
A fully integrated and automated microsystem for rapid pharmacogenetic typing of multiple warfarin-related single-nucleotide polymorphisms
Bin
Zhuang†
acd,
Junping
Han†
b,
Guangxin
Xiang
acd,
Wupeng
Gan
acd,
Shuaiqin
Wang
a,
Dong
Wang
cd,
Lei
Wang
cd,
Jing
Sun
e,
Cai-Xia
Li
e and
Peng
Liu
*a
aDepartment of Biomedical Engineering, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, School of Medicine, Tsinghua University, Beijing, 100084, China. E-mail: pliu@tsinghua.edu.cn; Fax: +86 10 62798732; Tel: +86 10 62798732
bChinese People's Public Security University, Beijing, 100038, China
cCapitalBio Corporation, Beijing, 102206, China
dNational Engineering Research Center for Beijing Biochip Technology, Beijing, 102206, China
eKey Laboratory of Forensic Genetics, Institute of Forensic Science, Ministry of Public Security, Beijing, 100038, China
First published on 2nd November 2015
Abstract
A fully integrated and automated microsystem consisting of low-cost, disposable plastic chips for DNA extraction and PCR amplification combined with a reusable glass capillary array electrophoresis chip in a modular-based format was successfully developed for warfarin pharmacogenetic testing. DNA extraction was performed by adopting a filter paper-based method, followed by “in situ” PCR that was carried out directly in the same reaction chamber of the chip without elution. PCR products were then co-injected with sizing standards into separation channels for detection using a novel injection electrode. The entire process was automatically conducted on a custom-made compact control and detection instrument. The limit of detection of the microsystem for the singleplex amplification of amelogenin was determined to be 0.625 ng of standard K562 DNA and 0.3 μL of human whole blood. A two-color multiplex allele-specific PCR assay for detecting the warfarin-related single-nucleotide polymorphisms (SNPs) 6853 (–1639G>A) and 6484 (1173C>T) in the VKORC1 gene and the *3 SNP (1075A>C) in the CYP2C9 gene was developed and used for validation studies. The fully automated genetic analysis was completed in two hours with a minimum requirement of 0.5 μL of input blood. Samples from patients with different genotypes were all accurately analyzed. In addition, both dried bloodstains and oral swabs were successfully processed by the microsystem with a simple modification to the DNA extraction and amplification chip. The successful development and operation of this microsystem establish the feasibility of rapid warfarin pharmacogenetic testing in routine clinical practice.
Introduction
The traditional “one-size-fits-all” approach in drug prescriptions may well be reaching its end days as more is learned about diseases and genes, and more drugs are developed using a biomarker-based method.1–3 The adoption of pharmacogenetic testing in clinical practice will enable the use of medications based on patients' genetic information (i.e. personalized medicine), resulting in improved safety and efficiency.4,5 One of the well-known examples is warfarin, which is the most widely prescribed anticoagulant drug in the US.6,7 Due to a narrow therapeutic window and large inter-patient variability in drug sensitivity, thrombosis and bleeding caused by inappropriate warfarin dosages have been identified as the second leading cause of drug-related emergency room visits.8,9 Although many factors, such as age and lifestyle, influence the warfarin response,9 single-nucleotide polymorphisms (SNPs) in the CYP2C9 (cytochrome P450, family 2, subfamily C, polypeptide 9) and VKORC1 (vitamin K epoxide reductase, complex 1) genes account for approximately 40% of the inter-patient variability in the warfarin dosage.10 Several independent studies have found that consideration of the patients' CYP2C9 and VKORC1 genotypes when prescribing the drug significantly reduced hospitalizations up to 43% compared to the non-genotyped groups.6,11,12 Despite compelling findings and clinical potential, incorporation of pharmacogenetic testing into routine clinical practice to guide treatments is still restricted mainly due to accessibility and the time-consuming nature of current existing diagnostic methods for probing these genetic variations.13The current pharmacogenetic testing for detecting SNPs or mutations is mainly constrained by sample preparation, which usually includes nucleic acid extraction from clinical samples and amplification of target sequences by polymerase chain reaction (PCR).14,15 Even though detection approaches, such as microarrays, capillary electrophoresis (CE), real-time PCR and DNA sequencing, have been significantly improved in sensitivity and automation, the entire analytical process starting from a raw sample to a result is still time-consuming, labor-intensive and high-priced.13,16 Microfluidic technology holds great promise to address these problems by integrating together the sample preparation steps with the detection to form a micro total analysis system (μTAS).14,17,18 In past decades, a variety of fully integrated microsystems based on different detection methods have been developed. For instance, the Cepheid GeneXpert system (Sunnyvale, CA) that integrated sample preparation with real-time PCR was developed for the diagnosis of tuberculosis. However, in the case of SNP and mutation detection, the capillary electrophoresis-based system is believed to be one of the best choices due to its multi-locus detection capability, the degree of total integration and the technology readiness level.
The first sample-in–answer-out microsystem based on electrophoretic detection was reported by Landers' group, in which a solid-phase extraction (SPE) column for DNA extraction was integrated with PCR and CE into a single device for pathogen detection.19 Meanwhile, Mathies' group has successfully demonstrated PCR-CE glass devices for pathogen detection and forensic DNA typing.20–22 Later, they introduced template purification and post-PCR purification structures into the microsystem for automated DNA typing from buccal swabs.23,24 In collaboration with the Forensic Science Service of UK, Zenhausern's group from the University of Arizona described a prototype instrument that consisted of a plastic extraction and amplification cartridge coupled with a glass CE microchip for rapid forensic short tandem repeat (STR) analysis from pre-prepared lysates of buccal swab samples.25 More recently, IntegenX (Pleasanton, CA) and GE Healthcare (Pittsburgh, PA) released their fully automated RapidHIT™ and DNAscan™ systems, respectively.26–28 Both instruments can complete STR typing in less than 90 minutes. In addition, MicroLab Diagnostics in collaboration with Landers' group reported a new prototype that is capable of performing automated DNA typing from buccal swabs or FTA® paper.29,30 While the emergence of so many new systems and products clearly indicates that the era of the “sample-in–answer-out” genetic analysis is approaching, the assays performed on these systems are still focused on forensic human identification by far, and the cost per assay is still quite expensive (>$250 per sample). With further improvement in cost and versatility, we believe that this extraordinary technology will play an important role in the field of pharmacogenetics.
Here, we present a fully integrated and automated microsystem consisting of disposable plastic chips for DNA extraction and amplification coupled with a 4" reusable glass array-CE chip in a modular-based integration format, operated on a compact control and detection instrument for rapid pharmacogenetic testing. Filter paper-based DNA extraction followed by “in situ” PCR amplification was conducted in the DNA extraction and amplification chip.31,32 After that, amplicons were driven into an injection chamber, where a novel needle-shaped injection electrode was employed to mix and deliver the amplicons with sizing standards to the CE chip for separation and detection. To demonstrate the potential application of the system, we performed the detection of three single-nucleotide polymorphisms, in CYP2C9 and VKORC1 from whole blood, bloodstains and buccal swabs. This work will promote the future use of rapid genetic analysis in clinical practice for guiding the prescription of drugs according to patients' genetic information.
Experimental section
Microdevice design and microfabrication
The fully integrated microchip assembly is composed of three separate microdevices: two identical plastic DNA extraction and amplification chips (DEA chip) and one glass capillary array electrophoresis chip (CAE chip), which are combined together in a modular-based integration format. As shown in Fig. 1A, the structure of the DEA chip is similar to that of the sample preparation chip developed previously by our group.32 Briefly, the DEA chip consists of two PMMA layers with sandwiched PDMS membrane discs (BISCO® HT-6240, Rogers, Woodstock, CT) for valve actuation, filter paper discs (Fusion 5, GE Healthcare, Pittsburgh, PA) for DNA extraction, and sealing tapes for chamber isolation. Two DNA extraction and amplification units, each of which contains an extraction inlet, a shared PCR inlet, a waste outlet, a loading chamber, a reaction chamber, an injection chamber and three mechanically actuated diaphragm microvalves, are symmetrically arranged on the DEA chip. The loading chamber is a 3 mm-diameter well for accommodating a variety of raw sample materials. According to different sample types, a piece of filter net with a mesh size of 80 μm is embedded into the chamber for carrying whole blood or a simply empty well is designed for housing dried bloodstains and buccal swabs. The 15 μL reaction chamber with a 3 mm-diameter piece of Fusion 5 filter paper disc is designed for DNA extraction and “in situ” PCR amplification. Three microvalves (valves 1, 2 and 3) are employed to switch flows on/off to the reaction chamber during extraction and to seal the chamber completely during thermal cycling. The injection chamber is constructed by aligning two 3 mm-diameter via holes together, which are located in the upper and lower PMMA layers, respectively. A piece of sealing tape (Microseal® B adhesive tape, Bio-Rad, Hercules, CA) is sandwiched between the two via holes to divide the injection chamber into two compartments: the upper compartment for receiving PCR products transferred from the reaction chamber and the lower one for storing pre-mixed sizing standards and formamide, which are preloaded into the DEA chip and later co-injected with PCR products into the CAE chip. Two pieces of BarSeal™ tape (Thermo Fisher, Waltham, MA) are applied to enclose the injection chamber from both sides of the chip.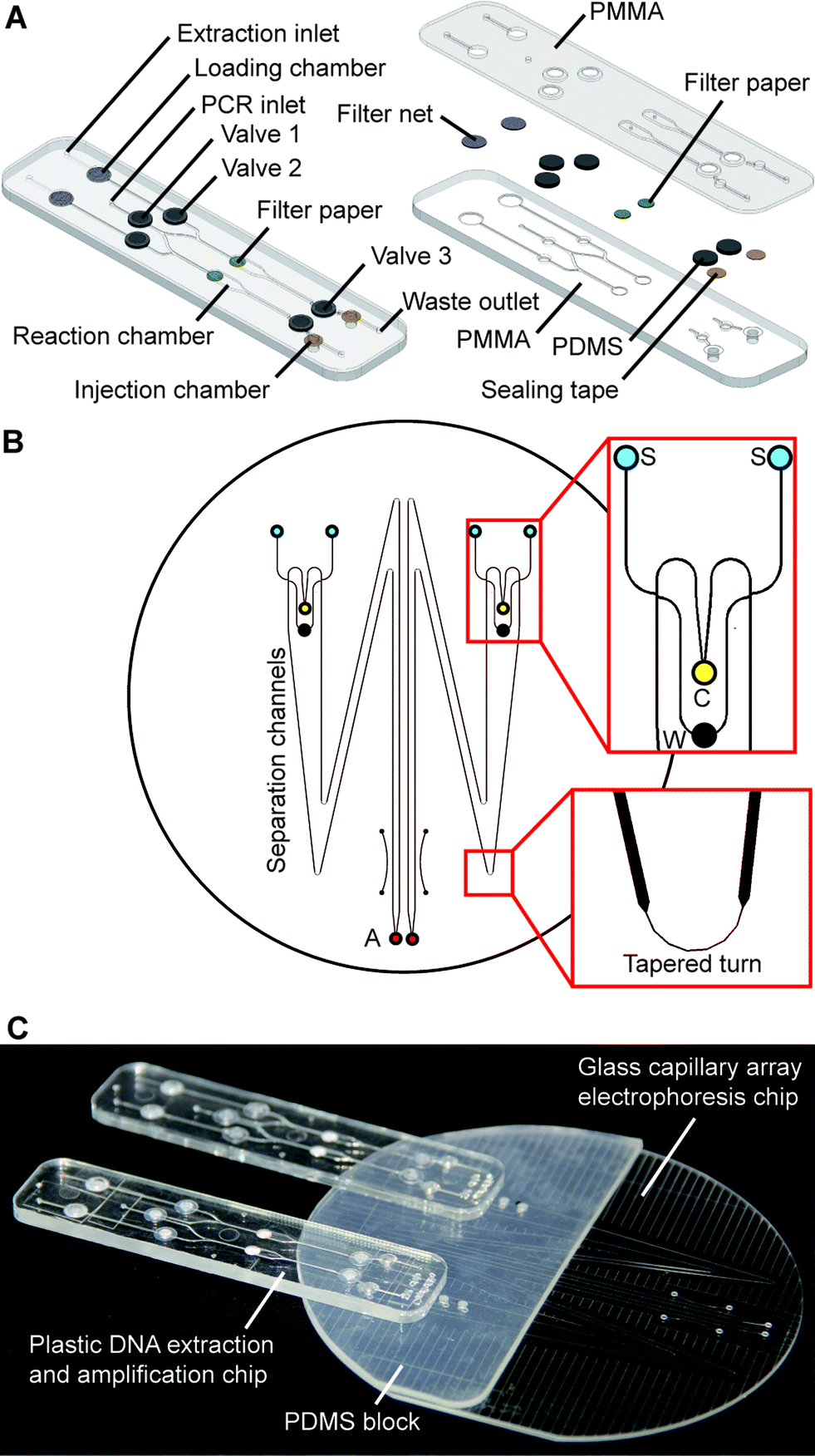 | ||
| Fig. 1 Schematic of fully integrated microchip assembly for warfarin pharmacogenetic testing. (A) Disposable plastic microchip for DNA extraction and PCR amplification. Left: The microchip contains two DNA extraction and amplification units, each of which contains an extraction inlet, a shared PCR inlet, a waste outlet, a loading chamber, a reaction chamber, an injection chamber and three microvalves. Right: Exploded view of the microchip structure. (B) Layout of the 4 inch glass capillary array electrophoresis chip. Upper expanded view shows the structure of the injection channels. Lower expanded view is a tapered turn incorporated into the separation channel. S: sample well (blue), C: cathode well (yellow), W: waste well (black) and A: anode well (red). (C) Photograph of the microchip assembly. | ||
The design of the CAE chip, as shown in Fig. 1B, is similar to those that have been published elsewhere.23,33 On a 4′′ glass wafer, four 16 cm-long separation channels with a cross-section of 200 × 40 μm are grouped into two doublets. Each doublet contains two double-T injectors with an offset of 200 μm, two sample wells, a cathode well, a waste well and an anode well. Due to the space limit of the chip, tapered turns are employed to extend the separation channels up to 16 cm with minimized diffusion disturbance during electrophoresis. Fabrication of the glass chip follows procedures previously described.23,33 Two 3 mm-thick PDMS blocks with punched buffer reservoirs are permanently bonded to the glass CAE chip using a plasma treatment method. To assemble the fully integrated microchip system, as shown in Fig. 1C, two DEA chips are reversibly attached to the PDMS blocks on the CAE chip by aligning four injection chambers with the corresponding sample wells. Non-leaky contact between the PDMS and the plastic DEA chips can be easily achieved by simple pre-cleaning of the PDMS surface.
As shown in Fig. 2, a novel injection electrode coupled with the injection chamber of the DEA chip was developed to enable the sample transfer from the DEA chip to the capillary electrophoresis chip, as well as the mixing of PCR products with the injection reagents. The steel electrode with a cross groove is coated with a layer of platinum to prevent electrochemical reactions. The injection is realized by actuating the electrode to punch through three layers of tape in the injection chamber completely. Due to the capillary action generated by the groove, the PCR products as well as the sizing standards and formamide are aspirated into the electrode, where all the solutions are well mixed together. After that, an electrical field is applied between the injection and the waste electrode to deliver the prepared samples into the injection channel for electrophoresis.
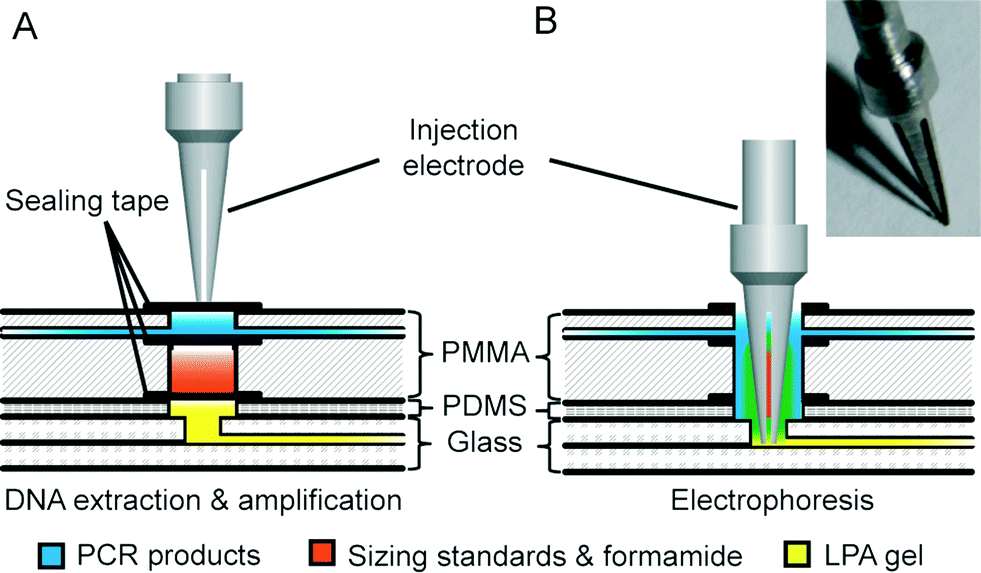 | ||
| Fig. 2 Schematic of injection electrode coupled with injection chamber. (A) The injection electrode with a cross groove is positioned on the top of the injection chamber, which is divided into two compartments: the upper for PCR products and the lower for sizing standards and formamide. (B) The electrode is actuated to penetrate through the injection chamber for injection. The insert is a photograph of the injection electrode. | ||
Instrumentation for microchip control and detection
A photograph of the instrument used to perform fully automated pharmacogenetic testing is shown in Fig. 3A. The system with dimensions of 48 × 35 × 35 cm contains a 488 nm diode laser (100 mW, Sapphire 488, Coherent, Santa Clara, CA), a confocal optical system with a linear-motion objective for detecting four different fluorescence signals, four high-voltage power supplies for electrophoresis, a fluidic control system, a microchip fixture and all the electronics for system operation. A custom-made LabVIEW program (National Instruments, Austin, TX) running on a laptop is used to control the system through a NI 6259 OEM multifunction DAQ board (National Instruments). | ||
| Fig. 3 Photographs of the compact control and detection instrument. (A) Image of the instrument. (B) The fluidic control system installed in the front of the system. (C) The microchip fixture located on the top of the instrument for accommodating the microchip assembly. (D) The structure of the manifold. | ||
The design of the optical system is identical to that developed previously by our group.32 Briefly, the laser beam is reflected by two mirrors, and then transmitted through a dichroic mirror into a linear-motion assembly that is mounted on a motorized scanning stage. The laser beam is further reflected by a prism in the assembly and focused to the microchip by an objective. Excited fluorescence is collected by the objective, and then reflected by the prism and the dichroic mirror into a four-color confocal system, where the light is directed into a multi-channel PMT (photomultiplier tube) with four detection wavelength ranges centered at 517, 549, 576 and 605 nm. The converted electrical signals are collected at a rate of 20 kHz by the DAQ board and processed using a 10 Hz low-pass filter in the LabVIEW program. The fluidic control system has also been described in detail previously.32 In the instrument, a tube rack that provides sodium hydroxide (NaOH) for cell lysis, hydrochloric acid (HCl) for neutralization, and 1× TE (Tris EDTA) and DI water for rinsing is installed in the front side of the instrument (Fig. 3B). A syringe pump (PSD/4, Hamilton, Bonaduz, Switzerland) and a modular valve positioner (MVP, Hamilton) are employed to realize the fluid pumping and switching functions, respectively. The fluids are routed to the microchip fixture located on the top of the system via PTFE tubing.
The microchip fixture functioning as the interface of the system to the microchip consists of a chip platform and a manifold, as shown in Fig. 3C. The frames of the fixture are made of Teflon, providing excellent chemical resistance and thermal insulation. The microchip assembly is placed into a recessed area on the chip platform and held in place with the manifold using screws. Underneath the glass CAE chip is an aluminum plate with a thin film heater controlled by a commercial PID module (XMT-624-SSR, HBKJ, Beijing, China) for heating the chip during electrophoresis. As shown in Fig. 3D, the chip manifold contains two portions: first, above the DEA chips, fluidic microports connected to the fluidic system, heating blocks for PCR thermal cycling, valve plungers and injection electrodes with self-locking solenoids are installed in the positions corresponding to the structure of the DEA chips; second, above the CAE chip, platinum electrodes supplying voltages to the corresponding buffer reservoirs are installed in the manifold.
Procedure of “sample-in–answer-out” genetic analysis
To prepare the CAE microchip, a dynamic coating solution (DEH-100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 1:1, The Gel Company, San Francisco, CA) was first injected into the channels using a syringe and incubated for 1 min, followed by washing with DI water. A sieving matrix, 4.5% (w/v) linear polyacrylamide (LPA) with 6 M urea in 1× Tris TAPS EDTA (TTE), was then loaded into the channels from the anode wells using a custom-made loading machine with a driving motor. After extra 1× TTE buffer was pipetted into the PDMS reservoirs, the CAE chip was placed onto the microchip platform and aligned with the detection window. Next, raw samples, such as whole blood, bloodstains and buccal swabs, were loaded into the loading chambers of the DEA chips, which were then sealed with a piece of adhesive tape. Two DEA chips were placed onto the platform in positions where the injection chambers of the chips were aligned with the corresponding sample reservoirs in the CAE microchip. The chip manifold was closed to press the microchip assembly from the top. Due to the natural adherence of PMMA to PDMS, non-leaky sealing between the DEA and CAE chips was achieved with the simple press by the manifold. Two Eppendorf tubes containing freshly prepared PCR Master Mix were inserted into the manifold and connected to the fluidic control system via PTFE tubing.
:methanol = 1:1, The Gel Company, San Francisco, CA) was first injected into the channels using a syringe and incubated for 1 min, followed by washing with DI water. A sieving matrix, 4.5% (w/v) linear polyacrylamide (LPA) with 6 M urea in 1× Tris TAPS EDTA (TTE), was then loaded into the channels from the anode wells using a custom-made loading machine with a driving motor. After extra 1× TTE buffer was pipetted into the PDMS reservoirs, the CAE chip was placed onto the microchip platform and aligned with the detection window. Next, raw samples, such as whole blood, bloodstains and buccal swabs, were loaded into the loading chambers of the DEA chips, which were then sealed with a piece of adhesive tape. Two DEA chips were placed onto the platform in positions where the injection chambers of the chips were aligned with the corresponding sample reservoirs in the CAE microchip. The chip manifold was closed to press the microchip assembly from the top. Due to the natural adherence of PMMA to PDMS, non-leaky sealing between the DEA and CAE chips was achieved with the simple press by the manifold. Two Eppendorf tubes containing freshly prepared PCR Master Mix were inserted into the manifold and connected to the fluidic control system via PTFE tubing.
Once the preparation procedure was finished, the instrument was turned on and the entire genetic analysis was conducted by the instrument automatically under the control of the LabVIEW program without any human intervention. First, DNA extraction was carried out following a similar protocol developed previously.31,32 Briefly, 200 μL of distilled water was aspirated through the DEA chip to rinse the raw samples and bring cells to the downstream filter disc in the reaction chamber, where cells were lysed completely with 100 μL of NaOH (20 mM) within 5 min. After that, diluted HCl (1.8 mM) was loaded into the chip followed by 1× TE buffer. This extensive rinsing step will stabilize the pH value in the reaction chamber. The thermal cycling process was started while all the on-chip valves were closed.
After the thermal cycles were completed, PCR products in the reaction chamber were pumped into the upper compartment of the injection chamber. The injection electrodes with a cross groove were actuated by the solenoids to punch through all the sealing tapes of the injection chambers. The amplicons, together with the DNA markers and formamide in the lower compartment, were transferred to the sample reservoirs of the CAE chip. CE separation was performed after the temperature of the microchip platform reached 60 °C. The samples were electrophoretically injected toward the waste by applying an electric field of 100 V cm−1 for 90 s while floating the anode and cathode. A separation field of 180 V cm−1 was then applied between the cathode and anode to start DNA migration along the 16 cm-long separation channel. The entire “sample-in–answer-out” analysis took 2 hours to complete without any manual operations. Once the genetic analysis was finished, the whole microchip assembly was taken out from the instrument. The two plastic DEA chips were disposed, while the glass CAE chip was cleaned for the next use.
Singleplex PCR system and sample preparation
A set of primers (forward: TAMRA-5′-CCCTGGGCTCTGTAAAGAA-3′, reverse: 5′-ATCAGAGCTTAAACTGGGAAGCTG-3′, Sangon Biotech, Shanghai, China) for amplification of a 106 bp and a 112 bp fragment from the amelogenin gene in X and Y chromosomes, respectively, is used for system characterization. The 100 μL Master Mix consists of 10 μL of PCR buffer (10×, Roche, Indianapolis, IN), 3 μL of dNTP (10 mM each), 8 μL of MgCl2 (25 mM), 5 μL of Roche FastTaq polymerase, 10 μL of bovine serum albumin (BSA, 50 μg μL−1, Sigma-Aldrich, St. Louis, MO), 3 μL of PEG10000 (160 μg μL−1, Sigma-Aldrich), 2 μL of each primer (10 μM each) and 57 μL of DI water. Standard K562 genomic DNA (Roche) was employed to test the performance of the sample-in–answer-out microsystem. Human whole blood was collected from a volunteer with informed consent. Dried bloodstains were prepared by pipetting 2 μL of blood on a piece of 903® paper. After being dried completely, 3 mm-diameter blood discs were punched off and kept at 4 °C until use. Buccal swab samples were prepared by gently scraping the inside of a volunteer's cheek for 30 s using Whatman sterile Omni swabs (GE Healthcare). The tips of the swabs (about 3 mm in length) were cut off with a razor blade and stored at 4 °C. The PCR protocol includes the initial activation of polymerases at 95 °C for 5 min, followed by 35 cycles at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s, and a final extension step for 10 min at 72 °C.Multiplex two-color allele-specific PCR for warfarin pharmacogenetic testing
Multiplex two-color allele-specific PCR for detecting the VKORC1 6853 (–1639G>A, rs9923231), VKORC1 6484 (1173C>T, rs9934438) and CYP2C9*3 (1075A>C, rs1057910) SNPs was developed in collaboration with CapitalBio Corporation (Beijing, China). All the primer sequences were provided by CapitalBio. To amplify each locus, the primer set consists of one shared reverse primer and two forward primers which specifically match two variant alleles and are labeled with FAM and TAMRA, respectively. The sizes of all the amplicons can be found in Table 1. Whole blood samples from five donors were kindly provided by CapitalBio with informed consent. The 100 μL Master Mix for multiplex allele-specific PCR is composed of 10 μL of PCR buffer (10×, Roche), 3 μL of dNTP (10 mM each), 8 μL of MgCl2 (25 mM), 5 μL of Roche FastTaq polymerase (Roche), 10 μL of BSA (50 μg μL−1), 3 μL of PEG10000 (160 μg μL−1), 22 μL of primer mix and 39 μL of DI water. The thermal cycling protocol is the same as that in the singleplex amplification.| VKORC1 6484 (1173T>C) | CYP2C9*3 (1075A>C) | VKORC1 6583 (–1639G>A) | |
|---|---|---|---|
| Wild type (FAM) | 152 | 182 | 234 |
| Mutant (TAMRA) | 150 | 183 | 234 |
Results and discussion
Integration of DNA extraction, PCR and CE
Although the translation of individual analytical steps into chip formats, such as DNA extraction, PCR and capillary electrophoresis, has been extensively published, the development of fully integrated microsystems for “sample-in–answer-out” genetic analysis is still a daunting task. It is because the integration is much more than the simple combination of several stand-alone functional units. Efficient and reproducible sample/product transport between functional units is the key to forming a seamlessly integrated system that possesses the advantages of high sensitivity, reproducibility and reliability. Here, we successfully incorporated several key technologies to achieve the goal of “sample-in–answer-out”. First, the integration of DNA extraction and PCR amplification is realized by employing an “in situ” amplification concept.31,32 In contrast to the most common bind–wash–elute DNA extraction,34,35 “in situ” amplification combines DNA extraction and PCR in a single reaction chamber, where amplification is performed directly without elution. This method not only dramatically simplifies the structures of the integrated system, but also ensures the entire DNA extract being mixed with PCR reagents without timing issues, leading to more sensitive detection.Second, a novel injection electrode coupled with an injection chamber was developed for the integration of the sample preparation and the detection unit. This new mechanism has three functions: (1) acting as a valve to isolate the PCR and CE structures; (2) mixing PCR products with sizing standards and formamide; (3) transferring the mixture from the DEA chip to the CAE chip. A piece of tape on the bottom of the injection chamber functions as a valve to isolate the sample preparation and the capillary electrophoresis chip in this system. To open this “tape valve”, the electrode is actuated to penetrate all the way through the DEA chip so that the tip of the electrode is brought into contact with the bottom of the sample reservoir of the CAE chip. Due to the capillary action, the PCR products and the injection reagents stored in the injection chamber are drawn into the cross groove of the electrode, where the solutions are thoroughly mixed together. After that, an electrical field is applied between the electrode and the waste reservoir to electrophorese the mixture from the groove to the injection channel for analysis. To verify the functions of this mechanism, we first loaded blue and red ink into the upper and lower compartments of the injection chamber, respectively (shown in Fig. 4A). The electrode was manually punched through the injection chamber and then immediately taken out to dispense all the absorbed solution into a series of spots on a piece of paper. As shown in Fig. 4B, all the spots show a uniform purple color, illustrating that the blue and red inks were mixed very well in the groove. To further verify the electrical injection of this mechanism, we conducted the electrophoretic separation of the samples that were injected conventionally and by the mechanism. Both injection methods can produce similar electropherograms (Fig. 4C) with balanced peak heights in the range of 100–300 bp, demonstrating the effectiveness of the injection. This simple integration and injection method further simplified the structures of the microchip assembly.
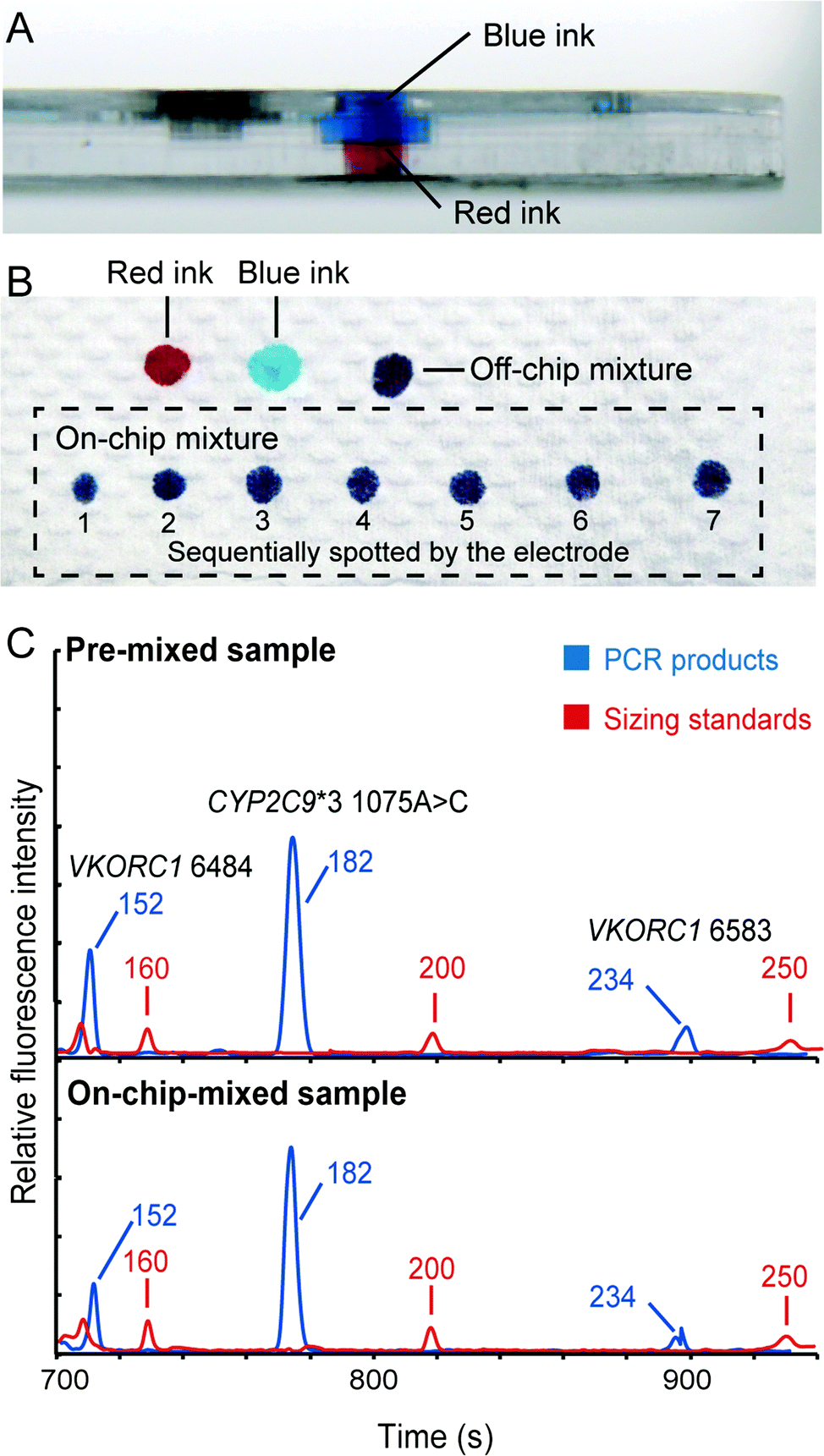 | ||
| Fig. 4 Verification of the injection electrode for sample mixing and injection to electrophoresis. (A) Side view of the injection chamber in the DNA extraction and amplification chip. The upper and lower compartments of the loading chamber were filled with blue and red ink, respectively. (B) Colors of the blue, red and mixed inks on a piece of paper. The ink in the loading chamber was absorbed into the electrode and then dispensed into a series of spots on a piece of paper. All the spots in the dotted box show a uniform purple color, illustrating that the blue and red inks were mixed immediately in the groove. (C) Comparison of the electropherograms generated by the conventional and injection electrode methods. | ||
Third, we adopted a modular-based integration approach to construct our “sample-in–answer-out” microsystem. This strategy provides two key advantages to our system: versatility and low cost. Different DEA chips for accommodating different types of samples can all be combined with the same CAE chips via the same integration mechanism. As a result, the assembled microchips are capable of processing multiple sample types, such as whole blood, bloodstains and buccal swabs, without any changes to the major structures of the instrument. In addition, only the low-cost, plastic DEA chips in the microchip assembly are for one-time use, while the glass CAE chip can be used multiple times. Thus, the cost of the genetic analysis could be significantly reduced. Overall, by employing the above three strategies, diverse samples can be efficiently processed and transferred in the microsystem without any complicated structures and delicate operations.
System characterization using singleplex PCR
We first employed standard K562 genomic DNA to verify the integration of the DNA extraction and amplification chip and the capillary array electrophoresis chip. Premixed PCR solutions containing 0.625, 1.25, 2.5 and 5 ng of K562 DNA were loaded into the DEA chips and then analyzed in the instrument. Following 35 cycles of PCR amplification, the amplicons were co-injected with ROX-labeled sizing standards into the CE channels for detection using the on-chip injection electrode. As demonstrated in Fig. 5A, the 106 bp fragments amplified from the amelogenin marker can be successfully obtained with as low as 0.625 ng of input DNA. Multiple primer-dimer peaks were also generated without any interference in the assay. In parallel, the negative control containing DI water instead of genomic DNA shows no peaks in the profile, demonstrating that the operations were contamination-free. Next, to verify the complete integration of the entire genetic analysis from DNA extraction to CE detection, human whole blood samples were processed and analyzed in the system. 0.3, 0.5 and 1 μL of blood samples were pipetted onto the filter nets in the loading chambers of the DEA chips, and then the chambers were sealed completely with pieces of tape. After the microchip assembly was loaded into the instrument, the automated process was conducted by the system. Fig. 5B reveals that, even with only 0.3 μL of input blood, the 106 bp amplified fragments can still be successfully obtained together with co-injected sizing standards. The complete procedure for analyzing human whole blood takes 2 hours, including 15 min for DNA extraction, 80 min for PCR and 25 min for electrophoresis. | ||
| Fig. 5 Limit of detection of the microsystem for the amelogenin marker. (A) PCR amplification and separation performed using standard K562 genomic DNA on the system. The detection limit was determined to be 0.625 ng of input DNA templates. (B) Genetic analyses of human whole blood from DNA extraction to electrophoretic detection. A complete trace was obtained from 0.3 μL of blood. | ||
Pharmacogenetic testing for guiding warfarin dosage
Pharmacogenetic testing is one of the essential methodologies for implementing the concept of precision medicine.2 However, the current genotyping process is still expensive and labor-intensive, and requires dedicated laboratories. Our “sample-in–answer-out” platform with the advantages of short turnaround time, low cost and complete automation may provide a promising tool towards the integration of genetic testing in routine clinical diagnosis. Hence, to demonstrate and verify the application of our system, we developed a multiplex two-color allele-specific PCR assay for detecting warfarin-related single-nucleotide polymorphisms in collaboration with CapitalBio Corporation. Three SNP loci, including VKORC1 6853 (–1639G>A), VKORC1 6484 (1173C>T) and CYP2C9*3 (1075A>C), were chosen due to their significant impact on warfarin dosing algorithms. First, the limit of detection of the system for this three-plex genetic analysis was explored using human blood that carries wild-type alleles. Different amounts of whole blood were loaded into the microchip, and then the automated analyses were conducted by the instrument following the same protocol as that in the singleplex test. Fig. 6 shows that a full profile containing three FAM-labeled wild-type amplicons (152 bp at VKORC1 6484, 182 bp at CYP2C9*3 (1075A>C) and 234 bp at VKORC1 6583) can be successfully obtained with as low as 0.5 μL of input blood. A blank profile of the negative control confirms that contamination-free operations were achieved.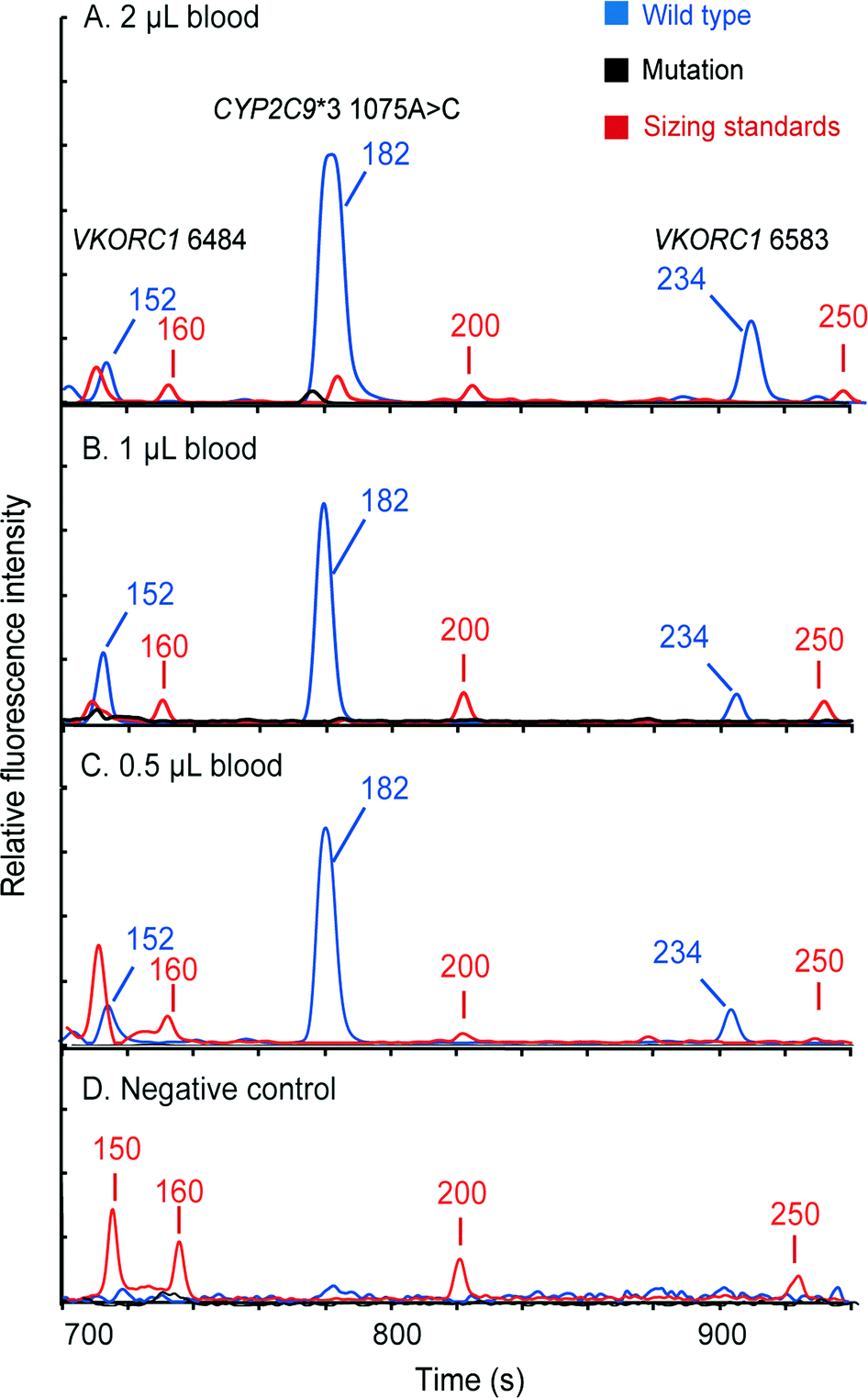 | ||
| Fig. 6 Detection limit of the microsystem for analyzing three SNP loci in VKORC1 and CYP2C9. A full profile was obtained from as low as 0.5 μL of input whole blood. A negative control experiment was conducted to confirm the absence of carryover. | ||
Following the limit-of-detection analysis, we then employed blood samples obtained from volunteers carrying various SNP genotypes to test the typing accuracy of the microsystem. In each assay, 2 μL of whole blood was processed by the instrument automatically. The entire analytical process including DNA extraction, amplification and CE detection was performed under the control of the LabVIEW program within two hours. Panel A in Fig. 7 presents a profile obtained from a control sample without any mutations, showing three FAM-labeled peaks. Panel B shows the result from a patient carrying homozygous mutants in all three loci, which produced three TAMRA-labeled peaks, including 150 bp at VKORC1 6484 (1173T>C), 183 bp at CYP2C9*3 (1075A>C) and 234 bp at VKORC1 6583 (–1639G>A). Due to the color change in the PCR products, the genotyping results can be read out immediately. Panel C presents the result of the genotyping from a sample with heterozygous alleles in the CYP2C9*3 SNP locus. Two peaks, one labeled with FAM from the wild-type allele and the other labeled with TAMRA from the mutant allele, were generated. Since the electrophoresis performed in our system can achieve a single-base separation resolution, the size and the color differences in these two peaks clearly illustrate the genotype of the blood sample. Next, the electropherogram shown in panel D contains a 150 bp TAMRA peak, a 182 bp FAM peak and a 234 bp TAMRA peak, illustrating that the blood carries two homozygous SNPs at VKORC1 6484 and VKORC1 6583. Finally, the sample with two heterozygous SNPs at VKORC1 6484 and CYP2C9*3 was analyzed, producing two peaks in each locus (panel E). All of the genetic profiles obtained from the fully integrated and automated system were in 100% accordance with those obtained using conventional methods, demonstrating that our microsystem has the capability of detecting SNPs correctly in a timely manner.
 | ||
| Fig. 7 Three-plex pharmacogenetic analyses of patients' blood samples with various genotypes. (A) Analysis conducted using 2 μL of the control sample, showing three FAM-labeled peaks (152 bp at VKORC1 6484, 182 bp at CYP2C9*3 and 234 bp at VKORC1 6583). (B) Genetic analysis of a patient carrying homozygous mutations in all three loci, which produced three TAMRA-labeled peaks (150 bp at VKORC1 6484, 183 bp at CYP2C9*3 and 234 bp at VKORC1 6583). (C) Analysis of a blood sample with a heterozygous mutant at CYP2C9*3. One peak labeled with FAM from the wild-type allele and the other labeled with TAMRA from the mutant were shown in the trace. (D) Testing of a blood sample obtained from a patient carrying two homozygous mutants in the 6484 and 6583 SNP loci of the VKORC1 gene. (E) Detection of a sample with two heterozygous mutations at VKORC1 6484 and CYP2C9*3, showing two peaks in each heterozygous SNP locus. | ||
Genetic testing from bloodstains and oral swabs
Since multiple types of samples may be encountered in clinical diagnosis, we further evaluated our system's versatility using dried bloodstains and oral swabs. In these experiments, the DEA chip in the microchip assembly was swapped with the one that has an empty loading chamber without the filter net. In the bloodstain test, a 3 mm-diameter disc of Whatman 903® paper that contains 2 μL of human whole blood was sealed into the loading chamber with a piece of tape. Once the chips were assembled and placed into the instrument, the automated analytical process was conducted by the instrument following a similar protocol to that in the whole blood analysis. The electropherogram showing three FAM-labeled peaks at the VKORC1 6484, CYP2C9*3 and VKORC1 6583 loci is presented in Fig. 8A. To type oral swab samples using our instrument, the tip (about 3 mm in length) of a Whatman sterile Omni swab was cut off with a razor blade and then placed into the loading chamber of the microchip assembly. After that, the rest of the analysis is the same as that for bloodstains. Fig. 8B shows that a full profile with three FAM-labeled peaks was successfully obtained. These results clearly indicate that the fully integrated microsystem is capable of conducting automated genetic analysis of multiple sample types with simple modifications to the disposable DEA microchip. | ||
| Fig. 8 Pharmacogenetic analyses conducted for (A) a dried bloodstain and (B) a buccal swab. Full profiles could be automatically obtained by the microsystem in two hours. | ||
The conventional genotyping methods based on the allele-specific PCR assay usually rely on several bulky instruments, have a lengthy process, and are prone to contamination. For example, the procedure of the microarray-based method includes DNA extraction, PCR, single-strand DNA preparation, hybridization, washing and detection, which takes about 7–8 hours to complete and needs a lot of manual operations. Real-time PCR detection is much faster and the operation is relatively simple, but the typing accuracy is often not adequate for clinical diagnosis. In contrast, our microfluidic system integrates the entire procedure of genetic testing into a single modular-based microdevice and the total analysis time is reduced to two hours. As a result, sample handling by users in our system is significantly reduced, eliminating the risk of cross-contamination and simplifying the operations. The electrophoretic separation reveals the sizing information of the PCR products, providing an additional check of the typing accuracy. In the future, it is possible to extend the use of our system to more directions, such as point-of-care diagnosis, forensic human identification, food safety testing and environmental monitoring.
Conclusions
The fully integrated “sample-in–answer-out” microsystem for pharmacogenetic analysis successfully combines microchip sample preparation and capillary electrophoresis into a modular-based microdevice operated in a compact control and detection instrument. The “in situ” PCR amplification enabled by the filter paper-based DNA extraction method dramatically simplifies the structure of the microchip. In addition, by employing a novel injection electrode, the plastic DEA chips were integrated with the glass CAE chip in a modular format, which facilitate the analysis of various sample types. The sensitivity of the microsystem was determined to be 0.625 ng of standard K562 DNA or 0.3 μL of human whole blood for the singleplex detection of the amelogenin marker, and 0.5 μL of blood for the genotyping of three warfarin-related SNPs. The entire genetic analysis from a sample to a profile can be completed in two hours without any human intervention. The successful development of this fully integrated and automated microsystem is a significant advance towards rapid, sensitive and reliable pharmacogenotyping for guiding patients' warfarin dosing in clinical practice. In the future, since the extraction–amplification–electrophoresis process is a common protocol for genetic analysis, our instrument can be extensively used for many purposes, such as forensic DNA typing, food safety, environmental monitoring, etc., without any major modifications to the system.Acknowledgements
We thank Prof. Jing Cheng, Prof. Wanli Xing and Prof. Guoliang Huang at the Department of Biomedical Engineering, Tsinghua University for their valuable discussions. This research was supported by the National Instrumentation Program (No. 2013YQ190467) of the Ministry of Science and Technology of China.References
- E. H. Rubin, J. D. Allen, J. A. Nowak and S. E. Bates, Clin. Cancer Res., 2014, 20, 1419–1427 CrossRef CAS PubMed.
- S. D. Mooney, Hum. Genet., 2015, 134, 459–465 CrossRef CAS PubMed.
- E. A. Ashley, JAMA, J. Am. Med. Assoc., 2015, 313, 2119–2120 CrossRef CAS PubMed.
- J. D. Roberts, G. A. Wells, M. R. Le May, M. Labinaz, C. Glover, M. Froeschl, A. Dick, J. F. Marquis, E. O'Brien, S. Goncalves, I. Druce, A. Stewart, M. H. Gollob and D. Y. F. So, Lancet, 2012, 379, 1705–1711 CrossRef CAS.
- A. Katsnelson, Nat. Med., 2013, 19, 249 CrossRef CAS PubMed.
- D. A. Flockhart, D. O'Kane, M. S. Williams, M. S. Watson, D. A. Flockhart, B. Gage, R. Gandolfi, R. King, E. Lyon, R. Nussbaum, D. O'Kane, K. Schulman, D. Veenstra, M. S. Williams and M. S. Watson, Genet. Med., 2008, 10, 139–150 CrossRef CAS PubMed.
- J. A. Johnson and L. H. Cavallari, Trends Cardiovasc. Med., 2015, 25, 33–41 CrossRef CAS PubMed.
- S. B. Daniel, M. C. Lovegrove, N. Shehab and C. L. Richards, N. Engl. J. Med., 2011, 365, 2002–2012 CrossRef PubMed.
- M. T. M. Lee and T. E. Klein, J. Hum. Genet., 2013, 58, 334–338 CrossRef CAS PubMed.
- J. A. Johnson, L. Gong, M. Whirl-Carrillo, B. F. Gage, S. A. Scott, C. M. Stein, J. L. Anderson, S. E. Kimmel, M. T. M. Lee, M. Pirmohamed, M. Wadelius, T. E. Klein and R. B. Altman, Clin. Pharmacol. Ther., 2011, 90, 625–629 CrossRef CAS PubMed.
- M. H. Eckman, J. Rosand, S. M. Greenberg and B. F. Gage, Ann. Intern. Med., 2009, 150, 73–83 CrossRef PubMed.
- R. S. Epstein, T. P. Moyer, R. E. Aubert, D. J. O'Kane, F. Xia, R. R. Verbrugge, B. F. Gage and J. R. Teagarden, J. Am. Coll. Cardiol., 2010, 55, 2804–2812 CrossRef CAS PubMed.
- B. L. Poe, D. M. Haverstick and J. P. Landers, Clin. Chem., 2012, 58, 725–731 CAS.
- P. Liu and R. A. Mathies, Trends Biotechnol., 2009, 27, 572–581 CrossRef CAS PubMed.
- S. J. Reinholt and A. J. Baeumner, Angew. Chem., Int. Ed., 2014, 53, 13988–14001 CrossRef CAS PubMed.
- S. Q. Wang, Y. J. Sun, W. P. Gan, Y. Liu, G. X. Xiang, D. Wang, L. Wang, J. Cheng and P. Liu, Biomicrofluidics, 2015, 9 CAS.
- L. Chen, A. Manz and P. J. R. Day, Lab Chip, 2007, 7, 1413–1423 RSC.
- A. Arora, G. Simone, G. B. Salieb-Beugelaar, J. T. Kim and A. Manz, Anal. Chem., 2010, 82, 4830–4847 CrossRef CAS PubMed.
- C. J. Easley, J. M. Karlinsey and J. P. Landers, Lab Chip, 2006, 6, 601–610 RSC.
- E. T. Lagally, J. R. Scherer, R. G. Blazej, N. M. Toriello, B. A. Diep, M. Ramchandani, G. F. Sensabaugh, L. W. Riley and R. A. Mathies, Anal. Chem., 2004, 76, 3162–3170 CrossRef CAS PubMed.
- P. Liu, T. S. Seo, N. Beyor, K. J. Shin, J. R. Scherer and R. A. Mathies, Anal. Chem., 2007, 79, 1881–1889 CrossRef CAS PubMed.
- N. Thaitrong, N. M. Toriello, N. Del Bueno and R. A. Mathies, Anal. Chem., 2009, 81, 1371–1377 CrossRef CAS PubMed.
- P. Liu, J. R. Scherer, S. A. Greenspoon, T. N. Chiesl and R. A. Mathies, Forensic Sci. Int.: Genet., 2011, 5, 484–492 CrossRef CAS PubMed.
- P. Liu, X. Li, S. A. Greenspoon, J. R. Scherer and R. A. Mathies, Lab Chip, 2011, 11, 1041–1048 RSC.
- A. J. Hopwood, C. Hurth, J. Yang, Z. Cai, N. Moran, J. G. Lee-Edghill, A. Nordquist, R. Lenigk, M. D. Estes, J. P. Haley, C. R. McAlister, X. Chen, C. Brooks, S. Smith, K. Elliott, P. Koumi, F. Zenhausern and G. Tully, Anal. Chem., 2010, 82, 6991–6999 CrossRef CAS PubMed.
- B. L. LaRue, A. Moore, J. L. King, P. L. Marshall and B. Budowle, Forensic Sci. Int.: Genet., 2014, 13, 104–111 CrossRef CAS PubMed.
- S. Jovanovich, G. Bogdan, R. Belcinski, J. Buscaino, D. Burgi, E. L. R. Butts, K. Chear, B. Ciopyk, D. Eberhart, O. El-Sissi, H. Franklin, S. Gangano, J. Gass, D. Harris, L. Hennessy, A. Kindwall, D. King, J. Klevenberg, Y. Li, N. Mehendale, R. McIntosh, B. Nielsen, C. Park, F. Pearson, R. Schueren, N. Stainton, C. Troup, P. M. Vallone, M. Vangbo, T. Woudenberg, D. Wyrick and S. Williams, Forensic Sci. Int.: Genet., 2015, 16, 181–194 CrossRef CAS PubMed.
- E. Tan, R. S. Turingan, C. Hogan, S. Vasantgadkar, L. Palombo, J. W. Schumm and R. F. Selden, Invest. Genet., 2013, 4, 16 CrossRef PubMed.
- D. Le Roux, B. E. Root, C. R. Reedy, J. A. Hickey, O. N. Scott, J. M. Bienvenue, J. P. Landers, L. Chassagne and P. de Mazancourt, Anal. Chem., 2014, 86, 8192–8199 CrossRef CAS PubMed.
- D. Le Roux, B. E. Root, J. A. Hickey, O. N. Scott, A. Tsuei, J. Y. Li, D. J. Saul, L. Chassagne, J. P. Landers and P. de Mazancourt, Lab Chip, 2014, 14, 4415–4425 RSC.
- W. Gan, B. Zhuang, P. Zhang, J. Han, C. Li and P. Liu, Lab Chip, 2014, 14, 3719–3728 RSC.
- B. Zhuang, W. P. Gan, S. Q. Wang, J. P. Han, G. X. Xiang, C. X. Li, J. Sun and P. Liu, Anal. Chem., 2015, 87, 1202–1209 CrossRef CAS PubMed.
- S. H. I. Yeung, S. A. Greenspoon, A. McGuckian, C. A. Crouse, C. A. Emrich, J. Ban and R. A. Mathies, J. Forensic Sci., 2006, 51, 740–747 CrossRef CAS PubMed.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 RSC.
- C. W. Price, D. C. Leslie and J. P. Landers, Lab Chip, 2009, 9, 2484–2494 RSC.
Footnote |
| † Joint first authors with equal contributions. |
| This journal is © The Royal Society of Chemistry 2016 |