
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic etching of soluble surface layers with on-line inductively coupled plasma mass spectrometry detection – a novel approach for determination of complex metal oxide surface cation stoichiometry
Andreas
Limbeck†
*a,
Ghislain M.
Rupp†
*a,
Markus
Kubicek
a,
Helena
Téllez
b,
John
Druce
b,
Tatsumi
Ishihara
b,
John A.
Kilner
bc and
Jürgen
Fleig
a
aInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9/164, 1060 Vienna, Austria. E-mail: A.Limbeck@tuwien.ac.at; ghislain.rupp@tuwien.ac.at; Tel: +43 1 58801 15180
bInternational Institute for Carbon-Neutral Energy Research, Kyushu University, 744, Motooka Nishi-ku, Fukuoka, 819-0395, Japan
cDepartment of Materials, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 28th June 2016
Abstract
In this work, an innovative approach for determining the surface stoichiometry of complex metal oxide (CMO) thin films is presented. The procedure is based on treatment of the sample surface with different etching solutions, followed by on-line analysis of the derived eluates using inductively coupled plasma – mass spectrometry (ICP-MS). Via consecutive treatment of the sample surface with water and diluted HCl, a differentiation between water soluble and acid soluble parts of near surface regions in thin films is enabled. The developed procedure was applied for the analysis of dense La0.6Sr0.4CoO3−δ (LSC) thin films indicating the presence of a water soluble Sr-rich phase with sub nm-thickness on top of the LSC films. The step-wise optimization of this technique is reported and the results are compared to measurements performed by low-energy ion scattering (LEIS). The detrimental effect of the water soluble Sr rich phase on the oxygen exchange activity of LSC thin film electrodes is verified by electrochemical impedance spectroscopy (EIS).
Introduction
Complex metal oxides (CMOs) are key materials in an increasing number of applications. Among the numerous crystal structures adopted by CMOs, those in the perovskite-type (ABO3) family have received the greatest attention followed by spinel-type (AB2O4) and pyrochlore-type (A2B2O7) structures. The growing interest over the past decades for these and other structures can be attributed to the functional diversity due to the wide range of properties that they can adopt.1 For example, in the fields of solid state electrochemistry and heterogeneous catalysis, complex metal oxides are attractive alternatives to expensive noble metal catalysts.2–4 For both disciplines, excellent knowledge of the structure and composition of the investigated material at the reaction sites, i.e. in most applications the solid/gas phase boundary (surface), is required for understanding reaction mechanisms and tailoring properties. Nanoscaled systems (e.g. nanoparticles, thin films) may not only improve functionalities but are also indispensable to increase the surface/bulk ratio for a detailed analysis of the region of interest. Particularly, thin film model systems, mostly deposited by physical and chemical vapor deposition techniques, offer the possibilities for using a large variety of analytical methods to characterize the surface and near surface region as well as depth profiling the layers beneath.Imaging methods, such as atomic force and scanning tunneling microscopy, are widely used to characterize the structure of the termination layer. For the analysis of the chemical composition, X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) or ion bombardment techniques such as secondary ion mass spectrometry (SIMS), Rutherford backscattering (RBS) and low energy ion scattering (LEIS) are often applied.5 However, quantification of the exact surface composition by these techniques is non-trivial. Alternative methods for quantitative analysis of thin films are glow-discharge mass spectrometry (GDMS)6,7 or laser-ablation inductively coupled plasma mass spectrometry (LA-ICP-MS).8,9 These and other well-established techniques also enable the analysis of depth profiles with sufficient sensitivity and depth resolution.
Application of these analytical tools has already shown that for complex metal oxides the structure and composition at and close to the surface frequently differ from the bulk. Elastic and electrostatic forces may drive segregation of dopants to the surface of CMOs,10,11 reactions between gas phase and solid may lead to the formation of secondary phases,12,13 and exsolution of metallic particles on the surface of perovskites is found under reducing conditions,14,15 to mention only a few examples. These changes are crucial for the (electro-)catalytic activity of the materials and their analysis requires methods that are specifically adapted to these complex phenomena. One example of a particularly challenging situation is given for the perovskite-type CMO La0.6Sr0.4CoO3−δ (LSC). LSC is a promising cathode material for intermediate temperature solid oxide fuel cells due its high activity for the surface oxygen exchange.16–18 However, degradation of the surface activity under various conditions is reported in the literature and several correlations to structural and/or compositional changes of the electrode surface have been reported.19,20 Chemical investigations on thin LSC films using XPS, SIMS and inductively coupled plasma – optical emission spectrometry (ICP-OES) already revealed an accumulation of Sr at the electrode surface, which may be responsible for the deactivation of the oxygen reduction reaction.21–23 Furthermore, Cai et al.23 proposed a secondary Sr phase such as SrO/Sr(OH)2 at the surface based on different binding energies found for Sr 3d electrons using XPS. Nonetheless, by XPS it is non-trivial to qualitatively (and almost impossible to quantitatively) deconvolute Sr surface species from Sr in the LSC lattice. Accordingly, additional analytical methods with complementary information content, e.g. not only on ionic binding situations but also on phases, are therefore highly desirable.
Recently, Kubicek et al.21 presented a novel tool for surface chemistry analysis to overcome limitations of conventional depth profiling techniques. There, the surface of LSC thin films was etched with dilute HCl, and the eluate containing the dissolved cations was then analyzed on-line by ICP-OES. In contrast to classic batch-wise extraction procedures, this approach provides time resolved information on the availability and solubility of any dissolved species. Application of a slightly modified etching procedure allowed the verification of a Sr enriched region at the surface of porous LSC thin films.24 However, limitations in sensitivity and reproducibility of analysis prevented the determination of surface cation stoichiometry as well as the measurement of dense LSC thin films. Thus, further methodological development of the dynamic etching procedure is compulsory; in particular, to enable the investigations required for a better understanding of Sr segregation and the underlying physical and chemical phenomena.
In the present study we show how this approach with dynamic etching can be continuously optimized. With the use of ICP-MS for element-selective on-line detection significant improvements in sensitivity were achieved. Adaption of the flow-injection system allowed consecutive etching of the sample with different solutions, thereby enabling a differentiation between water soluble and acid soluble LSC constituents. Moreover, improvements in accuracy and reproducibility of analysis could be obtained by changing material, design and volume of the etching cartridge. Achieved advancements allowed reliable measurement of the LSC thin film surface and near-surface cation composition, providing quantitative information about changes in the LSC stoichiometry. Applicability of the developed procedure for the analysis of dense LSC thin films was demonstrated by excellent agreement of the findings with results from low-energy ion scattering (LEIS) measurements.
Experimental
Reagents and standard solutions
High purity water was prepared using an Easypure water system (Thermo, USA, resistivity 18 MΩ cm) and used throughout the entire study. Nitric acid (65%) and hydrochloric acid (37%) was purchased from Merck (Darmstadt, Germany) with p.a. grade. Certified stock solutions (1000 mg L−1) of La, Sr and Co as well as Cu and Mn were procured from Merck (Darmstadt, Germany) and used for the preparation of calibration standards or as internal standards by dilution with 0.012 mol L−1 HCl.Sample synthesis and preparation
The LSC target for PLD was obtained from stoichiometric mixtures of La2O3, SrCO3, Co (99.995% Trace Select, Sigma Aldrich, Steinheim, Germany) powders and prepared by Pechini synthesis25 a described in ref. 24. LSC thin films with a thickness of 200 nm were deposited onto (100) oriented 9.5 mol% yttria stabilized zirconia (YSZ) substrates (5 × 5 × 0.5 mm3) the deposition procedure and parameters are given in ref. 26.Instrumentation
An iCAP 6500 series ICP-OES spectrometer (Thermo Scientific, USA) was used for method development. Sample introduction was performed using an APEX E high efficiency sample introduction system with a Meinhard concentric nebuliser and a cyclonic spray chamber (ESI Elemental Scientific, USA). Instrumental parameters used for ICP-OES measurement of thin surface layers are summarized in Table 1. ICP-MS analysis was conducted using quadrupole instrumentation (Thermo iCAP Qc, Thermor Scientific, Bremen, Germany). Sample introduction was accomplished employing a Peltier-cooled spray chamber equipped with a concentric quartz glass nebulizer. The instrumental parameters (Table 2) were optimized for maximum 115In signal and a 140Ce16O/140Ce ratio below 1.9% on a daily basis using a standard tuning solution. The amount of doubly charged ions was measured by the 137Ba++/137Ba+ ratio which was below 3% for all experiments. To minimize the influence of potential instrument induced artifacts, the sample signals were normalized using 55Mn as internal standard. Signal quantification was based on external calibration with aqueous standard solutions. Standards and blanks were measured at the beginning, during and at the end of an experimental session.| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Rf power | 1250 W | Nebulizer flow | 0.70 L min−1 |
| Total sample flow rate | 1.4 mL min−1 | Auxiliary flow | 0.8 L min−1 |
| Viewing height | 12 mm | Coolant flow | 12 L min−1 |
| Measurement mode: | transient signals with 1 s observation time per data point | ||
| Background correction: | constant shift from analytical line | ||
| Signal processing: | thermo ITEVA software | ||
| Element | λ 1 [nm] | λ 2 [nm] | λ 3 [nm] |
|---|---|---|---|
| La | 333.749 | 379.478 | 419.655 |
| Sr | 216.596 | 346.446 | 421.552 |
| Co | 228.696 | 238.892 | |
| Cu | 224.700 | 324.754 | |
| Mn | 257.610 |
| ICP-MS instrumentation | Thermo iCAP Q |
|---|---|
| Total sample flow rate | 0.8 mL min−1 |
| Nebulizer gas flow | 0.8 L min−1 |
| Aux. gas flow | 0.8 L min−1 |
| Cool gas flow | 14 L min−1 |
| RF power | 1550 W |
| Cones | Ni |
| Measured isotopes | 55Mn, 59Co, 63Cu, 65Cu, 86Sr, 87Sr, 139La |
| Dwell time per isotope | 0.010 s |
| Mass resolution | m/Δm = 300 |
For analysis of time resolved etching profiles a flow-injection (FI) system coupled to an ICP-OES or ICP-MS for element selective on-line detection was used. The applied FI-system consisted of a six port injection valve (VICI, Cheminert C22, USA) equipped with a sample cartridge for insertion of LSC samples. Arrangement of the applied FI-manifold was similar to the one recently described by Limbeck et al.27 for the dynamic extraction of water soluble trace metals in airborne particulate matter (Fig. 1). In the first part of the work, commercially available Chromafix® SPE columns (Macherey-Nagel, Germany, diameter 12 mm, length 14 mm) equipped with porous quartz frits were employed as sample cartridges. In the final procedure, homemade polytetrafluoroethylene (PTFE) micro-cartridges were used. In both cases the cartridges were connected to the FI-system using conventional luer-fittings, which allowed an easy and fast replacement of the etching compartment. The connections between the individual parts of the FI-system were made with PTFE tubes having an inner diameter of 0.7 mm.
 | ||
| Fig. 1 FI Manifold for dynamic etching of LSC thin film samples: P peristaltic pump, V 6-port valve. | ||
LEIS analysis of the LSC thin films were performed using a Qtac100 spectrometer (ION-TOF GmbH, Germany) fitted with a high brightness ion source to provide the primary beam (Hyperion, Oregon Physics, USA). Major advantage of LEIS is that the analyzer beam only probes the first atomic layer. The elemental composition of the surface was analyzed using a 6 keV Ne+ beam, which was directed normal to the surface and the kinetic energy distribution of the backscattered ions at 145° (collected over the entire azimuth) was analyzed to obtain the elemental composition of the first monoatomic layer. For depth profiling, a 1 keV Ar+ sputtering beam at a sputtering angle of 59 degrees was used. The sputtered area was 1.3 × 1.3 mm2, with the central region of 1 × 1 mm2 analyzed by the primary beam. The sputter rate for the depth calibration of the profiles was estimated by measuring the depth of a 300 × 300 μm2 crater using laser interferometry (LEXT OLS4000, Olympus, Japan), assuming that the rate was uniform for the LSC thin film. For the quantification of the cation surface coverage, the plateau signals at the end of the depth profiles were used as the reference value for the bulk stoichiometry, as described in ref. 28.
To measure the impedance of LSC electrodes, a second LSC thin film (same deposition conditions) was deposited on the back side of the YSZ substrate. The impedance was then measured by a Novocontrol Alpha A High Performance Frequency Analyzer with an alternating voltage of 10 mV (rms) in a frequency range of 106 to 10−2 Hz. A detailed description of the setup can be found in ref. 26.
Dynamic etching procedure with on-line ICP-OES or ICP-MS detection
The procedure starts with switching the valve in the load position, in this alignment the etching solution (water or dilute HCl including 1.0 mg L−1 Cu used as internal standard; flow rate 0.8 mL min−1) is directed straight to the mixing device, enabling the removal or insertion of a sample containing etching cartridge. Prior to sample insertion the cartridge was carefully cleaned in an ultrasonic bath of 0.24 mol L−1 HCl for 5 minutes to remove impurities remaining from the last measurement. After integration of a sample containing cartridge, the valve is switched back into the inject position and ICP-OES measurement is started. The etching solution introduced into the etching compartment initiates the etching process of the LSC film. The material dissolved by the etching agent is transported to the mixing device, where the eluate is combined with a make-up solution (0.24 mol L−1 HCl containing 0.5 mg L −1 Mn as additional internal standard; flow rate 0.6 mL min−1) and mixed thoroughly. The combined liquid flow is then directed into the sample introduction system of the ICP-OES instrument, where the transient elution profiles are measured.In case of more sensitive ICP-MS detection time resolved intensities for the investigated isotopes were recorded with Qtegra software and exported as .csv files for further data treatment, in particular internal standard correction to overcome potential non-spectral interferences. For interpretation and further discussion the intensity data derived from the etching experiments were converted into absolute masses, which have been removed from the sample surface per time unit. For this purpose calibration functions were determined by analyzing matrix adjusted standard solutions with the same instrumental setup. Standards containing defined amounts of the investigated elements (blank level up to contents of 1000 μg L −1) and 10 μg L −1 Cu (used as internal standard) were prepared just before use by appropriate dilution of respective stock solutions with 0.012 mol L−1 HCl. For analysis, the etching solution used as carrier was replaced with the standard solution to be investigated. Measurement of those standard solutions was performed in the load position solely. In analogy to the analysis of LSC samples, the liquid flow leaving the FI-manifold was mixed with make-up solution; and the combined flow introduced into the detection system. Prior to each ICP-MS analysis the system was purged with the examined standard solution for approximately 30 s, afterwards the signals of the selected isotopes were monitored for 120 s. The signal intensities (in counts per second) obtained for the investigated analytes were normalized using Mn as internal standard and correlated with the injected analyte masses, showing for all elements a linear behavior throughout the investigated mass range.
Results
Method development
Limitations in reproducibility and accuracy of analysis are considered the main drawbacks of the procedure reported in ref. 21. Typical sources for these problems are analyte losses and/or memory effects, changes in the carrier flow rate and/or composition, alterations in the nebulizer efficiency as well as variations in the plasma load, causing undesirable fluctuations in the measured signal intensities. Since the investigated elements provide an enhanced solubility at lower sample pH, adsorption losses and/or memory effects are only of concern when pure water is applied as etching agent. In the latter case, material dissolved from the LSC surface could be lost during transport from the sample cartridge into the plasma due to adsorption of dissolved material on different parts of the analytical system (tubings, nebulizer, spray chamber, torch), impeding an accurate measurement of the eluate composition. To minimize the risk of analyte losses and/or memory effects, the sample flow coming from the etching cartridge is mixed on-line with 0.24 mol L−1 HCl as a make-up solution. The combined liquid flow is then introduced into the nebulizer unit of the detecting ICP-OES, where the transient etching profiles are measured.To overcome the difficulties related with the mentioned non-spectral interferences, internal standards were applied. Whereas errors related to the sample introduction system could be compensated with any internal standard, the accurate correction of plasma related effects (e.g. energy transfer) requires the use of elements with excitation and ionization energies comparable to that of the target analytes. Thus, the elements Cu and Mn were selected as internal standards, having emission wavelengths close to that of the investigated LSC constituents. With the use of Mn (constituent of the make-up solution) changes in the sample introduction efficiency as well as plasma load could be monitored and corrected if necessary. Cu as constituent of the etching solution is used to control the liquid flow directed through the FI-manifold including the etching compartment. Moreover, the homogeneity of the solution leaving the mixing device could be controlled, a prerequisite for the accurate and reproducible measurement of etching profiles.
Beside improvement of the measurement step, a continuous optimization of the LSC thin film etching process is also required for reliable determination of distribution profiles. For this purpose an etching compartment is mandatory which delivers the eluent to the whole thin film surface uniformly, enabling homogeneous sample dissolution by the eluent. Moreover, the etching compartment should provide fast washout times to avoid mixing of sample eluates from successive sample depths, and the absence of any memory effects due to adsorption or release of analytes. The last issue is of special importance since the acidic make-up solution introduced to overcome the problem of analyte losses (see previous section) protects only the sample introduction system located behind the mixing device. Thus in the etching cartridge adsorption losses are still possible, since this part is positioned before the mixing device and therefore operated with etching solution only.
In the very first part of this work, where LSC thin films were simply sandwiched between two porous quartz frits and inserted into a SPE cartridge as reported by Kubicek et al.,21 differentiation between water soluble and acid soluble contents of LSC thin films was hampered. Memory-effects caused by the pH dependent adsorption/desorption of dissolved LSC constituents on the porous frits were found to be the main reason. To overcome the problems associated with the use of commercial SPE cartridges and porous frits as sample holders, a new etching compartment was manufactured, which allows fixation of the LSC sample without the use of any additional substrate. The material chosen for this application was PTFE, giving the advantage of being resistant to strong acids and acetone, which also enables an effective cleaning of the cartridge prior to next use. Particular emphasis was laid on the geometry of the etching compartment (for details see Fig. 2), which should prevent movement of the sample during the etching process. The square sample (5 × 5 mm2) is placed in the funnel shaped etching compartment (corners in contact with the wall) and the liquid can pass the sample along the edges. Moreover, the volume of the compartment was minimized from 984 μL to 289 μL to decrease washout time, hence lowering peak broadening. With this set-up reproducibility of analysis was improved significantly, since memory effects as well as unexpected fluctuations in the liquid flow could be completely avoided, enabling a reproducible determination of water and acid soluble LSC constituents.
 | ||
| Fig. 2 PTFE etching compartment. | ||
Element selective detection using ICP-MS
As demonstrated in ref. 21 the detection power of the ICP-OES employed as detection system is sufficient for the analysis of porous LSC films, but in case of dense LSC films with lower surface area limitations in sensitivity are expected. Compared to porous LSC films, the decreased surface area of dense LSC films results in a reduction of the etching rate, thus the amount of analyte introduced into the ICP per time unit is lower, impeding accurate analysis of the derived eluate. Insufficient detection power hampers also an appropriate characterization of the water soluble LSC phase, thus the stoichiometry determined for the water soluble phase could be erroneous. To overcome the problems related with ICP-OES detection, in all further experiments solely ICP-MS was used for element selective analysis. However, compared to ICP-OES the operating conditions of ICP-MS are different, thus, adaption of the dynamic etching procedure was necessary. In a first step the flow rates of etching and make-up solution were optimized, main purpose of this optimization step was to improve sample introduction efficiency without any deterioration of the etching profiles. Best results were achieved with a combined liquid flow of 1.0 mL min−1. For lower flow rates, a further increase in the sensitivity of ICP-MS analysis was observed, but the wash-out behavior of the etching compartment decreased distinctly. The internal standards selected for ICP-OES measurements were also applicable for detection with ICP-MS, since the ionization potentials of the target analytes are comparable (Co) or even lower (La, Sr) to that of the internal standard elements (Cu, Mn). Furthermore, ICP-MS analysis of etching and make-up solution delivered basically no signals on the m/z values used for detection of target analytes, indicating the absence of notable contaminations as well as spectral interferences. Concentrations of the applied internal standards were reduced to 10 μg L−1 (Cu) and 5 μg L −1 (Mn) only.With the optimized flow rate and the instrumental parameters described in Table 2 a significant increase in sensitivity could be achieved. For signal quantification matrix adjusted standard solutions and integration intervals of 1 s were applied, a more detailed description is presented in the experimental section. Compared to ICP-OES, the detection limits ranging from 7 (La at 379.478 nm) to 35 (Co at 228.696 nm) μg L−1 could be reduced by 2 magnitudes of order using the ICP-MS (0.03 μg L−1 for 139La, 0.04 μg L−1 for 59Co and 0.15 μg L−1 for 86Sr). With quadrupole ICP-MS instrumentation lower detection limits are usually possible, however, the operating conditions required in this special application (sample uptake rate of 1.0 mL min−1, and the measurement of transient signals with integration intervals of 1 s only) resulted in decreased sensitivity. The quality of the applied chemicals could also contribute to the achieved detection limits, since prevailing impurities might increase the background signals for the target analytes. Thus, with the use of ultrapure grade chemicals further improvements in sensitivity are expected. Nevertheless, the detection limits were low enough to quantify the transient data obtained during each step of the etching process. Fig. 3 shows element intensity/time profiles of an LSC thin film first etched with water and then with 0.012 mol L−1 HCl measured by ICP-MS. The 86Sr baseline signals were found to be in the order of 200 cps only, whereas the signal intensity increases to 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cps during water etching before it slowly diminishes to around 2000 cps and rises again to around 150000 cps after switching the eluent to diluted HCl. Using the determined calibration functions derived ICP-MS signals were converted into concentrations, which were found to range between some μg L−1 and several hundred μg L−1.
000 cps during water etching before it slowly diminishes to around 2000 cps and rises again to around 150000 cps after switching the eluent to diluted HCl. Using the determined calibration functions derived ICP-MS signals were converted into concentrations, which were found to range between some μg L−1 and several hundred μg L−1.
 | ||
| Fig. 3 Time resolved 59Co, 86Sr, 139La intensities of LSC thin films analyzed using PTFE compartment and ICP-MS. The eluent was switched from ultrapure H2O to 0.012 mol L−1 after ∼390 s. | ||
Analytical performance
As already mentioned in the introduction, changes of the surface cation stoichiometry are considered the main reason for degradation of the electrochemical performance of LSC. Thus, special emphasis was dedicated to the reliable analysis of surface and near-surface regions of LSC thin films. For this purpose, a set of LSC thin films was produced using identical PLD conditions. Analysis of water soluble parts of the films was performed with the use of water as etching solution; samples were subsequently treated with 0.012 mol L−1 HCl to continuously dissolve the remaining acid soluble LSC constituents. Progress of the etching process was monitored by ICP-MS measurement of the derived sample eluates. Obtained etching profiles do not reveal any obvious differences, indicating that the measurement of LSC thin films is highly reproducible. This is also confirmed by the small variations in the peak areas obtained for the water soluble LSC parts, showing relative standard deviations (n = 3) of 1.0% for Sr, 1.6% for La and 3.0% for Co. Similar findings can be found in literature for other FI-ICP-MS procedures reported for a variety of applications including on-line sample extraction,29 analyte enrichment30 and matrix or analyte separation.31Due to the higher sensitivity of the ICP-MS employed as detection system, experiments become feasible which were not possible with the setup described in ref. 21. One scientific task which could thus be addressed is the analysis of dense LSC thin films using water as etching solvent. This results in very low etching rates and thus sample eluates with concentrations not measurable with ICP-OES. To this end, a set of identical dense LSC thin film samples was investigated by means of both detection techniques and treated first with water and subsequently with etching solutions of increased acidity. For further discussion, the intensity/time data received by the detector was converted into molar ratios versus time profiles using the procedure developed for signal quantification. As expected, the ICP-OES signals were close to the respective detection limits, and do not allow a reliable differentiation between parts of LSC with different solubility. Detection by ICP-MS, on the other hand, leads to very precise and reliable profiles for all elements (Fig. 4a). From the ICP-MS results, it is evident that significant quantities of Sr but also Co and La were dissolved by the treatment with water. Moreover, the amount of water soluble species at the thin film surface decreased significantly with ongoing reaction time, see also Fig. 3. Changing the eluent to 0.012 mol L−1 HCl resulted in substantially increased signals for Sr, Co and La, until a horizontal plateau of signals (Fig. 3) as well as molar ratios (Fig. 4a) is observed, indicating a constant etching rate for all thin film constituents.
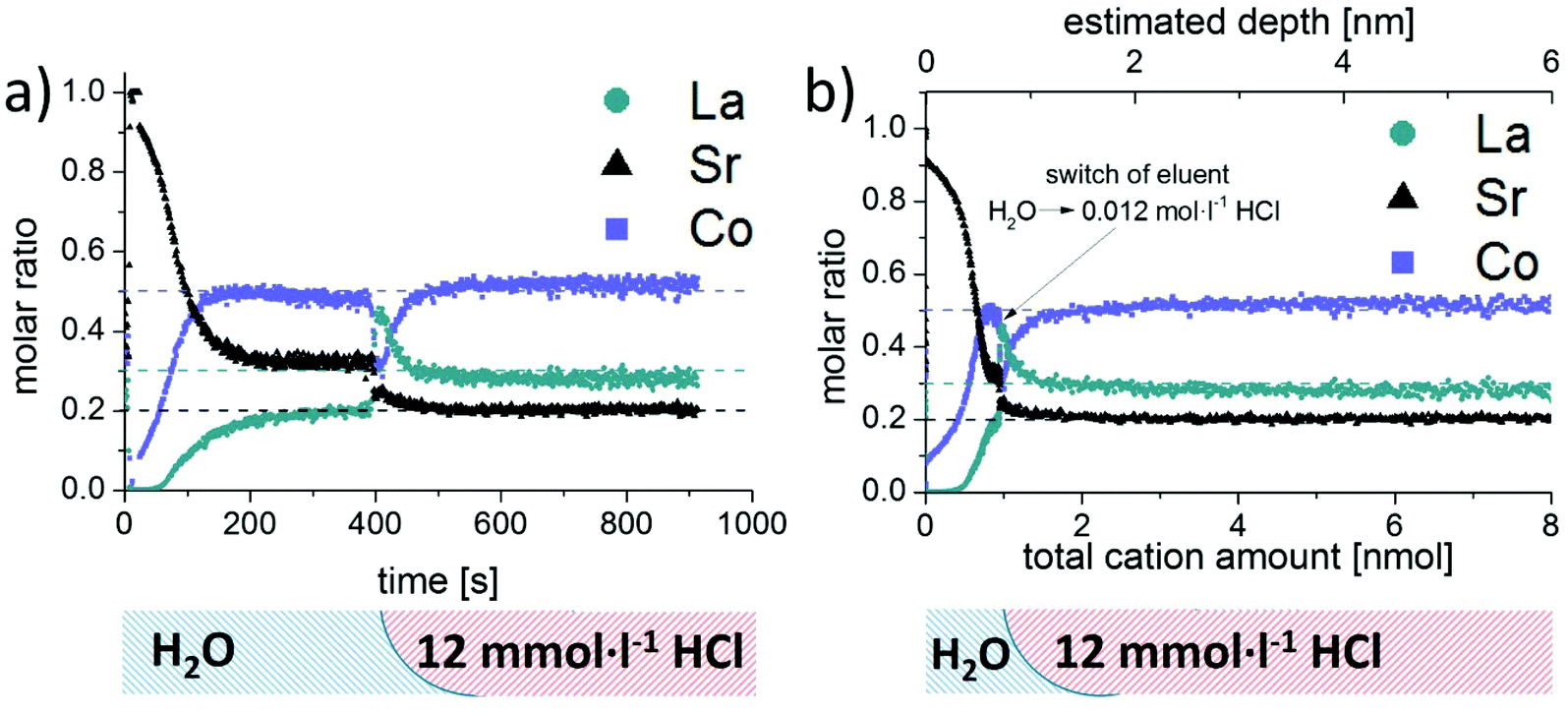 | ||
| Fig. 4 (a) shows the molar ratio of dense LSC thin films vs. measurement time during the dynamical etching procedure and analysis by ICP-MS, including description of applied eluent sequence below the profiles. In (b) the molar ratio is plotted vs. the total cation amount (similar to depth profiles). | ||
From the findings presented in Fig. 4a, it can be concluded that the near surface region of the LSC thin film contains mainly Sr, while Co and La were only detected in minor amounts. While with ongoing etching time and thus sample depth the absolute ICP-MS signals decreased drastically, the relative contribution of Co and La increased continuously, leading to a rather constant composition of the water soluble part of the LSC sample after approximately 200 s. Compared to the known LSC thin film stoichiometry (dotted line in Fig. 4a) this water soluble fraction (between 200–400 s) still exhibits significant differences – in particular a depletion of La and an enhanced contribution of Sr. However, only a very small amount of cations (<0.3 nmol) is dissolved during this time interval, which is also reflected by the tailing of the water-soluble peak in Fig. 3. With the use of 0.012 mol L−1 HCl as etching solution the detected molar ratio changed again, revealing practically the known composition of the LSC thin films.
Further information becomes available when relating the molar ratios to the total amount of eluted cations instead of time. It can be clearly deduced from the ICP-MS results (Fig. 4b), that the water soluble fraction represents only a minor part of the dissolved LSC thin film sample and is largely Sr enriched. Assuming that the LSC sample is dissolved homogeneously (layer by layer) the obtained concentration versus time plots could even be converted into depth profiles with knowledge of the respective etching rates. Application of this methodology would result in depth resolutions reaching the sub-nm range (see top abscissa in Fig. 4b) and thus be very competitive to traditional approaches such as GDMS,32 SIMS33 or LA-ICP-MS analysis.34 However, since preferential dissolution of the LSC thin film material on grain boundaries or defect rich regions will most probably occur, the depth scale should only be considered as a rough estimation.
For demonstration of accuracy and reproducibility four PLD deposition batches of LSC thin films, consisting of four individual samples each (i.e. 16 samples), were either analyzed using the procedure developed or in a conventional batch wise approach by dissolving the entire film. For both procedures, 0.012 mol L−1 HCl was chosen as etching solution. The analysis time for the dynamic measurements was extended accordingly to allow monitoring of the whole dissolution process, signal quantification was performed as mentioned before. For batch wise experiments the samples were treated with the etching solution in closed reaction tubes. After complete dissolution of the LSC thin films, the substrates were removed from the tubes and sample acidity of the derived solutions has been adjusted to 0.12 mol l−1 HCl prior to ICP-MS analysis. The results derived for the total cation amounts of the LSC samples were in excellent agreement, revealing only negligible differences between batch experiments and dynamic ICP-MS measurements. Furthermore, for quality assurance the samples derived from batch experiments were also measured using ICP-OES, delivering results practically identical to the ICP-MS findings. Thus, the applicability of the proposed dynamic etching procedure as well as the accuracy of the applied quantification approach is confirmed, in particular when considering that the production of LSC thin films can also generate minor variations in the composition of the investigated sample sets.
Analysis of the near surface region of different LSC thin films and comparison with LEIS measurements
The developed procedure was applied to compare the surface composition of different dense LSC thin films. In order to understand which parts of the films are water soluble and to compare these results to LEIS measurements, the following experiment was performed: three of six as-deposited LSC thin films were stirred ex-situ for 10 minutes in ultrapure H2O and blown dry with high purity N2. All six thin films were then analyzed by the dynamic ICP-MS etching procedure. Representative time-resolved concentration profiles for all elements are shown in Fig. 5a and b. The only significant difference between the as-deposited and H2O pretreated thin films is seen in the part of the profile resulting from H2O etching. | ||
| Fig. 5 Representative time-resolved ICP-MS concentration profiles for (a) as-deposited LSC thin films and (b) after ex situ H2O treatment. Inset in (b) directly compares the Sr concentration in the water-soluble part. | ||
The exact amount of cations etched by water were determined by integration (Fig. 5 beginning of the measurement up to the dashed line). The amount of water soluble Sr is decreased by 0.39 ± 0.02 nmol after the H2O pretreatment, while only minor changes are observed in the La and Co concentration. From the lattice parameter of SrO, it can be calculated that 0.33 nmol Sr would be needed to form one atomic layer of highly water-soluble SrO (100 direction) covering the entire 5 × 5 mm2 surface of the thin film. Accordingly, the additional 0.39 nmol Sr removed from LSC layers without H2O pretreatment corresponds to approximately 1.2 atomic layers of SrO. The remaining cations dissolved with as well as without H2O pretreatment would then sum up to about one to two slightly Sr enriched elementary cells of LSC, though inhomogeneous etching might play a role here. This result strongly suggests that the main difference between the surface composition of an as-deposited and a H2O treated layer is the absence of a strongly Sr-rich layer in the latter case. The fact that also after H2O pretreatment, in the beginning of the measurement, an increased amount of the LSC lattice is dissolved in water may be explained with the influence of trace gases, e.g. CO2, SO2 adsorbed at the sample surface or PTFE compartment.
To confirm the findings from ICP-MS measurements, further LSC thin films were prepared and analyzed by LEIS. Depth profiles over the first 5 nm of the thin films for the Sr/(Sr + La) and Co/(Sr + La) ratio are shown in Fig. 6a and b, respectively. The very first measurement points of the LEIS profiles represent the cation composition of the surface layer. For the as-deposited thin film it can be seen that most of the cation sites in the surface layer are occupied by Sr (81%), Co and La are thus strongly depleted. An enrichment in Sr and depletion of La is also determined for the first 2–3 nm of the LSC thin film, even though the sub-surface shows a Co/(Sr + La) ratio that is already very close to the bulk stoichiometry. After the ex-situ H2O treatment, the surface layer shows a decreased Sr/(Sr + La) ratio compared to the as-deposited sample, although Co is still depleted. A slight Sr enrichment is again observed beneath the surface.
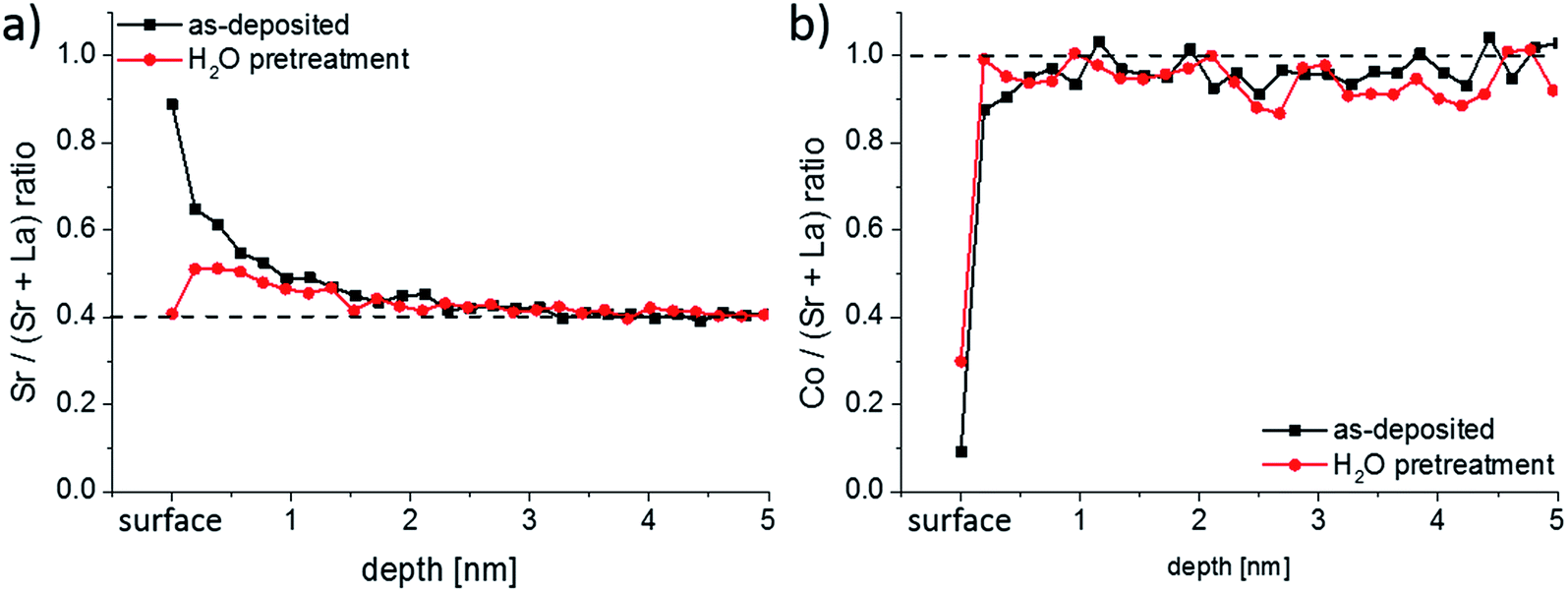 | ||
| Fig. 6 (a) Sr/(Sr + La) and (b) Co/(Sr + La) depth profiles measured by LEIS for as-deposited and ex situ H2O treated LSC thin films. | ||
First, a very good qualitative agreement is thus found between the LEIS results at the surface and the water-soluble amount of cations determined by ICP-MS. Both methods suggest that the LSC lattice has a very Sr rich termination layer. A Sr enriched near-surface region (1–3 nm information depth) was also confirmed by Cai et al.23 from XPS measurements on similar LSC thin films. In their work, they also proposed that the LSC lattice is SrO/Sr(OH)2 terminated, since evidence of SrCO3 species was not obtained. Also the La enriched and Co depleted surface composition measured by our LEIS study after the ex situ H2O treatment finds its counter-part in the ICP-MS experiment: a sharp peak of La together with a depletion of Co is observed after switching from H2O to diluted hydrochloric acid (Fig. 4a and b).
Quantitatively, LEIS results indicate a Sr coverage of 81% of the surface. ICP-MS, on the other hand, revealed 1.2 atomic layers of SrO. This difference may arise since LEIS probes a 2D projection of the surface and ICP-MS the true surface area (total solid–gas phase boundary) of the sample. The latter is larger due to cracks in the films. In a previous work we were able to show on microporous LSC thin films that the enhanced surface area from open porosity can be even quantified from the water-soluble amount of Sr.25
Finally, EIS measurements were conducted on as-deposited and H2O pretreated samples at 400 °C in 1 mbar pO2 in order to compare the electrochemical properties (O2 exchange kinetics); representative spectra are shown in Fig. 7. A detailed analysis of such impedance data is given in ref. 35 and showed that the low frequency arc can be correlated to the oxygen exchange reaction at the LSC surface. It can be seen that the surface treatment does not affect the electrolyte resistance but lowers the electrode resistance by about 50%. This confirms the detrimental effect of the water-soluble SrO top layer on the oxygen exchange activity.
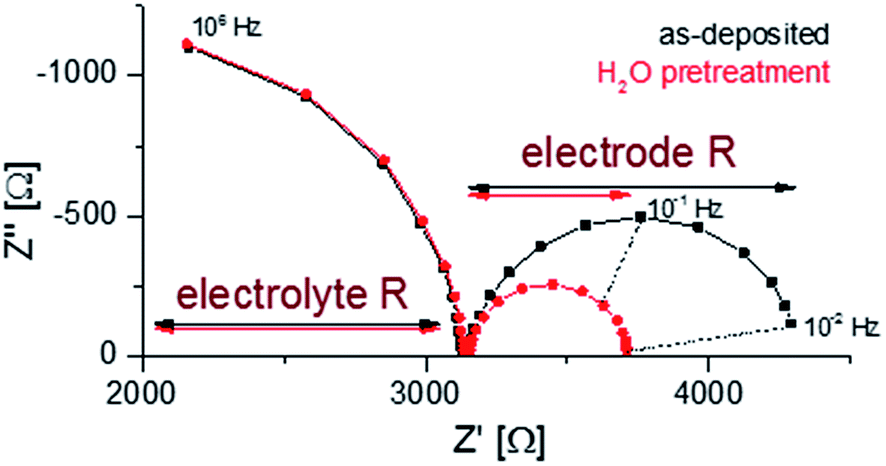 | ||
| Fig. 7 Nyquist plots of the impedance measurements performed at 400 °C in 1 mbar pO2 for as-deposited and H2O treated LSC. | ||
Conclusions
The analytical procedure presented in this work allows the chemical composition of surface and near surface regions of complex metal oxide thin films to be determined. Differentiation between water and acid soluble sample constituents was accomplished by preliminary treatment of the sample surface with water, followed by etching with diluted HCl, and element specific on-line analysis of the derived solutions. The required sensitivity was achieved with the use of ICP-MS as detection technique; non-spectral interferences during on-line ICP-MS measurement were corrected by means of internal standards. Stepwise optimization of the etching cartridge resulted in an improved washout behavior and the complete elimination of memory effects. Compared to conventional approaches used for surface analysis of solids such as XRF, SIMS, GDOES or GDMS, laser ablation combined with ICP-OES or ICP-MS, the necessity of appropriate matrix matched standards could be circumvented, since simple aqueous standards could be used for signal quantification. A further benefit of the proposed procedure is the ability to distinguish phases with different solubility, whereas common techniques allow only the measurement of total element contents.The developed procedure was applied for the analysis of LSC thin films. LEIS and on-line ICP-MS measurements revealed a water-soluble Sr-rich termination layer on freshly deposited dense LSC thin films. Qualitatively and quantitatively, a very good agreement between the results obtained by these two techniques was found. The oxygen exchange rates were measured by impedance spectroscopy and an improvement was determined after removing the water-soluble Sr-rich phase from the surface, suggesting that the LSC surface is blocked or inactivated by the formation of this water soluble Sr-rich phase.
Acknowledgements
The authors gratefully acknowledge funding by Austrian Science Fund (FWF) projects P21960-N17 and W1243. JAK, TI, HT and JD also acknowledge support from the International Institute for Carbon Neutral Energy Research (wpi-I2CNER), funded by the World Premier Research Center Initiative of the Ministry of Education, Culture, Sports, Science and Technology. HT thanks the financial support from the Japanese Society for Promotion of Science (JSPS postdoctoral fellowship and Kakenhi Grant-in-Aid P13770).References
- A. S. Bhalla, R. Guo and R. Roy, Mater. Res. Innovations, 2000, 4, 3–26 CrossRef CAS.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125 CAS.
- I. E. Wachs, Catal. Today, 2005, 100, 79–94 CrossRef CAS.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed.
- G. Friedbacher and H. Bubert, Surface and Thin Film Analysis: a Compendium of Principles, Instrumentation, and Applications, 2nd edn, 2011 Search PubMed.
- T. Nelis and J. Pallosi, Appl. Spectrosc. Rev., 2006, 41, 227–258 CrossRef CAS.
- B. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 2010, 679, 7–16 CrossRef PubMed.
- J. Pisonero, J. Koch, M. Wälle, W. Hartung, N. D. Spencer and D. Günther, Anal. Chem., 2007, 79, 2325–2333 CrossRef CAS PubMed.
- B. Hattendorf, J. Pisonero, D. Günther and N. Bordel, Anal. Chem., 2012, 84, 8771–8776 CrossRef CAS PubMed.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- S. B. Desu and D. A. Payne, J. Am. Ceram. Soc., 1990, 73, 3391–3397 CrossRef CAS.
- H. Wang, Y. Zhu, R. Tan and W. Yao, Catal. Lett., 2002, 82, 199–204 CrossRef CAS.
- A. Yan, V. Maragou, A. Arico, M. Cheng and P. Tsiakaras, Appl. Catal., B, 2007, 76, 320–327 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, M. Shah, L. D. Marks and S. A. Barnett, J. Electrochem. Soc., 2010, 157, B279–B284 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- R. M. Ormerod, Chem. Soc. Rev., 2003, 32, 17–28 RSC.
- F. Zhao, R. Peng and C. Xia, Fuel Cell. Bull., 2008, 2008, 12–16 CrossRef.
- H. Yokokawa, H. Tu, B. Iwanschitz and A. Mai, J. Power Sources, 2008, 182, 400–412 CrossRef CAS.
- V. I. Sharma and B. Yildiz, J. Electrochem. Soc., 2010, 157, B441–B448 CrossRef CAS.
- M. Kubicek, A. Limbeck, T. Fromling, H. Hutter and J. Fleig, J. Electrochem. Soc., 2011, 158, B727–B734 CrossRef CAS.
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Electrochem. Solid-State Lett., 2006, 9, A478–A481 CrossRef CAS.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- G. M. Rupp, A. Limbeck, M. Kubicek, A. Penn, M. Stoger-Pollach, G. Friedbacher and J. Fleig, J. Mater. Chem. A, 2014, 2, 7099–7108 CAS.
- M. P. Pechini, US3330697A, 1967.
- G. M. Rupp, A. Schmid, A. Nenning and J. Fleig, J. Electrochem. Soc., 2016, 163, F564–F573 CrossRef CAS.
- A. Limbeck, C. Wagner, B. Lendl and A. Mukhtar, Anal. Chim. Acta, 2012, 750, 111–119 CrossRef CAS PubMed.
- J. Druce, H. Tellez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 CAS.
- J. Buanuam, K. Tiptanasup, J. Shiowatana, M. Miro and E. Harald Hansen, J. Environ. Monit., 2006, 8, 1248–1254 RSC.
- D. Das, M. Dutta, M. L. Cervera and M. de la Guardia, TrAC, Trends Anal. Chem., 2012, 33, 35–45 CrossRef CAS.
- P. Galler, A. Limbeck, S. F. Boulyga, G. Stingeder, T. Hirata and T. Prohaska, Anal. Chem., 2007, 79, 5023–5029 CrossRef CAS PubMed.
- J. Pisonero, N. Bordel, C. Gonzalez de Vega, B. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Bioanal. Chem., 2013, 405, 5655–5662 CrossRef CAS PubMed.
- J. S. Fletcher and J. C. Vickerman, Anal. Chem., 2013, 85, 610–639 CrossRef CAS PubMed.
- A. Gutierrez-Gonzalez, C. Gonzalez-Gago, J. Pisonero, N. Tibbetts, A. Menendez, M. Velez and N. Bordel, J. Anal. At. Spectrom., 2015, 30, 191–197 RSC.
- G. M. Rupp, H. Tellez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, J. Mater. Chem. A, 2015, 3, 22759–22769 CAS.
Footnote |
| † A. Limbeck and G. M. Rupp contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |