
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic characterization of a low-temperature plasma ambient ionization probe operated with helium/nitrogen plasma gas mixtures
Andriy
Kuklya
*a,
Carsten
Engelhard
 *b,
Klaus
Kerpen
a and
Ursula
Telgheder
ac
*b,
Klaus
Kerpen
a and
Ursula
Telgheder
ac
aDepartment of Instrumental Analytical Chemistry, University of Duisburg-Essen (UDE), Universitätsstraße 5, 45141 Essen, Germany. E-mail: andriy.kuklya@uni-due.de; Fax: +49(0)201 183 6773; Tel: +49(0)201 183 6786
bDepartment of Chemistry and Biology, University of Siegen, Adolf-Reichwein Str. 2, D-57068 Siegen, Germany. E-mail: engelhard@chemie.uni-siegen.de; Fax: +49(0)271 740 2041; Tel: +49(0)271 740 4573
cIWW Water Centre, Moritzstr. 26, 45476 Mülheim a.d. Ruhr, Germany
First published on 16th June 2016
Abstract
In this study, a systematic spectroscopic characterization of a low-temperature plasma (LTP) probe operated with He/N2 gas mixtures is carried out. The influence of several experimental parameters (e.g., different He/N2 gas mixtures, discharge voltage, and gas flow rate) on the dielectric-barrier discharge afterglow was studied. It was found that an increase of the nitrogen concentration in the helium discharge gas (to values higher than 0.5% N2) leads to a significant decay of N2+, He, and O emission intensities. At 1% N2 and 99% He, oxygen emission bands were not detectable and intensity of He and N2+ emission bands were reduced by approximately five times compared to features in a 100% He discharge. Interestingly, the opposite trend was observed for NO, OH, and N2 species. Here, increasing the N2 fraction in the discharge gas mixture led to an enhancement of emission intensities. Maximum emission bands intensities of OH, NO, and N2 were detected at N2 concentrations of 0.5, 0.6, and 1.0%, respectively. A further increase of the N2 fraction leads to a decrease of emission intensities for all observed species (OH, NO, N2, He, and N2+). In general, an increase in discharge gas flow rate resulted in a significant increase of NO emission band intensities for all N2/He mixtures tested. However, only a minor effect was observed for N2 emission bands. Increasing the discharge voltage resulted in an increase of emission intensities of all detected species. Combined with spatially resolved investigations of the afterglow, these results are considered helpful to further optimize LTP performance. This is especially important for portable and direct analysis instrumentation, where LTP can be used as the ionization source and lower helium gas consumption is desirable because of cost and availability.
Introduction
Plasma-based ionization sources can be used for elemental and molecular mass spectrometry depending on their discharge characteristics. One variant of a non-equilibrium plasma that is currently used in molecular mass spectrometry is the low-temperature plasma (LTP) probe. The source is based on a dielectric-barrier discharge and was introduced by Harper et al. for its use in ambient desorption/ionization mass spectrometry (ADI-MS).1 In this configuration, a non-equilibrium low-temperature plasma is generated at atmospheric pressure within a glass tube and extends into the ambient environment (this region of the plasma being called the afterglow). The attractive feature of the LTP probe is the ability to perform desorption and ionization with a single probe. To achieve this, the afterglow directly interacts with the sample at adjustable angles. The temperature of the surface area in contact with the plasma plume is approximately 30 °C, and in turn, no significant heating of the sample occurs. In addition, the high-voltage electrode is electrically isolated from the direct discharge region, and therefore, the sample is not liable to electric shock. These features allow direct analysis of temperature-sensitive samples or even dried chemical residues on a human finger.1 Recently, we used a LTP successfully as an ionization source for differential ion mobility spectrometry.2In general, the LTP is considered a relatively soft ionization source. In a previous study by Albert et al. it was found that LTP shows comparable ionization characteristics to atmospheric pressure chemical ionization (APCI) for a class of amines, amides, and aldehydes.3 Also, LTP demonstrated higher relative ionization efficiencies compared to APCI for low molecular weight compounds and nonpolar to medium-polar compounds. Polycyclic aromatic hydrocarbons (PAHs) and imides were successfully ionized with LTP (in contrast to APCI). This general trend is attributed to the generation of several reagent ions (including O2+, O2−, N2+, NO+, NO2−, NO3− species and protonated water clusters) that are capable of ionizing a wide range of compounds.4,7 An additional source of chemical reactivity stems from plasma ultraviolet (UV) photons. It was demonstrated for dielectric-barrier discharges (DBDs) that most of the UV emission comes from excited NO molecules when the discharge is sustained in He with a 10% fraction of N2.5 It should also be noted that in many cases of LTP-ADI-MS application, additional heating of the sample (or sample holder) is required to enhance thermal desorption and maximize the signal (because of the low plasma temperature). However, because the probe operates at low power (<3 W) and low gas flow rates (typically between 0.08 and 1 L min−1 He) it is considered to have a great potential in portable, field-based applications.
With respect to portable and miniaturized instrumentation, the requirement to use helium may be a limitation. For example, instrumentation that is used for online (and on-site) monitoring should feature minimum helium consumption and operate with cheaper discharge gases. The use of alternative LTP discharge gases such as Ar, N2 and air, respectively was studied in the past.6 However, the overall sensitivity of the LTP coupled to a mass spectrometer was typically higher when helium was used as discharge gas.7 Despite the fact that some investigations on the formation of reactive species in the LTP probe4,7 and a DBD probe8 of similar construction operated with pure He (N2 concentration < 600 ppm) and N2 were performed, no systematic spectroscopic characterization of the LTP probe was carried out when the source is operated with He/N2 gas mixtures. Since nitrogen plays an important role in the soft ionization mechanism, the influence of the nitrogen fraction on the discharge and the afterglow should be studied in detail.
The influence of selected LTP probe parameters, such as probe geometry, electrode configuration, and tube dimensions was studied previously by Albert et al.3 In the present study, the influence of discharge voltage, discharge gas composition (N2 fraction in He), and discharge gas flow rate on the dielectric barrier discharge afterglow is characterized by means of optical spectroscopy. The spatial distribution of reactive species present in the afterglow was also determined. This information is essential not only for a better understanding of the mechanisms involved but also to further optimize the LTP performance for portable and on-site analysis applications.
Experimental
Low-temperature plasma probe
Configuration of the home-built LTP probe was similar to the geometry described in our previous publication.2 In brief, an inner solid stainless-steel electrode (0.7 mm diameter, 3 mm distance to capillary exit) and an outer copper-ring electrode (6 mm length, 3 mm distance to the capillary exit) were used to generate a dielectric-barrier discharge in a glass tube (4.0 mm outer diameter, 0.8 mm inner diameter). A relatively narrow gap between the glass tube and inner electrode (ca. 0.05 mm) was selected to achieve a high gas flow velocity and fast transport of the created excited species towards the exit of the probe. A high-voltage alternating current source (3–7 kVpp at 28–35 kHz, model PVM500, Information Unlimited, Amherst, USA) was applied between the outer copper-ring electrode and the inner pin electrode to generate the plasma. Voltage was measured using a portable wide-bandwidth high-voltage probe (≤60 kV, ≤80 MHz, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, PVM-1, North Star High Voltage, Marana, USA) and recorded using a digital storage oscilloscope (TDS 2012B, Tektronix, Beaverton, USA).
:1000, PVM-1, North Star High Voltage, Marana, USA) and recorded using a digital storage oscilloscope (TDS 2012B, Tektronix, Beaverton, USA).
Experimental setup
A schematic diagram of the experimental setup is presented in Fig. 1. Helium (99.999%, water <2.0 ppm, O2 < 2.0 ppm, N2 < 5.0 ppm, Air Liquide, Düsseldorf, Germany) and nitrogen (99.999%, water <2.0 ppm, O2 < 2.0 ppm, Air Liquide, Düsseldorf, Germany) were used to prepare discharge gas mixtures at flow rates between 100 and 600 mL min−1. Gas mixtures were prepared by mixing the flow of nitrogen, controlled by a mass flow controller (MFC, GFC17, 0–500 mL min−1 N2, Aalborg, USA), with an additional flow of helium in the desired ratio. Helium flow rate was controlled by a mass flow controller (MFC, GFC17, 0–500 mL min−1 He, Aalborg, USA). Because of the upper flow rate limit (of 500 mL min−1) of the available devices (MFC), the setup was slightly modified for measurements with a desired flow rate of, e.g., 600 mL min−1. Here, for a discharge in pure N2, the He-MFC was exchanged to a second N2 mass flow controller. Similarly, the N2-MFC was exchanged to a second He-MFC when a flow rate of 600 mL min−1 for a discharge in pure He was required.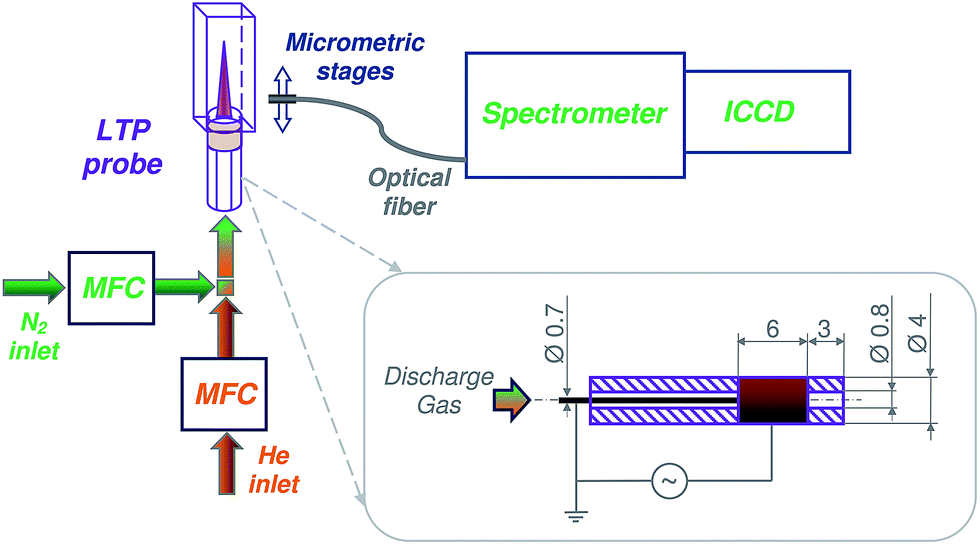 | ||
| Fig. 1 Schematic diagram of the experimental setup: intensified charge-coupled device (ICCD) camera, mass flow controller (MFC), low-temperature plasma probe (LTP). Dimensions of the LTP probe are depicted in the insert. | ||
For measurements with N2 fractions below 2.5% (see Fig. 3, bottom), a different MFC (GFC17, 0–20 mL min−1 N2, Aalborg, USA) was used for better precision. A perfluoroalkoxy (PFA) Swagelok® ¼ inch tee was used to couple the grounded stainless-steel electrode, tubing, and glass capillary. In order to minimize immediate diffusion of surrounding air into the discharge/afterglow, the outlet of the glass capillary was inserted into a quartz cuvette (approximately 23 mm to cuvette outlet; cuvette: QS 0.5 cm, Hellma Analytics, Müllheim, Germany) and sealed with PTFE-tape. The bottom of the cuvette was removed in order to provide an outlet for the discharge gas. In the current LTP setup, the concentration of surrounding oxygen and nitrogen at the capillary exit can be significantly reduced. The estimated concentration ranges for oxygen and nitrogen vary between 0.2–4.0% and 2.4–14.5%, respectively, depending on the discharge gas flow rates (100–600 mL min−1). The diffusion of surrounding oxygen and nitrogen into the discharge area is considered to be negligible.
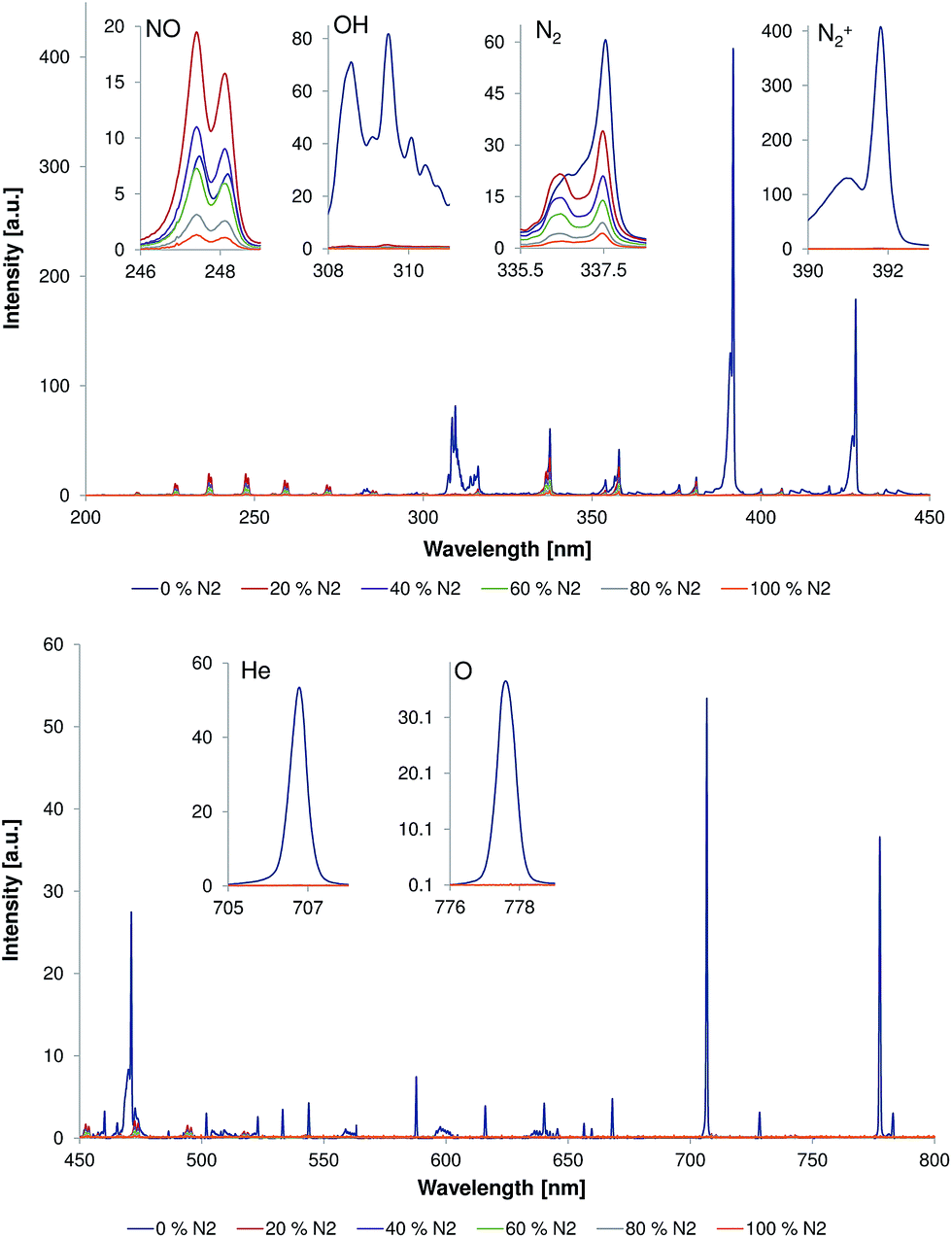 | ||
| Fig. 2 Emission spectra measured directly at the capillary outlet (d = 0 mm) within wavelength ranges of 200–450 nm (top) and 450–800 nm (bottom). The discharge gas flow rate was 500 mL min−1 and discharge voltage was 4 kVpp. Inserts show the strongest bands of NO (237 nm), N2 (337.1 nm), N2+ (391.4 nm), OH (308 nm), He (706.5 nm), and O (777 nm) measured under different He/N2 discharge gas compositions. | ||
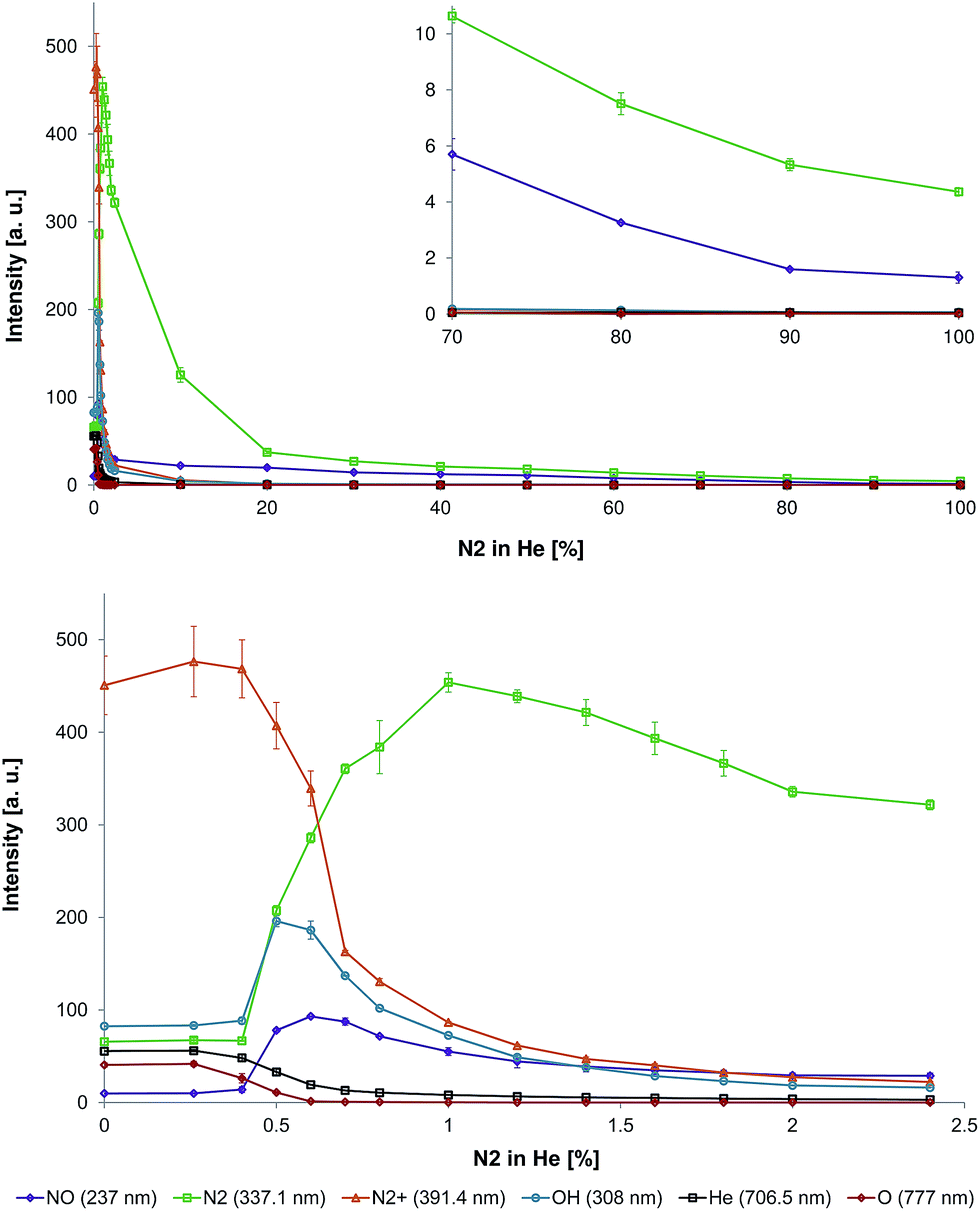 | ||
| Fig. 3 Top: Influence of N2 fraction in the He discharge gas on the emission intensities of NO (237 nm), N2 (337.1 nm), N2+ (391.4 nm), OH (308 nm), He (706.5 nm), and O (777 nm) in the LTP afterglow region (distance to capillary outlet d = 0 mm). Bottom: Zoom-in to lower nitrogen fractions to highlight the transition region. | ||
Spectroscopic measurements
Spectroscopic measurements were performed with an iStar ICCD camera (DH734-18U-03, Andor, Belfast, UK) connected to a Shamrock spectrometer (SR-303i, Andor, Belfast, UK). The readout mode was set to full vertical binning (FVB). Spectra were recorded in accumulating mode (100 accumulations) at a microchannel plate (MCP) gain level of 50, gate width of 50 ms, and an exposure time of 0.1 s. The spectrometer was equipped with a 1200.6 lines per mm grating and an entrance slit width of 100 μm. The wavelength resolution and accuracy are 0.186 nm and 0.0563 nm, respectively. Spectra could not be recorded simultaneously over the complete wavelength range of interest and were acquired in so-called step-and-glue mode (20% overlap). A UV-grade quartz optical fiber (numerical aperture is 0.12) was mounted on micrometric stages and positioned perpendicularly to the plasma afterglow/jet at a distance of 1 mm to the quartz cuvette. Taking into account the calculated spatial resolution of the used system (0.42 mm) and precision of the positioning system the mapping of the discharge was performed by shifting the optical fiber along the x-axis in 1 mm steps.For each sample, three single measurements were recorded. The mean values of peak intensities and the corresponding standard deviations were calculated using Microsoft Excel 2010 software.
The analyzed band heads from molecular and atomic species are as follows. The N2 band head of 337.1 nm corresponds to transition from the second positive band head: N2 (C 3Πu)ν′=0 → N2 (B 3Πg)ν′=0.7,9,23 The N2+ band head of 391.4 nm corresponds to transition from the first negative band head: N2+ (B 2Σ+u)ν′=0 → N2+ (X 2Σ+g)ν=0.7,9,23 The NO band head of 237 nm corresponds to transition: NO (A 2Σ+)ν′=0 → NO (X 2Π)ν′′=1.10 The OH band head of 308 nm corresponds to transition: OH (A 2Σ+)ν′=0 → OH (X 2Π)ν′′=0. The HeI band head of 706.5 nm corresponds to transition: 3 s 3S1 → 2p 3P0.7,23 The OI band head of 777 nm corresponds to transitions: 3p 5P1–3 → 3p 5S02.7
Results and discussion
Effect of the discharge gas composition
The possibility to reduce or even avoid the helium discharge gas consumption would help to reduce LTP operating costs and would make it easier to realize portable systems. Because the LTP probe is typically used as an open source, i.e. without a source housing (to be able to probe large objects etc.), diffusion of ambient air, e.g., some amount of nitrogen into the LTP discharge region is likely. Here, the influence of the nitrogen fraction on the emission pattern of the discharge and the afterglow was studied in a first experiment.Fig. 2 shows emission spectra (200–800 nm) measured directly at the capillary outlet (distance to capillary outlet d = 0 mm). The discharge gas flow rate was 500 mL min−1 and discharge voltage was 4 kVpp. The nitrogen fraction in the discharge gas was of 0, 20, 40, 60, 80, and 100%. Strong emission bands from molecular species, such as N2, N2+, NO, and OH, as well as from atomic species like He and O, were observed when the discharge gas was pure helium. These plasma species are quite similar to those observed by Chan et al. with other LTP-torch configurations in previous publications.4,7 However, an emission of low intensity from excited NO species, that was not reported by Chan et al., was observed in this study even when discharge was performed in pure helium. The reason for this difference is unclear today but presumably a result of different discharge parameters (power supply etc.), torch configurations (see Table 1), and a different sensitivity of the spectroscopic systems, respectively.
| V pp [kV] | f [kHz] | CS [mm2] | FR [L min−1] | GV [m s−1] | |
|---|---|---|---|---|---|
| a V pp is the discharge voltage, f is the discharge frequency, CS is the cross section between inner capillary wall and inner electrode, FR is the discharge gas flow rate, and GV is the gas velocity between the capillaries. GV was calculated as a ratio between FR and CS (in corresponding units). | |||||
| Ref. 4 | 11.8–14.8 | 1.2–2.4 | 37 | 0.4–1.6 | 0.2–0.7 |
| Ref. 8 | 6 | 20 | 0.8 | 0.3–1.0 | 6.3–20.8 |
| This study | 4 | 30 | 0.5 | 0.1–0.5 | 3.3–16.7 |
Presence of N2, N2+, and NO emission bands in the plasma afterglow during the discharge in pure helium can be explained by the diffusion of atmospheric nitrogen (and oxygen) into the discharge region. Exited N2+ can be generated by He metastables via Penning ionization and by He2+ ions via charge transfer, respectively.7,11 In the presence of oxygen, NO can be formed according to the Zeldovich reaction mechanism: N + O2 → NO + O followed by O + N2* → NO + N. However, a significant contribution to NO synthesis in non-thermal plasma is provided by the following molecular ion reactions: (1) N2+ + O2 → NO+ + NO; (2) O+ + N2 → NO+ + N; (3) O2+ + N2 → NO+ + NO.12 These reactions result in the formation of NO+. Interestingly, the positive NO+ ions can be also generated by He metastables via Penning ionization of NO.13,14 Despite the fact that formation of NO+ could not be confirmed spectroscopically with the current experimental setup, the formation of NO+ ions was previously reported when the LTP was coupled to a mass spectrometer15 and an ion mobility spectrometer,16 respectively.
N2+ and NO+ are considered to be important reactive species. N2+ is a reaction intermediate for the formation of other reagent ions such as protonated water clusters, (H2O)nH+. The following ionization of analytes proceeds via proton transfer and efficiency of ionization is dependent on proton affinity of analytes.17 NO+ as a reactant ion is important for monitoring of monoaromatic volatile organic carbons (VOCs, such as benzene, toluene, and xylene (BTX)),18 some explosive materials19 and various other compounds.20 The major reaction pathways observed for the NO+ reagent ion are ion–molecule association, electron transfer (by analytes with IE < 9.26 eV), and hydride abstraction.21,22 In addition, excited NO observed in the afterglow is a source of high energy photons (200–280 nm, see Fig. 2). Therefore, efficient formation and transport (within the afterglow) of these species is important for effective ionization of analytes.
Addition of 20% of nitrogen to the discharge gas resulted in almost complete disappearance of He, H, O, and N2+ emission bands. At this nitrogen concentration, NO and N2 emission bands were dominating over other emission bands. Interestingly, the intensity of these emission bands was changed differently with respect to the corresponding values in pure helium. Intensity of N2 emission bands was approximately 30% lower compared to that in pure helium; however, the intensity of NO emission bands increased approximately by a factor of two compared to that in pure helium (see inserts in Fig. 2). The following increase of the nitrogen fraction in discharge gas within the fractions range of 20–100% resulted in reduction of emission bands intensities of both NO and N2.
To investigate this phenomenon in more detail, the nitrogen fraction in the discharge gas was changed stepwise (in increments of 0.1–0.4% between 0% and 2.4% N2 in He and in increments of 7.6–10% between 2.4% and 100% N2 in He) and the following species were monitored: NO (237 nm), N2 (337.1 nm), N2+ (391.4 nm), OH (308 nm), He (706.5 nm), and O (777 nm). The corresponding data is depicted in Fig. 3.
It was found that a small amount of N2 in the discharge gas (0–0.3%) resulted only in a small increase in N2+ emission intensity. Also, emission intensities of other species did not significantly change as well (see Fig. 3, bottom). The increase of nitrogen fraction in discharge gas to 1% results in significant decrease of O, He, and N2+ emission bands intensity. At 1% N2 in He, emission bands of O were not detectable and emission intensities of He and N2+ were about five times lower compared to corresponding values in pure helium. A different relationship between emission bands intensity and nitrogen concentration in the discharge gas was observed for NO, OH, and N2. Increasing the nitrogen concentration in the discharge gas resulted in an increase of emission intensities. The maximal emission intensities of OH, NO, and N2 were achieved at a nitrogen concentration of 0.5%, 0.6%, and 1.0%, respectively. The increase of nitrogen concentration in discharge gas within the fractions range from 1 to 100% leads to a decrease of emission intensities for all three species. At a nitrogen concentration higher than 20%, emission bands of NO and N2 are dominating over emission from other species (emission of other species is negligible). Increasing the nitrogen fraction in the discharge gas from 20% to 100% resulted in continuous decrease of NO and N2 emission intensities (see Fig. 3, top).
These observations are in agreement with results from Olenici-Craciunescu et al., who investigated the influence of the N2 fraction in He on DBD characteristics (studied range 0–600 ppm N2 in He).23 Within the measured concentrations range, they observed an initial increase of N2+ emission intensities with a maximum between 140 and 450 ppm followed by a slight decay within the range of 450 to 600 ppm. However, helium (706.5 nm) intensities were decreasing with increasing nitrogen concentrations. The fact that an increase of N2+ intensities was only observed up to nitrogen concentrations of 450 ppm was explained by a limited amount of available helium metastables, which deliver the energy for the Penning ionization of nitrogen. However, the influence of higher nitrogen concentrations was not investigated in their study.
The influence of the nitrogen content on broadband UV emission was previously studied by Lu et al. for a pulsed DBD (with a different geometry).5 Here, it was observed that the increase of the nitrogen fraction (up to ∼5%) in helium resulted in an increase of broadband emission intensities between 165 nm and 310 nm. NO was found to be the main UV emission source. In our study, however, the maximum NO emission was found at a nitrogen concentration of 0.6% (see Fig. 3, bottom). The following increase of nitrogen concentration resulted in a reduction of UV emission intensity.
Effect of the discharge gas flow rate
To investigate the effect of flow rate on the emission spectra in the afterglow, experiments with flow rates within the range of 100 to 600 mL min−1 were performed. This effect was investigated for gas mixtures containing 0, 33, 50, 67, and 100% of N2 in He. These experiments demonstrated a significant influence of discharge gas flow rate on the observed emission spectra in the afterglow.At a discharge gas flow rate of 100 mL min−1, emission bands of N2 are dominating over emission from other species for most analysed discharge gases (33–100% N2). The analysis in a pure helium at discharge gas flow rates lower than 300 mL min−1 was not possible because of an observed electrical breakdown inside the capillary. A possible reason for the absence of an electrical breakdown at helium flow rates higher than 300 mL min−1 is a slightly increased pressure within the discharge region. According to Paschen's law, an increase in pressure results in a higher breakdown voltage. An increase of the discharge gas flow rate results in a much stronger increase of NO emission as compared to the observed increase of N2 emission. This tendency was observed for all analysed discharge gas compositions (see Fig. 4, bottom).
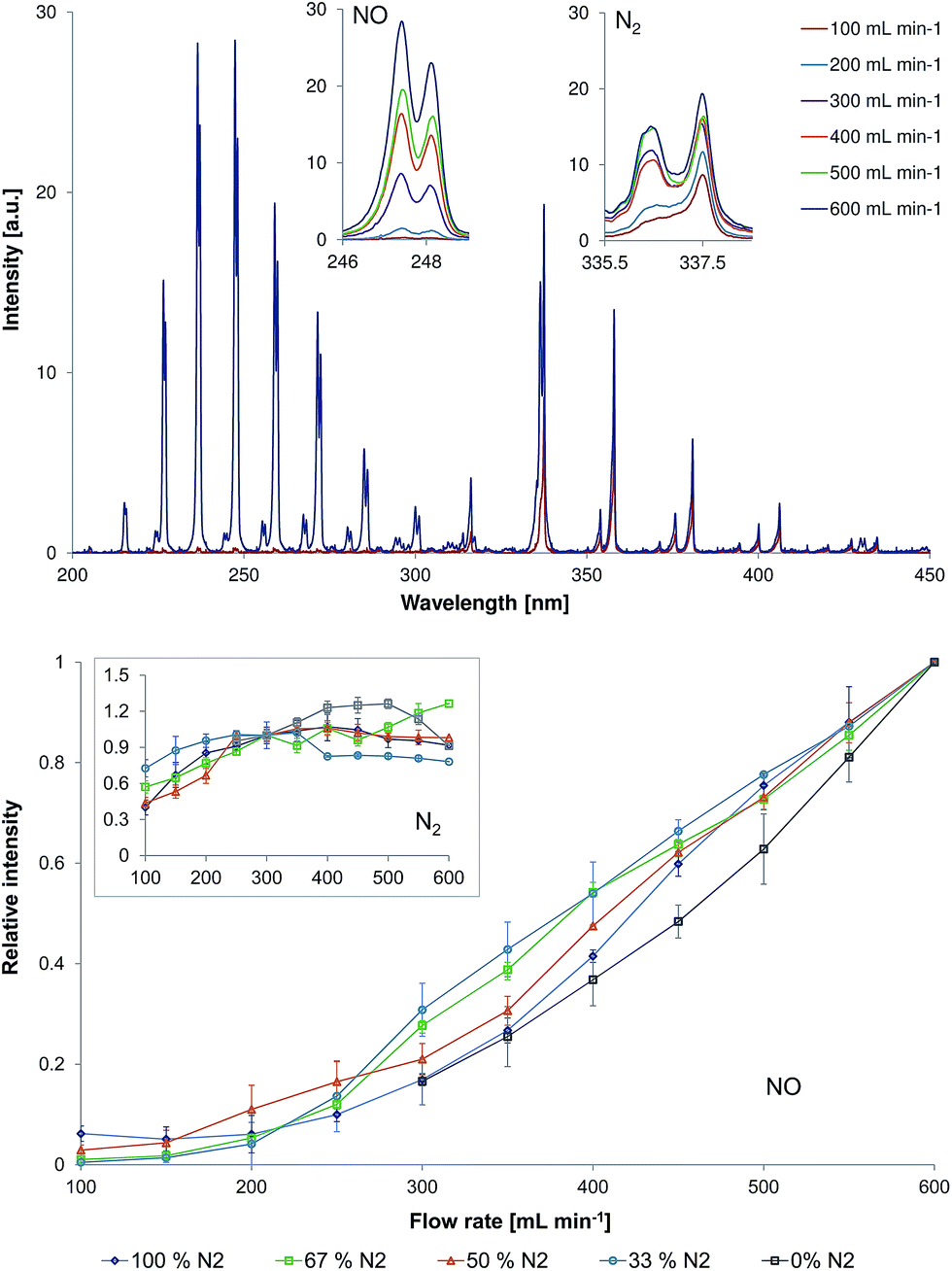 | ||
| Fig. 4 Top: Representative emission spectra (200–450 nm) measured directly at the LTP capillary outlet (d = 0 mm) for a 67%N2/33%He discharge and different gas flow rate (100–600 mL min−1) at a discharge voltage of 4 kVpp. The inserts show the strongest bands for NO (237 nm) and N2 (337.1 nm). Bottom: Influence of gas flow rate on the normalized emission intensity of NO (237 nm) and N2 (337.1 nm, insert) in the LTP afterglow operated with different N2/He discharge gas compositions. Note that NO and N2 emission intensity values are normalized by dividing the measured emission intensity values with those at a flow rate of 600 mL min−1 for NO and 300 mL min−1 for N2. | ||
One of the possible reasons for this observation can be a faster transport of ions with a gas flow. When increasing the flow rate, the time that is required for the transport of species from the plasma region to the capillary outlet is shortened. Thus, plasma species created at higher flow rates have less time for the gas chemistry as compared to the experiments at lower flow rates. Increase of the carrier gas flow rate from 100 to 600 mL min−1 results in a decrease of the residence time of the species in the region between the active plasma part and the measurement position from 9.29 × 10−4 to 1.55 × 10−4 s. The calculated lifetimes for excited NO and N2 species were determined to be approximately 1.2 × 10−4 and 3.0 × 10−4 s, respectively. The estimated lifetime of excited NO species is by 2.5 times shorter as compared to the one of excited N2 species. Therefore, an increase of the carrier gas flow rate by six times and, thus, a decrease of the residence time by six times results in a significant increase of excited NO species in the afterglow.
Emission spectra recorded within the wavelength range of 200–450 nm in discharge gas containing 67% of nitrogen in helium are exemplarily demonstrated in Fig. 4 (top, discharge voltage = 4 kVpp). The strongest emission of NO (237 nm) and N2 (337.1 nm) are highlighted with inserts in Fig. 4. The change of NO (237 nm) and N2 (337.1 nm, inset) emission intensities in different gas mixtures and flow rates (100–600 mL min−1) is presented in Fig. 4 (bottom).
At a helium flow rate of 300 mL min−1, the strongest emission was observed from N2+. Nevertheless, the relationship between NO and N2 emission intensity and discharge gas flow rate was similar to those observed for nitrogen containing mixtures.
Effect of the discharge voltage
Fig. 5 shows the influence of the discharge voltage on relative emission intensities of N2 (337.1 nm), NO (237 nm), N2+ (391.4 nm), OH (308 nm), He (706.5 nm), and O (777 nm) in helium discharge gases containing different nitrogen fractions. Experiments were performed at a fixed discharge gas flow rate (500 mL min−1) within varying discharge voltage (4–7 kVpp). However, only N2 and NO emission bands were observed when the measurements were performed in nitrogen containing mixtures. Increasing the discharge voltage resulted in an increase of the emission intensities for all species in all discharge mixtures. The strongest effect was observed for NO and N2 emission bands when the discharge was sustained in pure helium. Here, an increase of emission intensities in the afterglow by 8 and almost 12 times was observed, for NO (237 nm) and N2 (337.1 nm) emission bands respectively, when the discharge voltage was increased from 4 to 7 kVpp. The observed effect was less significant in nitrogen containing mixtures. Nevertheless, an intensity increase by a factor of 3–5 was achieved under the same conditions.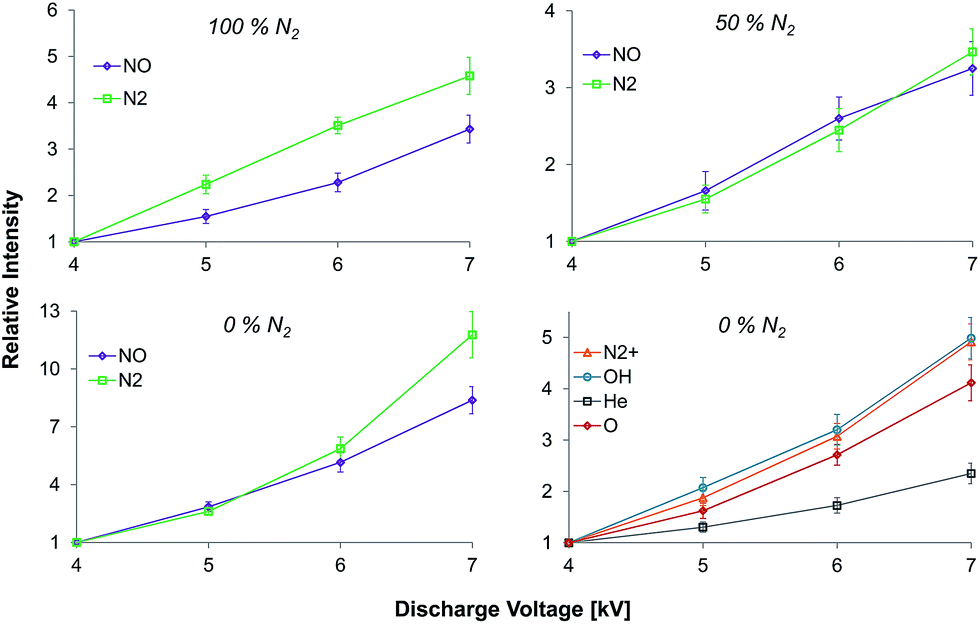 | ||
| Fig. 5 Influence of the discharge voltage on the relative intensities of N2 (337.1 nm), NO (237 nm), N2+ (391.4 nm), OH (308 nm), He (706.5 nm), and O (777 nm) (normalized to 4 kV) measured in the LTP afterglow with a discharge gas that contained different nitrogen fractions. | ||
Spatially resolved emission measurements
Spectroscopic plasma diagnostic, including spatially resolved measurements, were performed in the past (for comparable DBD-based systems in helium) by Chan et al.4 (LTP, one dielectric layer between the electrodes) and by Olenici-Craciunescu et al.8 (DBD-microplasma, two dielectric layers between the electrodes). Despite the fact that the source design used by Chan et al. is similar to the torch used in this study, the capillary/electrode dimensions and the discharge parameters used in this study are much closer to those used by Olenici-Craciunescu et al. (see Table 1).Fig. 6 shows normalized emission maps of selected molecular and atomic species observed in the LTP afterglow at different gas flow rates. In these figures, the vertical axis refers to the distance from the end of the LTP outlet (at 0 mm). In contrast to Chan et al. and Olenici-Craciunescu et al. no emission maxima outside the torch were observed for N2 and N2+, even when the measurements were performed in pure helium. The reason for this is still under investigation, but based on the obtained data is difficult to discuss the mechanistic aspects of plasma ionization. Nevertheless, a few interesting observations were made. As was partially demonstrated in Fig. 4, the intensity of NO emission demonstrates a significant dependence on the discharge gas flow rate. A flow rate decrease from 500 to 100 mL min−1 results in a decrease of NO emission intensity by more than five times in all discharge gases. However, the dependence of N2 and N2+ emission lines on the discharge gas flow rate was found to be not as severe, especially at higher helium fractions (50–100%). Emission of all observed species strongly decreases with increasing distance from the LTP outlet. At a distance of 3 mm the emission intensity of NO (237 nm) is reduced by almost 10 times as compared to its intensity measured directly at the outlet. This effect was found to be even stronger for N2 and N2+ emission bands (10% at a distance of 1.5–2 mm). Also, this decay was found to be relatively stronger compared to results reported earlier with similar DBD discharge parameters.8 The reason for this fast decay is not clear today and further studies should be performed to clarify this phenomenon.
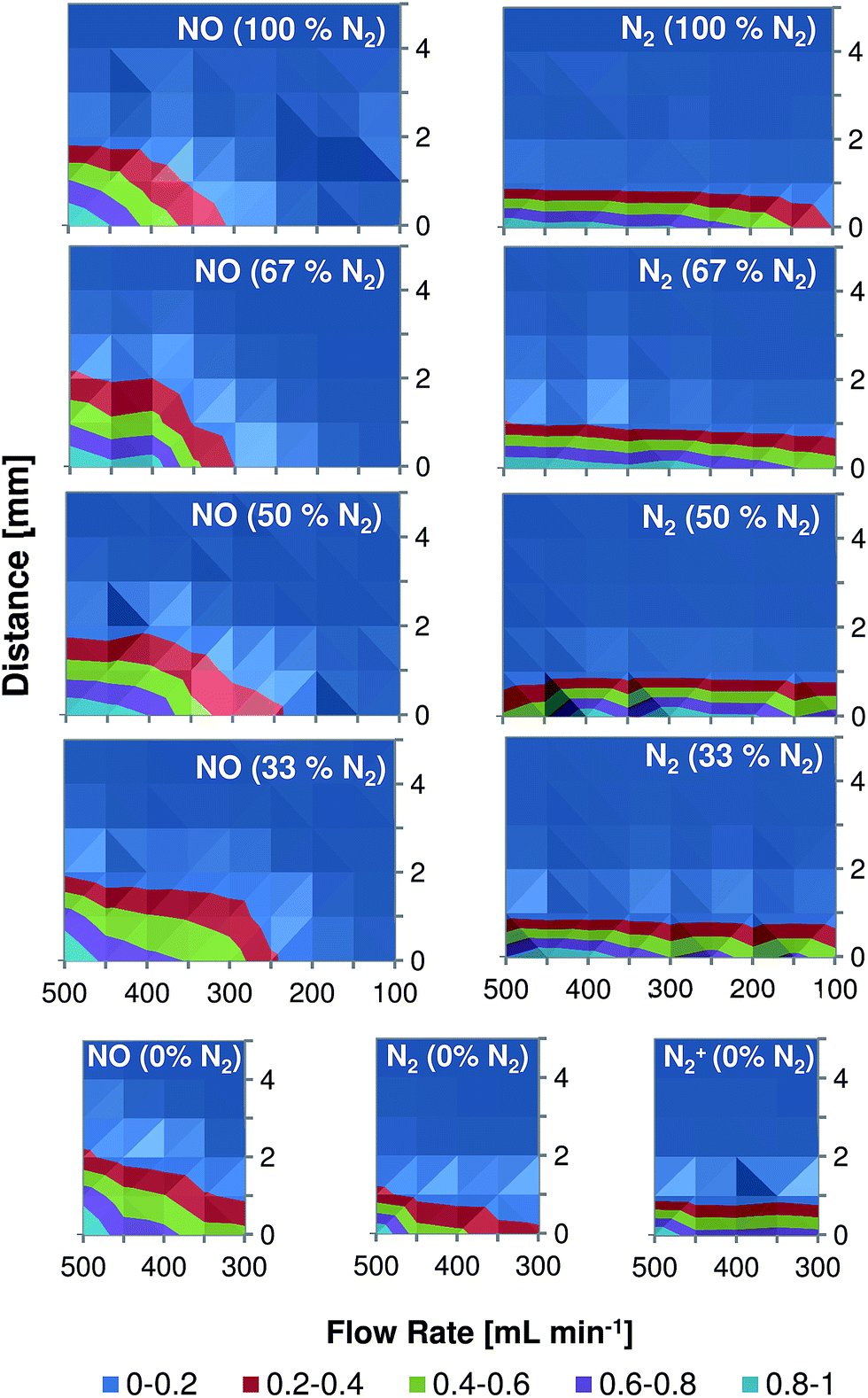 | ||
| Fig. 6 Two-dimensional contour plots demonstrating the dependence of relative intensities of NO (237 nm), N2 (337.1 nm), and N2+ (391.4 nm) emission in the LTP afterglow on the distance from the capillary outlet and the discharge gas flow rate. Measurements were performed in mixtures with different nitrogen content (0–100% of N2). | ||
Conclusions
A low-temperature plasma probe operated with helium/nitrogen mixtures was characterized by means of optical spectroscopy. Selected reactive species that are involved in analyte ionization processes (e.g., N2+, NO) were studied. A significant decrease of emission intensity (and to a first approximation also density) of N2+ in the afterglow was observed for nitrogen concentrations in the discharge gas higher than 0.4%. This is important because N2+ is generally considered to be a reaction intermediate that is also responsible for the formation of protonated water clusters.24 Therefore, the observed decrease of N2+ density can potentially limit the application of a N2/He driven LTP source for the analysis of analytes that are ionized via proton transfer reactions. In discharge gas mixtures that contained 0.4–50% nitrogen, however, the emission intensity of NO in the afterglow was higher compared to a 100% helium LTP. The maximum emission intensity was achieved in a mixture with a relatively low nitrogen fraction (0.6% N2 in He). Here, NO emission increased approximately ten times compared to that in a 100% helium discharge. Penning ionization of NO results in formation of NO+. This ion is an important reactant ion because may offer alternative routes to analyte ionization (e.g. electron transfer, ion–molecule association, and hydride abstraction mechanisms) as compared to ionization routes which involve N2+ intermediate (proton transfer mechanisms). In addition, excited NO is a source of high energy photons which can provide additional plasma reactivity. Thus, the ionization mechanism can be driven by a proper choice of the LTP discharge gas.Discharge gas flow rate is another parameter, which affects the intensity (and to a first approximation also density) of reactive species in afterglow. An increase of the discharge gas flow rate results in a much stronger increase of NO emission as compared to the observed increase of N2 emission. Thus, it is assumed that high discharge gas flow rates will be preferable for the analysis of analytes that can be ionized via NO+ species in the afterglow. However, investigation of the influence of the discharge gas composition and the flow rate on ionization mechanisms in the afterglow was not systematically investigated in this study and, therefore, should be investigated in detail in the future.
Acknowledgements
The glassblower and machine shop services at the University of Münster and the University of Siegen, as well as optical workshop and machine shop services at the University of Duisburg-Essen are gratefully acknowledged. This work was partly funded by the German Research Foundation through grant no. EN 927/1-1.Notes and references
- J. D. Harper, N. A. Charipar, C. C. Mulligan, X. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 9097–9104 CrossRef CAS PubMed.
- A. Kuklya, C. Engelhard, F. Uteschil, K. Kerpen, R. Marks and U. Telgheder, Anal. Chem., 2015, 87, 8932–8940 CrossRef CAS PubMed.
- A. Albert and C. Engelhard, Anal. Chem., 2012, 84, 10657–10664 CrossRef CAS PubMed.
- G. C. Y. Chan, J. T. Shelley, A. U. Jackson, J. S. Wiley, C. Engelhard, R. G. Cooks and G. M. Hieftje, J. Anal. At. Spectrom., 2011, 26, 1434–1444 RSC.
- X. Lu and M. Laroussi, J. Appl. Phys., 2005, 98, 023301 CrossRef.
- A. Albert, J. T. Shelley and C. Engelhard, Anal. Bioanal. Chem., 2014, 406, 6111–6127 CrossRef CAS PubMed.
- G. C. Y. Chan, J. T. Shelley, J. S. Wiley, C. Engelhard, A. U. Jackson, R. G. Cooks and G. M. Hieftje, Anal. Chem., 2011, 83, 3675–3686 CrossRef CAS PubMed.
- S. B. Olenici-Craciunescu, S. Müller, A. Michels, V. Horvatic, C. Vadla and J. Franzke, Spectrochim. Acta, Part B, 2011, 66, 268–273 CrossRef CAS.
- A. Qayyum, S. Zeb, M. A. Naveed, S. A. Ghauri and M. Zakaullah, J. Appl. Phys., 2005, 98, 103303 CrossRef.
- M. Tsuji, H. Ishimi and Y. Nishimura, Chem. Lett., 1995, 24, 873–874 CrossRef.
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2646–2653 CrossRef CAS PubMed.
- A. Fridman, Plasma Chemistry, Cambridge University Press, 2012 Search PubMed.
- W. P. West, T. B. Cook, F. B. Dunning, R. D. Rundel and R. F. Stebbings, J. Chem. Phys., 1975, 63, 1237 CrossRef CAS.
- J. A. Coxon, M. A. A. Clyne and D. W. Setser, Chem. Phys., 1975, 7, 255–266 CrossRef CAS.
- J. T. Shelley, A. Stindt, J. Riedel and C. Engelhard, J. Anal. At. Spectrom., 2014, 29, 359–366 RSC.
- M. T. Jafari, Low-Temperature Plasma Ionization Ion Mobility Spectrometry, Anal. Chem., 2011, 83, 797–803 CrossRef CAS PubMed.
- R. S. Blake, P. S. Monks and A. M. Ellis, Chem. Rev., 2009, 109, 861–896 CrossRef CAS PubMed.
- M. Sabo and Š. Matejčík, Analyst, 2013, 138, 6907–6912 RSC.
- P. Sulzer, B. Agarwal, S. Jürschik, M. Lanza, A. Jordan, E. Hartungen, G. Hanel, L. Märk, T. D. Märk, R. González-Méndez, P. Watts and C. A. Mayhew, Int. J. Mass Spectrom., 2013, 354–355, 123–128 CrossRef CAS.
- A. G. Harrison, Chemical Ionization Mass Spectrometry, CRC Press, 2nd edn, 1992 Search PubMed.
- D. Smith and P. Spanel, Mass Spectrom. Rev., 2005, 24, 661–700 CrossRef CAS PubMed.
- G. J. Francis, V. S. Langford, D. B. Milligan and M. J. McEwan, Anal. Chem., 2009, 81, 1595–1599 CrossRef CAS PubMed.
- S. B. Olenici-Craciunescu, A. Michels, C. Meyer, R. Heming, S. Tombrink, W. Vautz and J. Franzke, Spectrochim. Acta, Part B, 2009, 64, 1253–1258 CrossRef.
- A. Good, D. A. Durden and P. Kebarle, J. Chem. Phys., 1970, 52, 212–221 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |