, 117 and 136.
Cho et al. demonstrated a droplet-based platform for high-throughput screening of photosensitizer activity, that is, a chemical compound that undergoes a controlled light-induced change from a non-toxic to a toxic state for bacterial cells.137
Droplet microfluidics enables investigation of antibiotic susceptibility also at the single cell level. This technique is useful for analysis of heterogeneous bacterial populations. In a study presented by Eun et al., E. coli cells were encapsulated in agarose microparticles with various concentrations of rifampicin and analyzed using flow cytometry.61 The MIC of rifampicin was determined, and spontaneous mutants that had developed resistance to the antibiotic were isolated via FACS and subsequently characterized by DNA sequencing. The subunit or RNA polymerase was confirmed to be the antibiotic target as several mutations were found there, resulting in resistance.61 In another work, Liu et al. used the droplet platform to screen for antibiotic-resistant bacteria.124 They isolated fusidic acid-resistant mutants and estimated the frequency of resistance among a population of E. coli.
Droplets can be used as micro-reactors to study the effect of antibiotics over time in quasi-stable nutrient conditions. Jakiela et al. designed a system supporting micro-droplet chemostat reactors.7 The system allowed for incubation of bacteria in droplets in chemical environments that could be changed and controlled over time in a pre-programmed fashion. Droplets with saturated culture could be split and fused with fresh medium in order to provide stable conditions for bacterial growth. They demonstrated the response and adaptation of bacterial growth to changing antibiotic concentrations over time.7
Microbial physiology and interactions between bacteria
One of the great potential uses of droplet microfluidics in microbiology is the isolation of rare or slow-growing species from complex environments. The first to show a proof of concept for this approach were Grodrian et al., who used droplet plugs to isolate different bacterial monocultures from an environmental soil sample.138 Liu et al. used a similar approach to demonstrate that slow-growing Paenibacillus curdlanolyticus can easily be separated from fast-growing E. coli that otherwise would dominate and outnumber the slow-growing species in mixed bulk culture.100 Isolated single cells were further analysed after the incubation with cellulase assay in droplets as well as using Gram staining and FISH analysis.100One of the main challenges in current microbiology is the identification of conditions that support the growth of so far “unculturable” bacteria.139 Droplet microfluidics can provide advantageous features in determining such conditions by single cell isolation and low consumption of cultivation medium that is usually supplemented with the liquid substrate obtained from the environment of the investigated organism. Ma et al. presented a modified SlipChip device for targeted cultivation of bacteria.140 In the first step, single cells from complex environmental samples were stochastically confined in an array of nanoliter droplets in order to eliminate competition during growth of various species. After the incubation, each droplet was split into two microcompartments – each on a separate slide of a SlipChip. Subsequently, one ‘half’ of the droplet can be analysed via a destructive assay (e.g. PCR), while the positive hits (viable colonies) on the corresponding slide can be inoculated for macroscale culture.140 In the accompanying study, researchers used this device for the first successful cultivation of one of the species from the Human Microbiome Project's “Most Wanted” list.141
Droplets can also provide a suitable environment to demonstrate and investigate interactions between bacteria. Park et al. used droplet microfluidics to encapsulate symbiotic pairs of bacterial strains that both lacked the ability to synthesise one specific amino acid and therefore could not grow alone (Fig. 4A).142 When encapsulated together with a complementary strain, they could compensate for each other, live long and prosper.142 Cross-kingdom communication was demonstrated by Jarosz et al. who co-encapsulated yeast and bacteria, showing that it takes just a few bacterial cells to switch yeast metabolism from using glucose to other carbon sources via prion-based transformation.143 This phenomenon is mutually beneficial as yeast cells make less alcohol that is harmful for bacteria and is useful for yeast because their growth and long-term viability is improved with more complex carbon sources.143
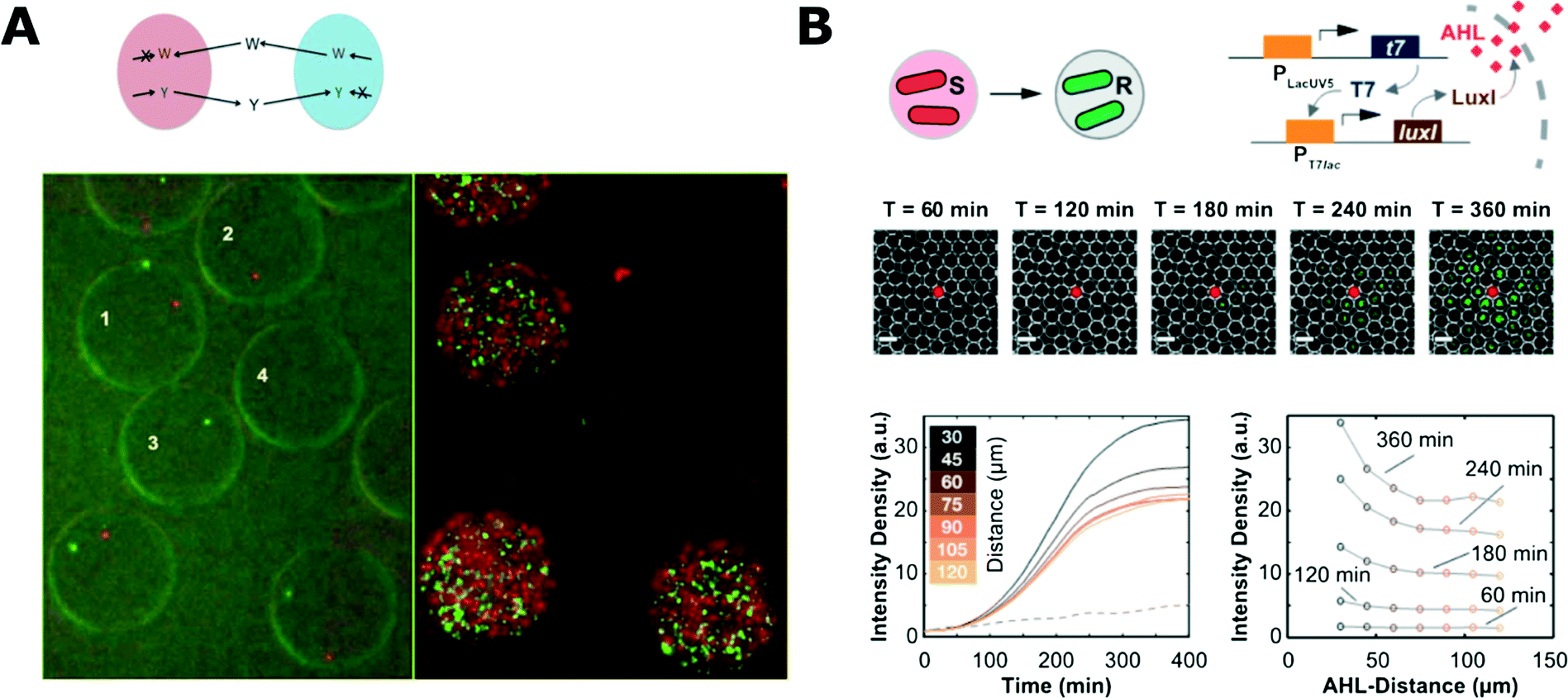 | ||
| Fig. 4 Microbial interaction studies in droplets. A) Co-encapsulation of symbiotic bacteria in droplets. Both strains are unable to synthesize one amino acid (W, tryptophan; Y, tyrosine) and they are unable to proliferate when encapsulated alone in separate droplets (droplets 2 and 3; 0 h left, 18 h right). Co-encapsulation allows bacteria to grow symbiotically (droplet 1) (figures adapted from ref. 142). B) Communication of bacteria between droplets. The system consisted of inducer sending (S) and receiving strains (R) that were encapsulated separately (top left). Inducer (N-acyl-L-homoserine lactone, AHL) diffuses from one droplet to another where it initiates expression of fluorescent reporter proteins. Fluorescence microscopy time-series showing propagation of the signal from the droplet with sender bacteria (middle). Evolution of the average fluorescence intensity for various distances from sender droplet (bottom left) and intensity profiles at different time-points (bottom right) (adapted with permission from ref. 147. Copyright 2013 American Chemical Society).147 | ||
Huang et al. used trapped droplets for long-term (10 day) monitoring of population dynamics in individual droplets93 and demonstrated the oscillating growth pattern of bacteria. After reaching a certain population density, a part of the bacterial population died that led to a sharp decrease in droplet fluorescence intensity. They also investigated and demonstrated the inoculum effect (IE) in their system. IE is a population-dependent phenomenon exhibiting resistance to intermediate antibiotic stress by colonies of high initial densities and susceptibility of populations at low densities.93 Droplets were also used as 3D microenvironments to investigate the biofilm formation of B. subtilis. A dual-labelling system was implemented so that the bacteria expressed one fluorescent protein while swimming and another protein when forming a matrix. Biofilm formation was explored upon specific interfaces in double and triple emulsions and upon negative and positive radii of droplet curvature.144
Droplet systems can also be engineered to allow for chemical signals to pass into the droplet bioreactors. Quorum sensing (QS) initiation was demonstrated with bacteria encapsulated in droplets held in traps for time-lapse analysis. The porous structure of the PDMS microfluidic chip allowed for diffusion of the QS trigger molecule into droplets from a nearby reservoir.145 Bai et al. trapped two droplets containing different bacterial strains and proved that the diffusion of a QS inducer from one droplet to another can initiate QS in a neighbouring droplet with other complementary bacterial strains.146 In a similar work, Weitz et al. used droplets with different bacterial strains and inducer molecules to show various chemical communication patterns between droplets (Fig. 4B).147 This approach enabled the demonstration of different interaction patterns: inducer reservoir(s)-to-bacteria, and bacteria-to-bacteria.147,148
Droplet microfluidic techniques for microbial biotechnology
Perhaps one of the most obvious and powerful applications of the features offered by droplet microfluidics is in high-throughput experiments for directed evolution. In their seminal work, Agresti et al. transformed members of a library of randomly mutated genes (coding horseradish peroxidase) into yeast cells and then enzymes were expressed and presented on the cell surface.149 The yeast cells were then co-encapsulated with non-fluorescent substrate into droplets. After a short incubation the droplets were sorted on a chip and as a result, selected droplets with higher fluorescence contained the most efficient variants of the enzyme. A tenfold increase in enzymatic activity was achieved after four rounds of selection. As the authors stated, droplet technology provided a 1000-fold increase in speed and a 1-million-fold reduction in cost of the assay compared to modern robotic screening platforms.149Kintses et al. used droplet microfluidics to perform directed evolution for enhanced hydrolytic activity of arylsulfatase from Pseudomonas aeruginosa (PAS).152 Three rounds of evolution led to enrichment of bacterial clones with a 6-fold increase in both enzyme activity and expression.152 In the follow-up experiment, the authors combined double emulsion droplets with FACS to demonstrate a 100000-fold enrichment of droplets containing active arylsulphatases.60 UV-mutagenesis was used to create spontaneous mutations in a library comprising 105 yeast cells in order to screen for improved secretion of industrially relevant enzymes. As a result, several mutant strains were isolated with increased α-amylase production rates (Fig. 5A).153 Whole genome sequencing revealed mutations among genes related to protein secretion as well as in genes that have not been linked to secretion before.150 In another study, Larsen et al. used a droplet platform to evolve polymerase capable of synthesizing nucleic acid polymers with unnatural backbone structures.154 Droplet microfluidic technology has also been used to perform directed evolution of other enzymes such as CotA laccase155 and phosphotriesterase.62
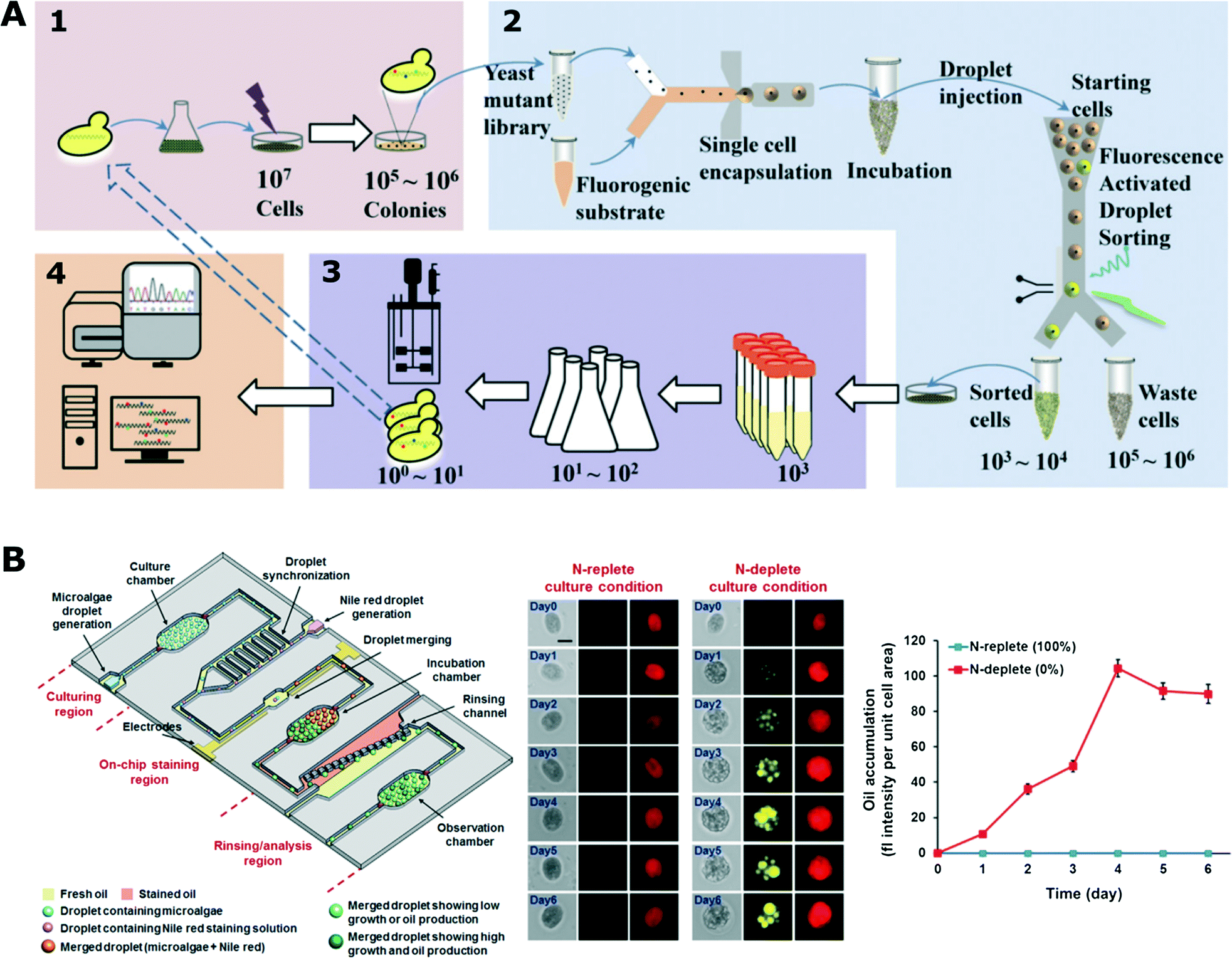 | ||
| Fig. 5 Biotechnological applications using droplet microfluidic platform. A) High-throughput screening for enhanced secretion of enzyme (α-amylase). The mutant library was generated by UV irradiation and then randomly encapsulated with fluorogenic substrate chemical so that each droplet contained a maximum of a single cell. Droplets with enhanced fluorescent signals were sorted and recovered for further analysis (adapted with permission from ref. 150. Copyright 2015 National Academy of Sciences).150 B) Droplet platform for microalgae screening for their growth and oil production. The platform comprised three functional parts (left): 1) droplet generation and incubation, 2) module for addition of fluorescent dye and 3) section for analysis and quantification of oil production rate. (Middle) Comparison between stressed (N-deplete) and non-stressed (N-replete) growth conditions for oil production (red – chlorophyll auto-fluorescence, yellow – labelled oil). (Right) Average fluorescence intensity of stained algae cells (adapted from ref. 151. Copyright 2016 John Wiley & Sons, Inc.).151 | ||
The ability to consume xylose by microorganisms is important in research on biofuels. Microdroplet technology has been used in a proof-of-principle study that demonstrated isolation of xylose-overconsuming yeast strains initially outnumbered 10 thousand times in the background of native cells.156 A similar technology was applied for the enrichment of cellulose-producing yeast cells157 and lactate-producing E. coli clones.156 In a different study, uncultured bacteria from a wheat stubble field were screened directly in droplets containing a fluorogenic cellobiohydrolase substrate for their ability to hydrolyse cellulosic biomass. Remarkably, sorting of droplets depending on cellobiohydrolase activity resulted in the selection of a bacterial population with respectively 17- and 7-fold higher cellobiohydrolase and endogluconase activity.158 Jiang et al. developed a microfluidic device for the formation and streaking of over a thousand droplets on a Petri dish.159 The microfluidic streak plate (MSP) approach enabled the analysis of microbial consortia from soil samples and led to the isolation and discovery of species with a high efficiency of degradation of polycyclic aromatic hydrocarbons (PAHs) or fluoranthene.159 Dong et al. presented a system for enrichment of chemotactic bacteria from a mixture of microbes.160 First, chemotaxis assay was performed in single-phase parallel-flow wide channel; next, separated single bacterial cells were encapsulated in droplets for further cultivation.160 Jang et al. presented a trapped droplet array to enrich the sample for bacteria that produce higher levels of L-tryptophan.161
In another work, a metagenomic DNA library was cloned into a vector that was expressed in E. coli hosts while encapsulated in droplets together with the respective enzyme substrates. The droplet platform enabled the discovery of new enzymes with catalytic activities (hydrolases for sulfate monoesters and phosphotriesters) that could not have been predicted using genomic tools.162 A similar metagenomic screening approach was also used for finding new lipolytic enzymes.163 Also, a metagenomic library was used for the production of antimicrobial compounds. An E. coli library was co-encapsulated with pathogenic S. aureus to screen for compounds that are lytic towards the pathogen. A SyTox® Orange viability probe was used to label the killed S. aureus as it stains only dead cells. A proof-of-principle demonstration was carried out with a metagenomic library from bacteria that naturally compete with S. aureus. Metagenomic inserts carrying bacteriolytic genes were successfully detected with the described platform.164
Microbes are often encapsulated within microgel particles or droplets coated with extra protective layers in order to enhance their stability over time and during handling of emulsions. One of the common applications of droplet microfluidic systems is encapsulation of the microbe of interest in alginate165,166 or agarose61,164,167microgel and also coating them with additional protective polyelectrolyte layers.62,167 Alternatively, droplet microgels can be used as a substrate to generate cage-like organic membrane structures168 or yeastosome – structures covered with single layers of cells that can be used for tissue engineering purposes.169
Algae have been increasingly investigated as potential bio-producers in biotechnology. Not surprisingly, droplet microfluidic platforms have also been applied for research on algae. Pan et al. encapsulated individual cells of different algae species in droplets and monitored their growth in various conditions over a period of up to 10 days.170 In a different experiment, the growth kinetics of single algae cells was monitored for a longer 17 day time period in a droplet array under changing light conditions mimicking natural environment.171 Lagus and Edd induced gametogenesis in algae cells and encapsulated different mating types together in droplets.172 Cells were ordered in microfluidic channels using inertial effects prior to droplet generation to increase the co-encapsulation rate. They showed that gametes retained their mating ability as they fused to form zygospore. Algae were able to continue their vegetative growth once the droplets were deposited on a standard agar plate; next, the emulsion was broken and the content of the droplet was released.172 Algae cells from an isogenic population demonstrate cell-to-cell heterogeneity in growth dynamics as shown by Damodaran et al. Most of the synchronized cells show fast growth, while there is always a sub-population of slow-growing cells. This difference in growth rates seemed to be a stochastic phenomenon with a currently unknown mechanism.5 In another work, a droplet platform was used to encapsulate algae cells to monitor their growth and oil production rate (Fig. 5B) under a range of culture conditions (nitrogen-limited and nitrogen-rich).151
Uniform amplification of a mixture of phage clones is crucial for selection of peptides and proteins presented on the phage capsid (technology known as phage display). Derda et al. used droplet microfluidics to co-encapsulate phages with the bacterial host for phage amplification and then separate individuals exhibiting a slower rate of amplification from the background of strains that amplify faster but do not display proteins of interest.173 Optimized inoculation and amplification conditions as well as library retrieval technology were described in a follow-up work.174 Bacteriophages can also be used for specific detection and enumeration of their host bacteria. Encapsulation of reporter phages in droplets with their host helps to decrease the detection time comparing to conventional agar plate or bulk culturing. Detection can be done by measurement of fluorescence175 or using label-free optofluidic technology.176
Outstanding challenges and opportunities
As we reviewed above, the ability to encapsulate microorganisms in droplets of controlled chemical composition and to screen large numbers of such liquid bioreactors brings in new possibilities in diagnostics and research in microbiology across multiple fields – from pharma to biotechnology.As the reactors are liquid and bounded by a liquid–liquid interface, they are quite sensitive to mechanical perturbation and to the mass transfer across the interface. This poses a number of technical challenges associated with their making, manipulation and execution of experimental protocols. The challenge is in achieving complementarity of the chemistry of the process with the mechanics of the droplet handling protocols. The bio-processes inside the droplets may require a specific chemistry or may themselves generate chemistries that in turn can alter the surface tension or mass transfer rates across the interface or in the liquids, or other properties that may be crucial either to the mechanics of handling the droplets or to the physico-chemistry of the assays.
We list below the issues of prevention of wetting and cross-contamination, monitoring and controlling the level of oxygen in the droplets, and providing stable and reliable detection chemistries. These form the standard checklist for experimenters. Advancements in each of these areas poses a good chance of broadening the use of droplet systems in microbiology. We also point out two subjects belonging to physical microfluidic engineering, i.e. the challenge to widen the portfolio of automated techniques for manipulation droplets in situ in the microfluidic chips. The second great challenge is in making the pivotal techniques (such as generation of droplets, detection, or sorting) more easily available to users having no background in microfluidics. Finally, we list a number of exciting opportunities for the field.
Wetting and cross-contamination
As the droplets are aqueous and the continuous liquids are immiscible organic liquids (either hydrocarbon, silicone or fluorinated oils) a default intuition is that microorganisms will not be able to pass between the droplets. Cross-contamination between the droplets may occur in three different ways: i) generation of micelles or small droplets sheared off from large plugs and picked up by others, ii) direct contact and coalescence, and iii) wetting of the walls of the channels by the droplets and leaving residues on the walls that are picked up by successive drops.Exchange of material between the droplets via the continuous liquid has gained some attention in the past few years. The recent hypothesis is that the transfer occurs via micellar or vesicular structures and not via larger droplets sheared off from the plugs.178 This effect must be checked in all the experiments that either use multiple droplets or manipulate them over extended periods of time.
The problem of direct coalescence can be solved with the appropriate choice of surfactants or with microfluidic engineering, such as controlling the distances between the droplets,7 or introduction of additional immiscible spacers between the droplets.5,128
Perhaps the most challenging is the problem of wetting of the droplet phase on the walls of the channels. It has been practically solved in the case of generation of droplets in the most common combination of PDMS chip, fluorinated oil and off-chip storage of droplets as dense emulsion in tubes155,179 or in syringes.150,158,162 Efficient methods of PDMS modifications using fluorosilanes (also in commercial mixtures such as Aquapel or HFE-1720) were developed during the past several years.179–181 However, the high elasticity of PDMS makes it difficult to precisely control the flow of long sequences of droplets due to slow flow caused by relaxation of pressure. Recent approaches proposed the use of chips and tubing made of stiff materials such as polycarbonate,7,105,113 poly(tetrafluoroethylene) (PTFE, Teflon)100,112 or fluorinated ethylene propylene (FEP).5,6 New methods of fabrication and modification of stiff polymers should be developed for systems for long-term culturing of bacteria without wetting or excessive emulsification. For instance, polycarbonate was modified hydrophobically for use with hydrocarbon oils,182 but there is a need for both stable hydrophobic and fluorophilic modification. Chips made of stiff polymers are usually fabricated via processes (milling, moulding) that introduce roughness of the surface that can lead to break-up of droplets via pinning and emulsification by shading small drops from a receding contact line. Other tricks can also help with the problem. For example, Bibette's group proposed to keep the droplets moving at all times in order to preserve a dynamic film of the continuous liquid between the droplets and the tubing.4,5 In general, new materials, new microfluidic modules and methods that would warrant the ability to handle large numbers of droplets in tubes or channels over extended periods of time are needed for further development of the field.
Controlling the concentration of oxygen and other gases in droplet bioreactors
The concentration of oxygen is one of the crucial parameters in most microbial cultures.183,184 It is important to balance between aerobic, micro-aerobic and anaerobic conditions depending on the cultured species. Control of the concentration of gases in droplet microfluidic systems is still challenging. For example, maintaining stable oxygen concentration throughout the duration of the whole assay was not achieved so far. The availability of gases can be partially controlled by the selection of liquids for the continuous phase. Gases are hardly soluble in mineral oils that support only anaerobic or micro-aerobic growth of bacteria in droplets.7,113 In contrast, fluorinated liquids (especially FC-40 and HFE-7500) present a large solubility of oxygen and support aerobic growth.185,186 A high initial concentration of oxygen in the continuous phase can be achieved by enriching of the oil before the reaction137 or by using a gas-permeable tubing that guides the oil from a container to the chip, through a controlled oxygen atmosphere.187 Still, some bacteria require a continuous supply of oxygen during their growth. Mahler et al. presented a device for dynamic incubation of organisms in droplets with a continuous exchange of fluorinated continuous liquid (Fig. 6A).111 Their strategy resulted in increased biomass yields comparable to that of classical systems such as microbial cultures in shaking flasks and multi-well plates.111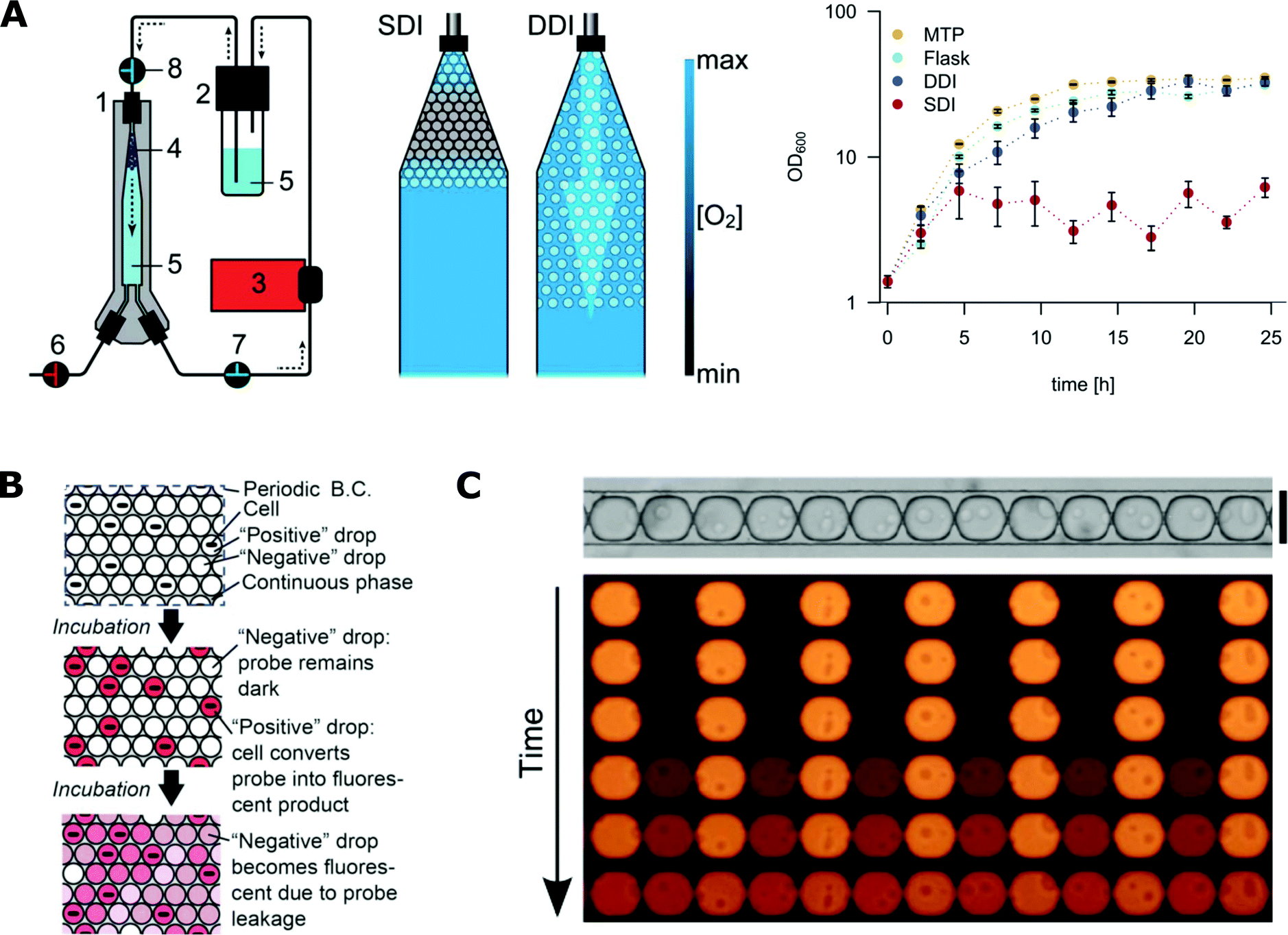 | ||
| Fig. 6 Challenges with microfluidics for microbiological applications. A) Oxygen availability during incubation in microfluidic droplets. On the left, droplet incubation setup with different oxygen availability rates during static (SDI) and dynamic (DDI) incubation. The numbers stand for 1) droplet incubator, 2) oil reservoir, 3) peristaltic pump, 4) droplet population, 5) perfluorinated oil, 6–8) valves. On the right, comparison of E. coli growth kinetics in cultures grown in 96-well plates (MTP), shaking flask, SDI and DDI (adapted from ref. 111 with permission from The Royal Society of Chemistry).111 B) The scheme for the dye leakage model in a 2D array of drops (B.C. stands for boundary condition). A positive drop contains one cell and a negative drop contains no cells. The drops are immersed in and separated by a continuous phase. Cells in positive drops convert the probes from the dark state into a fluorescent state (denoted by the pink colour), increasing the fluorescence intensity of positive drops. The fluorescent probe can also leak into the neighbouring negative drops and increase their intensity (adapted from ref. 177 with permission from The Royal Society of Chemistry).177 C) Experimental studies of the leakage process of fluorescent dye into neighbouring droplets that are assembled in traps in a row. (Figures adapted from ref. 178. Copyright 2016 Macmillan Publishers Limited).178 | ||
Similarly to oxygen, the concentration of other gases like methane has been investigated in the droplet platform. The fluorinated oil phase surrounding the water/media droplets has also a higher solubility for methane. That makes the droplet platform a useful system for conducting methanotrophic fermentation.188
The current challenge is to implement a strategy of continuous phase exchange to millifluidic systems6,7,128 in which droplets are transported in networks of long channels. The development of an effective strategy for controlling the concentration of oxygen (or other gases) during culturing of microorganisms in droplets is an outstanding problem, and solving it would have a high impact on the field.
Detection and analysis of microbes in droplets
One area where strong improvements are needed is the detection and analysis of cells and their metabolism in droplets. Current state-of-the art label-free technologies like measurement of turbidity,7 analysis of differences in light scattering124 or bright-field microscopy with image processing123 require a large number of bacteria or long incubation, respectively, to reach detectable levels suitable for analysis. Possible label-free technologies that have been suggested include optofluidic imaging of changes in refractive index,176 magnetic bead rotation-assisted detection117 or analysis of population dynamics with an intrinsic magnetic field.189Most of the research regarding droplet microfluidics in microbiology has been conducted using fluorescent labelling because this technology is accessible and already well established in conventional experiments in microbiology. Still, fluorescent labelling has drawbacks. It is comfortable and popular to use fluorescent protein expressing bacteria,96,145–147 yet this approach is limited to the specifically engineered strains and cannot be easily translated to other microorganisms.
Marker dyes for microbial metabolism or enzymatic activity can be used for a wider range of species and strains. A common problem associated with their use is the possible “leakage” of fluorescent signal molecules from droplet to carrier oil and to neighbouring droplets (Fig. 6B and C).99,107,177,190,191 The main mechanisms of leakage can be diffusion into oil or formation of micellar or vesicular structures that act as carriers between droplets.107,178,192 Decrease and averaging of the measured signal may compromise the sensitivity and resolution of analysis of the reactions occurring inside the droplets. Suggested solutions include replacement of the most common HFE-7500 liquid with-FC-40 fluorocarbon oil,190 decreasing the surfactant concentration,107,192 and addition of bovine serum albumin (BSA)107 or sugar additives to the water phase.193 Also, less leaky dyes like coumarin-dyes101 or dodecylresorufin114,191 can be used as reporter dyes. The most interesting and so far the most promising approach is using nanoparticles for the stabilization of droplets and prevention of leakage of hydrophobic molecules.90 Recently this method has been supplemented with coating of the nanoparticles with bovine serum albumin to increase the compatibility of the interface with enzymes.194
An interesting area for further developments is finding new fluorescent dyes and metabolic markers that are stably maintained in droplets. Alternatively, there could be advancements in complementary technologies that prevent such leakage of small molecules from droplets. We also foresee a surge in labelling technologies that are designed with a specific target in mind like DNAzymes108 or species/strain specific enzymes similar to BlaC.116 Definitely, the development of a sensitive label-free detection technology for small (nano-liter and smaller) droplet reactors, similar to the large-scale conventional OD readouts, would provide a great new thrust for the droplet culturing systems.
Engineering challenges in automation and simplification of droplet microfluidic systems
There are two major directions for further development of microfluidic systems for handling droplet bioreactors for experimentation in microbiology – further automation of complex liquid handling protocols and increasing the accessibility of the basic technologies for users without expertise in microfluidics.Control of the concentration of reagents during the assay is a very important aspect in microbial cultures. Microfluidic systems provide many approaches for the generation of sets of droplets with arbitrary combinations of reagents.80,113,195 Still, manipulating the concentration of reagents during the assay is very challenging. Usually this task requires multiple valves and optical feedback loops.7 The introduction of hydrodynamic and passive geometries can reduce the number of valves needed.79,94,161 Long-term cultures with on-line monitoring of growth typically use microliter scale droplets containing up to tens of thousands of microbes.7,128 Although a technique for splitting microliter plugs into libraries of nanoliter droplets has been demonstrated,196 there is still a great outstanding challenge of integrating long-term culturing in controlled chemical environments and monitoring of the resulting single-cell distributions of growth and other phenotypic factors.
The second important challenge is to make the droplet microfluidic technology more widely available for the microbiologists and research communities in general. Currently the technology is mostly used by engineering laboratories that design the microfluidic systems and “early adopters” in life sciences who collaborate with these engineering groups.197 In order to be truly successful and popular any novel and useful technology has to be widely available and cost-effective.
The situation is promising for droplet technologies as the first commercial devices are already available for nucleic acid research (droplet digital PCR). There is also a number of companies that already provide basic droplet microfluidic tools and custom chip development options that are suitable for research on microbiology as well as in life sciences in general. We believe that the most pivotal technologies could be adopted to simplistic and inexpensive formats. For example, the recent revival of interest in passive techniques for generation of droplets71,73,198 should lead to the development of chips that allow the transformation of a suspension of bacteria into a monodisperse emulsion simply by injecting it with an automatic pipette. Supplementing this with inexpensive optical detectors and sorters could boost the use of emulsion technologies in microbiology.
Opportunities
The ability to encapsulate single cells or small colonies in massively large numbers of bioreactors for growth, monitoring and selection provides numerous opportunities in tackling the most important problems in fundamental and applied microbiology.Currently, the most obvious and important problem is the massive build-up of resistance to antibiotics that already presents a global threat to public health. The imminent threat of transiting into the “post-antibiotic era” – in which common infections and minor injuries can kill – is calling for immediate and drastic measures.199 In 2015 the World Health Organization endorsed a global action plan on antimicrobial resistance to combat this grim apocalyptic vision.200 Among other suggestions, the plan calls for the development of new technologies for tackling the full extent of the problem.
Improved and widely accessible methods for screening of resistance to antibiotics are needed. Droplet technologies can certainly be used both for simple manual antibiotic susceptibility tests81 and for automated analyses. While the use of cocktails of antibiotics (or antibiotics and adjuvants) is one of the primary strategies for tackling bacteria,201–203 in the current state of the art there are practically no standardized methods for rapid evaluation of susceptibility to combinations. We believe that both passive and automated droplet techniques could offer solutions allowing the establishment of sensitivity to multiple combinations (e.g. 10 × 10) of drugs, each combination tested across a range of concentrations. Providing access to such technologies would surely have a major impact on tracking the epidemiology of resistance and understanding the evolution and patterns in spreading the mechanisms of resistance. It could also aid directly the clinical practices in choosing effective combinations in critical infections.
Recent studies have demonstrated that use of a specific combination of drugs can invert or modulate the selective advantage of resistant bacteria competing with sensitive strains and reverse the evolution of antibiotic resistance.201,204–206 The automated droplet microfluidic systems could also be engineered for massive in vitro screening of combinations of drugs for use in designing new combination treatments. The technological need in this field is truly outstanding and the ability to culture bacteria constitutes a technical bottleneck in research. Providing a technology that would allow screening, e.g. double or triple combinations of a library of a hundred drugs (i.e. 1002 or 1003 times a matrix of dilutions for each combination) or e.g. a pool of 30 drugs in combination, each mixed with one of a 10 thousand member library of candidate adjuvants per day would pose a high chance of finding new, unexpected, combinations of drugs and probably also elucidate the biomolecular mechanisms behind resistance.207–209 Also, the long-term culturing systems7 may be engineered to manipulate i) the chemical environment in time in each of hundreds of colonies, ii) change this environment in response to the observed growth patterns, and iii) transfer bacteria between the droplets. All these features provide unique opportunities to i) uncover the dynamics of development of resistance in the space of chemical composition of media and ii) program and test various ‘algorithms’ of administration of drugs aimed at promoting the desired evolutionary traits.
Classical diagnostics realized by in vitro culturing can be assisted or sometimes replaced by modern methods of detection of bacteria and evaluation of their antibiotic resistance using genetic analysis such as real-time PCR.210,211 Also in this field, droplet microfluidics offers a great opportunity by implementation of digital PCR for identification and precise quantification of bacteria and sample preparation for genome amplification. Digital PCR offers multiple advantages over real-time PCR and the technology is starting to find a position in analytical microbiology.212–214 We envisage that the ddPCR technique can be implemented as a module in microfluidic systems for studies of bacterial physiology and resistance to antibiotics. Detection of pathogens using ddPCR can also be improved in order to avoid steps of DNA isolation. Direct lysis of single bacteria followed by PCR in droplets has already been presented for Gram-negative bacteria;118 however, this task might be more challenging for G-positive bacteria cells that are much more stable against thermal lysis. Application of enzymatic lysis might disturb emulsion stability due to the presence of surfactants in lysis buffer. Recent methods using bulk emulsification proposed lysis in gelled droplets in the intermediate step before single-cell PCR amplification.17 Alternatively, cells can be lysed by exposing them to an electric field immediately prior to encapsulation in droplets, as was demonstrated for E. coli bacteria in a recent study presented by de Lange et al.102 However, that method still requires the addition of lytic enzyme (lysozyme) to obtain sufficient efficiency of lysis. The development of new efficient methods for the isolation of genetic material from single cells encapsulated in droplets should bring in new possibilities both for diagnostics and for research.
There are also fascinating opportunities in biotechnology. Droplet microfluidic systems have already been demonstrated as a powerful tool in directed evolution.60,62,149,152 These technologies are oriented towards screening of large (106–109) pools of mutants, yet do not support automated iteration of the process. We envision that further development of automated droplet systems should allow the construction of systems that i) incubate multiple mutants separately, ii) apply arbitrary selection criteria onto the pool, iii) select positives, collect them and re-grow, iterating the whole process automatically. The ability to freely tune the number of iterations and thresholds for selection could open the way to new understanding of the evolutionary trajectories215–217 and provide a new quality in applied research in biotechnology.
Finally, the increasing development of droplet microfluidic tools would definitely increase the possibilities of understanding the microbiome and elucidating the interactions that compose it. Droplet microfluidics might be useful for investigation of uncultured organisms – so-called microbial dark matter139 – in order to optimize the set of nutrients enabling growth of these species and their further extensive off-chip characterization.141,159 Unified Microbiome Initiative seeks to develop and apply new tools that enable understanding of the microbiomes of humans and other animals, plants, the earth, the oceans, and the atmosphere. The opportunities, technical needs, and potential approaches for this initiative were recently extensively reviewed.218,219 We expect that the ability to encapsulate small colonies and the use of stochastic confinement will have a significant influence in meta-genome screening for new antimicrobials and other bioactive agents.
Droplet microfluidics could also be a highly informative technology in the dynamically emerging new field of single-cell immunology.220,221 Droplets can be used for massively parallel studies of infection and interactions between pathogenic bacteria and eukaryotic cells, so far demonstrated at single cell level using classical methods.222 Microdroplet technology might combine high-throughput and parallelization with access to single cell profiles of transcriptome223,224 and possibly also for proteome.225
We foresee that the awareness and availability of droplet technology has a chance to rapidly increase in the near future mostly via collaborations between engineering and biology oriented laboratories. In the mid- and long-term future we expect droplet microfluidics to become a common technology in microbiology due to the availability of commercial standardized devices for droplet generation, manipulation and analysis. We also hope that the exciting new toolset may provide breakthroughs in tackling the most important challenges in microbiology.
Acknowledgements
The writing of this review was supported by the European Research Council Starting Grant 279647 and National Science Centre funding based on decision number DEC-2014/12/W/NZ6/00454 (Symfonia). This work was also supported by the Ministry of Science and Higher Education through the scholarship for outstanding young researchers, agreement 0722/E-64/STYP/10/295 (to T. S. K.), by the Estonian Research Council grant PUTJD589 (to O. S.), Foundation for Polish Science MISTRZ fellowship (to P. G.). T. S. K. obtained the funds for the preparation of the doctoral dissertation from the National Centre for Science within the scholarship on the basis of the decision number DEC-2014/12/T/ST4/00649. The authors would also like to thank Dr. Liis Andresen (Umea University, Sweden), Dr. Jaanis Juhanson (Swedish University of Agricultural Sciences, Uppsala, Sweden), Dr. Izabela Sitkiewicz (National Medicines Institute, Poland) and Dr. David Wareham (Queen Mary, University of London, UK) for carefully reading the manuscript and for providing inspiring suggestions.Notes and references
- M. E. Vincent, W. Liu, E. B. Haney and R. F. Ismagilov, Chem. Soc. Rev., 2010, 39, 974–984 RSC.
- Z. Z. Chong, S. H. Tan, A. M. Ganan-Calvo, S. B. Tor, N. H. Loh and N.-T. Nguyen, Lab Chip, 2015, 16, 35–58 RSC.
- K. Churski, P. Korczyk and P. Garstecki, Lab Chip, 2010, 10, 816–818 RSC.
- L. Boitard, D. Cottinet, N. Bremond, J. Baudry and J. Bibette, Eng. Life Sci., 2015, 15, 318–326 CrossRef CAS.
- S. P. Damodaran, S. Eberhard, L. Boitard, J. G. Rodriguez, Y. Wang, N. Bremond, J. Baudry, J. Bibette and F.-A. Wollman, PLoS One, 2015, 10, e0118987 Search PubMed.
- L. Jiang, L. Boitard, P. Broyer, A.-C. Chareire, P. Bourne-Branchu, P. Mahé, M. Tournoud, C. Franceschi, G. Zambardi, J. Baudry and J. Bibette, Eur. J. Clin. Microbiol. Infect. Dis., 2016, 35, 415–422 CrossRef CAS PubMed.
- S. Jakiela, T. S. Kaminski, O. Cybulski, D. B. Weibel and P. Garstecki, Angew. Chem., Int. Ed., 2013, 52, 8908–8911 CrossRef CAS PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- F. K. Balagaddé, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef PubMed.
- J. Lederberg, J. Bacteriol., 1954, 68, 258–259 CAS.
- H. Bachmann, M. Fischlechner, I. Rabbers, N. Barfa, F. Branco dos Santos, D. Molenaar and B. Teusink, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14302–14307 CrossRef CAS PubMed.
- J. C. Weaver, G. B. Williams, A. Klibanov and A. L. Demain, Nat. Biotechnol., 1988, 6, 1084–1089 CrossRef CAS.
- K. Zengler, G. Toledo, M. Rappe, J. Elkins, E. J. Mathur, J. M. Short and M. Keller, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15681–15686 CrossRef CAS PubMed.
- R. Nir, Y. Yisraeli, R. Lamed and E. Sahar, Appl. Environ. Microbiol., 1990, 56, 3861–3866 CAS.
- R. Nir, R. Lamed, L. Gueta and E. Sahar, Appl. Environ. Microbiol., 1990, 56, 2870–2875 CAS.
- M. S. Fitzsimons, M. Novotny, C. C. Lo, A. E. K. Dichosa, J. L. Yee-Greenbaum, J. P. Snook, W. Gu, O. Chertkov, K. W. Davenport, K. McMurry, K. G. Reitenga, A. R. Daughton, J. He, S. L. Johnson, C. D. Gleasner, P. L. Wills, B. Parson-Quintana, P. S. Chain, J. C. Detter, R. S. Lasken and C. S. Han, Genome Res., 2013, 23, 878–888 CrossRef CAS PubMed.
- S. J. Spencer, M. V. Tamminen, S. P. Preheim, M. T. Guo, A. W. Briggs, I. L. Brito, D. A. Weitz, L. K. Pitkänen, F. Vigneault, M. P. J. Virta and E. J. Alm, ISME J., 2015, 1–10 Search PubMed.
- T. Pereira, T. J. Millar and J.-A. Chuck, J. Microencapsulation, 2005, 22, 787–792 CrossRef CAS PubMed.
- K. Bernath, M. Hai, E. Mastrobattista, A. D. Griffiths, S. Magdassi and D. S. Tawfik, Anal. Biochem., 2004, 325, 151–157 CrossRef CAS PubMed.
- Y. Akselband, C. Cabral, D. S. Shapiro and P. McGrath, J. Microbiol. Methods, 2005, 62, 181–197 CrossRef CAS PubMed.
- T. Katsuragi, S. Tanaka, S. Nagahiro and Y. Tani, J. Microbiol. Methods, 2000, 42, 81–86 CrossRef CAS PubMed.
- C. Ryan, B. T. Nguyen and S. J. Sullivan, J. Clin. Microbiol., 1995, 33, 1720–1726 CAS.
- Y. Akselband, C. Cabral, T. P. Castor, H. M. Chikarmane and P. McGrath, J. Exp. Mar. Biol. Ecol., 2006, 329, 196–205 CrossRef CAS.
- A. Manome, H. Zhang, Y. Tani, T. Katsuragi, R. Kurane and T. Tsuchida, FEMS Microbiol. Lett., 2001, 197, 29–33 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- A. Persat, M. E. Suss and J. G. Santiago, Lab Chip, 2009, 9, 2454–2469 RSC.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- Q. Zhang, G. Lambert, D. Liao, H. Kim, K. Robin, C. Tung, N. Pourmand and R. H. Austin, Science, 2011, 333, 1764–1767 CrossRef CAS PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS PubMed.
- Y. Wakamoto, N. Dhar, R. Chait, K. Schneider, F. Signorino-Gelo, S. Leibler and J. D. McKinney, Science, 2013, 339, 91–95 CrossRef CAS PubMed.
- E. A. Ottesen, J. W. Hong, S. R. Quake and J. R. Leadbetter, Science, 2006, 1464–1467 CrossRef CAS PubMed.
- Y. Marcy, C. Ouverney, E. M. Bik, T. Lösekann, N. Ivanova, H. G. Martin, E. Szeto, D. Platt, P. Hugenholtz, D. A. Relman and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11889–11894 CrossRef CAS PubMed.
- P. C. Blainey, FEMS Microbiol. Rev., 2013, 37, 407–427 CrossRef CAS PubMed.
- R. Rusconi, M. Garren and R. Stocker, Annu. Rev. Biophys., 2014, 43, 65–91 CrossRef CAS PubMed.
- F. J. H. Hol and C. Dekker, Science, 2014, 346, 438 CrossRef CAS PubMed.
- F. Wu and C. Dekker, Chem. Soc. Rev., 2016, 45, 268–280 RSC.
- T. Thorsen, R. W. Roberts, F. H. Arnold and S. R. Quake, Phys. Rev. Lett., 2001, 86, 4163–4166 CrossRef CAS PubMed.
- S. L. Anna, N. Bontoux and H. A. Stone, Appl. Phys. Lett., 2003, 82, 364–366 CrossRef CAS.
- M. De Menech, P. Garstecki, F. Jousse and H. A. Stone, J. Fluid Mech., 2008, 595, 141–161 Search PubMed.
- P. Garstecki, H. A. Stone and G. M. Whitesides, Phys. Rev. Lett., 2005, 94, 1–4 Search PubMed.
- V. Van Steijn, C. R. Kleijn and M. T. Kreutzer, Phys. Rev. Lett., 2009, 103, 1–4 CrossRef PubMed.
- P. Garstecki, M. J. Fuerstman, H. A. Stone and G. M. Whitesides, Lab Chip, 2006, 6, 437–446 RSC.
- A. Huebner, D. Bratton, G. Whyte, M. Yang, A. J. DeMello, C. Abell and F. Hollfelder, Lab Chip, 2009, 9, 692–698 RSC.
- L. Frenz, K. Blank, E. Brouzes and A. D. Griffiths, Lab Chip, 2009, 9, 1344–1348 RSC.
- C. H. J. Schmitz, A. C. Rowat, S. Köster and D. A. Weitz, Lab Chip, 2009, 9, 44–49 RSC.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503 CrossRef CAS PubMed.
- X. Niu, S. Gulati, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1837–1841 RSC.
- N. Bremond, A. R. Thiam and J. Bibette, Phys. Rev. Lett., 2008, 100, 1–4 CrossRef PubMed.
- N. Shembekar, C. Chaipan, R. Utharala and C. A. Merten, Lab Chip, 2016, 16, 1314–1331 RSC.
- K. A. Price and B. M. Paegel, Anal. Chem., 2016, 88, 339–353 CrossRef PubMed.
- T. M. Tran, F. Lan, C. S. Thompson and A. Abate, J. Phys. D: Appl. Phys., 2013, 46, 114004 CrossRef.
- R. Seemann, M. Brinkmann, T. Pfohl and S. Herminghaus, Rep. Prog. Phys., 2012, 75, 016601 CrossRef PubMed.
- J. Lim, O. Caen, J. Vrignon, M. Konrad, V. Taly and J.-C. Baret, Biomicrofluidics, 2015, 9, 034101 CrossRef PubMed.
- M. Kim, M. Pan, Y. Gai, S. Pang, C. Han, C. Yang and S. K. Y. Tang, Lab Chip, 2015, 15, 1417–1423 RSC.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- T. Franke, S. Braunmüller, L. Schmid, A. Wixforth and D. A. Weitz, Lab Chip, 2010, 10, 789–794 RSC.
- A. R. Abate, J. J. Agresti and D. A. Weitz, Appl. Phys. Lett., 2010, 96, 1–3 CrossRef.
- S. W. Lim and A. Abate, Lab Chip, 2013, 13, 4563–4572 RSC.
- A. Zinchenko, S. R. A. Devenish, B. Kintses, P. Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS PubMed.
- Y. Eun, A. S. Utada, M. F. Copeland, S. Takeuchi and D. B. Weibel, ACS Chem. Biol., 2011, 6, 260–266 CrossRef CAS PubMed.
- M. Fischlechner, Y. Schaerli, M. F. Mohamed, S. Patil, C. Abell and F. Hollfelder, Nat. Chem., 2014, 6, 791–796 CrossRef CAS PubMed.
- D. J. Collins, A. Neild, A. deMello, A.-Q. Liu and Y. Ai, Lab Chip, 2015, 3439–3459 RSC.
- Z. Z. Chong, S. H. Tan, A. M. Ganan-Calvo, S. B. Tor, N. H. Loh and N.-T. Nguyen, Lab Chip, 2016, 16, 35–58 RSC.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS PubMed.
- A. B. Theberge, G. Whyte and W. T. S. Huck, Anal. Chem., 2010, 82, 3449–3453 CrossRef CAS PubMed.
- N. Damean, L. F. Olguin, F. Hollfelder, C. Abell and W. T. S. Huck, Lab Chip, 2009, 9, 1707–1713 RSC.
- O. J. Miller, E. Harrak, T. Mangeat, J. Baret, L. Frenz, B. El Debs, E. Mayot, M. L. Samuels, E. K. Rooney, P. Dieu, M. Galvan, D. R. Link and A. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS PubMed.
- J. Wegrzyn, A. Samborski, L. Reissig, P. M. Korczyk, S. Blonski and P. Garstecki, Microfluid. Nanofluid., 2013, 14, 235–245 CrossRef CAS.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- R. Dangla, S. C. Kayi and C. N. Baroud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 853–858 CrossRef CAS PubMed.
- V. van Steijn, P. M. Korczyk, L. Derzsi, A. Abate, D. A. Weitz and P. Garstecki, Biomicrofluidics, 2013, 7, 24108 CrossRef PubMed.
- N. Mittal, C. Cohen, J. Bibette and N. Bremond, Phys. Fluids, 2014, 26, 082109 CrossRef.
- J. Clausell-Tormos, A. D. Griffiths and C. A. Merten, Lab Chip, 2010, 10, 1302–1307 RSC.
- M. Sun, S. S. Bithi and S. A. Vanapalli, Lab Chip, 2011, 11, 3949 RSC.
- S. S. Bithi, W. S. Wang, M. Sun, J. Blawzdziewicz and S. A. Vanapalli, Biomicrofluidics, 2014, 25, 034118 Search PubMed.
- P. Abbyad, R. Dangla, A. Alexandrou and C. N. Baroud, Lab Chip, 2011, 11, 813–821 RSC.
- E. Fradet, P. Abbyad, M. H. Vos and C. N. Baroud, Lab Chip, 2013, 13, 4326–4330 RSC.
- P. M. Korczyk, L. Derzsi, S. Jakieła and P. Garstecki, Lab Chip, 2013, 13, 4096–4102 RSC.
- X. Niu, F. Gielen, J. B. Edel and A. J. deMello, Nat. Chem., 2011, 3, 437–442 CrossRef CAS PubMed.
- L. Derzsi, T. S. Kaminski and P. Garstecki, Lab Chip, 2016, 16, 893–901 RSC.
- C. N. Baroud, F. Gallaire and R. Dangla, Lab Chip, 2010, 10, 2032–2045 RSC.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef CAS PubMed.
- J.-C. Baret, Lab Chip, 2012, 12, 422–433 RSC.
- P. Gruner, B. Riechers, L. A. Chacon Orellana, Q. Brosseau, F. Maes, T. Beneyton, D. Pekin and J. C. Baret, Curr. Opin. Colloid Interface Sci., 2015, 20, 183–191 CrossRef CAS.
- J. Clausell-Tormos, D. Lieber, J. C. Baret, A. El-Harrak, O. J. Miller, L. Frenz, J. Blouwolff, K. J. Humphry, S. Köster, H. Duan, C. Holtze, D. A. Weitz, A. D. Griffiths and C. A. Merten, Chem. Biol., 2008, 15, 427–437 CrossRef CAS PubMed.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- K. Martin, T. Henkel, V. Baier, A. Grodrian, T. Schön, M. Roth, J. Michael Köhler and J. Metze, Lab Chip, 2003, 3, 202–207 RSC.
- A. Huebner, M. Srisa-Art, D. Holt, C. Abell, F. Hollfelder, A. J. DeMello and J. B. Edel, Chem. Commun., 2007, 1218–1220 RSC.
- M. Pan, L. Rosenfeld, M. Kim, M. Xu, E. Lin, R. Derda and S. K. Y. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 21446–21453 CAS.
- Y. Bai, E. Weibull, H. N. Joensson and H. Andersson Svahn, Sens. Actuators, B, 2014, 194, 249–254 CrossRef CAS.
- Y. L. Chiu, H. F. Chan, K. K. L. Phua, Y. Zhang, S. Juul, B. R. Knudsen, Y. P. Ho and K. W. Leong, ACS Nano, 2014, 8, 3913–3920 CrossRef CAS PubMed.
- S. Huang, J. K. Srimani, A. J. Lee, Y. Zhang, A. J. Lopatkin, K. W. Leong and L. You, Biomaterials, 2015, 61, 239–245 CrossRef CAS PubMed.
- K. Leung, H. Zahn, T. Leaver, K. M. Konwar, N. W. Hanson, A. P. Pagé, C.-C. Lo, P. S. Chain, S. J. Hallam and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7665–7670 CrossRef CAS PubMed.
- S. M. Bjork, S. L. Sjostrom, H. Andersson Svahn and H. N. Joensson, Biomicrofluidics, 2015, 9, 044128 CrossRef PubMed.
- A. Huebner, L. F. Olguin, D. Bratton, G. Whyte, W. T. S. Huck, A. J. De Mello, J. B. Edel, C. Abell and F. Hollfelder, Anal. Chem., 2008, 80, 3890–3896 CrossRef CAS PubMed.
- M. Najah, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2012, 84, 1202–1209 CrossRef CAS PubMed.
- J. Lim, J. Vrignon, P. Gruner, C. S. Karamitros, M. Konrad and J. C. Baret, Appl. Phys. Lett., 2013, 103, 203704 CrossRef.
- P. R. Marcoux, M. Dupoy, R. Mathey, A. Novelli-Rousseau, V. Heran, S. Morales, F. Rivera, P. L. Joly, J. P. Moy and F. Mallard, Colloids Surf., A, 2011, 377, 54–62 CrossRef CAS.
- W. Liu, H. J. Kim, E. M. Lucchetta, W. Du and R. F. Ismagilov, Lab Chip, 2009, 9, 2153–2162 RSC.
- M. Najah, E. Mayot, I. P. Mahendra-Wijaya, A. D. Griffiths, S. Ladame and A. Drevelle, Anal. Chem., 2013, 85, 9807–9814 CrossRef CAS PubMed.
- N. de Lange, T. M. Tran and A. R. Abate, Biomicrofluidics, 2016, 10, 024114 CrossRef CAS PubMed.
- P. Mary, A. Chen, I. Chen, A. R. Abate and D. A. Weitz, Lab Chip, 2011, 11, 2066–2070 RSC.
- S. Abalde-Cela, A. Gould, X. Liu, E. Kazamia, A. G. Smith and C. Abell, Interface, 2015, 12, 20150216 Search PubMed.
- K. Churski, A. Ruszczak, S. Jakiela and P. Garstecki, Micromachines, 2015, 6, 1514–1525 CrossRef.
- J. U. Shim, L. F. Olguin, G. Whyte, D. Scott, A. Babtie, C. Abell, W. T. S. Huck and F. Hollfelder, J. Am. Chem. Soc., 2009, 131, 15251–15256 CrossRef CAS PubMed.
- F. Courtois, L. F. Olguin, G. Whyte, A. B. Theberge, W. T. S. Huck, F. Hollfelder and C. Abell, Anal. Chem., 2009, 81, 3008–3016 CrossRef CAS PubMed.
- D.-K. Kang, M. M. Ali, K. Zhang, S. S. Huang, E. Peterson, M. A. Digman, E. Gratton and W. Zhao, Nat. Commun., 2014, 5, 5427 CrossRef CAS PubMed.
- A. Funfak, J. Cao, O. S. Wolfbeis, K. Martin and J. M. Köhler, Microchim. Acta, 2009, 164, 279–286 CrossRef CAS.
- J. Cao, S. Nagl, E. Kothe and J. M. Köhler, Microchim. Acta, 2015, 182, 385–394 CrossRef CAS.
- L. Mahler, M. Tovar, T. Weber, S. Brandes, M. M. Rudolph, J. Ehgartner, T. Mayr, M. T. Figge, M. Roth and E. Zang, RSC Adv., 2015, 5, 101871–101878 RSC.
- J. Q. Boedicker, L. Li, T. R. Kline and R. F. Ismagilov, Lab Chip, 2008, 8, 1265–1272 RSC.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- O. Scheler, T. S. Kaminski, A. Ruszczak and P. Garstecki, ACS Appl. Mater. Interfaces, 2016, 8, 11318–11325 Search PubMed.
- D.-K. Kang, X. Gong, S. Cho, J.-Y. Kim, J. B. Edel, S.-I. Chang, J. Choo and A. J. DeMello, Anal. Chem., 2015, 87, 10770–10778 CrossRef CAS PubMed.
- F. Lyu, M. Xu, Y. Cheng, J. Xie, J. Rao and S. K. Y. Tang, Biomicrofluidics, 2015, 9, 044120 CrossRef PubMed.
- I. Sinn, P. Kinnunen, T. Albertson, B. H. McNaughton, D. W. Newton, M. A. Burns and R. Kopelman, Lab Chip, 2011, 11, 2604–2611 RSC.
- S. W. Lim, T. M. Tran and A. R. Abate, PLoS One, 2015, 10, e0113549 Search PubMed.
- A. M. Sidore, F. Lan, S. W. Lim and A. R. Abate, Nucleic Acids Res., 2015, gkv1493 Search PubMed.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- M. M. Kiss, L. Ortoleva-donnelly, N. R. Beer, J. Warner, C. G. Bailey, B. W. Colston, J. M. Rothberg, D. R. Link, H. Leamon, R. Technologies and H. Ave, Anal. Chem., 2008, 80, 8975–8981 CrossRef CAS PubMed.
- D. Pekin, Y. Skhiri, J.-C. Baret, D. Le Corre, L. Mazutis, C. Ben Salem, F. Millot, A. El Harrak, J. B. Hutchison, J. W. Larson, D. R. Link, P. Laurent-Puig, A. Griffiths and V. Taly, Lab Chip, 2011, 11, 2156–2166 RSC.
- E. Zang, S. Brandes, M. Tovar, K. Martin, F. Mech, P. Horbert, T. Henkel, M. T. Figge and M. Roth, Lab Chip, 2013, 13, 3707–3713 RSC.
- X. Liu, R. E. Painter, K. Enesa, D. Holmes, G. Whyte, C. G. Garlisi, F. J. Monsma, M. Rehak, F. F. Craig and C. A. Smith, Lab Chip, 2016, 16, 1632–1643 Search PubMed.
- L. Boitard, D. Cottinet, C. Kleinschmitt, N. Bremond, J. Baudry, G. Yvert and J. Bibette, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7181–7186 CrossRef CAS PubMed.
- T. W. Hofmann, S. Hänselmann, J.-W. Janiesch, A. Rademacher and C. H. J. Böhm, Lab Chip, 2012, 12, 916 RSC.
- H. N. Joensson, M. Uhlén and H. A. Svahn, Lab Chip, 2011, 11, 1305–1310 RSC.
- L. Baraban, F. Bertholle, M. L. M. Salverda, N. Bremond, P. Panizza, J. Baudry, J. A. G. M. de Visser and J. Bibette, Lab Chip, 2011, 11, 4057–4062 RSC.
- D. Cottinet, F. Condamine, N. Bremond, A. D. Griffiths, P. B. Rainey, J. A. G. M. de Visser, J. Baudry and J. Bibette, PLoS One, 2016, 11, e0152395 Search PubMed.
- A. Funfak, R. Hartung, J. Cao, K. Martin, K. H. Wiesmüller, O. S. Wolfbeis and J. M. Köhler, Sens. Actuators, B, 2009, 142, 66–72 CrossRef CAS.
- A. Funfak, J. Cao, A. Knauer, K. Martin and J. M. Köhler, J. Environ. Monit., 2011, 13, 410–415 RSC.
- J. Cao, D. Kürsten, S. Schneider, A. Knauer, P. M. Günther and J. M. Köhler, Lab Chip, 2012, 12, 474 RSC.
- J. Cao, J. Goldhan, K. Martin and J. M. Köhler, Green Processes Synth., 2013, 2, 591–601 CAS.
- K. Wetzel, J. Cao, E. Kothe and J. M. Köhler, Eng. Life Sci., 2015, 15, 327–332 CrossRef CAS.
- J. Cao, D. Kürsten, K. Krause, E. Kothe, K. Martin, M. Roth and J. M. Köhler, Appl. Microbiol. Biotechnol., 2013, 97, 8923–8930 CrossRef CAS PubMed.
- J. Cao, S. Schneider, R. Schultheiß, A. Schober, J. M. Köhler and G. A. Groß, Microsyst. Technol., 2015, 21, 539–548 CrossRef CAS.
- S. Cho, D. Kang, S. Sim, F. Geier, J. Kim, X. Niu, J. B. Edel, S. Chang, R. C. R. Wootton, K. S. Elvira and J. Andrew, Anal. Chem., 2013, 85, 8866–8872 CrossRef CAS PubMed.
- A. Grodrian, J. Metze, T. Henkel, K. Martin, M. Roth and J. M. Köhler, Biosens. Bioelectron., 2004, 19, 1421–1428 CrossRef CAS PubMed.
- C. Lok, Nature, 2015, 522, 270–273 CrossRef CAS PubMed.
- L. Ma, S. S. Datta, M. A. Karymov, Q. Pan, S. Begolo and R. F. Ismagilov, Integr. Biol., 2014, 6, 796–805 RSC.
- L. Ma, J. Kim, R. Hatzenpichler, M. A. Karymov, N. Hubert, I. M. Hanan, E. B. Chang and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9768–9773 CrossRef CAS PubMed.
- J. Park, A. Kerner, M. A. Burns and X. N. Lin, PLoS One, 2011, 6, e17019 CAS.
- D. F. Jarosz, J. C. Brown, G. A. Walker, M. S. Datta, W. L. Ung, A. K. Lancaster, A. Rotem, A. Chang, G. A. Newby, D. A. Weitz, L. F. Bisson and S. Lindquist, Cell, 2014, 158, 1083–1093 CrossRef CAS PubMed.
- C. B. Chang, J. N. Wilking, S. H. Kim, H. C. Shum and D. A. Weitz, Small, 2015, 11, 3954–3961 CrossRef CAS PubMed.
- J. Shim, S. N. Patil, J. T. Hodgkinson, S. D. Bowden, D. R. Spring, M. Welch, W. T. S. Huck, F. Hollfelder and C. Abell, Lab Chip, 2011, 11, 1132–1137 RSC.
- Y. Bai, S. N. Patil, S. D. Bowden, S. Poulter, J. Pan, G. P. C. Salmond, M. Welch, W. T. S. Huck and C. Abell, Int. J. Mol. Sci., 2013, 14, 10570–10581 CrossRef PubMed.
- M. Weitz, A. Mückl, K. Kapsner, R. Berg, A. Meyer and F. C. Simmel, J. Am. Chem. Soc., 2014, 136, 72–75 CrossRef CAS PubMed.
- M. Schwarz-Schilling, L. Aufinger, A. Mückl and F. C. Simmel, Integr. Biol., 2016, 8, 564–570 RSC.
- J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J. C. Baret, M. Marquez, A. M. Klibanov, A. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6550–6550 CrossRef CAS PubMed.
- M. Huang, Y. Bai, S. L. Sjostrom, B. M. Hallström, Z. Liu, D. Petranovic, M. Uhlén, H. N. Joensson, H. Andersson Svahn and J. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4689–E4696 CrossRef CAS PubMed.
- H. S. Kim, A. R. Guzman, H. R. Thapa, T. P. Devarenne and A. Han, Biotechnol. Bioeng., 2016 DOI:10.1002/bit.25930.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Lainé and F. Hollfelder, Chem. Biol., 2012, 19, 1001–1009 CrossRef CAS PubMed.
- S. L. Sjostrom, Y. Bai, M. Huang, Z. Liu, J. Nielsen, H. N. Joensson and H. Andersson Svahn, Lab Chip, 2014, 14, 806–813 RSC.
- A. C. Larsen, M. R. Dunn, A. Hatch, S. P. Sau, C. Youngbull and J. C. Chaput, Nat. Commun., 2016, 7, 11235 CrossRef CAS PubMed.
- T. Beneyton, F. Coldren, J.-C. Baret, A. D. Griffiths and V. Taly, Analyst, 2014, 139, 3314–3323 RSC.
- B. L. Wang, A. Ghaderi, H. Zhou, J. J. Agresti, D. A. Weitz, G. R. Fink and G. Stephanopoulos, Nat. Biotechnol., 2014, 32, 473–478 CrossRef CAS PubMed.
- R. Ostafe, R. Prodanovic, W. Lloyd Ung, D. A. Weitz and R. Fischer, Biomicrofluidics, 2014, 8, 041102 CrossRef PubMed.
- M. Najah, R. Calbrix, I. P. Mahendra-Wijaya, T. Beneyton, A. D. Griffiths and A. Drevelle, Chem. Biol., 2014, 21, 1722–1732 CrossRef CAS PubMed.
- C.-Y. Jiang, L. Dong, J.-K. Zhao, X. Hu, C. Shen, Y. Qiao, X. Zhang, Y. Wang, R. F. Ismagilov, S.-J. Liu and W. Du, Appl. Environ. Microbiol., 2016, 82, 2210–2218 CrossRef PubMed.
- L. Dong, D.-W. Chen, S.-J. Liu and W. Du, Sci. Rep., 2016, 6, 24192 CrossRef CAS PubMed.
- S. Jang, B. Lee, H.-H. Jeong, S. H. Jin, S. Jang, S. G. Kim, G. Y. Jung and C.-S. Lee, Lab Chip, 2016, 16, 1909–1916 RSC.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed.
- M. Hosokawa, Y. Hoshino, Y. Nishikawa, T. Hirose, D. H. Yoon, T. Mori, T. Sekiguchi, S. Shoji and H. Takeyama, Biosens. Bioelectron., 2015, 67, 379–385 CrossRef CAS PubMed.
- T. C. Scanlon, S. M. Dostal and K. E. Griswold, Biotechnol. Bioeng., 2014, 111, 232–243 CrossRef CAS PubMed.
- C. J. Martinez, J. W. Kim, C. Ye, I. Ortiz, A. C. Rowat, M. Marquez and D. Weitz, Macromol. Biosci., 2012, 12, 946–951 CrossRef CAS PubMed.
- K. Liu, Y. Deng, N. Zhang, S. Li, H. Ding, F. Guo, W. Liu, S. Guo and X. Z. Zhao, Microfluid. Nanofluid., 2012, 13, 761–767 CrossRef CAS.
- D. Luo, S. R. Pullela, M. Marquez and Z. Cheng, Biomicrofluidics, 2007, 1, 34102 CrossRef PubMed.
- Y. Morimoto, W. Tan, Y. Tsuda and S. Takeuchi, Lab Chip, 2009, 9, 2217–2223 RSC.
- Y. W. Chang, P. He, S. M. Marquez and Z. Cheng, Biomicrofluidics, 2012, 6, 24118 CrossRef PubMed.
- J. Pan, A. L. Stephenson, E. Kazamia, W. T. S. Huck, J. S. Dennis, A. G. Smith and C. Abell, Integr. Biol., 2011, 3, 1043 RSC.
- A. Dewan, J. Kim, R. H. Mclean, S. A. Vanapalli and M. N. Karim, Biotechnol. Bioeng., 2012, 109, 2987–2996 CrossRef CAS PubMed.
- T. P. Lagus and J. F. Edd, RSC Adv., 2013, 3, 20512 RSC.
- R. Derda, S. K. Y. Tang and G. M. Whitesides, Angew. Chem., Int. Ed., 2010, 49, 5301–5304 CrossRef CAS PubMed.
- W. L. Matochko, S. Ng, M. R. Jafari, J. Romaniuk, S. K. Y. Tang and R. Derda, Methods, 2012, 58, 18–27 CrossRef CAS PubMed.
- K. F. Tjhung, S. Burnham, H. Anany, M. W. Griffiths and R. Derda, Anal. Chem., 2014, 86, 5642–5648 CrossRef CAS PubMed.
- J. Q. Yu, W. Huang, L. K. Chin, L. Lei, Z. P. Lin, W. Ser, H. Chen, T. C. Ayi, P. H. Yap, C. H. Chen and A. Q. Liu, Lab Chip, 2014, 14, 3519–3524 RSC.
- Y. Chen, W. A. Gani and S. K. Y. Tang, Lab Chip, 2012, 12, 5093–5103 RSC.
- P. Gruner, B. Riechers, B. Semin, J. Lim, A. Johnston, K. Short and J. Baret, Nat. Commun., 2016, 7, 10392 CrossRef CAS PubMed.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- A. R. Abate, J. Thiele, M. Weinhart and D. A. Weitz, Lab Chip, 2010, 10, 1774–1776 RSC.
- A. R. Abate, A. T. Krummel, D. Lee, M. Marquez, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 2157–2160 RSC.
- P. Jankowski, D. Ogonczyk, W. Lisowski and P. Garstecki, Lab Chip, 2012, 12, 2580–2584 RSC.
- F. Garcia-Ochoa and E. Gomez, Biotechnol. Adv., 2009, 27, 153–176 CrossRef CAS PubMed.
- F. Garcia-Ochoa, E. Gomez, V. E. Santos and J. C. Merchuk, Biochem. Eng. J., 2010, 49, 289–307 CrossRef CAS.
- J. G. Riess and M. LeBlanc, Pure Appl. Chem., 1982, 54, 2383–2406 CrossRef CAS.
- S. K. O. Ntwampe, C. C. Williams and M. S. Sheldon, Afr. J. Biotechnol., 2010, 9, 1106–1114 CAS.
- P. Abbyad, P.-L. Tharaux, J.-L. Martin, C. N. Baroud and A. Alexandrou, Lab Chip, 2010, 10, 2505–2512 RSC.
- J. Myung, M. Kim, M. Pan, C. S. Criddle and S. K. Y. Tang, Bioresour. Technol., 2016, 207, 302–307 CrossRef CAS PubMed.
- D. Karnaushenko, L. Baraban, D. Ye, I. Uguz, R. G. Mendes, M. H. Rümmeli, J. A. G. M. de Visser, O. G. Schmidt, G. Cuniberti and D. Makarov, Sci. Rep., 2015, 5, 12878 CrossRef CAS PubMed.
- L. Mazutis, J.-C. Baret, P. Treacy, Y. Skhiri, A. F. Araghi, M. Ryckelynck, V. Taly and A. D. Griffiths, Lab Chip, 2009, 9, 2902–2908 RSC.
- J. A. Stapleton and J. R. Swartz, PLoS One, 2010, 5, e15275 CAS.
- Y. Skhiri, P. Gruner, B. Semin, Q. Brosseau, D. Pekin, L. Mazutis, V. Goust, F. Kleinschmidt, A. El Harrak, J. B. Hutchison, E. Mayot, J.-F. Bartolo, A. D. Griffiths, V. Taly and J.-C. Baret, Soft Matter, 2012, 8, 10618–10627 RSC.
- P. A. Sandoz, A. J. Chung, W. M. Weaver and D. Di Carlo, Langmuir, 2014, 30, 6637–6643 CrossRef CAS PubMed.
- M. Pan, F. Lyu and S. K. Y. Tang, Anal. Chem., 2015, 87, 7938–7943 CrossRef CAS PubMed.
- K. Churski, J. Michalski and P. Garstecki, Lab Chip, 2010, 10, 512–518 RSC.
- T. S. Kaminski, S. Jakiela, M. A. Czekalska, W. Postek and P. Garstecki, Lab Chip, 2012, 12, 3995–4002 RSC.
- T. A. Duncombe, A. M. Tentori and A. E. Herr, Nat. Rev. Mol. Cell Biol., 2015, 16, 554–567 CrossRef CAS PubMed.
- Z. Li, A. M. Leshansky, L. M. Pismen and P. Tabeling, Lab Chip, 2015, 15, 1023–1031 RSC.
- DOI:10.1038/nature.2014.15135, 2014.
- http://www.who.int/drugresistance/global_action_plan, 2015.
- M. Baym, L. K. Stone and R. Kishony, Science, 2016, 351, 40 CrossRef CAS PubMed.
- J. P. Torella, R. Chait and R. Kishony, PLoS Comput. Biol., 2010, 6, e1000796 Search PubMed.
- R. J. Worthington and C. Melander, Trends Biotechnol., 2013, 31, 177–184 CrossRef CAS PubMed.
- R. Chait, A. Craney and R. Kishony, Nature, 2007, 446, 668–671 CrossRef CAS PubMed.
- J.-B. Michel, P. J. Yeh, R. Chait, R. C. Moellering and R. Kishony, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14918–14923 CrossRef CAS PubMed.
- T. Bollenbach, Curr. Opin. Microbiol., 2015, 27, 1–9 CrossRef CAS PubMed.
- P. Yeh, A. I. Tschumi and R. Kishony, Nat. Genet., 2006, 38, 489–494 CrossRef CAS PubMed.
- G. Chevereau and T. Bollenbach, Mol. Syst. Biol., 2015, 11, 807 CrossRef PubMed.
- G. Chevereau, M. Dravecká, T. Batur, A. Guvenek, D. H. Ayhan, E. Toprak and T. Bollenbach, PLoS Biol., 2015, 13, 1–18 Search PubMed.
- A. van der Zee, L. Roorda, G. Bosman, A. C. Fluit, M. Hermans, P. H. M. Smits, A. G. M. van der Zanden, R. te Witt, L. E. S. van Coppenraet, J. Cohen Stuart and J. M. Ossewaarde, BMC Infect. Dis., 2014, 14, 1–5 CrossRef PubMed.
- L. Azimi, M. Talebi, P. Owlia, M.-R. Pourshafie, M. Najafi, E. R. Lari and A. R. Lari, Gene, 2016, 576, 166–170 CrossRef CAS PubMed.
- A. Klančnik, N. Toplak, M. Kovač, H. Marquis, B. Jeršek and J. Microbiol, Methods, 2015, 118, 37–41 Search PubMed.
- S. H. Te, E. Y. Chen and K. Y.-H. Gin, Appl. Environ. Microbiol., 2015, 81, 5203–5211 CrossRef CAS PubMed.
- Y. Cao, M. R. Raith and J. F. Griffith, Water Res., 2015, 70, 337–349 CrossRef CAS PubMed.
- E. Toprak, A. Veres, J.-B. Michel, R. Chait, D. L. Hartl and R. Kishony, Nat. Genet., 2012, 44, 101–105 CrossRef CAS PubMed.
- V. Mozhayskiy and I. Tagkopoulos, Integr. Biol., 2013, 5, 262–277 RSC.
- A. C. Palmer and R. Kishony, Nat. Rev. Genet., 2013, 14, 243–248 CrossRef CAS PubMed.
- J. S. Biteen, P. C. Blainey, Z. G. Cardon, M. Chun, G. M. Church, P. C. Dorrestein, S. E. Fraser, J. A. Gilbert, J. K. Jansson, R. Knight, J. F. Miller, A. Ozcan, K. A. Prather, S. R. Quake, E. G. Ruby, P. A. Silver, S. Taha, G. van den Engh, P. S. Weiss, G. C. L. Wong, A. T. Wright and T. D. Young, ACS Nano, 2016, 10, 6–37 CrossRef CAS PubMed.
- M. O. A. Sommer, Curr. Opin. Microbiol., 2015, 27, 127–132 CrossRef CAS PubMed.
- M. Junkin and S. Tay, Lab Chip, 2014, 14, 1246–1260 RSC.
- P. K. Chattopadhyay, T. M. Gierahn, M. Roederer and J. C. Love, Nat. Immunol., 2014, 15, 128–135 CrossRef CAS PubMed.
- R. Avraham, N. Haseley, D. Brown, C. Penaranda, H. B. Jijon, J. J. Trombetta, R. Satija, A. K. Shalek, R. J. Xavier, A. Regev and D. T. Hung, Cell, 2015, 162, 1309–1321 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed.
- C. Albayrak, C. Jordi, C. Zechner, C. A. Bichsel, M. Khammash and S. Tay, Mol. Cell, 2016, 61, 914–924 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |