
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-chip microtubule gliding assay for parallel measurement of tau protein species†
Subhathirai
Subramaniyan Parimalam
a,
Mehmet C.
Tarhan
bc,
Stanislav L.
Karsten
cd,
Hiroyuki
Fujita
c,
Hirofumi
Shintaku
a,
Hidetoshi
Kotera
a and
Ryuji
Yokokawa
*a
aDepartment of Micro Engineering, Kyoto University, Kyoto, Japan. E-mail: ryuji@me.kyoto-u.ac.jp
bLaboratory for Integrated Micro Mechatronic Systems (LIMMS), Institute of Industrial Science (IIS), The University of Tokyo, Tokyo, Japan
cCenter for International Research on Micronano Mechatronics (CIRMM), Institute of Industrial Science (IIS), The University of Tokyo, Japan
dNeuroInDx Inc., Signal Hill, CA, USA
First published on 23rd March 2016
Abstract
Tau protein is a well-established biomarker for a group of neurodegenerative diseases collectively called tauopathies. So far, clinically relevant detection of tau species in cerebrospinal fluid (CSF) cannot be achieved without immunological methods. Recently, it was shown that different tau isoforms including the ones carrying various types of mutations affect microtubule (MT)–kinesin binding and velocity in an isoform specific manner. Here, based on these observations, we developed a microfluidic device to analyze tau mutations, isoforms and their ratios. The assay device consists of three regions: a MT reservoir which captures MTs from a solution to a kinesin-coated surface, a microchannel which guides gliding MTs, and an arrowhead-shaped collector which concentrates MTs. Tau-bound fluorescently labeled MTs (tau-MTs) were assayed, and the increase in fluorescence intensity (FI) corresponding to the total number of MTs accumulated was measured at the collector. We show that our device is capable of differentiating 3R and 4R tau isoform ratios and effects of point mutations within 5 minutes. Furthermore, radially oriented collector regions enable simultaneous FI measurements for six independent assays. Performing parallel assays in the proposed device with minimal image processing provides a cost-efficient, easy-to-use and fast tau detection platform.
Introduction
An early and differential diagnosis of neurodegenerative diseases is essential for effective therapeutic intervention and for the management of the disease outcome. The presence of distinct tau pathology specific to each tauopathy makes this protein an important differential biomarker.1,2 Six tau isoforms are expressed in neurons, and they differ according to the number of microtubule binding repeats (MTBRs; R1–R4 repeats) at the C-terminal region and the length of projection domains (0N–2N) at the N-terminal region.3 The levels of cerebrospinal fluid (CSF) total tau,4 phospho-tau5 and the tau isoform ratio6 (3R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4R) are evaluated in order to characterise the intracellular pathology.
:4R) are evaluated in order to characterise the intracellular pathology.
Currently, immuno-based methods such as the enzyme-linked immuno sorbent assay (ELISA) have been used to clinically detect CSF-tau species.7,8 This immuno-based method has been further developed by combining it with the two-photon Rayleigh scattering technique, in order to demonstrate highly sensitive CSF-tau detection.9 Non-immuno detection, based on electrochemical impendence spectroscopy,10 and capillary electrophoresis-based enzymatic reaction have also been reported.11 However, since standardized clinical procedures have not been well developed and established for reliable CSF-tau detection,12 alternative methods are in high demand.
Recently, a microtubule (MT)–kinesin-based transport system has been successfully validated for tau detection in two assay geometries. (i) In a lab-on-a-chip compatible kinesin motility assay, kinesin velocity was assayed on tau-bound MTs (tau-MTs) to differentiate various tau species. To increase the assay's sensitivity, tau-MTs were suspended between micro-scale walls.13 However, this method had a complex experimental setup and a long turnaround time (TAT) for multiple data processing. (ii) In a MT gliding assay, tau-MTs were assayed over a kinesin-coated surface using three parameters: MT landing rate, density and gliding velocity were used to differentiate tau species. The MT landing rate and MT density were defined as the number of MTs that landed per unit time per unit area and the number of surface-attached MTs per unit area, respectively.14–17 We previously reported a MT gliding assay based on tau specific interference on the MT–kinesin interaction.18 In brief, the interference was mainly defined by their MT binding properties, such as binding affinity,3 the steric effect of MTBRs19 and the protein conformation of tau when it binds to a MT surface.20 We demonstrated that MT–kinesin interaction is inhibited by 4R tau isoforms, consequently lowering the affinity of MTs for the kinesin-coated surface, together with their gliding capacity. Based on this previously established role of tau proteins in MT gliding assays,15,18,21 we designed a microfluidic device in an attempt to efficiently differentiate various tau species.
Materials and methods
Design of the tau detection device
We designed a microfluidic device with an assay region composed of a MT reservoir22 (24 × 104 μm2 in area), an arrowhead-shaped MT collector23 (175 μm2 in area, angles a = 20° and b = 55°), and a microchannel connecting the reservoir and the collector24 (100 μm in length and 5 μm in width) (Fig. 1a). In addition, an overhang structure was designed, circumscribing the entire assay region to prevent MTs from leaving the kinesin-coated surface.25 A single chip has six assay units oriented radially with collectors pointing towards the centre, and a flow cell was constructed for injecting solutions (Fig. 1c).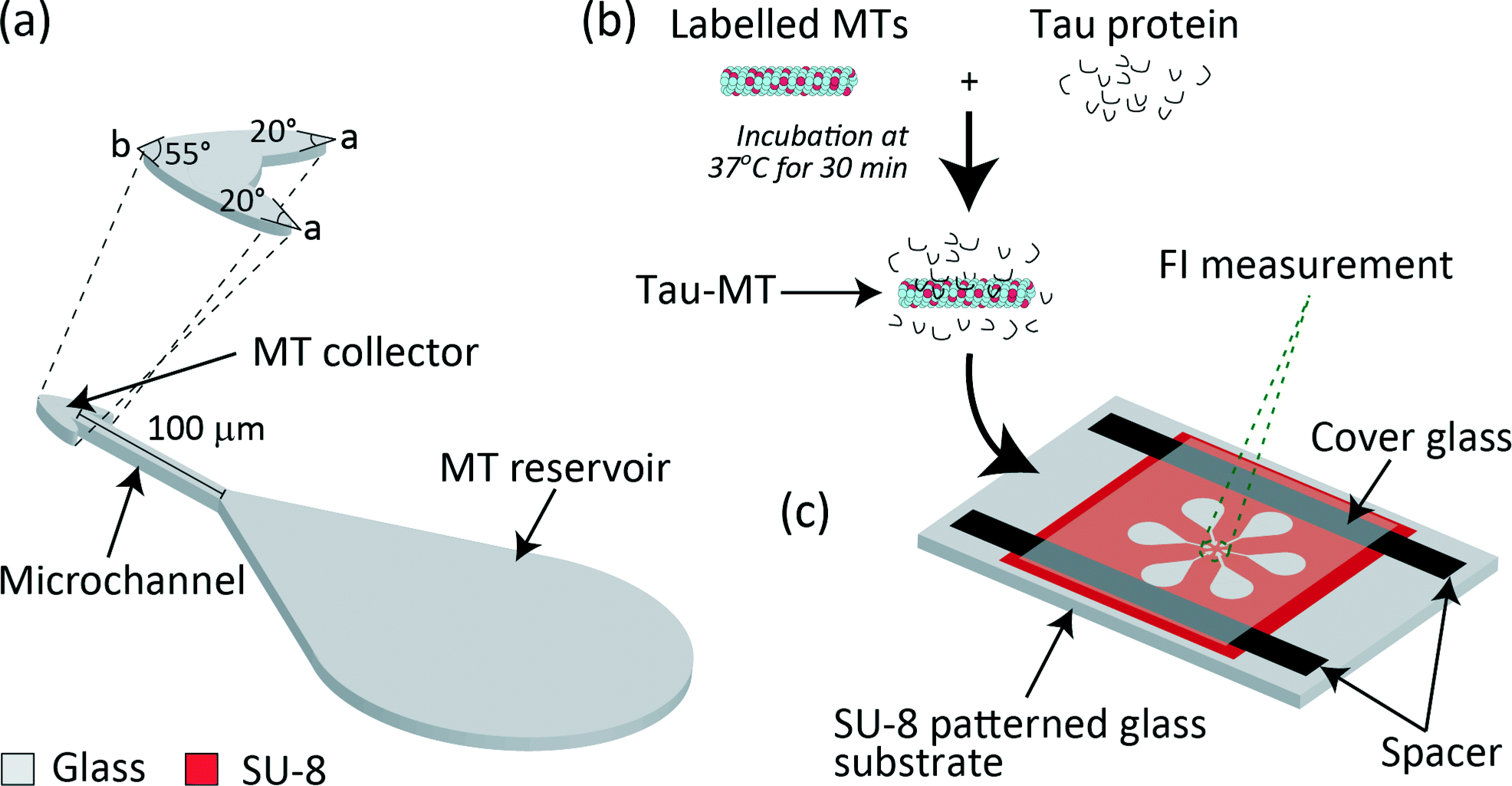 | ||
| Fig. 1 Schematic representation of (a) the microfludic device composed of a MT reservoir, microchannel and MT collector. Angles of the MT collector, a = 20° and b = 55°, are defined as shown in the enlarged schematic. (b) Preparation of tau-MT. (c) Overview of the six assay units radially patterned for simultaneous measurement of MT accumulation at all the six collectors. | ||
The reservoir should be as large as possible to capture many MTs on the kinesin-coated surface. However, owing to the limited area to locate six units radially, the area was designed as 24 × 104 μm2. This shape for guiding MTs to the microchannel was partly adopted from Lin et al. 2008.22
The arrowhead-shaped collector was designed to efficiently concentrate MTs. Previously, arrowhead-shaped structures were incorporated in parallel26 and circular microchannels27 as a rectifier to achieve unidirectional MT gliding. This idea was incorporated in our design, and only the inlet was connected to the microchannel to keep MTs gliding within the collector. To visualise all six collectors in a single frame under a fluorescent microscope and to detect them as six independent FIs, the collectors were separated by a distance of 15 μm and the area was designed as 175 μm2. The angle at the base of the arrowhead-shaped collector was designed as a = 20° and the tip angle was b = 55° for each collector.
The channel width was selected to be 5 μm (ref. 22 and 27) to prevent MTs from making U-turns28 and to guide them towards the collector. The length was designed as 100 μm for MTs to reach the collector within ∼100 s (gliding velocity of MT ∼1 μm s−1), which enabled an assay within a few minutes. In addition, the selected length enabled to enhance the differences in MT gliding velocities, which were measured along with the increase in FI.
Based on the device design, when a solution containing MTs was introduced, it was expected that kinesins immobilized in the MT reservoir would capture MTs from the solution.22 The capture efficiency of the reservoir would directly be affected by the amount and type of tau proteins bound to MTs owing to the differences in their landing rate and density.18 The captured MTs would glide through the microchannel towards the collector and be concentrated for FI measurement. Through MT gliding in the channel, velocity differences according to tau species would be enhanced.15,18 Therefore, the number of MTs reaching the collector would be determined by their capacity to bind and glide over the kinesin-coated surface. This results in differences in FIs after a given assay time.
Fabrication of the tau detection device
A glass substrate (24 mm × 36 mm, no. 1 thickness; Matsunami Glass) was cleaned using piranha solution (H2SO4:H2O2 = 3:1) at 80 °C for 20 min (Fig. 2). Then, 150 nm of aluminium (Al) was deposited on the substrate (VPC-260F, ULVAC). A negative photoresist, SU-8 3005 (Microchem), was spun (6000 rpm, 30 s, 1 μm-thick film) on the Al-coated glass substrate, exposed through a photomask to UV light at the optimum dose of 51 mJ cm−2, developed in a SU-8 developer and rinsed in isopropanol. The overhang structure was created by following a previously established fabrication process.28 In brief, following the removal of the Al layer from the assay region, Al underlying the SU-8 was over-etched for the overhang structure. Al etching was carried out just prior to the tau detection assay to preserve the clean glass surface.
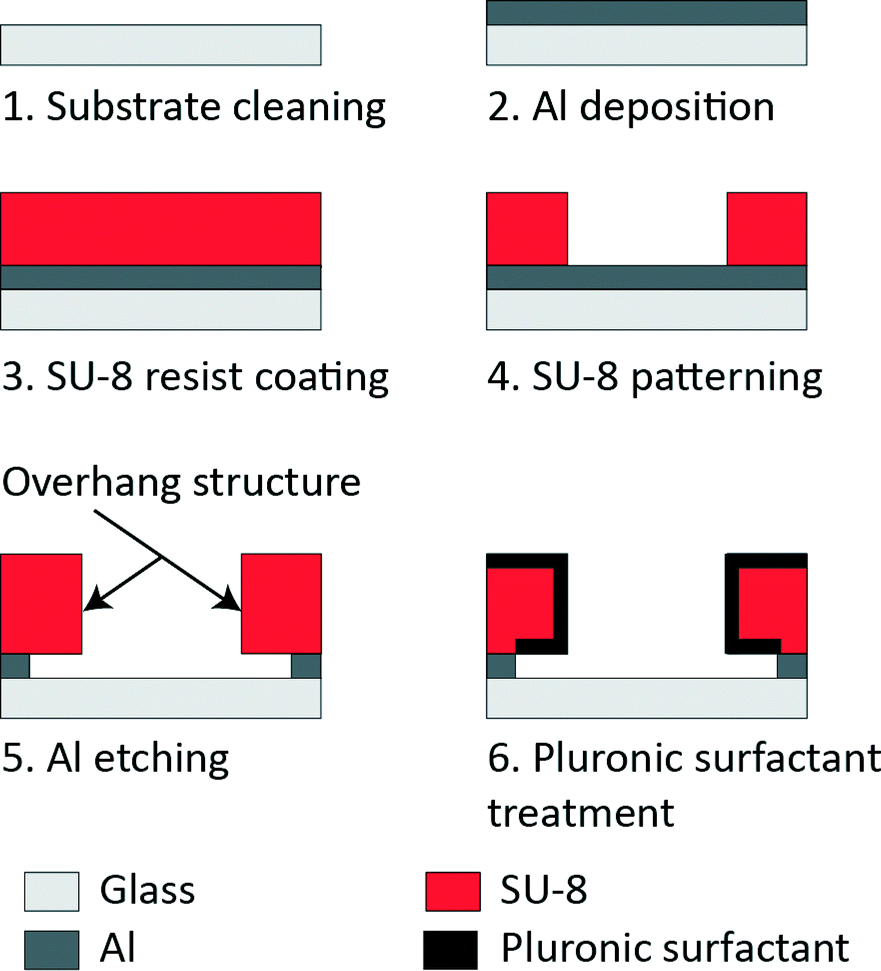 | ||
| Fig. 2 Microfluidic device fabrication and Pluronic surfactant treatment. | ||
Selective kinesin patterning in the microfluidic device
A flow cell (Fig. 1c) was created over the microfluidic device using a cover glass (12 mm × 18 mm; Matsunami Glass) and paraffin tape as spacers (12.7 μm in thickness; Bemis, Parafilm M). Two glasses were sandwiched together on a hot plate at 120 °C for 1 min to melt the paraffin and glue the glasses, leaving the other two sides open for fluid exchange. Before introducing the protein solutions into the device, nonspecific kinesin binding was eliminated by pre-treating the device with a Pluronic surfactant (2 mg ml−1), which is a triblock polymer consisting of poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO). The PEO chain formed a hydrophilic and protein repelling interface on the SU-8 (hydrophobic) surface. It blocked kinesin binding to the SU-8 surface.29 On the contrary, the glass surface (assay region) was left hydrophobic to immobilise kinesin. The microfluidic device was thoroughly rinsed with DI H2O and BRB80 before the assay.Preparation of proteins
Kinesin and tubulin protein preparations are described elsewhere.30,31 In brief, recombinant Homo sapiens kinesin (amino acid residues 1–573) was expressed, isolated and purified from Escherichia coli Rosetta (DE3), then stored in liquid nitrogen (LN2). BRB80 containing 80 mM KOH-PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid)), 1 mM MgCl2 and 1 mM EGTA (ethylene glycol tetraacetic acid) was used as a buffer solution for the entire experiments. Kinesin solution was prepared by diluting kinesin to 30 μg ml−1 in BRB80 containing 2.5 mg ml−1 casein, 1 mM ATP and 1 mM MgCl2. Tubulin was purified from porcine brains obtained from a local slaughterhouse (Ikeda Food, Kyoto, Japan) by two cycles of an assembly–disassembly procedure and phosphocellulose chromatography. A portion of the prepared tubulin was labeled with tetramethyl rhodamine (C1171, Molecular Probes) using standard protocols.32 Tubulin was stored in LN2 until use. MTs were polymerized from fluorescently labelled tubulin and unlabelled tubulin (1:10 molar ratio) at 37 °C for 30 min in BRB80 buffer containing 1 mM MgSO4 and 1 mM GTP. Polymerized MTs were stabilized by 40 μM paclitaxel and used within 1–2 days after polymerization. Recombinant tau proteins (lyophilized in 50 mM, MES buffer) purchased from rPeptide were resuspended in DI H2O and stored at −80 °C.
For the tau detection assay, paclitaxel (10 μM) stabilized MTs (5 μM) were prepared and sheared 30–35 times using a needle (22S gauge, 51 mm in length; Hamilton) to obtain a uniform distribution with a MT length of 8.5 ± 2.6 μm. Tau-MT solutions were prepared by incubating tau isoforms (2N4R, 2N3R) and 2N4R-mutants (V248L, G272V, P301L, V337M and R406W) at a final concentration of 1 μM with 0.5 μM MTs at 37 °C for 30 min for all the assays unless otherwise mentioned (Fig. 1b). Prior to the assay, the tau-MT solution was diluted 5-fold in the motility solution containing 10 μM paclitaxel, 1 mM MgCl2, 1 mM ATP, 25 mM D-glucose, 216 μg ml−1 glucose oxidase, 36 μg ml−1 catalase and 1% (v/v) of 2-mercaptoethanol in BRB80 to minimize the photobleaching effect in fluorescence imaging. MTs without tau incubation were taken as a control (no tau-MT). The respective tau-bound MTs, henceforth, are denoted as 2N4R-MT, 2N3R-MT, V248L-MT, G272V-MT, P301L-MTs, V337M-MTs and R406W-MT.
On-chip protein assay
The Pluronic surfactant-treated microfluidic device was flushed with casein solution (0.5 mg ml−1 casein in BRB80) and incubated for 5 min at room temperature. Next, the kinesin solution was introduced and incubated for 5 min. Finally, the diluted tau-MT solution (0.1 μM MT) was introduced and the flow cell was immediately sealed with vacuum grease (8009-03-8, APIEZON). Image acquisition was started after 5 min of incubation.Imaging and data processing
Fluorescence images were obtained using an inverted fluorescence microscope (IX71, Olympus, Japan) equipped with a 100× oil immersion objective lens (UPLSAPO 100XO, Olympus) and a charge-coupled device camera (ORCA-R2, Hamamatsu, Japan). MTs were observed by epifluorescence using a 100 W mercury short-arc lamp (USH-1030 L, Ushio, Japan) with a neutral density 12 filter and U-MWIG3 fluorescence filter cube. Image acquisition was performed with an exposure time of 500 ms in conjunction with the imaging system HDR-35 recording software (Hamamatsu). Images of 86.7 × 66.0 μm2 (1 pixel = 129 nm) were stored in a 12-bit TIFF format and processed using ImageJ (National Institute of Health, USA). The background was subtracted using the Rolling-Ball background correction plugin,33 then the region of interest enclosing a single collector was selected (220 μm2) and the average FI was measured. FI values measured in three independent devices (18 assay regions) for each type of tau-MT were averaged and normalized by the FI value obtained from the control device, i.e. no tau-MT assayed device. Student's t-test was performed to estimate the significance of the difference between the two experimental conditions. Analysis of variance (ANOVA) was applied to evaluate if the difference between the groups was statistically significant.Results and discussion
MT motility in the microfluidic device
The ability of the device to carry out MT gliding assay was firstly tested using no tau-MTs. All MTs that entered the microchannel reached the collector without any detected U-turns (Fig. 3a). Over 94% of MTs were kept in the collectors due to their arrowhead shape (Table S1†). MTs were concentrated within the MT collectors over time (Fig. 3b and c), reaching saturation approximately after 25 min (Fig. 3d). The image of the overhang structure created using the same fabrication process was previously shown in ref. 28, Fig. 2a by Fujimoto et al. Throughout the assay, the overhang structure and selective kinesin patterning efficiently kept MTs in the assay region. None of the MTs gliding along the periphery of the assay region were able to climb out of the overhang structure (Table S2†). Therefore, the conceptual device design and its application for MT gliding assay were demonstrated successfully.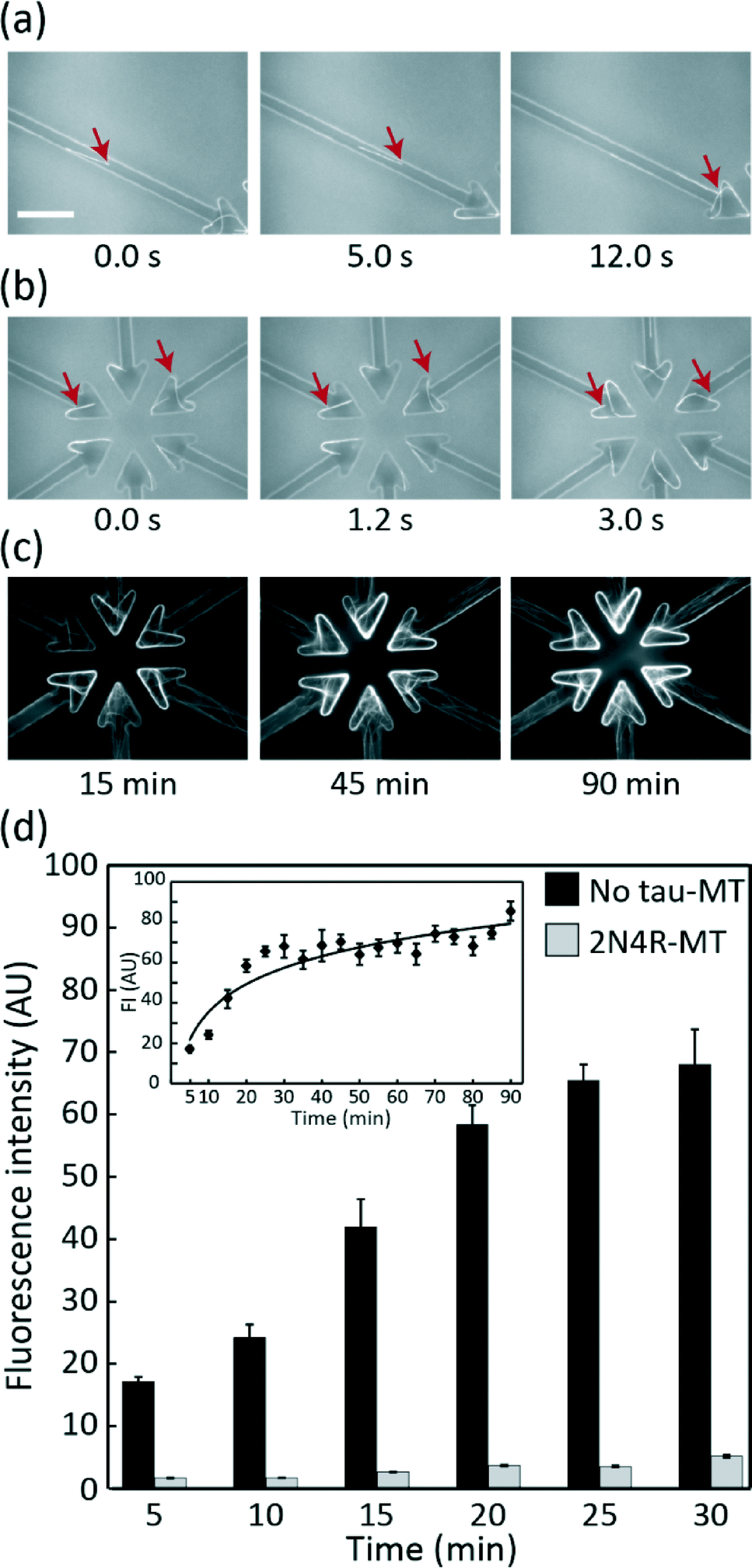 | ||
| Fig. 3 MT motility and accumulation in the collector region in the microfluidic device. (a) Sequential images of a MT gliding in a channel towards the collector. (b) MTs recirculated within collectors. (c) MTs concentrated in collectors at 15, 45 and 90 min. Scale bar, 20 μm. (d) FI of 2N4R-MT and no tau-MT. The 2N4R-MT collector showed a significantly lower FI (t-test: p < 0.001 mean ± SEM), and the difference was significant 5 min after introducing MTs. Inset represents the FI profile of no tau-MT at the collector for 90 min. | ||
MT accumulation depends on the binding and gliding capacity of MTs over a kinesin-coated surface
Measuring FI at the collectors was a simple but effective way of detecting how specific tau protein species affect MT–kinesin binding and gliding. The FI of 2N4R-MTs in the collector was significantly lower than that of the control even at 5 min after starting the assay (t-test, p < 0.001, Fig. 3d). The lower FI of 2N4R-MTs can be due to: (i) the lower landing rate and density of 2N4R-MTs (Fig. S1a and S1b†) over the kinesin-coated surface, as shown in our previous report18 resulting in fewer MTs available for transport to the collector, (ii) the lower gliding velocity of 2N4R-MTs,15,21 resulting in slower accumulation of MTs in the collector. Hence, all these parameters played their roles in regulating the amount of MTs accumulated in the collectors.Differentiation of 3R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :4R tau isoform ratios
:4R tau isoform ratios
The accumulation of tau-MTs with different ratios of 2N3R:2N4R, 1:0, 3:1, 1:1, 1:3 and 0:1 (with a total tau concentration of 1 μM), is shown in Fig. 4 as normalized FIs. MTs incubated with any of the 2N3R:2N4R ratios tested here demonstrated significantly lower FIs than the control (t-test, p < 0.001). Accumulation of MTs decreased with the increase in 2N4R. The FIs of 1:1, 1:3 and 0:1 were indistinguishable (p > 0.05) and were significantly lower than those of 3:1 and 1:0. Further, the FI of 3:1 was significantly lower than that of 1:0; therefore, we were able to differentiate 2N3R and 2N4R tau isoforms and their different ratios from the ratio of 1:1 in our microfluidic device.
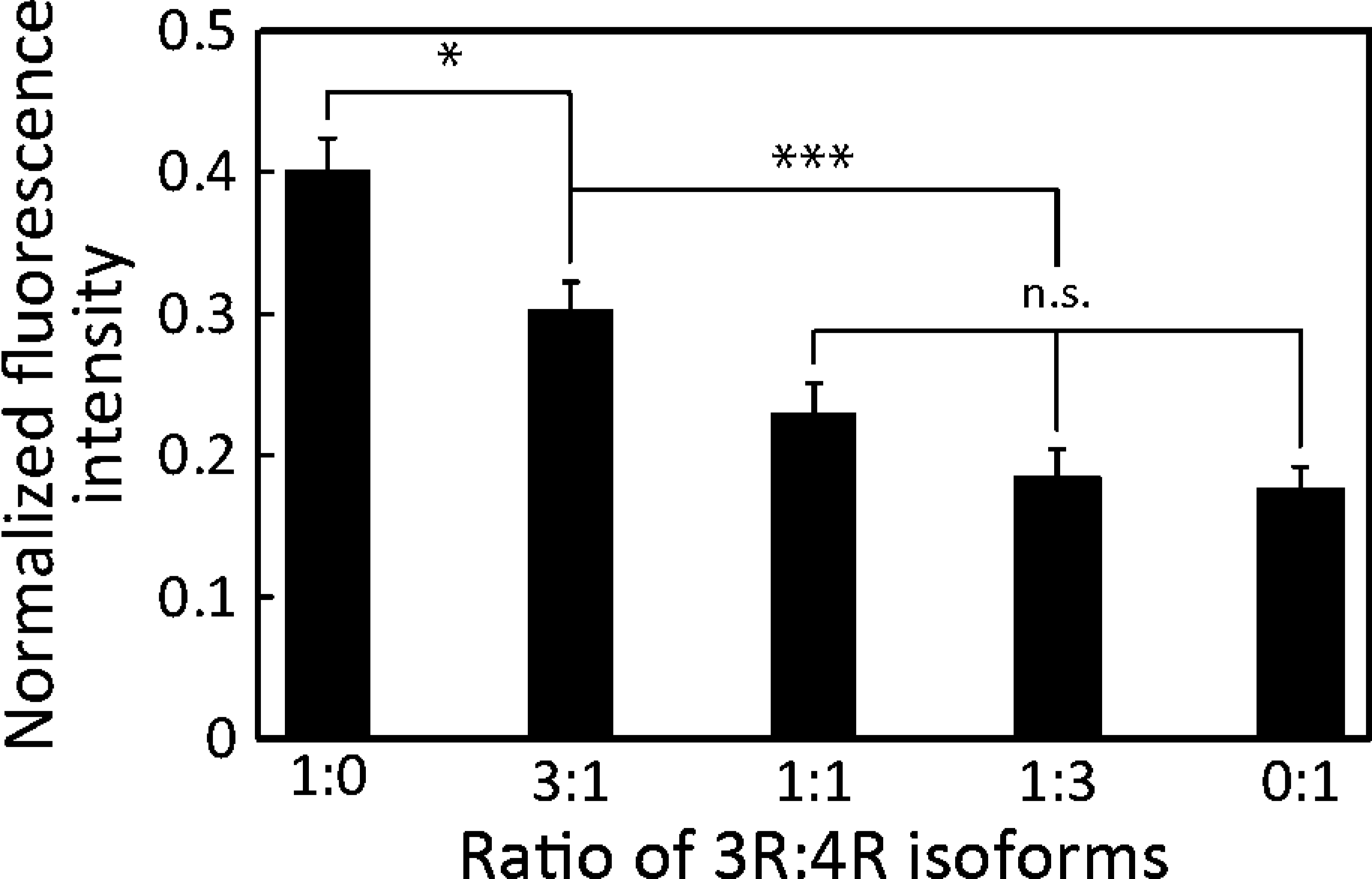 | ||
| Fig. 4 Differentiation of different 2N3R:2N4R isoforms. FIs decreased with the increase in 2N4R tau. Mean ± SEM; ***: p < 0.001; *: p < 0.05; n.s.: p > 0.05 (ANOVA); n ≥ 12. | ||
The effect of various 2N3R:2N4R ratios on MT accumulation can be seen, because 3R and 4R tau isoforms have different affinity to MTs due to the net charge of MTBRs: 4R tau isoforms have a higher net positive charge at MTBRs, resulting in higher affinity to MTs than 3R tau isoforms (Table S3†). Importantly, the higher net positive charge results in higher steric hindrance of MT–kinesin interaction,13 ultimately resulting in less efficient binding of MTs to the kinesin-coated surface. This leads to low MT landing rate, binding density and gliding velocity.15,18,21 Therefore, when the total charge of 4R tau exceeds that of 3R tau in the mixture, we were able to differentiate various 3R:4R tau ratios (Fig. 4). Certainly, the low FI in our on-chip assay recaptured the greater hindrance of 4R isoforms than that of 3R isoforms on the MT–kinesin interaction, as previously reported in other studies.13,15,18,21,34
Differentiation of MTBR mutations in the 2N4R tau isoform
All of the five mutant forms of 2N4R tau showed a significant increase in MT accumulation compared to wild 2N4R (p < 0.05, Fig. 5). We were able to differentiate the MTBR mutations (V248L, G272V, V337M and P301L) from the non-MTBR mutation R406W (Fig. 5; p < 0.05). Moreover, among the MTBR mutations, G272V was significantly different from V337M and P301L (Fig. 5; p < 0.05). MAPT mutations are known to alter the tau binding affinity to MTs with respect to their position in the tau structure: the affinity is decreased by mutations in MTBRs compared to those in other regions.35,36 According to their locations, P301L has the most deteriorating effect on the binding affinity, followed by V337M, G272V and R406W, although the effect of V248L has not been reported yet.37 Unlike MTBR mutations, the non-MTBR mutation, R406W, is known to affect the binding affinity to MTs only when it is phosphorylated.38 This might be a reason why our nonphosphorylated R406W showed lower FI compared to other MTBR mutations. Therefore, the decrease in tau binding affinity corresponding to the mutation position was captured in our on-chip assay.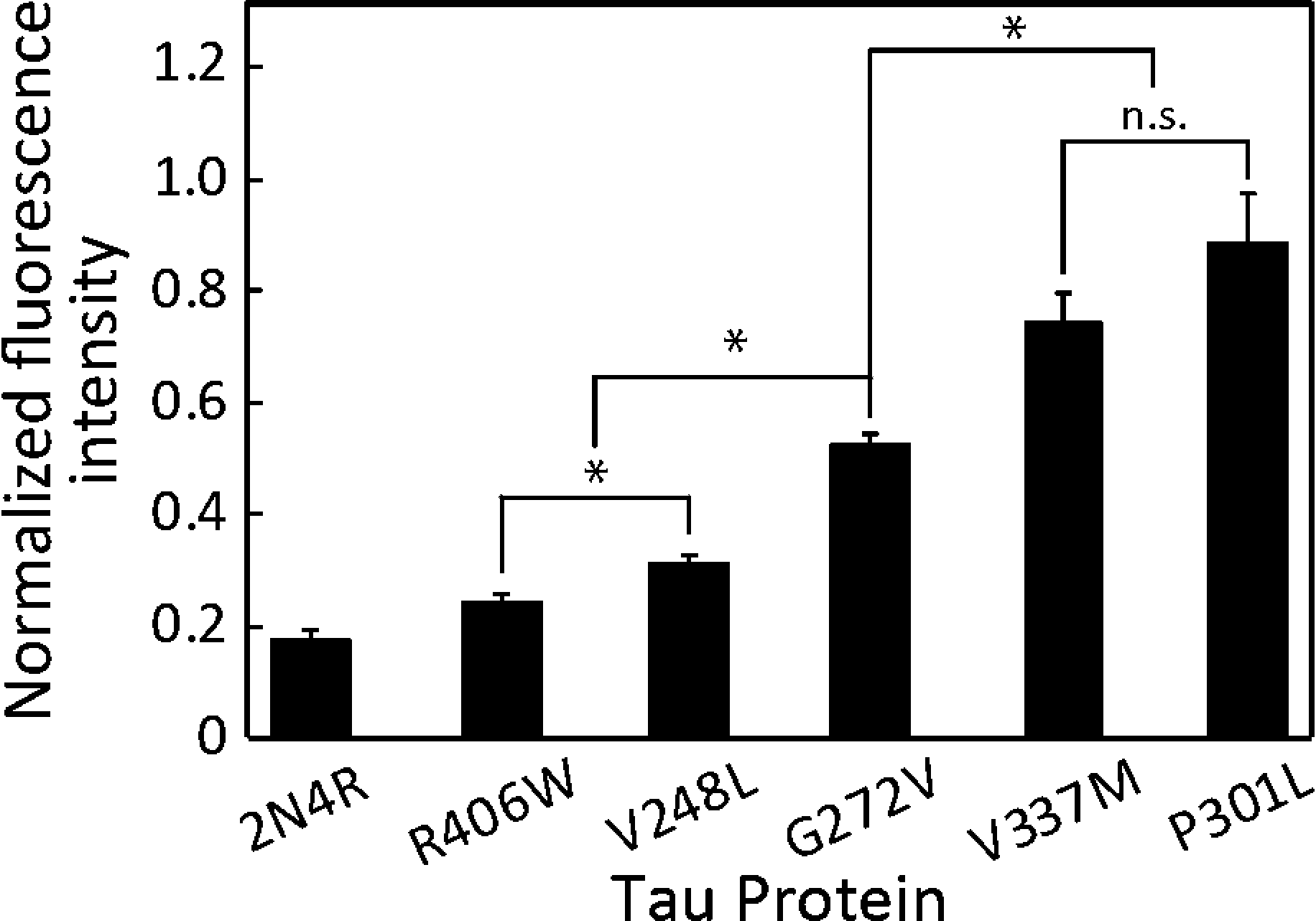 | ||
| Fig. 5 Various 2N4R tau mutations demonstrate distinct effects on FIs. MTs incubated with wild 2N4R showed consistently lower FI than any of the five mutants analyzed. The FI of R406W was significantly lower than those of V248L, G272V, V337M and P301L (p < 0.05). The FI of G272V was significantly lower than those of V337M and P301L (p < 0.05) and higher than those of V248L and R406W (p < 0.05). Mean ± SEM; *: p < 0.05; n.s.: p > 0.05 (ANOVA); n ≥ 16. | ||
Tau detection limit
To identify the detection limit of the proposed device, we assayed lower concentrations (0 and 100 nM) of 2N4R (Fig. 6). The lowest detectable concentration was 100 nM (p < 0.05, Fig. 6). Although our device was not optimized to detect CSF-tau (<14 pM), the detection limit of the 2N4R tau isoform falls into the ranges (0.2–1.0 μM) reported by another non-immuno tau detection method.10 A lower detection limit can be achieved by a trade-off with assay time. Longer assays emphasise the effect of binding and gliding for extremely low concentrations. Moreover, performing assay in a longer microchannel results in an amplified effect of the gliding velocity, which requires a longer assay time. As a result, faster or more sensitive detection can be optimized depending on targeted applications.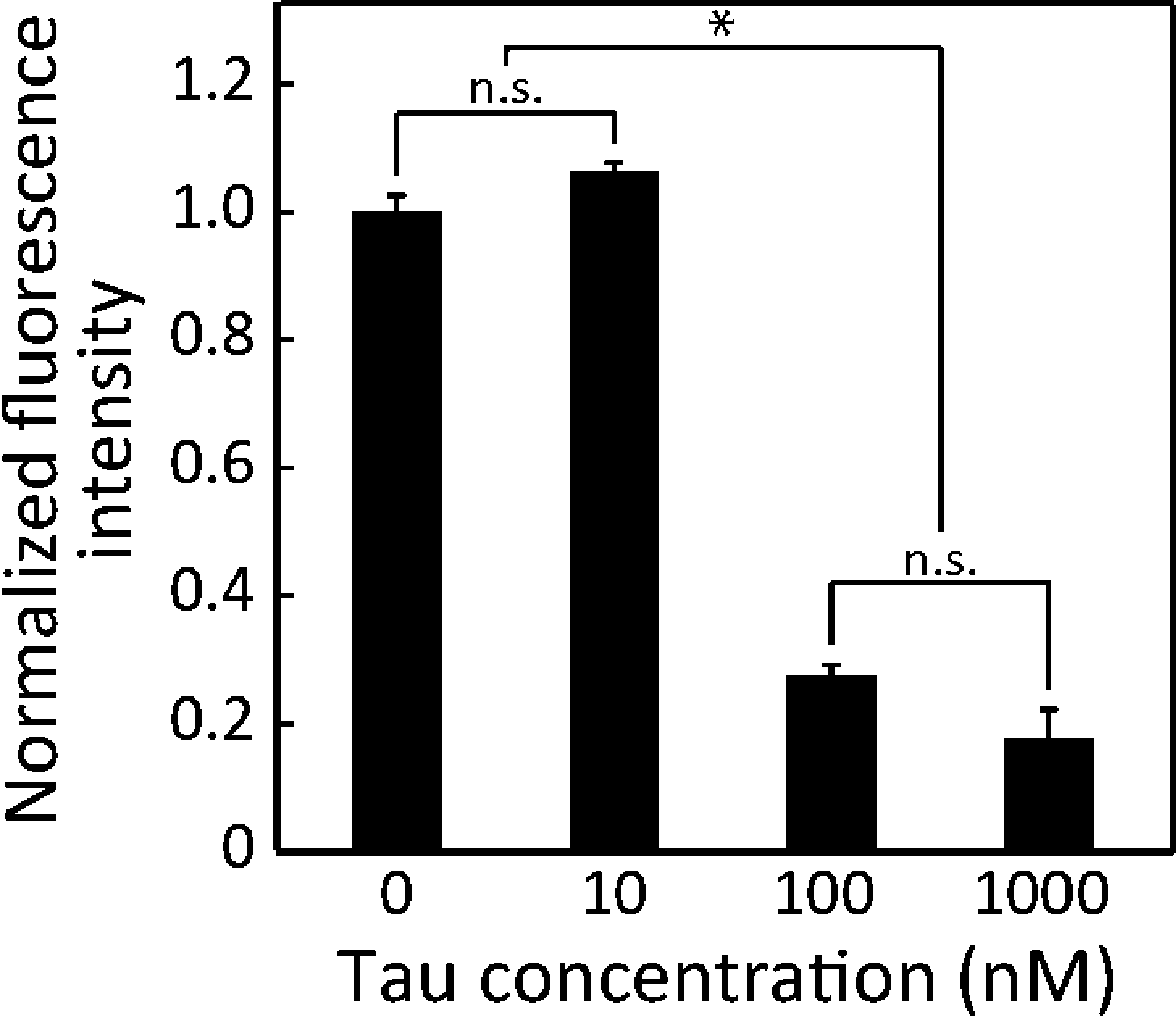 | ||
| Fig. 6 Detection limit of the device. Significantly lower MT accumulation became apparent above 100 nM 2N4R. No significant difference with the control (no tau-MT) was noticed for 10 nM 2N4R. Mean ± SEM; *: p < 0.05; n.s.: p > 0.05 (ANOVA); n ≥ 16. | ||
Conclusion
We developed a single detection unit facilitating the analysis of the tau effect on MT–kinesin binding13,18 and MT gliding15,18,21 towards on-chip tau detection. The device was designed such as to emphasize the differences in MT affinity in the reservoir, MT motility in the microchannel and the arrowhead-shaped collector to effectively capture the additive outcome from both these parameters. The combined effect of these parameters resulted in higher sensitivity (∼10% higher) than those obtained by off-chip measurements.18 The TAT (∼5 min) is shorter than in conventional tau protein detection methods (4–48 h).8,39 Although the sensitivity is lower than those of conventional immuno-based methods,7,8 it is relatively better than those of other non-immuno assays.10,13 This method requires only a single image to determine the effect of a particular tau species, which can be more widely used than kinesin motility-based detection that necessitates velocity measurement.13The overall alteration of the 3R:4R tau isoform ratio irrespective of the type of projection domain (0N, 1N or 2N) is attributed to various neurodegenerative disorders.40 Because the binding affinity of tau proteins is determined by the number of MTBRs, our assay has the potential to differentiate when the 3R:4R ratio is altered by diseases. However, the next challenge to apply our device to an actual CSF sample will be designing a method to eliminate the influence of other coexisting proteins such as other MAPs and mutants. Thus, the current setup is still in the preclinical stage, but has the potential to be a future antibody-free detection method owing to its short assay time and easy readout. In addition, the device may also be applied for elucidation of the effect of other microtubule associated proteins (MAPs).41
Acknowledgements
This work is supported by Nakatani foundation (RY), Japan Society for the Promotion of Science (JSPS) (06035-123332) (RY), JSPS and NSF under the Japan U.S. Cooperative Science Program (11033011-000121) (RY), NIH/NIMH 2R44MH091909 (SLK), JSPS L-15536 (SLK), Grant-in-Aid for Scientific Research (KAKENHI) 26790030 (MCT), Kyoto University Supporting Program for Interaction-Based Initiative Team Studies (SPIRITS) (SPS, RY) as part of the Program for Promoting the Enhancement of Research Universities and the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.References
- H. V. Vinters, Annu. Rev. Pathol.: Mech. Dis., 2015, 10, 291–319 CrossRef CAS PubMed.
- S. Schraen-Maschke, N. Sergeant, C.-M. Dhaenens, S. Bombois, V. Deramecourt, M.-L. Caillet-Boudin, F. Pasquier, C.-A. Maurage, B. Sablonniere, E. Vanmechelen and L. Buee, Biomarkers Med., 2008, 2, 363–384 CrossRef CAS PubMed.
- B. L. Goode, M. Chau, P. E. Denis and S. C. Feinstein, J. Biol. Chem., 2000, 275, 38182–38189 CrossRef CAS PubMed.
- K. Blennow, NeuroRx, 2004, 1, 213–225 CrossRef PubMed.
- H. Hampel, K. Blennow, L. M. Shaw, Y. C. Hoessler, H. Zetterberg and J. Q. Trojanowski, Exp. Gerontol., 2010, 45, 30–40 CrossRef CAS PubMed.
- C. M. Karch, A. T. Jeng and A. M. Goate, J. Biol. Chem., 2012, 287, 42751–42762 CrossRef CAS PubMed.
- M. Vandermeeren, M. Mercken, E. Vanmechelen, J. Six, A. Vandevoorde, J. J. Martin and P. Cras, J. Neurochem., 1993, 61, 1828–1834 CrossRef CAS PubMed.
- D. Wagshal, S. Sankaranarayanan, V. Guss, T. Hall, F. Berisha, I. Lobach, A. Karydas, L. Voltarelli, C. Scherling, H. Heuer, M. C. Tartaglia, Z. Miller, G. Coppola, M. Ahlijanian, H. Soares, J. H. Kramer, G. D. Rabinovici, H. J. Rosen, B. L. Miller, J. Meredith and A. L. Boxer, J. Neurol., Neurosurg. Psychiatry, 2015, 86, 244–250 CrossRef PubMed.
- A. Neely, C. Perry, B. Varisli, A. K. Singh, T. Arbneshi, D. Senapati, J. R. Kalluri and P. C. Ray, ACS Nano, 2009, 3, 2834–2840 CrossRef CAS PubMed.
- J. O. Esteves-Villanueva, H. Trzeciakiewicz and S. Martic, Analyst, 2014, 139, 2823–2831 RSC.
- H. Nehme, S. Chantepie, J. Defert, P. Morin, D. Papy-Garcia and R. Nehme, Anal. Bioanal. Chem., 2015, 407, 2821–2828 CrossRef CAS PubMed.
- N. A. Verwey, W. M. van der Flier, K. Blennow, C. Clark, S. Sokolow, P. P. De Deyn, D. Galasko, H. Hampel, T. Hartmann, E. Kapaki, L. Lannfelt, P. D. Mehta, L. Parnetti, A. Petzold, T. Pirttila, L. Saleh, A. Skinningsrud, J. C. Swieten, M. M. Verbeek, J. Wiltfang, S. Younkin, P. Scheltens and M. A. Blankenstein, Ann. Clin. Biochem., 2009, 46, 235–240 CrossRef CAS PubMed.
- M. C. Tarhan, Y. Orazov, R. Yokokawa, S. L. Karsten and H. Fujita, Lab Chip, 2013, 13, 3217–3224 RSC.
- H. Hagiwara, H. Yorifuji, R. Satoyoshitake and N. Hirokawa, J. Biol. Chem., 1994, 269, 3581–3589 CAS.
- A. Peck, M. E. Sargin, N. E. LaPointe, K. Rose, B. S. Manjunath, S. C. Feinstein and L. Wilson, Cytoskeleton, 2011, 68, 44–55 CrossRef CAS PubMed.
- A. Seitz, H. Kojima, K. Oiwa, E. M. Mandelkow, Y. H. Song and E. Mandelkow, EMBO J., 2002, 21, 4896–4905 CrossRef CAS PubMed.
- C. T. Lin, M. T. Kao, E. Meyhofer and K. Kurabayashi, Appl. Phys. Lett., 2009, 95 Search PubMed.
- S. Subramaniyan Parimalam, M. C. Tarhan, S. L. Karsten, H. Fujita, H. Shintaku, H. Kotera and R. Yokokawa, Micro Electro Mechanical Systems (MEMS), IEEE 27th International Conference on, 26–30 Jan. 2014, pp. 314–317 Search PubMed.
- B. Trinczek, A. Ebneth, E. M. Mandelkow and E. Mandelkow, J. Cell Sci., 1999, 112(Pt 14), 2355–2367 CAS.
- K. A. Butner and M. W. Kirschner, J. Cell Biol., 1991, 115, 717–730 CrossRef CAS PubMed.
- D. Yu, N. E. LaPointe, E. Guzman, V. Pessino, L. Wilson, S. C. Feinstein and M. T. Valentine, J. Alzheimer's Dis., 2014, 39, 301–314 CAS.
- C. T. Lin, M. T. Kao, K. Kurabayashi and E. Meyhofer, Nano Lett., 2008, 8, 1041–1046 CrossRef CAS PubMed.
- Y. Hiratsuka, T. Tada, K. Oiwa, T. Kanayama and T. Q. Uyeda, Biophys. J., 2001, 81, 1555–1561 CrossRef CAS PubMed.
- Y. M. Huang, M. Uppalapati, W. O. Hancock and T. N. Jackson, Biomed. Microdevices, 2007, 9, 175–184 CrossRef PubMed.
- J. Clemmens, H. Hess, R. Doot, C. M. Matzke, G. D. Bachand and V. Vogel, Lab Chip, 2004, 4, 83–86 RSC.
- L. Jia, S. G. Moorjani, T. N. Jackson and W. O. Hancock, Biomed. Microdevices, 2004, 6, 67–74 CrossRef CAS PubMed.
- C. T. Lin, M. T. Kao, K. Kurabayashi and E. Meyhofer, Small, 2006, 2, 281–287 CrossRef CAS PubMed.
- K. Fujimoto, M. Kitamura, M. Yokokawa, I. Kanno, H. Kotera and R. Yokokawa, ACS Nano, 2013, 7, 447–455 CrossRef CAS PubMed.
- C. H. Ho, L. Limberis, K. D. Caldwell and R. J. Stewart, Langmuir, 1998, 14, 3889–3894 CrossRef CAS.
- R. Yokokawa, M. C. Tarhan, T. Kon and H. Fujita, Biotechnol. Bioeng., 2008, 101, 1–8 CrossRef CAS PubMed.
- R. C. Williams, Jr. and J. C. Lee, Methods in Enzymology, 1982, 85(Pt B), 376–385 CrossRef.
- A. Hyman, D. Drechsel, D. Kellogg, S. Salser, K. Sawin, P. Steffen, L. Wordeman and T. Mitchison, Methods Enzymol., 1991, 196, 478–485 CAS.
- S. R. Sternberg, Computer, 1983, 16, 22–34 CrossRef.
- M. Lu and K. S. Kosik, Mol. Biol. Cell, 2001, 12, 171–184 CrossRef CAS PubMed.
- M. van Slegtenhorst, J. Lewis and M. Hutton, Exp. Gerontol., 2000, 35, 461–471 CrossRef CAS PubMed.
- M. Hong, V. Zhukareva, V. Vogelsberg-Ragaglia, Z. Wszolek, L. Reed, B. I. Miller, D. H. Geschwind, T. D. Bird, D. McKeel, A. Goate, J. C. Morris, K. C. Wilhelmsen, G. D. Schellenberg, J. Q. Trojanowski and V. M. Y. Lee, Science, 1998, 282, 1914–1917 CrossRef CAS PubMed.
- N. Matsumura, T. Yamazaki and Y. Ihara, Am. J. Pathol., 1999, 154, 1649–1656 CrossRef CAS PubMed.
- P. K. Krishnamurthy and G. V. W. Johnson, J. Biol. Chem., 2004, 279, 7893–7900 CrossRef CAS PubMed.
- F. R. Petry, J. Pelletier, A. Bretteville, F. Morin, F. Calon, S. S. Hebert, R. A. Whittington and E. Planel, PLoS One, 2014, 9(1–12), e94251 Search PubMed.
- C. Luk, Y. Compta, N. Magdalinou, M. J. Martí, G. Hondhamuni, H. Zetterberg, K. Blennow, R. Constantinescu, Y. Pijnenburg, B. Mollenhauer, C. Trenkwalder, J. Van Swieten, W. Z. Chiu, B. Borroni, A. Cámara, P. Cheshire, D. R. Williams, A. J. Lees and R. de Silva, J. Neurochem., 2012, 123, 396–405 CrossRef CAS PubMed.
- A. Iyer, N. E. LaPointe, K. Zielke, M. Berdynski, E. Guzman, A. Barczak, M. Chodakowska-Zebrowska, M. Barcikowska, S. Feinstein and C. Zekanowski, PLoS One, 2013, 8(1–11), e76409 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc01486g |
| This journal is © The Royal Society of Chemistry 2016 |