
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous on-chip fluorescence labelling, free-flow isoelectric focusing and marker-free isoelectric point determination of proteins and peptides
Christin
Herzog
a,
Elisabeth
Poehler
a,
Andrea J.
Peretzki
a,
Sergey M.
Borisov
b,
Daniel
Aigner
b,
Torsten
Mayr
b and
Stefan
Nagl
 *a
*a
aInstitut für Analytische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: nagl@chemie.uni-leipzig.de; Web: http://research.uni-leipzig.de/nagl Tel: +49 341 97 36066
bInstitut für Analytische Chemie und Lebensmittelchemie, Technische Universität Graz, Stremayrgasse 9/III, 8010 Graz, Austria
First published on 24th March 2016
Abstract
We present a microfluidic platform that contains a micro flow reactor for on-chip biomolecule labelling that is directly followed by a separation bed for continuous free-flow electrophoresis and has an integrated hydrogel-based near-infrared fluorescent pH sensor layer. Using this assembly, labelling of protein and peptide mixtures, their separation via free-flow isoelectric focusing and the determination of the isoelectric point (pI) of the separated products via the integrated sensor layer could be carried out within typically around 5 minutes. Spatially-resolved immobilization of fluidic and sensing structures was carried out via multistep photolithography. The assembly was characterized and optimized with respect to their fluidic and pH sensing properties and applied in the IEF of model proteins, peptides and a tryptic digest from physalaemine. We have therefore realized continuous sample preparation and preparative separation, analyte detection, process observation and analyte assignment capability based on pI on a single platform the size of a microscope slide.
Introduction
Miniaturized lab procedures on microfluidic chips enjoyed good success and are now widely applied in certain areas such as flow reactors, PCR, electrophoresis or cytometry. A generation ago, the concept of a micro total analysis system (μTAS) was introduced, aiming at the integration of all required lab procedures on one single miniaturized chip platform.1Much progress has been achieved in this area, but in particular, for preparative multistep procedures to date, almost all platforms still fall short of being a satisfactory and economic solution for integration of all required laboratory working stages. Often, additional periods for sample preparation or postprocessing are required that are carried out outside chip platforms or only a small portion of the sample is transferred to the next step.
A promising method in this respect is micro free-flow electrophoresis (μFFE)2–4 because of its ability to continuously and preparatively separate and isolate biomolecules from mixtures on a small scale. Its flow rates that are compatible with reactors and other microfluidic structures, and its inherent compatibility with many matrices and buffers for biomolecules allow them to stay in their native conformation. FFE is often used for preseparation in advance of other procedures.5 In recent years, miniaturized integrated FFE assemblies have been presented by multiple groups in many variants.6–17
In some works, μFFE has been connected with other on- or off-chip functionalities. Geiger et al. have coupled an off-chip nano-LC column18 with chip-based FFE for the 2D separation of a tryptic digest, whereas Benz et al. have coupled μFFE with subsequent nanospray-MS for the mass analysis of organic separation products.19 Jezierski et al. have linked a flow reactor with micro free-flow zone electrophoresis on a single chip for the NBD-labelling and successive separation of amino acids.20
In order to gain real-time information about a preparative or analytical process on a microfluidic chip, an integrated sensing approach is needed. Many different methods have been pursued, but in particular, for the spatially-resolved observation of small molecules and process parameters, optical fluorescent and luminescent chemical sensors are very favorable.21,22 They may be assembled from molecular probes in suitable polymer matrices and integrated into microfluidic chips typically as a thin layer covering the whole microchannel network or only select areas.23
Fluorescent and luminescent pH sensors have been applied in microfluidic chips mainly for cell culture and monitoring of other bioprocesses and could be incorporated with a variety of different microfabrication techniques.24–32
As with other variations of electrophoresis, one mode of miniaturized FFE is microfluidic free-flow isoelectric focusing (μFFIEF), where compounds are separated in a pH gradient according to their isoelectric point (pI).33–37 IEF offers an alternative dimension for separation and the pI offers a means for determination or assignment of a particular compound. The pI may be used for identification of biological materials.38 So far, this is mostly performed using off-line methods and therefore needs additional laboratory working steps.39,40
We introduced a method for rapid determination of the isoelectric point (pI) of proteins and other (bio)-molecules during FFIEF using a fluorescent pH sensor layer in a separation bed.41 This assembly was able to determine the pI of proteins and other compounds on-chip with good precision and repeatability. Later, we extended this approach using inkjet printing of pH sensing matrices in select areas in a microfluidic chip42 and another readout method based on fluorescence lifetime measurements.43 However, in all cases, a fluorescence labelling step was still required prior to the chip-based FFIEF procedure.
To circumvent the off-chip labelling step in IEF, we introduced a semi-UV-transparent FFE platform along with a readout assembly that is capable of simultaneous monitoring of the intrinsic fluorescence of certain biomolecules when excited in the deep UV and near-infrared (NIR) monitoring of a pH sensor layer for pI determination.44 Therefore, unlabelled biomolecular mixtures could be separated and analysed. However, this system was limited to biomolecules that show sufficient UV fluorescence (mainly proteins and peptides with multiple tryptophans or tyrosines), required an elaborate and expensive optical setup and was lower in sensitivity than a detection based on label fluorescence.
Herein, we now demonstrate a generally applicable solution for integration of the working steps of analyte visualization, FFIEF separation, and determination of isoelectric points. We describe a combined miniaturized chip platform the size of a microscope glass slide that is able to perform continuous on-chip labelling of biomolecular samples and mixtures, their free-flow isoelectric focusing, observation of the pH gradient and assignment of isoelectric points (pI) based on an integrated pH sensor layer, all within a total processing time of about five minutes. This platform was successfully validated with mixtures of proteins, peptides and a tryptic digest of physalaemin.
Experimental section
Materials
Oligoethyleneglycol diacrylate (OEG-DA258, Mn ∼ 258), oligoethyleneglycol diacrylate (OEG-DA700, Mn ∼ 700), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (HHMP), 4-acryloylmorpholine (AM), 2,2-dimethoxy-2-phenylacetophenone (DMPA), (3-methacryloyloxypropyl)-trichlorosilane (TPM), trichloromethane, Tween 20, phosphate buffered saline (PBS), bovine serum albumin (BSA), conalbumin type I (chicken egg white), myoglobin (horse heart), lactalbumin type III (bovine milk), β-lactoglobulin B (bovine milk), ubiquitin (bovine erythrocytes), neurotensin, oxytocin, leucine-enkephalin, physalaemin and endothelin were purchased from Sigma-Aldrich (Steinheim, Germany). n-Heptane was bought from Roth (Karlsruhe, Germany). Dimethyl sulfoxide (DMSO), tris(hydroxymethyl)-aminomethane (TRIS), H3BO3, H3PO4, CH3COOH, NaOH and NaHCO3 were from Merck (Darmstadt, Germany). The ampholyte mixture pH 4–7 was purchased from AppliChem (Darmstadt, Germany) and the activated labelling dye Atto 425-NHS from Atto-Tec (Siegen, Germany). The NIR-fluorescent pH probe N-(3-([N-(3-(methacryloylamino)-propyl)amino]sulfonyl)-2,6-diisopropyl-phenyl)-N′-(4-([N-(3-(methacryloylamino)propyl)-amino]-sulfonyl)-2,6-diisopropylphenyl)-1-(4-methyl-1-piperazinyl)-6,7,12-trichloroperylene-3,4,9,10-tetracarboxylic bisimide (a perylene bisimide derivative, abbreviated as PBI herein) was synthesized by D. Aigner according to ref. 45.Britton–Robinson buffers (BRB) were made from 10 mM H3BO3, H3PO4 and CH3COOH, and the pH was adjusted by titration with 1 M NaOH solution and monitored using a Lab 850 pH meter (SI Analytics, Mainz, Germany).
Microchip fabrication
Fabrication of microfluidic chips with an integrated optical pH sensor layer was performed using multiple photopolymerization steps in a procedure related to and extended from that in ref. 41. Briefly, in the first step, the sensor layer was fabricated between two glass slides, one was with acrylate surface modification and one was untreated. We used a 60 μL mixture of 85.0% (w/w) acryloylmorpholine, 14.8% (w/w) oligoethylene glycol (OEG-DA700), 0.02% (w/w) PBI as a pH probe and 0.2% (w/w) photoinitiator. Photopolymerization was carried out using a 4′′ FE5 Flood Exposure (Süss MicroTec, Munich, Germany) device equipped with a Hg lamp (14 mW cm−2 at 365 nm) at an illumination time of 15 s. After the exposure, the unacrylated glass slide was removed.In the second step, the microfluidic structure was prepared on top of the sensing layer. 100 μL of OEG-DA258 with 1% (w/w) 2,2-dimethoxy-2-phenylacetophenone was spread over the sensing layer and covered with a microscope slide as a top plate. The top plate was equipped with access holes generated using a Point II powder blaster (Barth, Königsbach-Stein, Germany). To achieve defined channel heights of the fluidic structure, adhesive transfer foil (467MP adhesive transfer tape, 60 μm layer thickness, 3M, Neuss, Germany) was used as a spacer between the sensing layer and the top plate. The sandwich of glass slides, sensor layer and prepolymer was illuminated for 1.3 s using a photomask to define the microfluidic channels. The non-polymerized prepolymer was removed from the structure by aspiration. For complete curing, the whole microchip was illuminated for 1.3 s again.
Optical setup and sensor characterisation
The microchip was positioned on an IX 70 epi-fluorescence microscope (Olympus, Hamburg, Germany) equipped with a ProEM 512 CCD camera (Princeton Instruments, Acton, USA), a Fluar 2.5×/0.12 objective (Zeiss, Jena, Germany), two LEDs (420 nm: M420L3, 660 nm: M660L3, Thorlabs, Dachau, Germany) and filter sets (420 nm: λexc 412–438 nm, λem 470–550 nm, 660 nm: λexc 620–680 nm, λem 698–766 nm) for fluorescence detection during the FFIEF separation.The pH dependency of the integrated sensor layer was measured via flushing of the microchip with Britton–Robinson buffers (BRB) of different pH values (3–10). The flow rate was 50 μL min−1 and fluorescence intensity was recorded using the CCD camera. For flow stability, the microchip was flushed with pH 10 BRB and the fluorescence intensity was observed over time. Permanent illumination at λexc 660 nm using the LED was employed to investigate the photostability of the sensor layer. Fluorescence intensities were observed over 30 min and were normalized to the initial intensity value.
Response times were determined by flushing the microchip under the same conditions periodically with pH 3 and pH 10 BRBs using a photomultiplier tube (PMT) H7711 (Hamamatsu, Herrsching, Germany) as the detector.
Labelling reaction
The online labelling reaction was performed as follows: biomolecules were dissolved in ampholyte solution (0.1% ampholyte pH 4–7 and 0.05% Tween 20) to get a stock solution. This stock solution was diluted in labelling buffer (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) 200 mM sodium bicarbonate buffer pH 9 and 150 mM PBS pH 7.4 with an overall pH of 8.3) to obtain concentrations of 10 μM for proteins and 100 μM for peptides, except endothelin that had 40 μM.
:1 (v/v) 200 mM sodium bicarbonate buffer pH 9 and 150 mM PBS pH 7.4 with an overall pH of 8.3) to obtain concentrations of 10 μM for proteins and 100 μM for peptides, except endothelin that had 40 μM.
The labelling dye solution consisted of Atto 425 stock solution (1 mg of Atto 425-NHS in 100 μL of DMSO) in labelling buffer with a final concentration of 10 μM dye. The biomolecule solution and the labelling dye solution were continuously fed into the on-chip micro reactor using the flowing flow rates: 300 nL min−1 biomolecule solution and 100 nL min−1 dye solution for the protein and peptide separations (Fig. 2 and 3a–c), and 400 nL min−1physalaemin tryptic digest solution along with 200 nL min−1 dye solution (Fig. 3d).
Free-flow isoelectric focusing
To realize the isoelectric focusing in a microfluidic chip, syringe pumps (cetoni neMESYS low pressure, Korbussen, Germany) and a high voltage source (HCL 35-6500, FuG Elektronik, Rosenheim, Germany) were used. The observation of analytes and the pH sensor layer was accomplished in different spectral channels using distinct optical filter sets as mentioned above.An electrical contact was accomplished via external electrodes that were plugged into the anolyte and catholyte channels (see Fig. 1). Copper wires were put inside silicone tubes and fixated with Elastosil E43. A short piece of silicone tube was used as a sleeve and pulled over the pinching hole. It was also fixated with silicone glue. Therefore, an electrical contact from the outside of the tube to the inside, where the respective solution is pumped through, is established (Table 1).
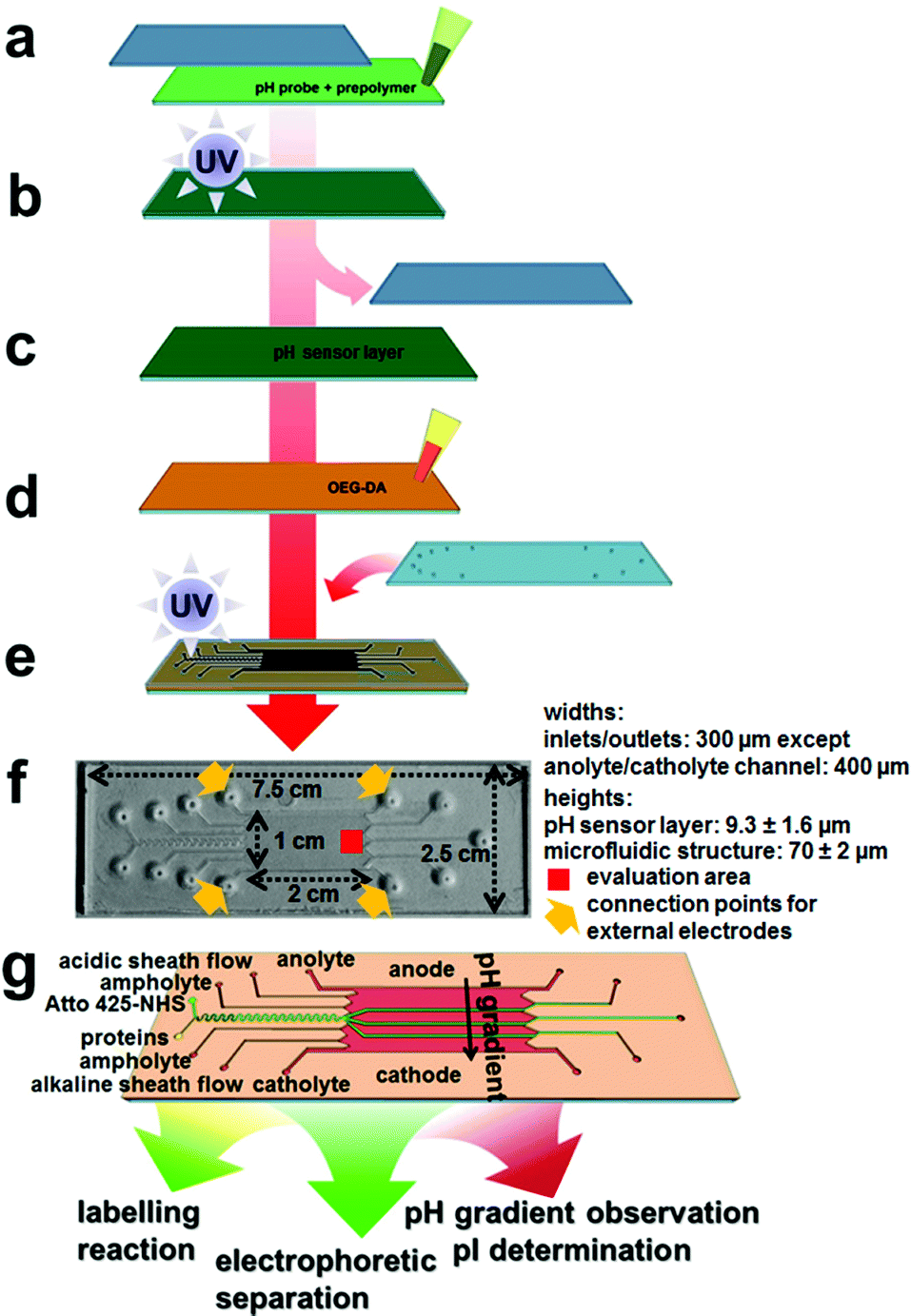 | ||
| Fig. 1 Fabrication of microfluidic chips for integrated labelling, IEF and pH sensing. (a) Application of pH probe and matrix prepolymer onto acrylated glass, sealing with cover glass, (b) photopolymerization of the pH sensor layer, (c) removal of cover glass, (d) application of OEG-DA onto the sensor layer, (e) addition of cover glass with access holes, masked photopolymerization of the microfluidic structure, (f) picture of a completed microfluidic chip with added relevant length scales. The evaluation area (red) and the connection points for external electrodes (orange) are indicated, and (g) schematic of the chip and channel layout. | ||
| Channel | Proteins (Fig. 2) | Peptides (Fig. 3c) | Proteins (Fig. 3d) | Digest (Fig. 3e) |
|---|---|---|---|---|
| a Flow rates are in μL min−1. b Linear velocities are in mm s−1. c Electric field strengths are in V cm−1. | ||||
| Anolytea | 7 | 4 | 7 | 7 |
| Acidic sheatha | 1 | 2 | 1 | 5 |
| Ampholytea | 1 | 1 | 1 | 3 |
| Labela | 0.1 | 0.1 | 0.1 | 0.2 |
| Proteinsa | 0.3 | 0.3 | 0.3 | 0.4 |
| Ampholytea | 1 | 1 | 1 | 3 |
| Alkal. sheatha | 1 | 2 | 1 | 5 |
| Catholytea | 7 | 4 | 7 | 7 |
| Linear velocityb | 0.44 | 0.34 | 0.44 | 0.73 |
| Electric fieldc | 40 | 20 | 300 | 17.5 |
For FFIEF, the pH sensor layer was illuminated using a 660 nm LED and the Atto 425-labelled compounds were excited using a 420 nm LED with a driving current of 1 A each using the abovementioned filter sets.
Tryptic digestion
The degradation of physalaemin was performed off-chip (Scheme 1). Therefore, a solution of 1.07 mM physalaemin was prepared. To 250 μL of this solution, 53 μL of a 100 μM trypsin solution was added, which corresponded to 2 mol% (w/w) with respect to the peptide. The reaction buffer was 100 mM TRIS buffer pH 8.2, and the final concentration of the peptide in the reaction solution was 966 μM. This solution was shook for 30 min at 37 °C and introduced onto the microchip subsequently.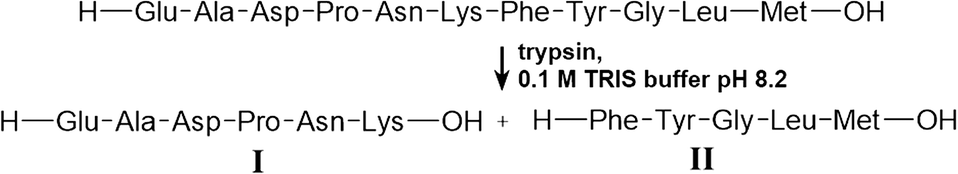 | ||
| Scheme 1 Tryptic digestion of physalaemin. | ||
Results and discussion
We present a continuously working process and analysis platform with integrated sample preparation, process observation and analyte assignment capabilities. Therefore, a micro flow reactor, a microfluidic channel structure for free-flow isoelectric focusing and a pH sensor layer were combined in one miniaturized chip platform.Microchip fabrication
The microfluidic chip containing multiple functionalities was fabricated from an acrylate-modified glass slide where the pH sensor prepolymer was applied and photopolymerized to a height of 9.3 ± 1.6 μm between the acrylated bottom slide and a plain borosilicate top glass slide with a visually very homogeneous distribution of the pH sensing matrix (Fig. 1a and b). The non-functionalized top slide did not bind the sensor matrix covalently and could be removed (Fig. 1c).The microfluidic structure could be manufactured on top of the pH sensor layer using the short-chain oligoethylene glycol acrylate prepolymer OEG-DA258 (Mn ∼ 258). This compound is moderately hydrophobic both in its initial and its polymerized form due to its molecular structure being dominated by acrylate groups.
Therefore, it does not mix well with the hydrogel sensor layer on the bottom and can be applied as a thin film on top of it (Fig. 1d). A top glass slide with powderblasted inlet and outlet holes and a printed photomask was added and the assembly was photopolymerized (Fig. 1e). Adhesive transfer tape (467MP from 3M) was used at the edges of the glass slide-based structure to obtain a structural height of ∼70 μm.
A picture of a completed platform is shown in Fig. 1f, and a schematic of the design is shown in Fig. 1g. The serpentine-shaped micro flow reactor, inlet and outlet channels were 300 μm in width, and the electrode channels were 400 μm in width. The microreactor was 50 mm in effective length and the separation bed for FFIEF was 10 mm in width and 20 mm in length. This layout allowed for straightforward introduction of all fluids and good reaction and separation characteristics, although only a fraction of the actual FFIEF bed width was employed for biomolecular separations. This microfluidic assembly could be fabricated in a regular laboratory environment within a couple of hours. A cleanroom environment is advantageous but not mandatory for chip fabrication.
Integrated pH sensor characteristics
The fluorescent pH sensor layer we used here is similar to that used an earlier study and characterized in more detail there.44 Its use stems from two main advantages with respect to the application presented here. (A) Due to its generation via photopolymerization from liquid precursors, it may readily be integrated into a microfluidic chip fabrication protocol, as shown in the preceding section. (B) The spectral position of its absorbance and emission maximum in the far-red/near infrared allows enough space to use a different fluorescent dye in the visible range as a label for biomolecules without interference by the sensor layer. When suitable light sources are provided, the two channels may be switched easily via the choice of filters in a standard inverted fluorescence microscope.Generally, the covalent immobilisation of the pH probe into the sensor matrix and of the sensing matrix on the glass slide allowed for high flow stability, with a decrease in fluorescence intensity of only 3.7 ppm s−1 upon application of a flow rate of 50 μL min−1 in the FFIEF separation bed. The photostability is also very good and in accordance with the values reported in the literature.13 An intensity decrease of ∼27 ppm s−1 at constant excitation with a 660 nm LED could be determined. Decreasing fluorescence intensity with higher pH was observed due to photoinduced electron transfer (PET) in the pH probe. The pKa was determined as 5.91 ± 0.09 and the working range is roughly between pH 4.7 and 7.2. The pH sensor showed fast response times (t95) of about 6 to 7 seconds upon changing the pH from 3 to 10 and back and therefore enabled almost real-time observation of the pH.
On-chip labelling
Herein, we selected the fluorescent dye Atto 425 in its reactive NHS-ester form for the on-chip conjugation of biomolecules. This compound was chosen for several reasons; it is brightly fluorescent with a high quantum yield and therefore enables sensitive detection. With its absorption (λex: 436 nm) and emission (λem: 484 nm) maximum in the blue spectral range, it is spectrally well-separated from the NIR pH sensor matrix and, therefore, both the label and the pH probe could be detected without virtually no interference from each other.Most importantly, however, due to its small molecular mass and its tertiary amino group, the bioconjugate with this label is expected to have a very similar isoelectric point (pI) than the unconjugated parent compound. Upon bioconjugation with Atto 425, one amino group is replaced with another and the small coumarin-based chromophore adds only insignificant molar mass e.g. to an average protein (Fig. 2a). This is in contrast to many other labels that introduce charges or significantly more mass to a conjugate. However, in principle, suitable labels could also be found from other compound classes.
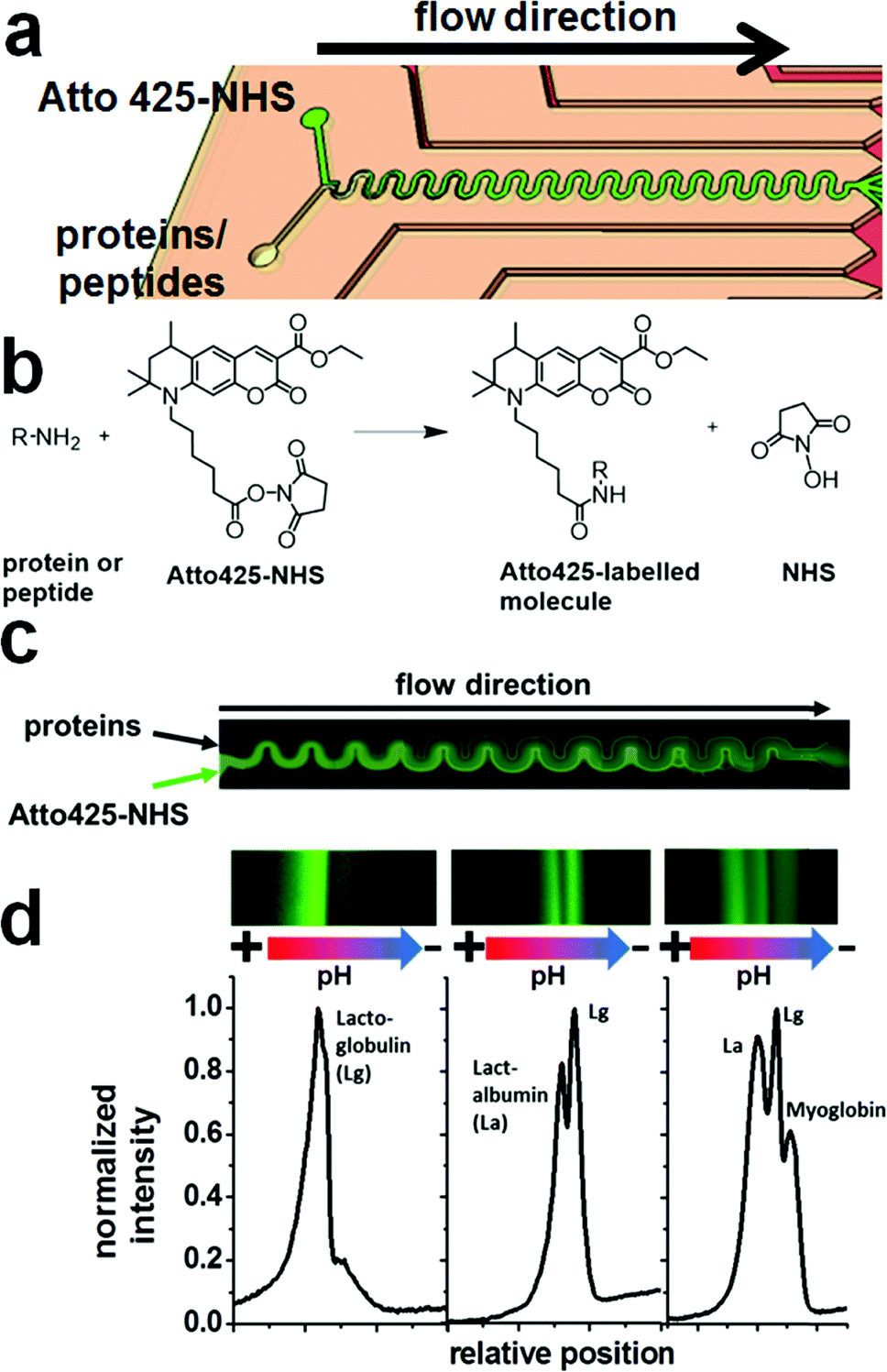 | ||
| Fig. 2 (a) Schematic of the microreactor part of the microfluidic chip, (b) reaction scheme for the labelling of a biomolecular amino group with the coumarin derivative Atto 425-NHS, (c) example flow reaction of a protein mixture in the on-chip microreactor (stitched from multiple false-coloured fluorescence images), and (d) fluorescence images (false-coloured) and resulting electropherograms from different reaction mixtures, isoelectrically focused after on-chip labelling in the microreactor, from left to right: lactoglobulin (Lg, left), lactalbumin (La) and Lg (middle), La, Lg and myoglobin (right). | ||
For the realization of an on-chip labelling reaction, sufficient mixing and fast reaction of substrates in a reactor channel is required. The mixing was investigated in the flow reactor of 50 mm in effective length with different flow rates and relative substrate flows. Up to a total flow velocity of 0.56 mm s−1 (0.8 μL min−1), complete mixing of the flows in the reactor was observed. (Fig. 2b) The mixing was found to be mostly independent of the ratio of the inlet flows.
To test the efficiency of the on-chip labelling reaction at the optimized settings (300 nL min−1 for the protein solution and 100 nL min−1 for the Atto 425-NHS solution, see the exp. section), a different number of proteins was introduced into the reactor and the reaction products were separated with FFIEF (Fig. 2c).
The number of bands was found to be equal to the number of different proteins in all cases, confirming the on-chip reaction of Atto 425-NHS with the model biomolecules. However, no bands of the unconjugated dye could be observed. It could be assumed that unconjugated dye was either present in too small concentration to be detected or attached non-covalently to the biomolecules or ampholytes or reacted with the latter also.
Protein and peptide separations
After the microreactor step, the reaction products were then introduced into the separation bed that had the same height of around 70 μm and contained a ca. 9 μm thick pH sensing hydrogel layer on the bottom surface (Fig. 3a). The fluorescence signal of the pH sensor layer was plotted in red in Fig. 3, whereas the Atto 425 label fluorescence of the focused bands is displayed in green.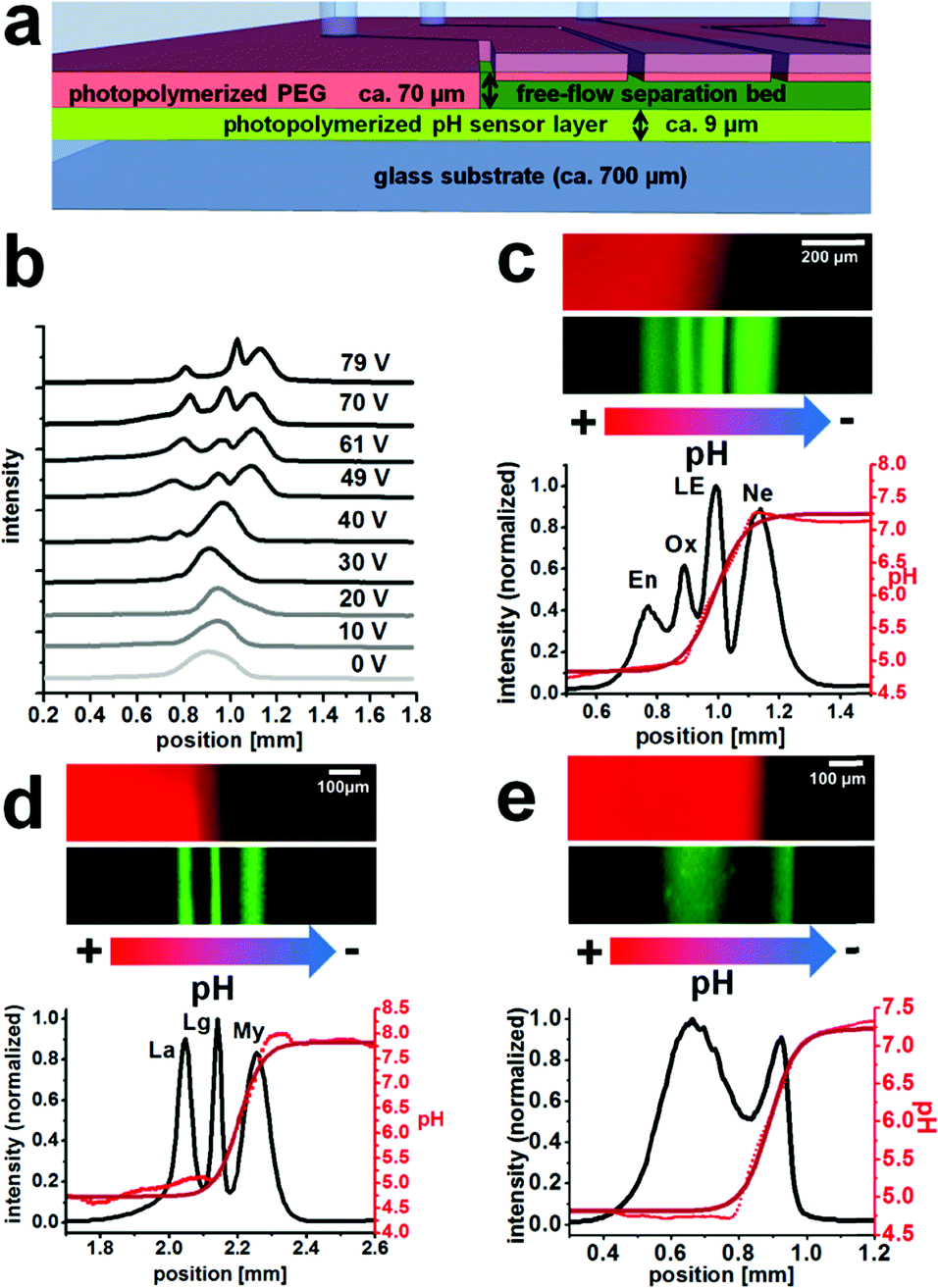 | ||
| Fig. 3 (a) Schematic cross-section through the microfluidic chip within the separation area (not to scale), (b) electropherograms of peptide IEF (En, Ox and Ne) depending on the applied electric potential, (c, d and e) IEF with simultaneous pH gradient observation (false-colored fluorescence images, green: labeled compounds, red: pH sensor layer), (c) separation of the peptides endothelin (En), oxytocin (Ox), leucine-enkephalin (LE) and neurotensin (Ne), (d) separation of the proteins lactalbumin (La), lactoglobulin (Lg) and myoglobin (My), and (e) separation of the products from a tryptic digest of physalaemin. The absolute positions displayed on the x-axis correspond to the evaluation area and were different for each experiment. | ||
The integral fluorescence at each point on the x-axis was used to create a graph of position versus fluorescence intensity (Fig. 3c–e).The black line is the intensity of the Atto 425 channel displaying the separation, the bright red dots are the individual pH values at each position obtained via entering the intensities into the pH calibration curve, and the dark red line is a fit of the pH curve. Regions where the pH curve is flat indicate the borders of the dynamic range of the employed sensor layer. The pH is not necessarily constant across these areas.
In the separation bed, the reactor outlet fluid was mixed with ampholyte pH 4–7 solution and further sheath flows and IEF was performed upon initiation of an electric field perpendicular to the flow regime. Application of a high voltage electric field may cause local heating that can disturb the separation. However, since we operated with relatively modest voltages and fast flow rates, we did not find this to be a major issue in the experiments shown herein. Fig. 3b shows an example optimization process of the operating voltage at an overall linear velocity of 0.86 mm s−1 for the IEF of the peptides En, Ox and NE with the optimum around 70 V.
The separation of four on-chip Atto 425-labelled neuropeptides (endothelin, oxytocin, leucine-enkephalin and neurotensin) could be demonstrated at a linear velocity of 0.34 mm s−1 and an electric field of 20 V cm−1, with chromatographic peak resolutions of 0.7, 0.8 and 0.9 (Fig. 3c). The integrated pH sensor layer allowed in situ determination of the pI of the separated compounds. For the neuropeptides oxytocin and leucine-enkephalin that are known to have a pI value inside the working range of the pH sensor (approx. from 4.7 to 7.2), good results with less than 0.1 pH unit difference with respect to values in the literature were obtained (Table 2).
| Compound | Lit. pIa | Exp. pI | pI difference |
|---|---|---|---|
| a Protein pKa values are from ref. 46, theoretical peptide pKa values were calculated using ref. 47. | |||
| Lactalbumin (La) | 4.5–4.8 | 4.63 ± 0.18 | 0.0 |
| Lactoglobulin (Lg) | 5.2–5.4 | 5.35 ± 0.15 | 0.0 |
| Myoglobin (My) | 6.5–7.2 | 7.41 ± 0.24 | 0.2 |
| Endothelin (En) | 4.3 | 4.79 ± 0.06 | 0.5 |
| Oxytocin (Ox) | 5.0–5.2 | 5.01 ± 0.06 | 0.0 |
| Leucine-Enkephalin (LE) | 5.9–6.0 | 6.08 ± 0.13 | 0.1 |
| Neurotensin (Ne) | 9.52 | 7.18 ± 0.10 | 2.3 |
| Physalaemin, fission product I | 3.99 | 4.82 ± 0.04 | 0.8 |
| Physalaemin, fission product II | 6.00 | 6.06 ± 0.06 | 0.1 |
Endothelin and neurotensin showed a larger deviation but have a pI value that is outside the pH sensor fitting range (Fig. 3c). It should also be mentioned that an ampholyte with a pH of 4 to 7 was used, which is not well-suited for neurotensin with a pI of around 9.5. The reproducibility of pI determination was generally very good and low standard deviations were obtained from repeated pI determination.
Equally, the labelled proteins lactalbumin, lactoglobulin and myoglobin could be separated continuously using 300 V cm−1 at 0.44 mm s−1 with a good chromatographic resolution of 1.0 between either of the bands (Fig. 3d).
Their bands could easily be assigned based on the measured isoelectric points and showed good agreement with values in the literature (Table 2). It would be possible to confirm the assignment by guiding fractions to distinct outlets and analyzing their content, but that was not pursued within the course of this work.
Finally, a tryptic digest of the peptide physalaemin was investigated. The digest itself was executed off-chip in a reaction vial and was described to lead to two fission products (Scheme 1). The resulting mixture was introduced into the microchip, labelled with Atto 425 and electrophoretically separated (Fig. 3e) at a field strength of 17.5 V cm−1 and a linear velocity of 0.73 mm s−1. Two bands could be identified and separated with a chromatographic resolution of 0.9. They may be assigned to the fission products, possibly with a background of unreacted peptide in the left band.
The unreacted peptide and one fission product are expected to have a pI value of around 4.0. The pI value determined for this band is around 4.8 which is at the edge of the working range of the pH sensor (Fig. 3e). Although this value is different from the expected value of 4.0 for the first fission product, the integrated pH sensor correctly assigns a pI value of 4.8 or less to this band. The pI value of the second band was within the working range of the sensor and determined as 6.06. This is very close to the calculated value of 6.0 for this compound. A trypsin band could not be observed, presumably due to its low concentration or degradation.
By translating the flow rates and linear velocities in the chip into residence times, it may be stated that all the three biomolecule mixtures could be continuously labelled and separated, and that the isoelectric point values could be determined with the overall residence times in the microreactor and subsequently in the μFFE, all within around four to six minutes. This impressively demonstrates the capability of miniaturized, microfluidic chip-based systems to perform multiple laboratory working steps using only a fraction of the time and sample amounts of conventional bench-scale procedures.
Conclusions
We introduced a multifunctional microfluidic chip that allowed the combination of many processing steps on one single platform. Biomolecules could be successfully labelled in an on-chip micro flow reactor that led to a micro free-flow isoelectric focusing system where the compounds could be continuously separated and detected via label fluorescence.Additionally, the integrated pH sensor layer allowed the observation of the pH gradient in IEF and the quantification of the pI value of focused bands. Since the pI is a characteristic value of each biomolecule, it allows mapping and potential assignment of separated molecules. The working principle could be successfully shown with peptides, proteins and a tryptic digest.
In the future, this work and the functionalities that are present on this single continuous labelling, separation and sensing platform could be further extended, e.g. by integration of a second reactor, other pH sensor matrices that cover one another and a wider working range, or microfluidic chip integration of cell lysis or alternative detection modalities in either processing step.
Acknowledgements
Financial support of this work by the German Research Foundation (DFG, NA 947/1-2 and 2-1) is gratefully acknowledged.References
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- D. Kohlheyer, J. C. T. Eijkel, A. van den Berg and R. B. M. Schasfoort, Electrophoresis, 2008, 29, 977–993 CrossRef CAS PubMed.
- R. T. Turgeon and M. T. Bowser, Anal. Bioanal. Chem., 2009, 394, 187–198 CrossRef CAS PubMed.
- F. J. Agostino and S. N. Krylov, TrAC, Trends Anal. Chem., 2015, 72, 68–79 CrossRef CAS.
- A. Rizzi, C. Gerner, M. Islinger, C. Eckerskorn and A. Völkl, Electrophoresis, 2010, 31, 1754–1763 CrossRef PubMed.
- D. Janasek, M. Schilling, J. Franzke and A. Manz, Anal. Chem., 2006, 78, 3815–3819 CrossRef CAS PubMed.
- D. P. de Jesus, L. Blanes and C. L. do Lago, Electrophoresis, 2006, 27(24), 4935–4942 CrossRef PubMed.
- B. R. Fonslow and M. T. Bowser, Anal. Chem., 2008, 80, 3182–3189 CrossRef CAS PubMed.
- D. Kohlheyer, J. C. T. Eijkel, S. Schlautmann, A. van den Berg and R. B. M. Schasfoort, Anal. Chem., 2008, 80, 4111–4118 CrossRef CAS PubMed.
- Y. S. Huh, T. J. Park, K. S. Yang, E. Z. Lee, Y. K. Hong, S. Y. Lee, D. H. Kim and W. H. Hong, Ultramicroscopy, 2008, 108, 1365–1370 CrossRef CAS PubMed.
- M. Becker, A. Mansouri, C. Beilein and D. Janasek, Electrophoresis, 2009, 30, 4206–4212 CrossRef CAS PubMed.
- P. Wang, L. Zhang, Y. Shan, Y. Cong, Y. Liang, B. Han, Z. Liang and Y. Zhang, J. Sep. Sci., 2010, 33, 2039–2044 CrossRef CAS PubMed.
- S. Koehler, C. Weilbeer, S. Howitz, H. Becker, V. Beushausen and D. Belder, Lab Chip, 2011, 11, 309–314 RSC.
- S. Podszun, P. Vulto, H. Heinz, S. Hakenberg, C. Hermann, T. Hankemeier and G. A. Urban, Lab Chip, 2012, 12, 451–457 RSC.
- H. Ding, X. Li, X. Lv, J. Xu, X. Sun, Z. Zhang, H. Wang and Y. Deng, Analyst, 2012, 137, 4482–4489 RSC.
- J. E. Prest, S. J. Baldock, P. R. Fielden, N. J. Goddard, R. Goodacre, R. O'Connor and B. J. Treves Brown, J. Chromatogr., B, 2012, 903, 53–59 CrossRef CAS PubMed.
- F. J. Agostino, L. T. Cherney, V. Galievsky and S. N. Krylov, Angew. Chem., Int. Ed., 2013, 52, 7256–7260 CrossRef CAS PubMed.
- M. Geiger, N. W. Frost and M. T. Bowser, Anal. Chem., 2014, 86, 5136–5142 CrossRef CAS PubMed.
- C. Benz, M. Boomhoff, J. Appun, C. Schneider and D. Belder, Angew. Chem., Int. Ed., 2015, 54, 2766–2770 CrossRef CAS PubMed.
- S. Jezierski, V. Tehsmer, S. Nagl and D. Belder, Chem. Commun., 2013, 49, 11644–11646 RSC.
- C. McDonagh, C. S. Burke and B. D. MacCraith, Chem. Rev., 2008, 108, 400–422 CrossRef CAS PubMed.
- M. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102–3114 RSC.
- S. A. Pfeiffer and S. Nagl, Methods Appl. Fluoresc., 2015, 3, 034003 CrossRef.
- W. Zhan, G. H. Seong and R. M. Crooks, Anal. Chem., 2002, 74, 4647–4652 CrossRef CAS PubMed.
- W.-G. Koh and M. Pishko, Sens. Actuators, B, 2005, 106, 335–342 CrossRef CAS.
- P. Mela, S. Onclin, M. H. Goedbloed, S. Levi, M. F. Garcia-Parajo, N. F. van Hulst, B. J. Ravoo, D. N. Reinhoudt and A. van den Berg, Lab Chip, 2005, 5, 163–170 RSC.
- N. Klauke, P. Monaghan, G. Sinclair, M. Padgett and J. Cooper, Lab Chip, 2006, 6, 788–793 RSC.
- A. R. Thete, G. A. Gross, T. Henkel and J. M. Koehler, Chem. Eng. J., 2008, 135, 327–332 CrossRef.
- A. Funfak, J. Cao, O. S. Wolfbeis, K. Martin and J. M. Köhler, Microchim. Acta, 2009, 164, 279–286 CrossRef CAS.
- L. Florea, C. Fay, E. Lahiff, T. Phelan, N. E. O'Connor, B. Corcoran, D. Diamond and F. Benito-Lopez, Lab Chip, 2013, 13, 1079–1085 RSC.
- S. G.-D. Pedro, A. Salinas-Castillo, M. Ariza-Avidad, A. Lapresta-Fernández, C. Sánchez-González, C. S. Martínez-Cisneros, M. Puyol, L. F. Capitan-Vallvey and J. Alonso-Chamarro, Nanoscale, 2014, 6, 6018–6024 RSC.
- S. Krabbe, D. E. Achatz, T. Nieradzik, C. Gerhardy and W. K. Schomburg, Procedia Eng., 2015, 120, 598–601 CrossRef CAS.
- Y. Xu, C.-X. Zhang, D. Janasek and A. Manz, Lab Chip, 2003, 3, 224–227 RSC.
- D. Kohlheyer, G. A. J. Besselink, S. Schlautmann and R. B. M. Schasfoort, Lab Chip, 2006, 6, 374–380 RSC.
- Y.-A. Song, M. Chan, C. Celio, S. R. Tannenbaum, J. S. Wishnok and J. Han, Anal. Chem., 2010, 82, 2317–2325 CrossRef CAS PubMed.
- B. Walowski, W. Hüttner and H. Wackerbarth, Anal. Bioanal. Chem., 2011, 401, 2465–2471 CrossRef CAS PubMed.
- L.-J. Cheng and H.-C. Chang, Lab Chip, 2014, 14, 979–987 RSC.
- J. Kiraga, P. Mackiewicz, D. Mackiewicz, M. Kowalczuk, P. Biecek, N. Polak, K. Smolarczyk, M. R. Dudek and S. Cebrat, BMC Genomics, 2007, 8, 163 CrossRef PubMed.
- P. G. Righetti, J. Chromatogr. A, 2004, 1037, 491–499 CrossRef CAS PubMed.
- J. Wen, E. W. Wilker, M. B. Yaffe and K. F. Jensen, Anal. Chem., 2010, 82, 1253–1260 CrossRef CAS PubMed.
- S. Jezierski, D. Belder and S. Nagl, Chem. Commun., 2013, 49, 904–906 RSC.
- C. Herzog, E. Beckert and S. Nagl, Anal. Chem., 2014, 86, 9533–9539 CrossRef CAS PubMed.
- E. Poehler, C. Herzog, M. Suendermann, S. A. Pfeiffer and S. Nagl, Eng. Life Sci., 2015, 15, 276–285 CrossRef CAS.
- E. Poehler, C. Herzog, C. Lotter, S. A. Pfeiffer, D. Aigner, T. Mayr and S. Nagl, Analyst, 2015, 140, 7496–7502 RSC.
- D. Aigner, S. M. Borisov, P. Petritsch and I. Klimant, Chem. Commun., 2013, 49, 2139 RSC.
- (a) P. G. Righetti and T. Caravaggio, J. Chromatogr., 1976, 127, 1–28 CrossRef CAS PubMed; (b) P. G. Righetti, G. Tudor and K. Ek, J. Chromatogr., 1981, 220, 115–194 CrossRef CAS.
- Unknown authors, Protein/Peptide Property Calculator, http://www.lifetein.com/peptide-analysis-tool.html, accessed on October 20, 2015.
| This journal is © The Royal Society of Chemistry 2016 |