
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrosequencing on a glass surface†
Ana V.
Almeida
abd,
Andreas
Manz
abd and
Pavel
Neužil
*acd
aKIST-Europe, Microfluidics Group, Campus E7.1, 66111 Saarbrücken, Germany. E-mail: ana.almeida@kist-europe.de; manz@kist-europe.de; pavel.neuzil@gmail.com
bMechatronics Department, Universität des Saarlandes, Campus A5, 66123 Saarbrücken, Germany
cDepartment of Microsystem Engineering, School of Mechanical Engineering, Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi'an Shaanxi, 710072, PR China
dCentral European Institute of Technology (CEITEC), Brno University of Technology (BUT), Technická 3058/10, CZ-616 00 Brno, Czech Republic
First published on 16th February 2016
Abstract
We demonstrate the use of open-surface microfluidics to sequence DNA by pyrosequencing at the plain hydrophobically coated surface of a microscope glass cover slip. This method offers significant advantages in terms of instrument size, simplicity, disposability, and functional integration, particularly when combined with the broad and flexible capabilities of open-surface microfluidics. The DNA was incubated on superparamagnetic particles and placed on a hydrophobically coated glass substrate. The particles with bound DNA were moved using magnetic force through microliter-sized droplets covered with mineral oil to prevent water evaporation from the droplets. These droplets served as reaction “stations” performing pyrosequencing as well as washing stations. The resequencing protocol with 34-mer single-stranded DNA (ssDNA) was used to determine the reaction performance. The de novo sequencing was performed with 51-mer and 81-mer ssDNA. The method can be integrated with previously shown sample preparation and PCR into a single sample-to-answer system on a plain glass surface.
Introduction
Deoxyribonucleic acid (DNA) molecules carry the majority of genetic information for all living species as well as viruses. This information is coded in a sequence of nucleotides in the DNA1 and the process to determine the precise order of nucleotides is referred to as sequencing.2 Classical Sanger sequencing is based on selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication.3,4 This original method, as well as others developed later, has become an essential tool for many basic and applied research applications.5 Sequencing instruments were developed during the mapping of the entire human genome, which was completed in 2003 (ref. 6). Since then, a number of methods have been implemented in commercial DNA sequencers2,7 and they are now capable of rapidly sequencing the whole genome. These are routinely used in microbial,8–10 DNA methylation,11 and single nucleotide polymorphism (SNP) analyses.12 However, these systems are complex, typically bulky, and require qualified personnel to operate them.A frequently used sequencing method is pyrosequencing. This is a real-time sequencing-by-synthesis approach based on the detection of the inorganic pyrophosphate (PPi) released during the DNA polymerization reaction.13–15 This cascade of three enzymatic reactions is shown below:
 | (1) |
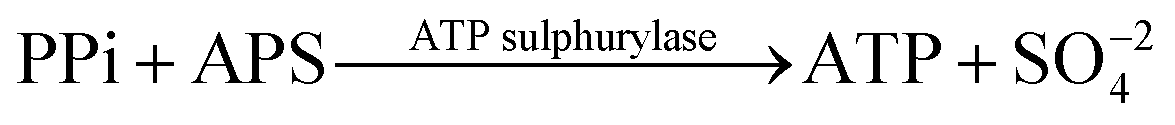 | (2) |
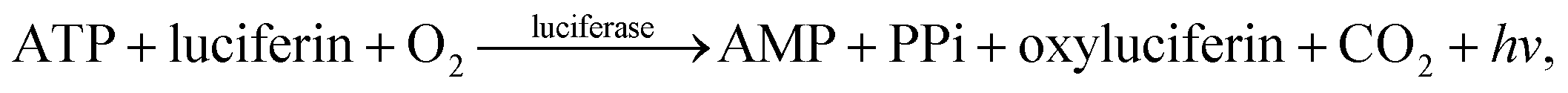 | (3) |
Microfluidics emerged as a powerful new field in 1993, following its demonstration in miniaturized capillary electrophoresis19 and then by a flow-through polymerase chain reaction (PCR).20 Nevertheless, both simple microfluidics systems required vast external instrumentation. Gradually, the instrumentation was optimized and new, simplified approaches developed21,22 and microfluidics-based systems started to play an increasingly important role in addressing the demand for faster, more accessible and easier methods for applications, including DNA sequencing technologies.23–25 Traditionally, microfluidics devices were based on closed-channel systems fabricated by lithography, etching, and bonding processes. The process was complex and devices suffered from the formation of air bubbles and their trapping. Motivated by advancements in point-of-care (POC) and home-care systems, open-surface microfluidics (OSM) for biotechnology has been recently gaining attention.26 This technique requires simpler fabrication than traditional microfluidic chips, which eliminates cleanroom processes, suppresses air bubble-related problems, and offers user-friendly operation due to the device simplicity.
Sample manipulation and preparation were demonstrated using magnetic particles in a closed system by integrating various functions from a biological sample to DNA extraction and purification.27 The same magnetic particle manipulation was used with OSM, where undesirable water evaporation was eliminated by sample encapsulation with mineral oil, forming a “virtual reaction chamber” (VRC). A proof-of-concept of this type of microfluidics was demonstrated by PCR and sample preparation strategies.28–30 A number of complex bioassays have been shown where droplets were self-contained and operated as reaction chambers as well as sample transportation units.31 The mineral oil encapsulation effectively prevented sample evaporation for hours even at elevated temperature.28 Extreme cases were demonstrated earlier with water samples heated to ≈200 °C (ref. 32 and 33), where no water evaporation from the encapsulated sample was observed.
Here, we present an approach to perform three-enzyme pyrosequencing using magnetically actuated VRCs by means of the OSM concept. The reactions are conducted on a plain microscope glass cover slip, thereby eliminating all microfabrication steps involved in other microfluidic approaches. These are performed on a disposable substrate, which makes the platform significantly simpler than conventional microfluidics with closed channels and pumps, OSM-type with surface acoustic waves (SAW),34,35 or using electro-wetting techniques.36
Materials and methods
Materials
For pyrosequencing preparation, ≈100 μL of paramagnetic particles M280 Dynabeads from Life Technologies, GmbH (Germany) was washed three times and resuspended in 100 μL of binding buffer (≈10 mM Tris buffer pH ≈7.6, ≈2 M NaCl, ≈1 mM EDTA solution and ≈0.1% Tween 20). All chemicals were provided by Sigma-Aldrich, GmbH (Germany). Amounts of ≈4 μg of biotinylated PCR amplicon in ≈100 μL of H2O were added to the particles and incubated at ≈65 °C for ≈15 min with periodic mixing for binding of the DNA. The double-stranded amplicon was denatured by exposing the beads to ≈100 μL of ≈0.5 M NaOH for ≈1 min. The beads were then washed by NaOH followed by three additional washing steps in magnesium annealing buffer (≈20 mM Tris acetate, pH ≈7.6, ≈5 mM magnesium acetate) to obtain ssDNA. The beads were resuspended in ≈100 μL of magnesium annealing buffer prior to adding ≈5 μL of ≈10 μM sequencing primer for hybridization of the primer. The solution was heated to ≈80 °C for ≈2 min and cooled back to room temperature using a heat block to allow the primer to hybridize. The bead/template/primer complex was washed three times in pyrosequencing wash buffer as described below; then, ≈15 μg of single-stranded binding protein (SSB) by Promega, GmbH (Germany) was added and the mixture incubated at room temperature for ≈10 min. The beads were washed with pyrosequencing wash buffer and resuspended in ≈200 μL of pyrosequencing wash buffer to achieve a final bead mass concentration of ≈5 μg μL−1. On the basis of the manufacturer's specified binding capacity, it is estimated that the ≈5 μL aliquot used in each sequencing reaction contained between ≈150 and ≈460 pmol of template DNA.Separate stock solutions were prepared for pyrosequencing: washing buffer, enzymes, dNTPs, and DNA. Pyrosequencing washing buffer was prepared by mixing 100 mM Tris acetate pH ≈7.6 with ≈0.5 mM ethylenediaminetetraacetic acid (EDTA) pH ≈ 8.0, ≈5 mM magnesium acetate, and ≈0.01% Tween 20. Enzyme solution was prepared with pyrosequencing washing buffer by adding ≈13.5 mU μL−1 ATP sulfurylase by New England BioLabs Inc. (Germany), ≈1.5 μg μL−1 luciferase and ≈1.8 μg μL−1D-luciferin by Sigma-Aldrich, GmbH (Germany), ≈6.9 U μL−1 Klenow (exo-) fragment by ThermoFisher Scientific, GmbH (Germany), and ≈0.01% Tween 20, ≈18 μM stock solutions of each dNTP by Thermoscientific, GmbH (Germany) was added to pyrosequencing washing buffer with ≈15 μM APS by Sigma-Aldrich (Germany), ≈3 mM dithiothreitol (DTT) by Sigma-Aldrich, GmbH (Germany), ≈120 ng μL−1 single-stranded binding protein (SSB), and ≈4.6 U μL−1 Klenow (exo-) fragment.
All primers and synthetic sequences were purchased from Eurofins MWG Operon, GmbH (Germany).
Methods
Prior to pyrosequencing, DNA has to be extracted from the sample, purified, and amplified by PCR. Most of these steps are either labor-intensive or require complex robotic systems. Except for pyrosequencing, all other necessary steps have been previously integrated using the OSM concept.29,30 The system proposed here is fully compatible with previous work, thus, it is possible to be integrated later on into a single “sample-to-answer” device.The pyrosequencing experiments were conducted on a glass cover slip as shown schematically in Fig. 1A. The ssDNA bound to SPP was placed into a first station as shown in (a), with a magnet underneath it. The particles are then dragged (b) using magnetic force into the first (E) enzyme-containing droplet. This droplet is in close vicinity with the (S + G) substrate and nucleotide dGTP-containing droplet, thus, the droplets merged and the pyrosequencing reaction sequence was initiated. The particles are then dragged into the washing droplet. This washing droplet is at a larger distance from the other droplets, so no merging of droplets occurs. The washing steps were designed as the major determinant of maximum read length via complete removal of unincorporated or excess nucleotides at each step. We have proposed washing the bead/template/primer complexes using washing buffer to remove nucleotides as well as other reaction byproducts between nucleotide additions. Subsequently, the particles are dragged into another station (c) with two droplets, one again with enzyme and the other one with substrate and another nucleotide (dTTP).
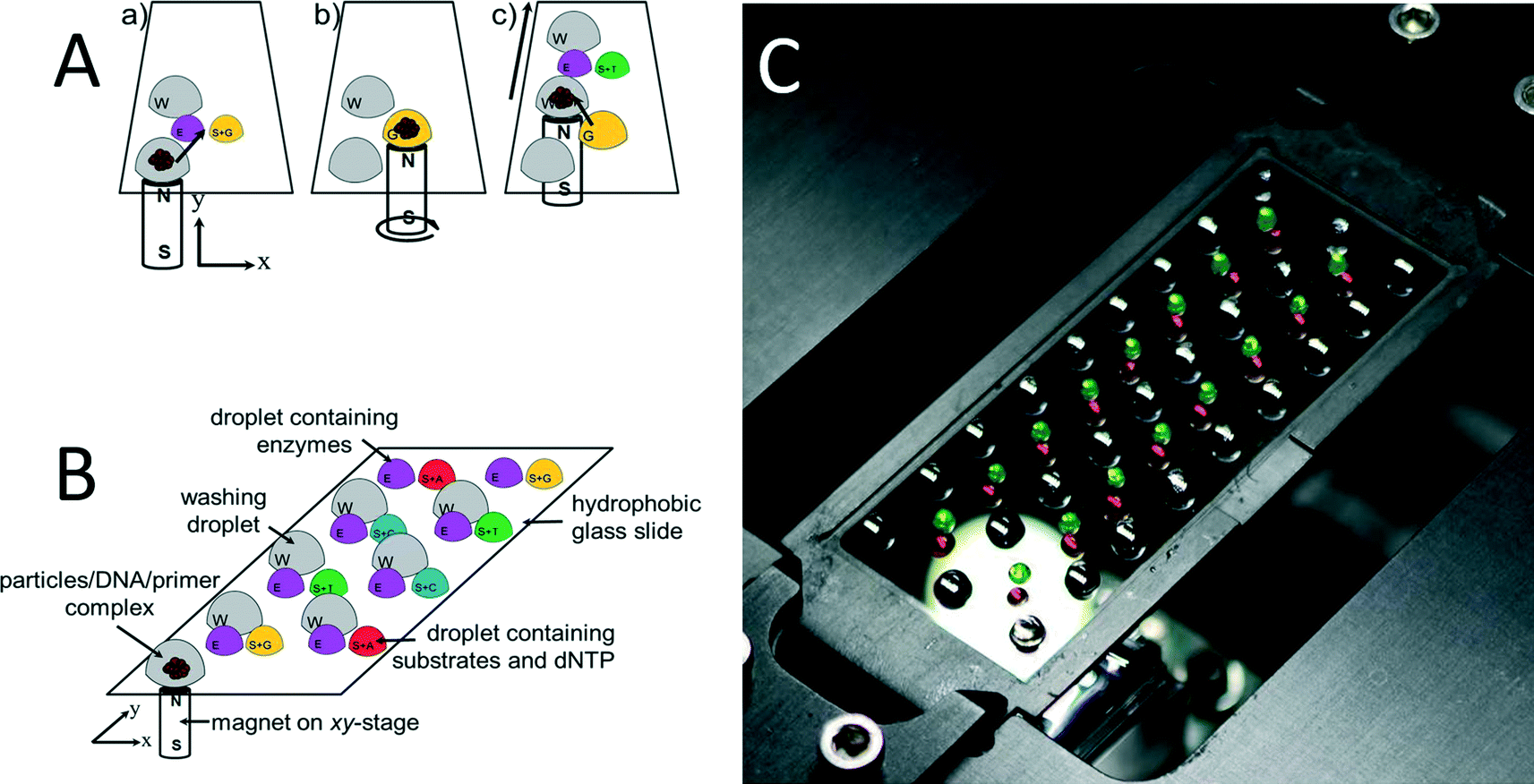 | ||
| Fig. 1 (A) Representation of a typical nucleotide addition cycle. All droplets are covered with oil, thereby effectively preventing evaporation of water from the sample as well as reagent degradation by oxidation. (a–c) Three phases of the pyrosequencing reaction. (B) Principle of the experiment setup; (W) washing droplets; (E) droplets containing all the needed enzymes for the pyrosequencing reaction (S + G − T − C − A). (C) Photograph of the setup consisting of the hydrophobically coated glass cover slip mounted onto a motorized X–Y translation stage with a neodymium–iron–boron magnet fixed beneath the cover slip. The droplets contained color ink to increase the contrast. | ||
A glass containing eight reaction stations is schematically shown in Fig. 1B and a photograph of an actual setup with 16 stations in Fig. 1C as well as a single-loading and unloading droplet containing DNA/primer/SPP. The droplets were colored with ink to increase the contrast. The glass with the dimension of ≈60 × ≈20 mm and thickness of ≈170 μm was treated with 1H,1H,2H,2H-perfluorodecyltriethoxysilane (FAS17), making the surface highly hydrophobic, with a water contact angle of ≈95°. The glass cover slip was placed on a computer controlled X–Y stage. A disc-shaped neodymium magnet with a diameter of ≈3 mm, model N45 mm by Supermagnetic, GmbH (Germany) was placed underneath the glass cover slip.
The DNA/primer/SPP complex was immobilized on SiO2-covered SPP and moved through the individual reaction/washing stations in the form of droplets using magnetic force. These “stations” were formed by pipetting individual droplets onto the glass covered with a mineral oil M5904 by Sigma-Aldrich, GmbH (Germany), thereby preventing water evaporation from reaction droplets. After a series of experiments, the volumes of stations as well as oil covering them are summarized in Table 1.
| Washing droplet | Enzyme droplet | Substrate and nucleotide droplet | |
|---|---|---|---|
| Droplet volume (μL) | ≈10 | ≈3 | ≈2 |
| Oil volume (μL) | ≈2 | ≈1 | ≈1 |
We placed a photomultiplier tube (PMT) model H10492-001 from Hamamatsu Photonics K.K. (Japan) above the glass aligned with the magnet underneath. Since the sample was always attracted by the magnet and kept on the glass above it, it was also directly below the PMT; thus, all reactions were conducted at the sample location with respect to the PMT. With this setup, both the magnet and the PMT were stationary while the glass was movable. The stage with the PMT and magnet was placed in a black box to eliminate ambient light noise. The PMT was equipped with an internal current-to-voltage converter, with its gain adjusted to ≈0.7 V using a custom made PMT controller setting the gain value to ≈105 as interpolated from a graph supplied by the manufacturer. The PMT output was directly connected to the input of an oscilloscope model DPO 7054 provided by Tektronix, Inc. (USA) to record optical power as a function of time.
The PMT voltage output as function of time captured by an oscilloscope was processed using a custom written MATLAB script. First, the data histogram based on frequency data count was formed and its peak was used to determine the signal baseline, which was subsequently subtracted from the data. All peaks were identified and individually processed to extract the values of maximum amplitude as well as their areas under the curve (AUC). The AUC unit corresponds to the electrical charge C, as it is time integration of electrical current i from 0 s to t s:
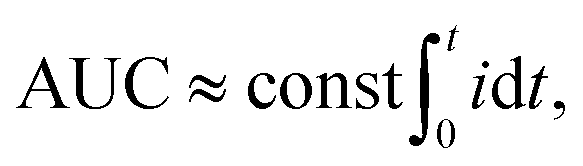 | (4) |
Results and discussion
An initial series of experiments with and without a washing step was conducted. Performing the pyrosequencing reactions without the washing step resulted in a 30% carry-over signal by the DNA/primer/SPP complex. Once the washing step was incorporated, the carry-over signal was eliminated (see Fig. S1 in ESI†); thus, each reaction was terminated with a single washing step. As one can see from the results, once the washing step was incorporated into the experiment design, no emitted light was observed from the sample, which allows us to conclude that no significant amount of chemicals were carried over.For each experiment, we used new (hydrophobically coated) glass cover slips to eliminate potential sample-to-sample cross contamination, which is an advantage of having a disposable and microfabrication-free platform.
We also tried to overcome the diffusion-dependent rate of the pyrosequencing process by moving SPP within the droplet during the reaction. The stage moved by ±≈2 mm along both X and Y axes at the speed of ≈2 mm s−1, which shortened the reaction time by a factor of almost 2 (Fig. S2 in ESI†). Initially, the signal peak amplitude was ≈1.00 V and its area was ≈0.32 mC. Stimulated by stirring, the peak amplitude increased to ≈1.99 V and the area dropped to ≈0.27 mC. The AUC amplitude dropped to ≈85% of its original value while the signal peak amplitude increased by factor of ≈2. The signal reached the baseline amplitude at time ≈275 s; thus, we decided that keeping the reaction under the PMT for ≈300 s should be sufficient.
Next, we performed a set of experiments to determine optimum DNA. We used ≈55 μg of beads and DNA concentration from ≈0.3125 pmol to ≈20 pmol. The DNA concentration of ≈1.25 pmol seemed to be optimum (Fig. S3–S7 in ESI†). This concentration resulted in well-distinguished groups for each number of nucleotide incorporation (Fig. S4D and E in ESI†). The peak voltage and AUC amplitude were (0.969 ± 0.227) V and (0.147 ± 0.021) mC (mean ± standard deviation) per single nucleotide incorporation, respectively, with correlation coefficient of 0.959 and 0.896 for peak voltage and AUC, respectively. This peak voltage signal was ≈100 times higher than the background noise while requiring minimum DNA consumption.
As mentioned earlier, each glass cover slip can accommodate up to 16 reaction sets (washing + enzyme and substrate droplets). Sequencing of more than 16 nucleotides then requires multiple glass cover slip and the sample has to be unloaded from one glass and loaded to another glass. These loading/unloading stations were designed not to contain matching nucleotides, thus, they also served as reference points to determine the background signal. We pipetted out the particles from the unloading station and transferred them to another glass slip, with the aid of a magnet at the surface of the pipette tip, into a loading station. Once the experiments were completed, we calculated the AUC and peak voltage amplitudes from 5 transfers in unloading and loading stations and compared them with each other. We found that the loading PMT peak and AUC amplitude were (0.79 ± 0.08) V and (0.094 ± 0.005) mC (mean ± standard deviation), respectively. Unloading PMT and AUC amplitude were (0.79 ± 0.11) V and (0.098 ± 0.014) mC (mean ± standard deviation), respectively. The results during loading and unloading are practically identical, proving successful beads transfer. This information is crucial to verify that we can transfer beads from glass to glass while maintaining the same amplitude of emitted light due to nucleotide incorporation during DNA sequencing.
We also tested the maximum number of homopolymeric nucleotide additions per single incorporation (Fig. S8 in ESI†). We observed that the amplitude of emitted light starts to drop after incorporations of more than seven nucleotides. We extracted the peak voltage and AUC amplitude of (1.451 ± 0.067) V and (0.175 ± 0.020) mC (mean ± standard deviation), respectively, per single nucleotide incorporation. Peak voltage amplitude provided more consistent signal than the one of AUC. The signal level after each successive wash cycle could also be diminished in the reaction complex because of the loss or denaturation of either the DNA template or the sequencing primer.
The system was calibrated with DNA concentration of ≈1.25 pmol by performing a re-sequencing experiment (Fig. 2). A glass cover slip was prepared with the dNTPs placed in predetermined order, which were incorporated into the complementary DNA strand resulting in a final 34-mer. The sequence consisted of 21 single, 3 double, 1 triple, and 1 quadruple nucleotide incorporations, and 4 mismatches. We had to use two glass cover slips with single sample transfer from the first glass to the second glass. This system allowed us to monitor emitted light of known nucleotide incorporation and to determine the reaction stability as well as reproducibility as shown in typical pyrogram in Fig. 2A. The DNA/primer/SPP complex was subsequently moved into a washing droplet. The resequencing experiment was repeated five times to suppress random errors and to demonstrate repeatability shown by error bars in Fig. 2B. A mismatched event was performed during the sample loading and unloading to determine the background emission as marked by blue ellipses (Fig. 2C). We found that both the peak voltage as well as the AUC amplitude can be used for identification of the number of incorporated nucleotides. This is confirmed by the histograms in Fig. 2D and E, each containing five groups separated from others. The PMT peak voltage and AUC amplitude are linearly proportional to number of incorporated nucleotides. This linear relationship and stable signal amplitude through the experiment demonstrated feasibility of the method as well as little or no loss of beads, DNA, or primers. Extracted peak voltage and AUC amplitudes for single nucleotide incorporation were (1.667 ± 0.140) V and (0.213 ± 0.026) mC (mean ± standard deviation), respectively (Fig. 2F). The background peak voltage value and AUC amplitudes were only (0.799 ± 0.079) AUC and (0.010 ± 0.012) mC (mean ± standard deviation), respectively, well below the signal for single nucleotide incorporation.
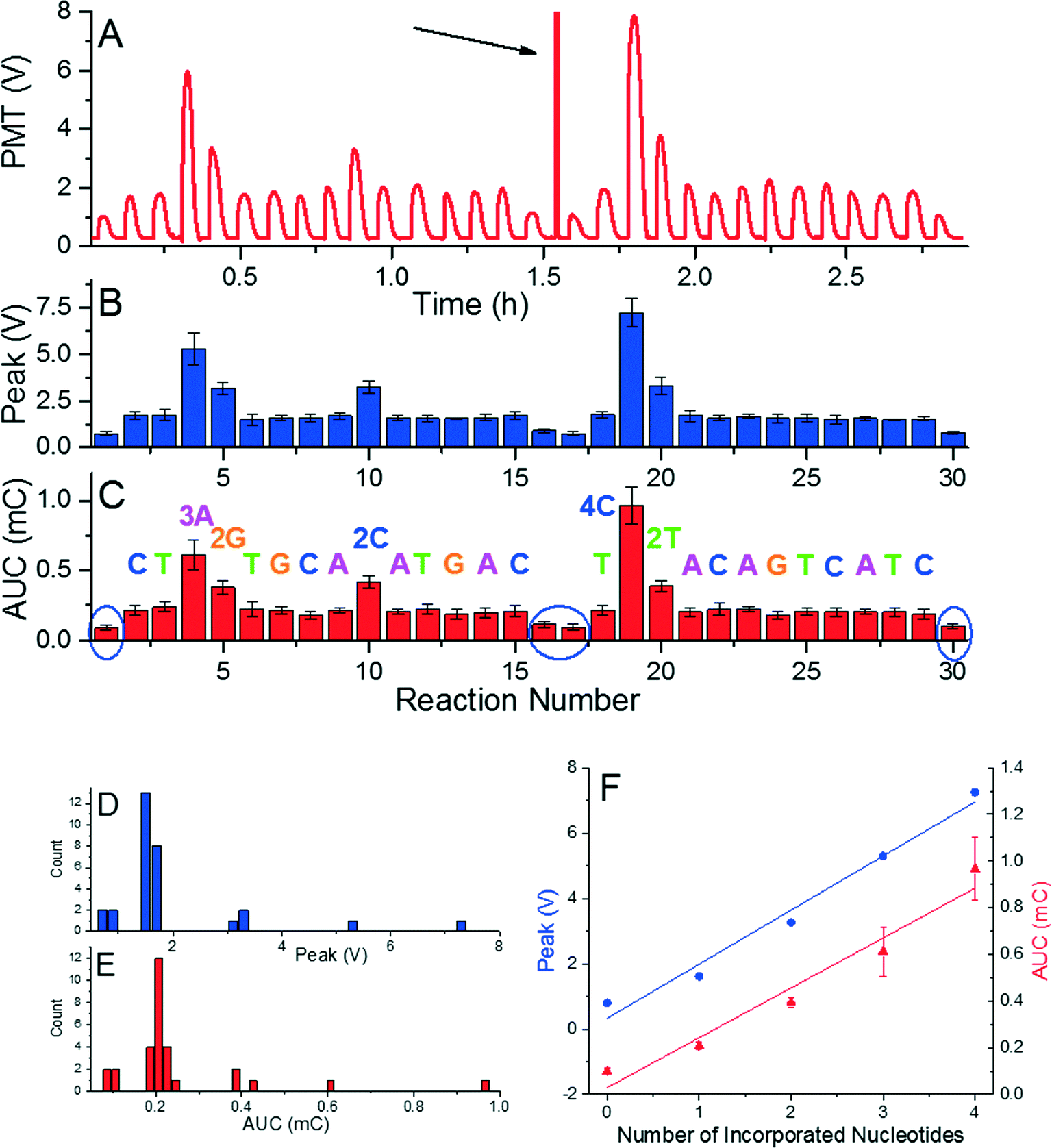 | ||
| Fig. 2 (A) A typical pyrogram or resequencing. (B) Mean value of peak voltage and its standard error as function of reaction number. A mismatched event was performed in the end of the run to determine the background emission (marked by blue ellipses). (C) Mean value of AUC and its standard error as the function of the reaction. Frequency counts of average peak values (D) and AUCs (E) distinguish five groups according to the number of incorporated nucleotides with a nearly perfect linear relationship. (F) Peak amplitude (blue circles – left axis) and AUC (red triangles – right axis) as a function of the number of incorporated nucleotides with slope of (1.657 ± 0.140) V and (0.214 ± 0.026) mC for peak voltage and AUC, respectively. The nonlinearity is small as the correlation coefficients are 0.972 and 0.944 for peak voltage and AUC, respectively. This linear relationship and stable signal amplitude through the experiment demonstrated feasibility of the method as well as little or no loss of beads, DNA, or primers. | ||
The order of reaction stations for resequencing is designed to match the sequence and incorporate nucleotide(s) in each reaction. Presence of a single nucleotide polymorphism (SNP) is detected as a missing reaction and there will be no nucleotide incorporation once the SNP occurs. The first and last reactions at each glass surface are performed with mismatched nucleotides during sample loading/unloading to detect a background signal and calibrate the system. Once there is a nucleotide mismatch detected during resequencing, the signal will be either stronger than expected (in the case of incorporation of an identical nucleotide), or as intense as the background signal with no incorporation. In the latter case, the pyrosequencing would stop there but the user would know that there is at least one SNP in the tested sequence.
Demonstrating the capability of pyrosequencing method at the glass surface was conducted by performing de novo sequencing experiment with a DNA template with the length of 51 bps. The sequence consisted of 16 single, 9 double, 2 triple, 1 quadruple, and 1 quintuple nucleotide incorporations, and 30 mismatches. We performed the experiment with four nucleotides added in a repeated cyclical fashion having 70 reaction stations. This experiment required five glass cover slips and took less than seven hours (Fig. 3A) and was conducted again five times to suppress random errors. The captured signals were processed the same way as in the previous experiments (Fig. 3B and C). In addition, histograms of both extracted values (Fig. 3D) show better grouping of the peak voltage amplitude than that for AUCs (Fig. 3E). The signals' peak voltage amplitudes in each group are relatively far from other groups, thus, distinguishing each group is an easy task. The extracted data presented here enabled an accurate sequence determination of all 51 bases. Extracted peak voltage and the AUC amplitude show they are linearly proportional to the number of incorporated nucleotides (Fig. 3F). The value of peak voltage and AUC amplitude are (1.605 ± 0.068) V and (0.213 ± 0.021) mC (mean ± standard deviation), respectively. Again, the peak voltage signal shows better stability than the AUC as marked by blue ellipse at Fig. 3F, where the signal for the four incorporated nucleotides is practically identical to the one for three incorporated nucleotides.
 | ||
| Fig. 3 (A) A typical pyrogram of de novo sequencing. (B) Mean value of peak voltage and its standard error as function of reaction number. A mismatched event was performed in the end of the run to determine the background emission (marked by blue ellipses). (C) Mean value of AUC and its standard error as function of reaction. Frequency counts of mean peak values (D) distinguish five groups according to the number of incorporated nucleotides. Mean values of AUC (E) are not as well defined as the ones for peak values. Amplitude of AUC for four nucleotide additions (marked by an ellipse) is higher than the one for five. (F) Peak amplitude (blue circles – left axis) and AUC (red triangles – right axis) as function of number of incorporated nucleotides with slope of (1.605 ± 0.068) V and (0.213 ± 0.022) mC for peak voltage and AUC, respectively. The peak amplitude is linearly proportional to the number of incorporated nucleotides with correlation coefficient of 0.991. The same coefficient for AUC is 0.953. The peak signal amplitude provides more consistent output than the one for AUC. | ||
As a final experiment, we tried to incorporate a sequence with length of 81 nucleotides performed in 104 reactions in a single measurement using de novo sequencing configuration. The sequence consisted of 35 single, 8 double, 4 triple, 2 quadruple, and 2 quintuple nucleotide incorporations, and 53 mismatches. The experiment required seven glass cover slips and took less than 10 h (Fig. 4A). Extracted peak voltage and AUC amplitudes were (1.495 ± 0.117) V and (0.182 ± 0.027) mC (mean ± standard deviation), respectively (Fig. 4B and C). The signal per incorporated nucleotide is again well above the background of (0.794 ± 0.117) V and (0.102 ± 0.021) mC (mean ± standard deviation) for peak voltage and AUC amplitudes, respectively (Fig. 4D). The method presented here is then capable of incorporating 81 nucleotides and, as the PMT signal is has consistent amplitude per nucleotide incorporation, there is high probability that even greater number of nucleotide can be incorporated. As observed in previous experiments, peak voltage provides more consistent data than the one of AUC amplitude. The amplitude of AUC for four incorporated nucleotides was higher than the one for 5 nucleotides as marked by arrows in the histograms in Fig. 4E and F.
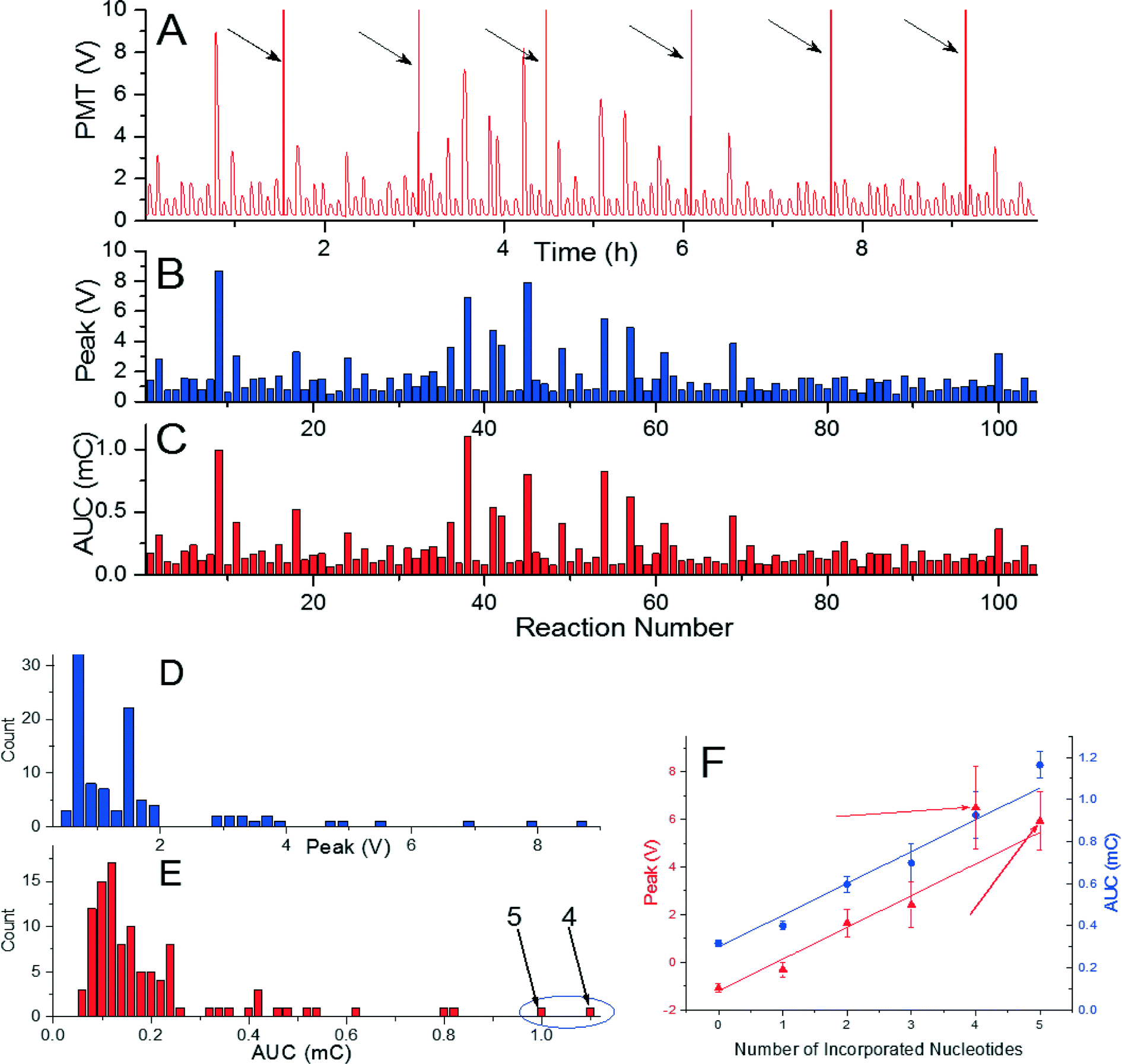 | ||
| Fig. 4 (A) A pyrogram of long de novo sequencing with 104 reactions. (B) Extracted value of peak voltage as function of reaction number. (C) Extracted mean value of AUC as function of reaction number. Frequency counts of mean peak values (D) distinguish six groups according to the number of incorporated nucleotides. (E) Frequency counts of the AUC are not well separated, with signal from four incorporated nucleotides at higher amplitude than the one for five (both marked by arrows). (F) Peak amplitude (blue circles – left axis) and AUC (red triangles – right axis) as function of number of incorporated nucleotides with slope of (1.339 ± 0.145) V and (0.151 ± 0.019) mC for peak voltage and AUC, respectively. The peak amplitude is linearly proportional to the number of incorporated nucleotides with correlation coefficient of 0.944. The same coefficient for AUC is 0.927. The peak signal amplitude then provides more consistent output than the one for AUC. | ||
Overall, we observed that the peak voltage amplitude provided more consistent data than the one of the AUC. The peak voltage amplitude per nucleotide incorporation varied between (1.354 ± 0.075) V, (1.657 ± 0.140) V, (1.605 ± 0.068) V and (1.339 ± 0.145) V (mean ± standard deviation) homopolymeric stretches (Fig. S8 ESI†), re-sequencing (Fig. 2), de novo sequencing of 51 (Fig. 3), and 81 nucleotides (Fig. 4), respectively. The differences are probably caused by slight uncertainty in concentration of chemicals due to manual pipetting as well as temperature fluctuation. Sample preparation was time-consuming and experiments were performed on different days.
Conclusion
We have demonstrated a simple pyrosequencing system for both de novo and re-sequencing using open surface microfluidics with magnetic force actuation on a microscope glass cover slip surface without any surface patterning. The droplets were covered with mineral oil, thereby forming virtual reaction chambers. An oil layer was able to sufficiently suppress water evaporation from the sample. As a proof of principle, we designed a robust system with a high signal-to-noise ratio of more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The ratio of an output signal from PMT to its noise is given by reagents, such as background reactions of substrates, as well as by the ambient light leakage into the PMT system. We observed small, yet constant, light leakage into the system, which affected the PMT output signal.
:1. The ratio of an output signal from PMT to its noise is given by reagents, such as background reactions of substrates, as well as by the ambient light leakage into the PMT system. We observed small, yet constant, light leakage into the system, which affected the PMT output signal.
The peak signal amplitude per nucleotide incorporation was between 1 and 2 V while noise superposed at the signal was below ≈4 mV. The PMT gain was nearly one hundred times lower than its maximum. This large margin in the testing setup suggests that the system has room for improvement, such as increasing its throughput by significantly decreasing the droplet size, probably down to nL level.
We showed a series of experiments starting with re-sequencing providing excellent stable signal with small standard error of the peak voltage amplitude of ≈8.4% per nucleotide incorporation. Loading and unloading stations with on purpose introduced mismatch provided signal with ≈50% amplitude lower than the one with nucleotide incorporation. The system might then become a suitable tool for SNP analysis using this re-sequencing strategy.
We also performed successfully a de novo pyrosequencing experiment with 51 and 81 nucleotide incorporations. The signal degradation with time of number of nucleotide incorporation was minimal; therefore, we can hypothesize that even greater numbers can be incorporated. Increasing the number of sequencing experiments to five suppressed random errors and improved reliability of the method.37,38
The system presented here has a flexible configuration that can be used for either de novo sequencing or re-sequencing. These applications can also be altered in real-time based on the instantaneous results. The presented method offers significant advantages in terms of instrument size, simplicity, disposability, and achievable levels of functional integration.
The system should be further optimized to increase the throughput and significantly shorten the time required for each reaction. The process is controlled by the diffusion of individual compounds assisted by simple mixing. Decreasing the sample volume as well as elevating the system's temperature would make the reaction faster and lead to more favorable time required for each pyrosequencing experiment.
An automated pipetting station followed by a lyophilization step also would make this method more user friendly as operators would only require to dispense an appropriate amount of de-ionized water at pre-defined reaction and washing locations and cover those droplets with mineral oil.
The system presented here is compatible with previously designed PCR as well as sample preparation systems. In future, one could create a single complex sample-to-answer system starting with a raw sample, DNA release, its purification, PCR, and pyrosequencing.
Acknowledgements
We would like to thank J. Pipper from Baldr Biotechnologies Pte Ltd, Singapore for useful discussion regarding the pyrosequencing experiment, as well as for his help with the experiment setup and Xun Wenpeng from Northwestern Polytechnic University, Xi'an, China, for his help with MATLAB script. We would like to also to thank Mark Tarn, University of Hull, J, Neužil from Czech Academy of Science and J. Drbohlavová from Brno University of Technology for their editorial help. P. Neužil acknowledges partial financial support by Central European Institute of Technology (CEITEC), grant number CZ.1.05/1.1.00/02.0068.References
- M. E. W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1st edn, 1984 Search PubMed.
- L. T. C. França, E. Carrilho and T. B. L. Kist, Q. Rev. Biophys., 2002, 35, 169–200 CrossRef.
- F. Sanger and A. R. Coulson, J. Mol. Biol., 1975, 94, 441–448 CrossRef CAS PubMed.
- F. Sanger, S. Nicklen and A. R. Coulson, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5463–5467 CrossRef CAS.
- S. D. Boyd, Annu. Rev. Pathol.: Mech. Dis., 2013, 8, 381–410 CrossRef CAS PubMed.
- M. V. Olson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4338–4344 CrossRef CAS.
- M. L. Metzker, Nat. Rev. Genet., 2010, 11, 31–46 CrossRef CAS PubMed.
- E. M. Harrison, G. K. Paterson, M. T. G. Holden, J. Larsen, M. Stegger, A. R. Larsen, A. Petersen, R. L. Skov, J. M. Christensen, A. Bak Zeuthen, O. Heltberg, S. R. Harris, R. N. Zadoks, J. Parkhill, S. J. Peacock and M. A. Holmes, EMBO Mol. Med., 2013, 5, 509–515 CrossRef CAS PubMed.
- M. Kato-Maeda, C. Ho, B. Passarelli, N. Banaei, J. Grinsdale, L. Flores, J. Anderson, M. Murray, G. Rose, L. M. Kawamura, N. Pourmand, M. A. Tariq, S. Gagneux and P. C. Hopewell, PLoS One, 2013, 8, e58235 CAS.
- C. U. Köser, J. M. Bryant, J. Becq, M. E. Török, M. J. Ellington, M. A. Marti-Renom, A. J. Carmichael, J. Parkhill, G. P. Smith and S. J. Peacock, N. Engl. J. Med., 2013, 369, 290–292 CrossRef PubMed.
- Y. Li and T. O. Tollefsbol, Methods Mol. Biol., 2011, 791, 11–21 CAS.
- A. Alderborn, A. Kristofferson and U. Hammerling, Genome Res., 2000, 10, 1249–1258 CrossRef CAS PubMed.
- P. Nyrén and A. Lundin, Anal. Biochem., 1985, 151, 504–509 CrossRef.
- M. Ronaghi, S. Karamohamed, B. Pettersson, M. Uhlén and P. Nyrén, Anal. Biochem., 1996, 242, 84–89 CrossRef CAS PubMed.
- E. D. Hyman, Anal. Biochem., 1988, 174, 423–436 CrossRef CAS PubMed.
- M. Ronaghi, M. Uhlen and P. Nyren, Science, 1998, 281, 363–365 CrossRef CAS PubMed.
- A. Ahmadian, M. Ehn and S. Hober, Clin. Chim. Acta, 2006, 363, 83–94 CrossRef CAS PubMed.
- F. Mashayekhi and M. Ronaghi, Anal. Biochem., 2007, 363, 275–287 CrossRef CAS PubMed.
- D. J. Harrison, K. Fluri, K. Seiler, Z. H. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CAS.
- M. U. Kopp, A. J. de Mello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- D. J. Boles, J. L. Benton, G. J. Siew, M. H. Levy, P. K. Thwar, M. A. Sandahl, J. L. Rouse, L. C. Perkins, A. P. Sudarsan, R. Jalili, V. K. Pamula, V. Srinivasan, R. B. Fair, P. B. Griffin, A. E. Eckhardt and M. G. Pollack, Anal. Chem., 2011, 83, 8439–8447 CrossRef CAS PubMed.
- E. F. Kirkness, Nat. Biotechnol., 2009, 27, 998–999 CrossRef CAS PubMed.
- A. Russom, N. Tooke, H. Andersson and G. Stemme, Anal. Chem., 2005, 77, 7505–7511 CrossRef CAS PubMed.
- B. P. Casavant, E. Berthier, A. B. Theberge, J. Berthier, S. I. Montanez-Sauri, L. L. Bischel, K. Brakke, C. J. Hedman, W. Bushman, N. P. Keller and D. J. Beebe, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10111–10116 CrossRef CAS PubMed.
- U. Lehmann, C. Vandevyver, V. K. Parashar and M. A. M. Gijs, Angew. Chem., Int. Ed., 2006, 45, 3062–3067 CrossRef CAS PubMed.
- P. Neuzil, J. Pipper and T. M. Hsieh, Mol. BioSyst., 2006, 2, 292–298 RSC.
- J. Pipper, M. Inoue, L. F. P. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS PubMed.
- J. Pipper, Y. Zhang, P. Neuzil and T.-M. Hsieh, Angew. Chem., Int. Ed., 2008, 47, 3900–3904 CrossRef CAS PubMed.
- Z. Long, A. M. Shetty, M. J. Solomon and R. G. Larson, Lab Chip, 2009, 9, 1567–1575 RSC.
- P. Neuzil, W. Sun, T. Karasek and A. Manz, Appl. Phys. Lett., 2015, 106 Search PubMed.
- M. O. Altmeyer, A. Manz and P. Neuzil, Anal. Chem., 2015, 87, 5997–6003 CrossRef CAS PubMed.
- L. Y. Yeo and J. R. Friend, in Annual Review of Fluid Mechanics, Vol 46, ed. S. H. Davis and P. Moin, Annual Reviews, Palo Alto, 2014, vol. 46, pp. 379–406 Search PubMed.
- Z. Guttenberg, H. Muller, H. Habermuller, A. Geisbauer, J. Pipper, J. Felbel, M. Kielpinski, J. Scriba and A. Wixforth, Lab Chip, 2005, 5, 308–317 RSC.
- M. G. Pollack, R. B. Fair and A. D. Shenderov, Appl. Phys. Lett., 2000, 77, 1725–1726 CrossRef CAS.
- D. Sims, I. Sudbery, N. E. Ilott, A. Heger and C. P. Ponting, Nat. Rev. Genet., 2014, 15, 121–132 CrossRef CAS PubMed.
- M. C. Schatz, A. L. Delcher and S. L. Salzberg, Genome Res., 2010, 20, 1165–1173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc00114a |
| This journal is © The Royal Society of Chemistry 2016 |