
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Update on the status of metrology for metalloproteins
Claudia
Swart
*a and
Norbert
Jakubowski
b
aPhysikalisch-Technische Bundesanstalt, Bundesallee 100, 38108 Braunschweig, Germany. E-mail: claudia.swart@ptb.de
bBundesanstalt für Materialforschung und -prüfung BAM, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: nobert.jakubowski@bam.de
First published on 3rd August 2016
Abstract
Metalloproteins, which represent about 30% of the total proteome, are often important markers for distinguishing between healthy and diseased states of patients. As such markers have become increasingly important in clinical diagnostics, some of these proteins are routinely analysed in clinical laboratories. Reliable and comparable results are the basis for the investigation of changes in the proteome due to different health conditions. Nevertheless, for many proteins the results achieved with different routine measurement procedures or in different laboratories vary widely, thus hampering medical insights and the development of treatments. Reference measurement procedures with results traceable to the International System of Units (SI) will also help to greatly improve the performance of routine measurement procedures and, this way, they support the understanding of changes in the metallome. This perspective will give an overview of the efforts during the last three years to achieve reliable quantification of metalloproteins. The analytes covered were chosen due to their importance in clinical diagnostics. Haemoglobin (HGB), transferrin (TRF), superoxide dismutase (SOD) or ceruloplasmin (CER), for example, can serve as markers for diseases such as Down's syndrome in prenatal diagnostics (e.g. SOD), inflammation (acute-phase proteins such as TRF) or deficiency diseases (e.g. HGB, TRF, and CER). Moreover, they are used for the control of treatment efficiency, e.g. total HGB as the most important marker for anaemia treatment or the glycosylated form of haemoglobin A (HBA1c) for the treatment of diabetes. On the other hand, selenoproteins, namely glutathione peroxidase (GPX), seem to play an important role in cancer prevention and in reducing the side effects of chemotherapies.
1. Introduction
As metalloproteins represent around 30% of the whole proteome they have become increasingly important in clinical diagnostics. A metalloprotein is “a protein whose function is conferred by a metal. […] Metal is usually bound as a hydrated ion or a metal-containing cofactor”.1 They have to be distinguished from metal-containing proteins which bind the metal nonspecifically, not in their active centre, and whose function is not dependent on the presence of this metal.In clinical diagnosis the comparability and reliability of the results for the determination of the respective analyte independent of time, place and equipment are crucial. However, the comparability of the results for many proteins obtained with different measurement procedures in different laboratories is still an issue in clinical chemistry.2 The best way to achieve this goal is to establish a traceability chain for each protein as already established for many inorganic and smaller organic analytes. For these analytes, a database for the calibration and measurement capabilities of various National Metrology Institutes (NMIs) exists and is hosted at the Bureau International des Poids et Mesures (BIPM) (http://kcdb.bipm.org/). “The ideal aim of a metrological traceability chain is that measurement results produced by a routine measurement procedure are the same as if the quantity in the patient samples had been measured by the reference measurement procedure”.2 However, for many metalloproteins such a traceability chain is quite challenging to establish. Before a meaningful reference system can be set up, the analyte has to be defined unambiguously. Considering, for example, that about 750 different genetic and posttranslational variants are known for haemoglobin (HGB) the task is enormous. A practical compromise is often that a group of modifications, which are clinically indistinguishable, is used as the analyte in question. So first of all analytical chemists, biologists, and physicians together have to agree on the clinically relevant form(s) of the protein in question. For the glycosylated form of HGB the definition is that all the forms of haemoglobin A (HBA0) which are glycosylated at the N-terminal valin of the β-chain, independently of other possible modifications in the protein, are summarised as HBA1c. Only for HBA1c a reference system is provided by the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC), but its implementation worldwide was met with unexpected resistance.3 Due to the lack of a reference system, harmonisation of the results obtained with the most commonly used routine methods can only be achieved by using a common calibrator. However, the provision of a reference system is preferable whenever possible as it guarantees long-term stability.4 Methods used for harmonisation are mainly immunoassays such as enzyme linked immunosorbent assays (ELISAs), immunonephelometry, and immunoturbidimetry or optical methods used in routine analysis, while for standardisation mass spectrometry (MS) or optical methods combined with chromatographic separation are used to establish a reference system and certify reference materials. In the best of cases, the traceability chain allows us to trace the results back to the International System of Units (Système international d'unités, SI) via a pure substance reference material whose purity has to be well known.2 However, even in the rare cases such a material is available in clinical diagnostics thorough control of the fate of the analyte has to be ensured regarding losses or changes of the analyte during sample preparation, separation and detection. In principle, this can best be achieved with an internal standard behaving as similarly to the analyte as possible, preferably identically, which is best fulfilled when the analyte is applied in an isotopically enriched form. This is called species specific isotope dilution (ID). This isotopically labelled protein is added to the sample prior to any sample treatment and, thus, shares the fate of the analyte. This way it can be accounted for possible losses or species transformation. In the case of many metalloproteins, the metal incorporated into the protein can be exchanged by its isotopically enriched form for the labelling. Fig. 1 shows a possible approach to produce such a species specific spike material. By detecting the isotope ratio, the metal content and, thus, the protein content can be determined. However, quantifying metalloproteins via their metal content assumes that the metal is preserved in the protein structure during sample preparation and separation. For detection and quantification ICP-MS can be used which is a selective and sensitive elemental detector. Furthermore, as only the metal is detected, it has to be ensured that all other compounds carrying the same metal are separated from the analyte.
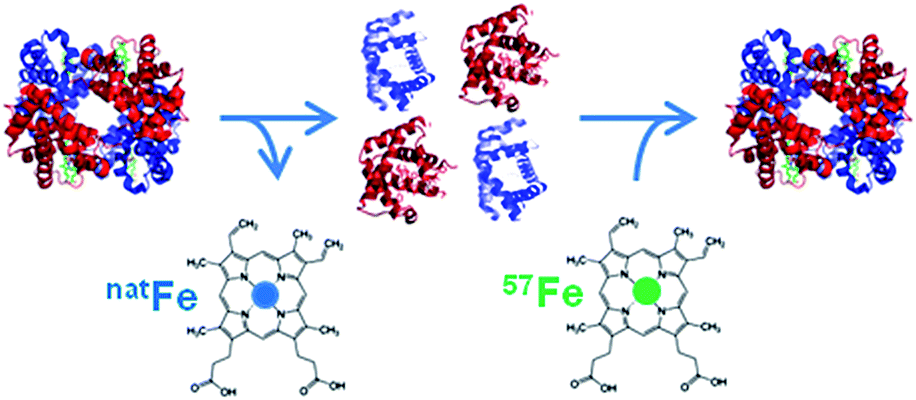 | ||
| Fig. 1 Example of the production of a species-specific spike material for the quantification of metalloproteins using ID (reproduced from ref. 8 with permission from the Royal Society of Chemistry). | ||
To improve the situation, the project Metrology for Metalloproteins (HLT05) was launched in the framework of the European Metrology Research Programme (EMRP)5 dealing with the iron (Fe) containing proteins transferrin (TRF) and HGB, the copper (Cu) containing proteins Cu, Zn-superoxide dismutase (SOD1) and ceruloplasmin (CER) and the selenoproteins glutathione peroxidase (GPX1) and selenoprotein P (SEPP). A short outline was given in an editorial of the RSC journal “Metallomics”.6 Besides, several efforts were made worldwide to improve the situation in clinical routine analysis regarding the sensitivity, reliability and comparability of results. As an extensive review was compiled and published in 2013,7 only a perspective is given here for each of the covered metalloproteins and the main focus will be on achievements since the end of 2012.
2. Quantification of selected metalloproteins
2.1. Transferrin
TRF is an Fe transport protein synthesised in the liver which can bind up to two Fe ions.9 TRF has a varying degree of glycosylation resulting in so called sialoforms. Nine different sialoforms are known depending on the number of glycan branches with terminal sialic acids bound to the protein. The natural pattern of these sialoforms is changed in the case of some diseases. The most prominent one is chronic alcohol abuse which results in an increase of the ratio of TRF with a low glycosylation level to total TRF. The content of the so called S4 form (two biantennary N-glycans with four sialic acids), usually the most abundant form, is shifted to a higher content of S2 (two sialic acid residues) and even S0 (no sialic acid), which is called carbodeficient TRF (CDT), in the case of alcohol abuse.10Besides the methods used for quantification already described elsewhere,7 some new approaches were developed in the last few years. For total TRF quantification the focus of the last few years was on the optimisation of existing assays. Gao et al. used chemiluminescence resonance energy transfer (CRET) based competitive immunoassay and enhanced the quenching effect by using amorphous carbon nanoparticles (ACNPs) bound to TRF.11 For this, a 96-well microplate was coated with anti-TRF and horseradish peroxidase. Without the addition of TRF bound to ACNPs as a fluorescence quencher the intensity of the luminol emission catalysed by the peroxidase reached a maximum. When adding the sample containing TRF and the TRF bound to ACNPs the intensity of the luminescence decreases with increasing TRF concentration in the sample as more quencher can bind to the antibody in the assay. The linear range of the method was (20–400) ng mL−1, with a limit of detection (LOD) of 20 ng mL−1. As the concentration in real samples is much higher, the samples have to be diluted. The relative standard deviation (RSD) for the determination of TRF in real samples is below 7.84%. A similar approach was taken by Kong et al. who developed a label-free electro-chemiluminescence immunosensor.12 The label-free approach was chosen, as labelling carries the risk of damaging the protein. Furthermore, it is time- and labour-intensive and the quantitative conversion has to be guaranteed. Their goal was to develop a disposable, simple, low cost and sensitive sensor for total TRF quantification based on a commercially available ELISA kit. The silver (Ag) immunosensor consisted of an Ag/AgCl reference electrode, a carbon auxiliary electrode and a carbon working electrode which was modified with gold (Au) nanoparticles coated with luminol and chitosan. The TRF antibody was then immobilised on the surface of the dried unfunctionalised nanoparticles. After adding the sample, the sensor was immersed in buffer/H2O2 solution to detect the resulting electro-luminescence. The LOD achieved with this method is 0.033 ng mL−1 TRF with a RSD of 5.8%. Although these methods achieved low LODs, they are hardly fit to serve as reference measurement procedures as many factors such as unblocked unspecific binding sites and interferences are difficult to control. Besides no information was given by the authors concerning the reproducibility of the production of the sensor and, thus, nothing is known about the comparability of results from different devices.
A completely different approach was investigated by Esteban-Fernández et al. who separated TRF and human serum albumin (HSA) from human serum samples with gel electrophoresis (GE) after labelling with metal coded affinity tag (MeCAT).13 As the tag, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) loaded with ytterbium (Yb) including an iodoacetamide cysteine-reactive group was used. TRF and HSA labelled with isotopically enriched 171Yb were added to the sample prior to separation and ID was performed by means of laser ablation (LA)-ICP-MS detection and also by direct infusion after spot mineralization. A requirement for this method is that the analyte labelled with isotopically enriched and natural MeCAT behaves identically on the gel and during LA. To determine the mass bias, TRF and HSA labelled with natural Yb MeCAT were always run on a separate lane in each gel. As the protein is not distributed homogeneously over the whole spot, LA was performed at the centre of the spot. An RSD of 4.2% could be achieved. In Fig. 2a, the signal intensity of the isotopes 171Yb and 172Yb from a transversal ablation of spots is shown containing different amounts of protein but always with the same natural![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enriched proportion. Fig. 2b demonstrates that the 172Yb:171Yb ratio is constant for all spots even when the protein distribution inside the spot is not homogeneous. However, to be used as a reference method, a complete labelling has to be ensured for every sample. Due to the fact that biological samples show a wide variety regarding fat content, high albumin (ALB) content, etc. and the label binds to every protein containing cysteins (Cys), this is hard to achieve. Another way of labelling was applied by Bustos et al.14 using CdSe/ZnS quantum dots conjugated with a polymeric layer, streptavidin and biotinylated antibodies. A second antibody was then bound to the heavy chain of first antibody. The quantification was performed either with spectroscopy using ultraviolet or visible light excitation (UV/Vis) (565 nm) or by detecting cadmium (Cd) or selenium (Se) with ICP-MS. The latter showed one order of magnitude lower LOD (0.15 ng mL−1) and a far larger linear range but the RSD was twice as large as with UV/Vis detection (14% versus 7%). However, as the number of atoms per quantum dot has to be known and might vary from dot to dot and the stability of the functionalised quantum dots influences the result, the use of this method as a reference method is limited.
:enriched proportion. Fig. 2b demonstrates that the 172Yb:171Yb ratio is constant for all spots even when the protein distribution inside the spot is not homogeneous. However, to be used as a reference method, a complete labelling has to be ensured for every sample. Due to the fact that biological samples show a wide variety regarding fat content, high albumin (ALB) content, etc. and the label binds to every protein containing cysteins (Cys), this is hard to achieve. Another way of labelling was applied by Bustos et al.14 using CdSe/ZnS quantum dots conjugated with a polymeric layer, streptavidin and biotinylated antibodies. A second antibody was then bound to the heavy chain of first antibody. The quantification was performed either with spectroscopy using ultraviolet or visible light excitation (UV/Vis) (565 nm) or by detecting cadmium (Cd) or selenium (Se) with ICP-MS. The latter showed one order of magnitude lower LOD (0.15 ng mL−1) and a far larger linear range but the RSD was twice as large as with UV/Vis detection (14% versus 7%). However, as the number of atoms per quantum dot has to be known and might vary from dot to dot and the stability of the functionalised quantum dots influences the result, the use of this method as a reference method is limited.
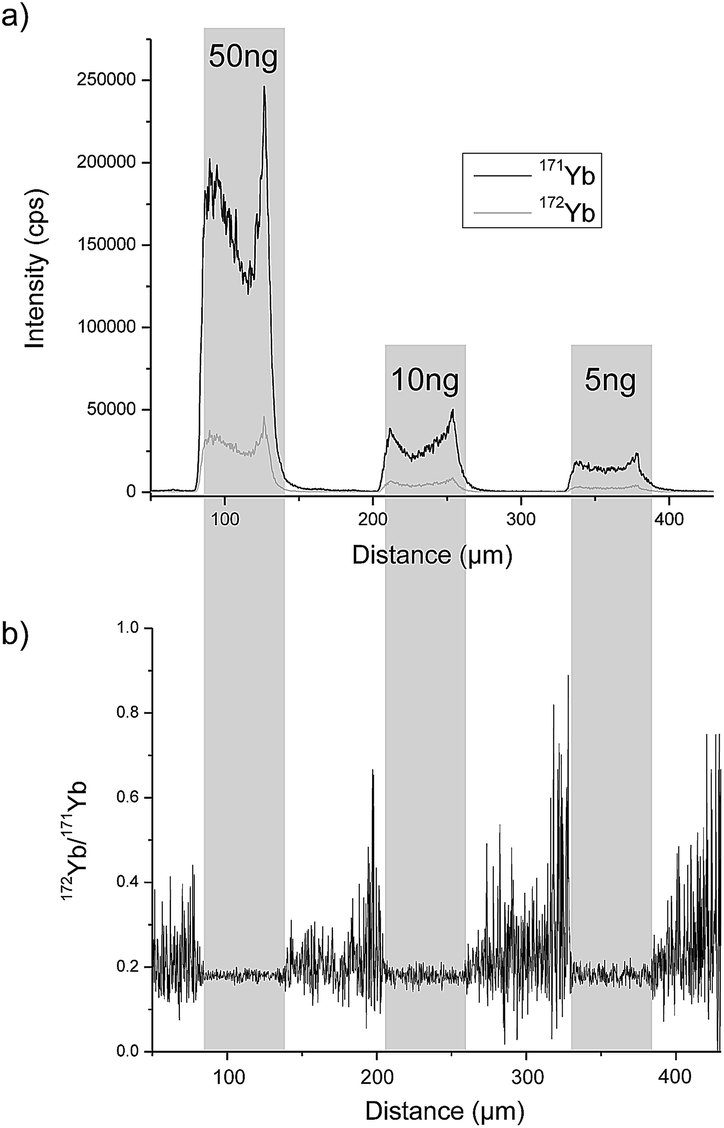 | ||
| Fig. 2 LA-ICP-MS analysis of gel electrophoresis spots containing 50 ng, 10 ng and 5 ng of MeCAT(Yb)-IA labeled HSA spiked with isotopically enriched MeCAT(171Yb)-IA labeled HSA in a proportion 1:2: (a) signal intensities of isotopes 171Yb and 172Yb. (b) Isotope ratio 172Yb/171Yb (reproduced from ref. 8 with permission from the Royal Society of Chemistry). | ||
A measurement procedure for the quantification of total TRF content via its Fe content, first developed by del Castillo Busto et al.15 using HPLC-ICP-MS, was improved and validated within the EMRP project HLT05 to serve as a candidate reference measurement procedure. A species specific spike material was produced and thoroughly characterised regarding its protein structure, Fe load and isotopic composition.16,17 A similar approach was developed by Ordonez et al., who besides total TRF also quantified the sialoforms using postcolumn ID-ICP-MS.18 Both methods shall be used for the certification of the SRM-909c reference material in future. To reduce the influence of Fe background and low isotopic enrichment in the spike material, triple isotope dilution mass spectrometry (IDMS) was used for the quantification of total TRF.19
Besides the determination of total TRF, the quantification of the various sialoforms is important as described above. One method to separate the various sialoforms of TRF in serum is the use of capillary electrophoresis (CE) coupled with UV/Vis or MS detection after purification of the sample using an immunoaffinity cartridge. MS enables the identification and quantification of proteins with less risk of interferences compared to UV/Vis. However, the coatings normally used in CE show considerable bleeding which interferes with MS detection. Medina-Casanellas et al. developed a coating based on a physically adsorbed polyacrylamide monolayer which shows a lower bleeding and can be applied for MS and prepared a microcartridge for immunoaffinity purification of TRF in serum.20 The disadvantage of the MS detection was that the sialoforms are not resolved properly as the ionisation source of the MS disturbed the CE separation due to a sucking effect of the nebulizer gas flow. Furthermore, it turned out that a small amount of ALB was un-specifically bound in the cartridge. As the concentration of ALB in serum is significantly higher than that of TRF, it interfered with the UV/Vis detection of TRF. A similar approach is the use of capillary zone electrophoresis (CZE). To avoid the adsorption of proteins to the walls, a coating is needed to cover reactive –SiOH groups. Again bleeding of the coating of the capillary walls poses serious problems in connecting CZE to MS. Kohler et al. tested various possible coatings and found that anionic coatings such as hexadimethrine bromide–dextran sulphate (each 10%) in combination with 20 mM ammonium acetate (pH 8.5) as a background electrolyte was the best compromise between the resolution of the sialoforms, protein adsorption and coating stability.21 With UV/Vis detection, the system achieved good separation and stability. However, when the CZE is coupled to a time-of-flight MS with electrospray ionisation (ESI-TOF-MS), signal suppression and loss of separation capability were observed. Using two routine methods based on CZE with UV/Vis detection Caslavska et al. monitored genetic variants in 7290 patient samples.22 They found that the separation of sialoforms and of genetic variants is possible. However, subtypes of the same phenotype such as C1C2 could not be resolved and the two assays gave different results for the genetic subtypes. Without a reference, there is no way to decide which assay (if any) gives the right results.
As the results of the determination of CDT as a marker for chronic alcohol abuse might also have legal consequences and the comparison of results obtained with three different measurement kits revealed significant differences;23 the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) established a working group for the standardisation of CDT measurements in 2005.4 The goal was a reference system based on a primary reference measurement procedure with S2-sialoform of TRF as the calibrator. It turned out that, as the candidate reference measurement procedure measures S2 in relation to the sum of all sialoforms and every sialoform has a different absorption coefficient, this was not feasible. Furthermore, the S2 form is not available in the purity required for calibration purposes. Therefore, only a comparison measurement procedure was developed using HPLC with UV/Vis detection at 470 nm which can be used for the harmonization of routine measurement procedures. Serum samples spiked with S2 and lyophilised serum samples were tested as candidate reference materials, but they turned out to be unsuitable. Finally, a candidate reference material was prepared using patient samples which is stable for at least 3 years when frozen at −70 °C. The commutability was tested for the reference measurement procedure and the routine measurement procedures. The relationship between the reference measurement procedure and the routine measurement procedures was found to be linear so that a harmonization is feasible. However, some routine methods are interfered by high S3 concentrations. This shows once again that actual standardisation is preferable to harmonization due to the long-term stability of the reference system. Landberg et al. investigated the cases where the proposed HPLC reference method cannot separate S2 and S3 sialoforms due to a shortened retention time of the S3 form (0.6% of all samples). First the CDT ratio was determined with the proposed HPLC-UV/Vis and total TRF with immunonephelometry. Then the glycans were released from the protein and these oligosaccharides were analysed with matrix assisted laser desorption ionisation (MALDI) TOF-MS. It turned out that patients with TRF containing the inseparable S2 and S3 forms have more triantennary branches and partly fucosylated antennae. They could not find any genetic reasons for this anomaly, but suggested a possible association with liver diseases. As S3 is not related to alcohol abuse but an interference with S2 would increase the peak height, and, thus the result for the relevant S2 form, this shows how important a reliable reference measurement procedure is.
In these procedures every step should be under control to avoid losses or transformations. For most methods for TRF discussed here this is difficult either because they contain a separation step in which TRF can be adsorbed to the wall of capillaries or columns or they contain a labelling step where 100% conversion cannot be guaranteed in all samples. Furthermore, some methods show matrix effects whether from the sample or from the separation phase. In the case of CDT, an additional challenge is the separation of the relevant sialoforms to avoid interferences. A solution would be to add TRF in an isotopically enriched form (either in the amino acid backbone or in the metal) to the sample directly after sampling so that this ideal internal standard has the same fate as the analyte. However, this still leaves the challenge of incomplete separation to deal with.
2.2. Haemoglobin
Haemoglobin (HGB) is an Fe-containing protein responsible for oxygen transport in vertebrates, which consists of four subunits, each containing a haeme group which can bind one O2 molecule.24 The most abundant form in humans with (96–98)% is HBA0 which consists of two α-chains and two β-chains. Total HGB serves as a marker for anaemia while the glycosylated form HBA1c is used for the long-term monitoring of diabetes.24 The determination of HGB can also be used to monitor the effectiveness of cleaning procedures for reusable devices in hospitals using, for example, fluorescence detection.25 For HBA1c it was recognised early that standardisation is necessary and different regional systems were implemented, for example the National Glycohemoglobin Standardization Program (NGSP) system in the USA, Mono-S in Sweden (named after the separation column used for separation) and the system of the Japan Society of Clinical Chemistry (JSCC). An IFCC working group was established in 1995 to implement a universal reference system. The first step was to define the analyte as HBA0 glycosylated at the N-terminal valine of the β-chain. This definition still includes a number of variants as it says nothing about further posttranslational changes, but, nevertheless, it is a clinically relevant parameter. The next step was the implementation of a reference system including reference measurement procedures and reference materials.26As an IFCC reference measurement procedure exists for HBA1c (ref. 27) the number of new approaches is quite limited. Besides reference measurement procedures, point-of-care testing devices for HBA1c are still rare. One cheap approach is the lateral flow immunoassay developed by Ang et al.28 For the preparation of the capillary flow kit, detection strips containing the capturing antibodies were mounted on a nitrocellulose membrane. Au colloids modified with antibodies were adsorbed on a conjugate pad. After the sample passed the capturing antibody the modified colloids were mobilised to form a sandwich immunoassay. The assay showed an enhanced sensitivity compared to others without the use of Au nanoparticles. Interferences from other glycated HBA0 forms and the colour of the blood could not be detected. The kit consists of three test lines which indicate the HBA1c concentration for untrained users, namely the patient, depending on how many of these strips change their colour. After calibration of the intensity the kit can also be used for quantification of HBA1c. The linear range for the ratio HBA1c/HBA0 was (4–12)% in quantitative mode which covers most of the clinically relevant range. It would have been interesting to see a comparison of the results obtained with this method and the IFCC reference method to ensure the traceability of the proposed point-of-care kit and the comparability with other methods in use.
Besides the main form of HGB, HBA0, various other genetic variations are known such as HBA2 and HBS. Tang et al. developed a novel GE method using the concept of moving reaction boundaries.29 The different buffer systems deployed resulted in sharp protein zones with a coefficient of variation of below 9%. The identification of the HGB forms was achieved using MALDI-TOF-MS while the quantification was performed optically.
For total HGB, the World Health Organisation (WHO) recommended a reference method based on the conversion of HBG to haemiglobincyanide (HiCN) already in 1965.30 However, due to the toxicity of the reagents used in the method, it cannot be applied in many countries with restrictions on the use of potassium cyanide. Therefore, possible replacements of this method were investigated avoiding toxic reagents. Balderas-López et al. used a photoelectric approach without any conversion of HGB necessary at all.31 When comparing their method to the reference method they found higher concentrations of HGB in blood which might hint at an influence of the conversion of HGB to HiCN on the results. An electrochemical detection method for HGB was developed by Prasad et al. using a pencil lead modified with L-Cys bound to quantum dots adsorbed to multi-walled carbon nanotubes, which were then coated with a polymer imprinted with HGB on the surface.32 They achieved a LOD of 7 ng mL−1 with a RSD of 1%. However, blood samples have to be diluted 105 to be in the linear range and avoid matrix effects, the stability of the film and the binding sites may vary from sensor to sensor and some other proteins were found to interfere with HGB. Neither the reproducibility for the production of the sensor nor possible memory effects or the number of samples which can be measured with one sensor are given, which makes the assessment of its potential as a reference method difficult.
Further on the way to become a reference measurement procedure is the approach using the alkaline haematin detergent (AHD) for the conversion of HGB before UV/Vis detection.33 In 2015, it was also implemented, besides the established HiCN method, in an improved version of the German standard DIN 58931 “Haematology – Determination of haemoglobin concentration in blood – Reference method” as a routine measurement procedure for total HGB in clinical laboratories with results traceable to the SI via comparison with IDMS methods.34,35 In the AHD method, the haemolysate is treated with sodium hydroxide and Triton X-100 and haemin chloride is used as the calibrator for UV/Vis detection.
As described in the Introduction, species specific IDMS is a promising approach for the development of reference measurement procedures. This requires the protein to be in an isotopically enriched form. Recently, Brauckmann et al. developed an elegant method to produce HGB containing isotopically enriched Fe as a spike for IDMS allowing species specific ICP-IDMS to be used for the quantification of total HGB via Fe.8 They applied this spike material for the quantification of total HGB and HBA0 in the reference material IRMM/IFCC-467 certified for total HGB. The resulting value of (122.1 ± 1.8) mg g−1 was in good agreement with the certified value of (0.956 ± 0.005) mol mol−1 corresponding to (119.7 ± 3.7) mg g−1. For HBA0 (117.0 ± 1.6) mg g−1 was found using ion exchange chromatography to separate the various HGB variants. The low expanded uncertainty of 1.6% and the addition of isotopically labelled HGB at the beginning of the sample preparation as an ideal internal standard make this approach perfectly suitable as a reference measurement procedure. Furthermore, compared to the HiCN or AHD approach no conversion of the analyte, which is always subject to kinetic control, is necessary. As it uses the whole analyte as spike, it also has the advantage compared to the IFCC reference method that no quantitative release of the signal peptide has to be ensured.
2.3. Cu, Zn-superoxide dismutase
Superoxide dismutase 1 (SOD1) is a homodimeric enzyme containing one Cu and one zinc (Zn) ion in each subunit.36,37 The structure of SOD1 is highly conserved in eukaryotes. It is responsible for the conversion of the superoxide radical (O2−) to oxygen (O2) and hydrogen peroxide (H2O2). As an acute phase protein, it is a marker for inflammatory processes. Furthermore, reactive oxygen species are also suspected to be involved in neurological disorders such as Alzheimer's and Parkinson's diseases.38 Therefore, the distribution of SOD in neurons and astrocytes would be enlightening which was investigated by Hare et al. using size exclusion chromatography (SEC) coupled with ICP-MS.39 However, the quantification was performed using external calibration which cannot account for analyte losses in sample preparation and separation. Furthermore, quantitative results are only given for the amount of metal in the fractions not for metalloproteins.In routine analysis the enzyme is usually measured using enzymatic activity assays. Already in 2006 Attar et al. demonstrated that different kits show significantly different activities.40 However, there is still no reference method or material available for the harmonisation or even standardisation of SOD1 measurements. As the active form of SOD1 necessarily contains Cu and Zn, the quantification of the enzyme can also be performed via the metal content. As described above species specific IDMS is the most reliable method provided that a species specific spike material exists. In the case of SOD1 the metals can be chemically removed41,42 and replaced with their isotopically enriched form.43,44 This method is a good starting point for the development and validation as a reference measurement procedure for SOD1 in human erythrocyte lysate which was successfully conducted within the EMRP project HLT05 by Gleitzmann et al.45 As discussed before this approach is less prone to matrix effects and interferences compared to external calibration or activity assays and is, therefore, more promising than other approaches.
2.4. Ceruloplasmin
Ceruloplasmin (CER) is a blue α2-glycoprotein which is synthesized in the liver and serves as the Cu storage protein. It binds (90–95)% of the whole amount of Cu in the body and is involved in the Fe metabolism as ferroxidase catalysing the oxidation of Fe(II) to Fe(III), which can then be incorporated into TRF. CER contains six tightly bound Cu ions and one or two labile Cu ions.46,47 CER is also discussed to be involved in neurodegenerative diseases.48 As CER is mainly produced in the liver, the correct synthesis of CER and its activity can be influenced by liver diseases. Siotto et al. showed that for some diseases it might be useful to determine the ratio of intact CER to the total concentration of CER rather than only the total concentration.49 Nevertheless, the methods on the market and under development are mainly for the quantification of total CER. A potential method for the separation and detection of CER is the use of native polyacrylamide GE (PAGE). However, as Cu is a ubiquitous element the Cu background in the gel and in the applied buffers has to be controlled carefully.49 A new approach for the quantification of CER is the use of an electrochemical magneto-immunosensor based on magnetic nanoparticles functionalised with anti-CER developed by Ojeda et al.50 They compared two different sensors, both based on competitive immunoreactions with alkaline phosphatase labelled CER conjugates and the detection of the enzymatic reaction, regarding the dynamic range, LOD and RSD. They found that the sensor using streptavidin and biotinylated anti-CER for the immobilisation of the antibody was more fit-for-purpose for real samples after 1:1000 dilution of the samples to reduce matrix effects than the one based on protein A used for immobilisation. The developed assay showed a LOD of 0.018 μg mL−1 with a RSD of about 9% in serum samples and a recovery of spiked CER of (90–100)%. The modified magnetic nanoparticles were stable for up to 42 days when stabilised with phosphate buffered saline with Tween20®. However, all these assays have still to be investigated carefully regarding possible interferences and the reproducibility between the various batches of antibodies and assays.
For all methods it has to be taken into account that CER is highly sensitive especially to protease induced degradation,51,52 which may hamper the reliability of some methods considerably. The addition of isotopically labelled CER directly after sampling would reveal such changes in the sample and would allow the correction to some extent. Again the labelling is possible both via the amino acid sequence and via the metal. In the case of the latter the Cu background has to be controlled carefully due to the ubiquity of Cu.
2.5. Selenoproteins
Se is an essential element for humans found in so called selenoproteins. In contrast to Se containing proteins, which incorporate selenomethionine (Se-Met) unspecifically instead of methionine (Met), they contain the genetically encoded amino acid selenocysteine (Se-Cys). This amino acid is considered to be the 21st amino acid, encoded genetically with UGA (otherwise used as a stop codon) and a Se-Cys incorporation sequence (SECIS).53,54 The selenoproteins investigated so far are GPX, responsible for the reduction of peroxides, and SEPP, the Se storage protein in the body containing 10 Se-Cys. As selenoproteins, namely GPX, seem to play an important role in cancer prevention and in reducing the side effects of chemotherapy, Se-supplementation is often used in parallel to chemotherapy. However, the range between deficiency (daily intake of less than 30 μg for adults) and toxicity (daily intake of more than 700 μg for adults) is rather narrow.55Recent developments in measurement procedures for selenoproteins are mainly based on the detection of Se with ICP-MS. Gómez-Espina et al. used SEC-ICP-MS with post-column IDMS for the separation and quantification of GPX1 in red blood cells.56 Before separation HGB was precipitated. As the selectivity of SEC is rather limited, the peak purity was verified by collecting the relevant fraction, which was then tryptically digested and identified with ESI-Q-TOF-MS. As post-column IDMS was used, the column recovery as well as the amount of GPX1 co-precipitated with HGB has to be known. This was investigated by measuring the enzymatic activity of GPX1 and total Se in the samples before and after column passage and resulted in an overall recovery of about 90%. To be able to determine a number of Se-species in the corresponding serum and cerebrospinal fluid (CSF) samples, Solovyev et al. used an orthogonal approach with both SEC and strong anion exchange (SAX) chromatography coupled with ICP-MS or SAX and CE coupled with ICP-MS.57 For the quantification a SEPP standard, produced by purifying the protein from human plasma with AFC, was proposed. Rhodium (Rh) was added as an internal standard either directly to the samples for analysis of total Se or to the post-column for the separation methods. Se containing ALB (Se-ALB), SEPP, GPX1, and thioredoxin reductase (TRXR) as well as inorganic Se could be detected in the samples. An even more elaborate approach was taken by García-Sevillano et al. who used a number of SEC, SAX and AF columns (up to five columns) in-line to separate selenoproteins and Se-metabolites in human serum.58 The recovery along such a sophisticated separation line might be an issue, but the sum of all Se containing peaks was within the uncertainty of the certified value for Se when analysing the reference material BCR-637. The quantification was achieved with post-column IDMS. The standard deviation in real samples was up to 10%. For speciation of selenoproteins in cell cultures, Bianga et al. used a combination of isoelectrofocusing (IEF) on electrophoretic strips with LA-ICP-MS.59 After protein extraction and precipitation the selenoproteins were separated by IEF. Afterwards, the Se containing proteins were localised with LA-ICP-MS and the protein was then identified using ESI-Orbitrap-MS/MS. No quantification was performed although the determination of total Se in the single spots should be possible with LA-ICP-MS. However, matrix matched standardisation is difficult.
The presented measurement procedures are all convincing approaches to quantify selenoproteins in biological samples. However, as they use external calibration or post-column IDMS, they have insufficient control over possible sample losses or conversions. This problem can only be solved using species-specific IDMS. A special challenge here is the production of the species specific spike material. As Se is covalently bound in selenoproteins, it is not possible to remove and replace it. The isotopic label has to be introduced already during the biosynthesis of the protein itself which can elegantly be done using a method developed by Konopka et al.60 They introduced simultaneously isotopically labelled selenomethionine and stable isotope-labelled amino acids in a cell-free protein synthesis.
An alternative and novel strategy for the absolute quantification of selenium included in SEPP1 was presented very recently by Deitrich et al.61 This strategy is based on the use of species specific double IDMS by using protein specific peptides [namely ENLPSLCSUQGLR and AEENITESCQUR (where U is SeCys)] which are custom synthesised and enriched in the 76Se isotope. These peptides have been spiked to blood plasma samples. After spiking a tryptic digestion of the plasma proteins and alkylation of the resulting peptides have been performed, followed by isotope ratio determinations using HPLC-ICP-MS/MS. Protein bound Se down to the peptide level in a complex plasma matrix was accurately detected with a total content of Se of 105.5 μg kg−1. The method enabled the selective Se speciation analysis of human plasma samples without the need of extensive clean-up or pre-concentration steps. For the assessment of accuracy of the method, two plasma reference materials (BCR-637 and SRM1950) were analysed using hyphenated methods and the species specific approach. The Se mass fractions obtained via the isotopic ratios of 78Se/76Se and 82Se/76Se for each of the Se-peptides were found to agree within 2.4% with the literature data of the reference material. This work represents a systematic approach to the accurate quantification of plasma SEPP1 at clinical levels and is pioneering the concept of species specific isotope dilution for quantitative metalloproteomics, which can be applied for other metalloproteins as well, as soon as protein specific peptides are synthesised. However, the quantitative release of the signal peptides has to be ensured. Furthermore, peptides may behave differently during sample preparation and may, thus, degrade during digestion. This has to be checked carefully.
3. Conclusions
During the last three years many efforts were made in international projects and committees to develop and validate candidate reference measurement procedures and protocols for various clinical parameters. However, the main efforts in method development are still focused on the development of methods which are more sensitive and easier to handle and can be used for point-of-care diagnostics and, thus, need to be robust as well. An overview of all methods and their figures of merit is summarised in Table 1. However, especially those instruments need to deliver reliable results to enable the medical practitioner/patients to act on them and this requires a reference system to which those methods can be linked. Nevertheless, many methods are too complex and time consuming to be used as routine methods. In particular the methods involving several separation steps are too elaborate to be used in routine laboratories. Furthermore, the MS technique is too expensive and is hardly used in routine analysis. However, they allow insight into different aspects of protein behaviour in a research context.| Method | Analyte | LOD | RSD | Linear range | Reference |
|---|---|---|---|---|---|
| a u: uncertainty calculated with k = 1; U: uncertainty calculated with k = 2; n.g.: not given in original publication. | |||||
| CRET based competitive immunoassay enhanced by using ACNP bound to TRF | Total TRF | 20 ng mL−1 | 7.84% | (20–400) ng mL−1 | Gao et al.11 |
| Label-free electro-chemiluminescence immunosensor | Total TRF | 0.033 ng mL−1 | <5.8% | (0.1–18) ng mL−1 | Kong et al.12 |
| GE-LA-ICP-MS with MeCAT labelling | Total TRF | 50 μmol L−1 | 4.2% | n.g. | Esteban-Fernández et al.13 |
| GE-LA-ICP-MS with CdSe/ZnS quantum dots | Total TRF | 0.15 ng mL−1 | 14% | n.g. | Bustos et al.14 |
| HPLC-ICP-MS | Total TRF | n.g. | U = 3.5% | n.g. | Castillo Busto et al.;14,15 Frank et al.17,19 |
| Postcolumn ID-ICP-MS | Total TRF, sialoforms | n.g. | <6% | n.g. | Ordonez et al.18 |
| CE-MS | Sialoforms | 50 μg mL−1 | 5% | n.g. | Medina-Casanellas et al.20 |
| CZE-MS | Sialoforms | n.g. | n.g. | n.g. | Kohler et al.21 |
| HPLC-MS (IFCC reference measurement procedure) | HBA1c/HBA0 | n.g. | u = 0.8% | (0–20)% | C. Weykamp et al.27 |
| Lateral flow immunoassay | HBA1c/HBA0 | n.g. | n.g. | (4–12)% | Ang et al.28 |
| GE method using moving reaction boundaries | HBG isoforms | Only qualitative | n.g. | n.g. | Tang et al.29 |
| Photoelectric detection | Total HGB | n.g. | n.g. | n.g. | Balderas-López et al.31 |
| Electrochemical detection using L quantum dots | Total HGB | 7 ng mL−1 | 1% | (27.8–444.0) ng mL−1 | Prasad et al.32 |
| AHD method | Total HGB | n.g. | U = 0.5% | (0–200) g L−1 | Heuck et al.,33 K. Witt et al.34 |
| Species specific ICP-IDMS | Total HGB | n.g. | U = 1.6% | n.g. | Brauckmann et al.8 |
| Species specific ICP-IDMS | SOD1 | n.g. | U = 1.5% | n.g. | Gleitzmann et al.45 |
| PAGE | CER | 11.58 IU L−1 | u = 3.2% | n.g. | Siotto et al.49 |
| Electrochemical magneto-immunosensor | CER | 0.018 μg mL−1 | 9% | (0.05–1.04) μg mL−1 | Ojeda et al.50 |
| SEC-ICP-MS with post-column IDMS | GPX1 | n.g. | 4% | 0–20 ng mL−1 | Gómez-Espina et al.56 |
| SAX-ICP-MS | Se-ALB, SEPP, GPX1, TRXR | 0.032 μg L−1 | n.g. | n.g. | Solovyev et al.57 |
| SEC, SAX and AF column ICP-MS | GPX | 0.2 ng g−1 | 21% | n.g. | García-Sevillano et al.58 |
| SEPP | 0.7 ng g−1 | 12% | |||
| Se-ALB | 0.9 ng g−1 | 9% | |||
| SeO32− | 1.1 ng g−1 | 2% | |||
| SeO42− | 1.3 ng g−1 | n.g. | |||
| IEF-LA-ICP-MS | SEP15, GPX1, GPX4, TRXR1, TRXR2 | Only qualitative | n.g. | n.g. | Bianga et al.59 |
| Species specific HPLC-ICP-IDMS/MS | SEPP1 | 1.38 μg kg−1 | U = 5% | n.g. | Deitrich et al.61 |
In this perspective, new approaches based on species specific ID for the quantification of metalloproteins via the metal are critically highlighted. These approaches are coming close to a fully transparent traceability chain, because primary traceable standards can already be obtained for elements such as Cu. Cell-free protein synthesis is a promising approach for the production of isotopically enriched proteins or peptides in case the metal is covalently bound and cannot be exchanged by its isotopically enriched form in the native protein. Such approaches offer new opportunities also for the synthetic production of proteins where elements such as carbon (C), nitrogen (N), oxygen (O) and hydrogen (H) are fully replaced by enriched isotopes for instance by 13C, 12N, 17O and 2D. Novel organic MS can already analyse intact proteins with all their posttranslational modifications leading to the vision that in future ID analysis of intact proteins will become the gold standard to either produce novel certified reference materials or to develop standard protocols or even quantitative results in clinical diagnosis. All the concepts are developed already for inorganic standards and protocols but joint interdisciplinary efforts are still needed to reach this goal also for proteins. To quantify whole proteins reliably is not only a challenge in proteomics research, but also more and more important in clinical diagnosis. Therefore, joint efforts of analytical and clinical chemists as well as physicians are of paramount importance. In the case of the quantification of metalloproteins atomic spectroscopy, which allows the sensitive and reliable quantification of the metal and thus the corresponding protein, can take an important place. It is for the sake of everybody's health!
Acknowledgements
The work of this study is part of the project HLT05, which was funded within the framework of the EMRP. The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union.References
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC.
- G. H. White, Ann. Clin. Biochem., 2011, 48, 393–409 CrossRef CAS PubMed.
- W. Hoelzel, C. Weykamp, J.-O. Jeppsson, K. Miedema, J. R. Barr, I. Goodall, T. Hoshino, W. G. John, U. Kobold, R. Little, A. Mosca, P. Mauri, R. Paroni, F. Susanto, I. Takei, L. Thienpont, M. Umemoto and H.-M. Wiedmeyer, Clin. Chem., 2004, 50, 166–174 CAS.
- C. Weykamp, J. Wielders, A. Helander, R. F. Anton, V. Bianchi, J.-O. Jeppsson, C. Siebelder, J. B. Whitfield and F. Schellenberg, Clin. Chem., 2014, 60, 945–953 CAS.
- Physikalisch-Technische Bundesanstalt, http://www.ptb.de/emrp/metallomics.html, accessed 2012-11-09, 2012.
- C. Swart, P. Fisicaro, H. Goenaga-Infante and S. Zakel, Metallomics, 2012, 4, 1137–1140 RSC.
- C. Swart, Anal. Bioanal. Chem., 2013, 405, 5697–5723 CrossRef CAS PubMed.
- C. Brauckmann, C. Frank, D. Schulze, P. Kaiser, R. Stosch and C. Swart, J. Anal. At. Spectrom., 2016 10.1039/C6JA00028B.
- G. de Jong, J. P. van Dijk and H. G. van Eijk, Clin. Chim. Acta, 1990, 190, 1–46 CrossRef CAS.
- V. Sanz-Nebot, P. González, I. Toro, A. Ribes and J. Barbosa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 798, 1–7 CrossRef CAS.
- H. Gao, W. Wang, Z. Wang, J. Han and Z. Fu, Anal. Chim. Acta, 2014, 819, 102–107 CrossRef CAS PubMed.
- W. Kong, H. Zhou, H. Ouyang, Z. Li and Z. Fu, Anal. Methods, 2014, 6, 2959–2964 RSC.
- D. Esteban-Fernández, F. S. Bierkandt and M. W. Linscheid, J. Anal. At. Spectrom., 2012, 27, 1701–1708 RSC.
- A. R. Montoro Bustos, M. Garcia-Cortes, H. González-Iglesias, J. Ruiz Encinar, J. M. Costa-Fernández, M. Coca-Prados and A. Sanz-Medel, Anal. Chim. Acta, 2015, 879, 77–84 CrossRef CAS PubMed.
- E. del Castillo Busto, M. Montes-Bayón, J. I. Garcia Alonso, J. A. Caruso and A. Sanz-Medel, Analyst, 2010, 135, 1538–1540 RSC.
- M. E. del Castillo Busto, M. Montes-Bayón, J. Bettmer and A. Sanz-Medel, Analyst, 2008, 133, 379–384 RSC.
- C. Frank, O. Rienitz, R. Jährling, D. Schiel and S. Zakel, Metallomics, 2012, 4, 1239–1244 RSC.
- Y. N. Ordonez, R. F. Anton and W. C. Davis, Anal. Methods, 2014, 6, 3967–3974 RSC.
- C. Frank, O. Rienitz, C. Swart and D. Schiel, Anal. Bioanal. Chem., 2013, 405, 1913–1919 CrossRef CAS PubMed.
- S. Medina-Casanellas, F. Benavente, E. Giménez, J. Barbosa and V. Sanz-Nebot, Electrophoresis, 2014, 35, 2130–2136 CrossRef CAS PubMed.
- I. Kohler, M. Augsburger, S. Rudaz and J. Schappler, Forensic Sci. Int., 2014, 243, 14–22 CrossRef CAS PubMed.
- J. Caslavska, J. Joneli, U. Wanzenried, J. Schiess, C. Lanz and W. Thormann, J. Sep. Sci., 2014, 37, 1663–1670 CrossRef CAS PubMed.
- A. Helander, G. Eriksson, H. Stibler and J.-O. Jeppsson, Clin. Chem., 2001, 47, 1225–1233 CAS.
- Klinisches Wörterbuch - Pschyrembel®, Walter de Gruyter GmbH&Co.KG, Berlin, 261st edn, 2007 Search PubMed.
- J. Frey, A. Guan, Z. Li, S. Turtil and K. S. Phillips, Anal. Bioanal. Chem., 2015, 407, 6885–6889 CrossRef CAS PubMed.
- W. G. John, Clin. Biochem., 2012, 45, 1048–1050 CrossRef CAS PubMed.
- C. Weykamp, W. G. John, A. Mosca, T. Hoshino, R. Little, J.-O. Jeppsson, I. Goodall, K. Miedema, G. Myers, H. Reinauer, D. B. Sacks, R. Slingerland and C. Siebelder, Clin. Chem., 2008, 54, 240–248 CAS.
- S. H. Ang, C. Y. Yu, G. Y. Ang, Y. Y. Chan, Y. b. Alias and S. M. Khor, Anal. Methods, 2015, 7, 3972–3980 RSC.
- Y.-Y. Tang, H.-Y. Wang, L. Chen, S. Li, C.-G. Guo, H.-Z. Fan, C.-X. Cao and L.-Y. Fan, Anal. Bioanal. Chem., 2013, 405, 8587–8595 CrossRef CAS PubMed.
- World Health Organization (WHO), http://www.209.61.208.233/en/Section10/Section17/Section53/Section480_1728.htm, accessed 2012-11-06, 2012.
- J. A. Balderas-López, Y. M. Gómez y Gómez and M. E. Bautista-Ramírez, Int. J. Thermophys., 2015, 36, 844–848 CrossRef.
- B. B. Prasad, A. Prasad and M. P. Tiwari, Talanta, 2013, 109, 52–60 CrossRef CAS PubMed.
- C. C. Heuck, H. Reinauer and W. G. Wood, Clin. Lab., 2008, 54, 255–272 CAS.
- K. Witt, H. U. Wolf, C. Heuck, M. Kammel, A. Kummrow and J. Neukammer, Metrologia, 2013, 50, 539 CrossRef CAS.
- DIN 58931:2010–08, Haematology - Determination of haemoglobin concentration in blood - Reference method, DIN, 2010.
- Y. Yamazaki and T. Takao, Anal. Chem., 2008, 80, 8246–8252 CrossRef CAS PubMed.
- D. Chen, Q. Wang, H. Zhang, S. Mi, J. Wang, Q. Zeng and G. Zhang, Int. J. Quantum Chem., 2010, 110, 1394–1401 CAS.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2012, 120, 149–166 CrossRef CAS PubMed.
- D. J. Hare, A. Grubman, T. M. Ryan, A. Lothian, J. R. Liddell, R. Grimm, T. Matsuda, P. A. Doble, R. A. Cherny, A. I. Bush, A. R. White, C. L. Masters and B. R. Roberts, Metallomics, 2013, 5, 1656–1662 RSC.
- F. Attar, E. Keyhani and J. Keyhani, Appl. Biochem. Microbiol., 2006, 42, 101–106 CrossRef CAS.
- C. L. Deitrich, A. Raab, B. Pioselli, J. E. Thomas-Oates and J. Feldmann, Anal. Chem., 2007, 79, 8381–8390 CrossRef CAS PubMed.
- B. Sutter, P. L. Bounds and W. H. Koppenol, Protein Expression Purif., 2000, 19, 53–56 CrossRef CAS PubMed.
- C. Deitrich, S. Braukmann, A. Raab, C. Munro, B. Pioselli, E. Krupp, J. Thomas-Oates and J. Feldmann, Anal. Bioanal. Chem., 2010, 397, 3515–3524 CrossRef CAS PubMed.
- Y. Nuevo Ordóñez, C. L. Deitrich, M. Montes-Bayón, E. Blanco-González, J. Feldmann and A. Sanz-Medel, J. Anal. At. Spectrom., 2011, 26, 150–155 RSC.
- J. Gleitzmann, A. Raab, D. Schulze, H. Watzig, J. Feldmann and C. Swart, J. Anal. At. Spectrom., 2016 10.1039/C5JA00459D.
- G. Musci, S. D. Marco, G. C. Bellenchi and L. Calabrese, J. Biol. Chem., 1996, 271, 1972–1978 CrossRef CAS PubMed.
- V. Samygina, A. Sokolov, M. Pulina, H. Bartunik and V. Vasil’ev, Crystallogr. Rep., 2008, 53, 655–662 CrossRef CAS.
- J. Kristinsson, J. Snaedal, G. Tórsdóttir and T. Jóhannesson, Neuropsychiatr. Dis. Treat., 2012, 8, 515–521 CAS.
- M. Siotto, P. Pasqualetti, M. Marano and R. Squitti, J. Neural Transm., 2014, 121, 1281–1286 CrossRef PubMed.
- I. Ojeda, M. Moreno-Guzmán, A. González-Cortés, P. Yáñez-Sedeño and J. M. Pingarrón, Electroanalysis, 2013, 25, 2166–2174 CrossRef CAS.
- K. A. Moshkov, S. Lakatos, J. Hajdu, P. ZÁVodszky and S. A. Neifakh, Eur. J. Biochem., 1979, 94, 127–134 CrossRef CAS PubMed.
- R. Beetham, P. White, P. Riches, D. Bullock and F. MacKenzie, Clin. Chem., 2002, 48, 2293–2294 CAS.
- G. Ballihaut, L. E. Kilpatrick, E. L. Kilpatrick and W. C. Davis, Metallomics, 2012, 4, 533–538 RSC.
- J. Köhrle, R. Brigelius-Flohé, A. Böck, R. Gärtner, O. Meyer and L. Flohé, Biol. Chem., 2000, 381, 849–864 CrossRef PubMed.
- I. Heras, M. Palomo and Y. Madrid, Anal. Bioanal. Chem., 2011, 400, 1717–1727 CrossRef CAS PubMed.
- J. Gómez-Espina, E. Blanco-González, M. Montes-Bayón and A. Sanz-Medel, J. Anal. At. Spectrom., 2012, 27, 1949–1954 RSC.
- N. Solovyev, A. Berthele and B. Michalke, Anal. Bioanal. Chem., 2013, 405, 1875–1884 CrossRef CAS PubMed.
- M. A. García-Sevillano, T. García-Barrera and J. L. Gómez-Ariza, J. Chromatogr. A, 2013, 1318, 171–179 CrossRef PubMed.
- J. Bianga, Z. Touat-Hamici, K. Bierla, S. Mounicou, J. Szpunar, L. Chavatte and R. Lobinski, J. Proteomics, 2014, 108, 316–324 CrossRef CAS PubMed.
- A. Konopka, N. Zinn, C. Wild and W. D. Lehmann, in Shotgun Proteomics, Springer, New York, 2014, vol. 1156, pp. 337–363 Search PubMed.
- C. L. Deitrich, S. Cuello-Nuñez, D. Kmiotek, F. A. Torma, M.-E. Del Castillo Busto, P. Fisicaro and H. Goenaga-Infante, Anal. Chem., 2016, 88, 6357–6365 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |