
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombination of single particle ICP-QMS and isotope dilution analysis for the determination of size, particle number and number size distribution of silver nanoparticles
Carina A.
Sötebier
a,
Daniel J.
Kutscher
b,
Lothar
Rottmann
b,
Norbert
Jakubowski
a,
Ulrich
Panne
*ac and
Jörg
Bettmer
*d
aFederal Institute for Materials Research and Testing-BAM, Richard-Willstätter-Str. 11, D-12489 Berlin, Germany. E-mail: Ulrich.Panne@bam.de; Tel: +49-30-8104-1000
bThermo Fisher Scientific, Hanna Kunath Str. 11, D-28199 Bremen, Germany
cHumboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, D-12489 Berlin, Germany
dUniversity of Oviedo, Department of Physical and Analytical Chemistry, C/Julián Clavería 8, E-33006 Oviedo, Spain. E-mail: bettmerjorg@uniovi.es; Fax: +34-985-103125; Tel: +34-985-103000 ext. 5366
First published on 22nd August 2016
Abstract
Single particle ICP-MS (spICP-MS) has gained great influence in the analysis of engineered nanoparticles (NPs) due to its simplicity, speed and ability to obtain a particle number size distribution. Despite its many advantages, the method is hampered by matrix effects affecting the sensitivity of the instruments. Consequently, over- or underestimated particle sizes might be obtained. To overcome these challenges, we present in this work the detection of both Ag isotopes with a quadrupole mass spectrometer for the application of isotopic dilution analysis (IDA) in combination with spICP-MS. Here, the isotopes are measured sequentially using the conventional spICP-MS integration time of 10 ms. Citrate stabilized Ag NPs of a spherical shape with the nominal diameters of 30, 40, 50 and 80 nm have been investigated. The experimental concept of adding 109Ag+ solutions to the NP suspensions resulted in the NP spikes being only visible in the 107Ag trace. Therefore, a maximum of 45% of the particles was detected compared to that by conventional spICP-MS. A modified mass flow equation was applied to determine the particle sizes, particle size distributions and particle number concentrations of various Ag NPs. The addition of different spike concentrations between 0.5 and 4 μg L−1 109Ag resulted in similar particle diameters, suggesting that the calculated diameter might be independent of the spike concentration. This would have the advantage that no size information would be needed before the analysis. By analyzing Ag NP suspensions in a simulated seawater matrix, we demonstrate its significant influence on the particle size determination using conventional spICP-MS. A lower transport efficiency of 6.1% was found in the matrix compared to 7.3% without the matrix. In our approach, the addition of the matrix influenced the NP intensity stronger than the spike signal, resulting in slightly smaller diameters using IDA–spICP-MS with the matrix compared to the results without the matrix. On the other hand, the IDA–spICP-MS approach with the matrix can result in equivalent results for the particle sizes compared with conventional spICP-MS using suspensions without the matrix. Due to the lower instrument sensitivity in the matrix, a diameter of 30 nm was found to be close to the detection limit of the instrument.
Introduction
The widespread and continuously growing use of nanomaterials has led to a variety of regulations with a need for highly specific analysis methods. In 2011, the European Commission released a recommendation on the definition of nanomaterials. This definition states that at least 50% of the particles need to be in the range between 1 and 100 nm in at least one of their dimensions.1 As a consequence, highly sensitive analytical methods with the ability to determine the number-size distribution are needed. Another challenge is the wide variety of nanoparticle (NP)-containing products, for example creams, polymers, and dietary supplements. For their investigation, matrix-tolerant and -independent analysis methods need to be developed.Inductively coupled plasma-mass spectrometry (ICP-MS) is well recognized for its unique features in the trace and ultratrace determination of elements and elemental species. In recent years, this analytical technique has entered the field of nanomaterial analysis as a valuable tool for the detection of metal-containing NPs.2 Besides being able to determine the total metal concentration in fractions of NPs (e.g. after ultrafiltration or cloud point extraction),3,4 studies have demonstrated that hyphenated techniques with ICP-MS are a powerful tool. The on-line combination of ICP-MS as the detector and separation techniques such as (gel) electrophoresis,5–7 (hydrodynamic) liquid chromatography,5,8–11 and most frequently, field-flow fractionation techniques12–15 was applied to the characterization of NP-bound elements. Although these approaches principally allow the simultaneous detection of several elements bound to the particulate matter, they are laborious and time-consuming techniques with restricted detection limits, usually in the low μg L−1 range. Moreover, quantitative analyses are currently hampered by low recoveries during the separation and fractionation steps. This applies especially to field-flow fractionation techniques.16,17 In this regard, post-channel species-unspecific on-line isotope dilution approaches have been suggested to improve the quality of quantitative results.16,18
In 2003, Degueldre et al. suggested the use of short dwell times to monitor individual manufactured NPs using an ICP-MS.19 This methodology – called single-particle ICP-MS (spICP-MS) – has gained great importance in the characterization of metallic NPs.
Recent studies have investigated the fundamentals of spICP-MS and shown the main advantages of spICP-MS to be its simplicity and speed.20,21 Basically, three important pieces of information about a sample can be obtained: (i) the baseline signal is directly proportional to the soluble (non-particulate bound) fraction of the element of interest, (ii) the number of events (signals) can be correlated with the particle number concentration, and (iii) the signal intensity of each event can be converted into the mass fraction of the monitored element and allows the determination of its distribution within the NP sample. Using an external calibration and with the knowledge of the transport efficiency of the sample introduction system, the sample flow and the response factor, the observed signal intensities of the NP events can be converted into absolute masses. When the shape of the NP is known, the particle diameter can be calculated in a next step. In most cases, the shape is unknown. Typically, a solid, spherical shape is assumed.21,22 Several applications and improvements in terms of alternative sample introduction systems and data processing have been published recently.20,23–27 However, matrix effects can have an influence on the detection sensitivity and the nebulization efficiency, causing the particle mass fraction to be over- or underestimated.22 In order to improve the robustness against matrix effects, isotope dilution analysis (IDA) was – to the best of our knowledge – first suggested in 2013 (ref. 28) and later applied to the rapid size determination of Ag NPs in wastewater and river water.21
Based on the generally accepted methodology of spICP-MS, we present a proof-of-concept study on the combination of IDA and spICP-MS applied to the analysis of the size, size distribution and particle number concentration of Ag NPs as an example. The approach is described in detail for the use of a quadrupole mass spectrometer (QMS) as a mass analyzer with its well-known limitation of monitoring multiple isotopes sequentially. Following some basic considerations, the developed concept is compared to conventional spICP-MS for the determination of particle sizes, particle size distributions and particle number concentrations. Considering the example of a simulated seawater matrix, the results prove the matrix-independency of spICP-MS in combination with IDA. This approach is a promising tool for quantitative analysis of nanomaterials in complex matrices containing an element with minimum two isotopes detectable by ICP-MS.
Experimental
Materials
Ultrapure water (18.2 MΩ cm) was generated by a Millipore Element system (EMD Millipore, Billerica, MA, USA) equipped with a Quantum ICP Polishing Cartridge. A CertiPUR grade Ag elemental standard and an isotopically enriched CertiPUR spike solution Ag-109 were purchased from Merck (Darmstadt, Germany). Citrate-stabilized Ag NPs with the nominal diameters of 30, 40, 60 and 80 nm and the transmission electron microscopy (TEM) values of 32.7 nm ± 4.8 nm, 40.6 nm ± 3.0 nm, 60.8 nm ± 6.6 nm and 79.8 nm ± 5.4 nm, given by the manufacturer, were purchased from Nanocomposix Europe (Prague, Czech Republic). Within the text these materials are named NC30, NC40, etc. The reference material 8013 Au NPs with a nominal diameter of 60 nm was acquired from NIST (Gaithersburg, MD, USA). Sodium chloride (p. A. grade) was obtained from neoLab Migge (Heidelberg, Germany). The ICP-MS was operated using 99.999% Argon 5.0 (Linde AG, Pullach, Germany).Instrumentation
The experiments were carried out using an iCAP Q ICP-MS instrument (Thermo Fisher Scientific, Bremen, Germany). The experimental conditions are compiled in Table 1. A Bandelin RK 100 H (Bandelin, Berlin, Germany) ultrasonic bath was used for sample sonification.| Parameter | Value |
|---|---|
| a During the conventional spICP-MS experiments, only the isotope 107Ag was monitored. For the isotopic dilution experiments, both 107Ag and 109Ag were detected. | |
| Nebulizer | Concentric PFA nebulizer with 400 μL min−1 uptake rate |
| Spray chamber | Cyclonic spray chamber, quartz |
| Injector | 2.5 mm inner diameter, quartz |
| Sample cone | Ni cone with 1.1 mm orifice diameter |
| Skimmer cone | Ni cone with 0.5 mm orifice diameter and 2.8 mm insert |
| Power [W] | 1550 |
| Nebulizer gas flow rate [L min−1] | 1.09 |
| Aux gas flow rate [L min−1] | 0.65 |
| Cool gas flow rate [L min−1] | 14 |
| Sample flow rate [mL min−1] | 0.33 |
| Spray chamber temperature [°C] | 2.7 |
| Transport efficiency [%] | 6.3 |
| Dwell time [ms] | 10 |
| Isotopes monitoreda | 107Ag, 109Ag |
Sample preparation
The samples were prepared by dilution in Milli-Q water. The first dilution step was sonication in an ultrasonic bath for 5 min before further dilution. The samples were always prepared freshly before the analysis.For the imitation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 diluted seawater matrix, sodium chloride with a concentration of 30 mmol L−1 was added to the final dilution of the sample.
:20 diluted seawater matrix, sodium chloride with a concentration of 30 mmol L−1 was added to the final dilution of the sample.
An isotopically enriched 109Ag aqueous standard solution was added to the samples for the isotopic dilution experiments. The used concentrations can be found in Table 3. The Ag+ concentration of the isotopically enriched 109Ag standard solution was determined by inverse isotopic dilution analysis.
Determination of the transport efficiency
The transport efficiency was determined by using the particle number method described by Pace et al.29 First, the particle number concentration NP of the NIST reference material RM 8013, Au NPs with a diameter d of 56 nm ± 0.5 nm (TEM), was calculated. The particle mass flow concentration cP of the diluted particle suspension was calculated using the given total Au concentration of 51.86 mg kg−1 ± 0.64 mg kg−1. The particle number concentration (NP) is given by eqn (1).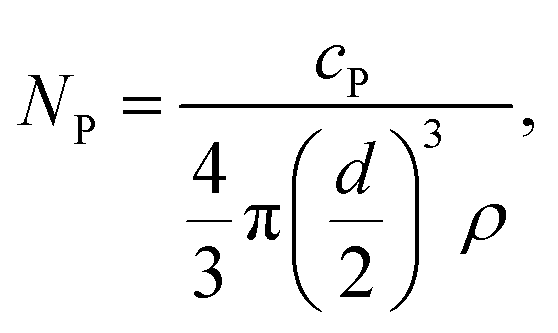 | (1) |
In a second step, the transport efficiency ηn was calculated as shown in eqn (2).
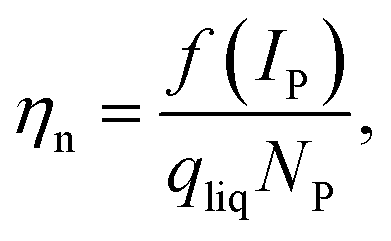 | (2) |
Results and discussion
General considerations
In general, spICP-MS equipped with a QMS is limited to monitoring one isotope per integration time30 as a single particle event has a duration of approximately 300–500 μs.19 The shortest dwell time available in the instrumentation applied here (iCAP Q) is 100 μs. This would result in 3–5 points per particle peak in conventional spICP-MS. As the goal of this work was to measure two isotopes sequentially, 2–3 points per NP peak on each isotope would be measured, which would not be sufficient and might result in a measurement of incomplete particle events. The latter is likely to result in false particle masses. Nowadays other QMS instruments can provide shorter integration times eventually useful for multiple isotope detection in spICP-MS.31 This limitation can be principally overcome in time-of-flight (ToF) instrumentations as demonstrated recently,32,33 but they might still lack sufficient sensitivity for monitoring smaller NPs. The remaining question, which we intended to explore, was whether a regular ICP-quadrupole-MS with typical conditions for spICP-MS can be used for the detection of two isotopes of the same element using conventional spICP-MS integration times.21The idea behind this work was to use an ICP-QMS in single particle mode in combination with the well-established concept of species-unspecific isotope dilution analysis.34,35 When a Ag NP enters the plasma, both isotopes (107Ag and 109Ag) can principally be used for its analysis thanks to the known natural isotopic abundance (and the determined mass bias). In QMS, the two isotopes will be measured sequentially, so that a single particle event – within the restricted time frame of e.g. 10 ms – can be only monitored in one of the two available isotopes. This increases the risk of incomplete particle events.30 Furthermore, the change between the two masses requires a short stabilization time of the QMS, which in the case of the iCAP Q instrument are 1.38 ms from mass 107 to 109 and 0.67 ms for the reverse mass jump. During this stabilization time, the detector does not register and events cannot be observed. Using shorter dwell times (≤5 ms) for both isotopes would result in a loss of particle events of at least 17%. As a result, 10 ms was the preferential dwell time (entire measurement cycle: 22 ms) in this study considering roughly 9% of undetectable particles.
In accordance with recent work,22 the matrix effects in spICP-MS using an external calibration cannot be neglected in order to provide reliable results in terms of particle mass (size). To our experience, an aqueous matrix containing 30 mmol L−1 NaCl can easily induce a signal depression of 30% in the observed particle events leading to a significant analytical error (data not shown). As matrix matching is not always possible, the use of an internal standard might provide more accurate results. In this work, isotope dilution analysis in combination with spICP-MS is applied to compensate the observed matrix effects. An isotopically enriched standard of 109Ag+ was added to the sample, both Ag isotopes were monitored at a dwell time of 10 ms (ref. 21) and the resulting data were treated corresponding to post-column or species-unspecific IDA.34,35 The use of a QMS required an approximation for the isotope ratio 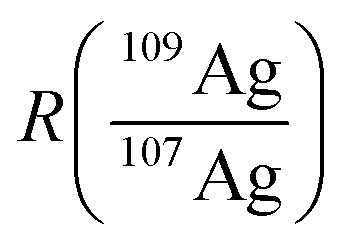 , as the signal intensities of the NPs cannot be measured simultaneously for both naturally occurring isotopes (I(107Agnat,NP) and I(109Agnat,NP)).
, as the signal intensities of the NPs cannot be measured simultaneously for both naturally occurring isotopes (I(107Agnat,NP) and I(109Agnat,NP)).
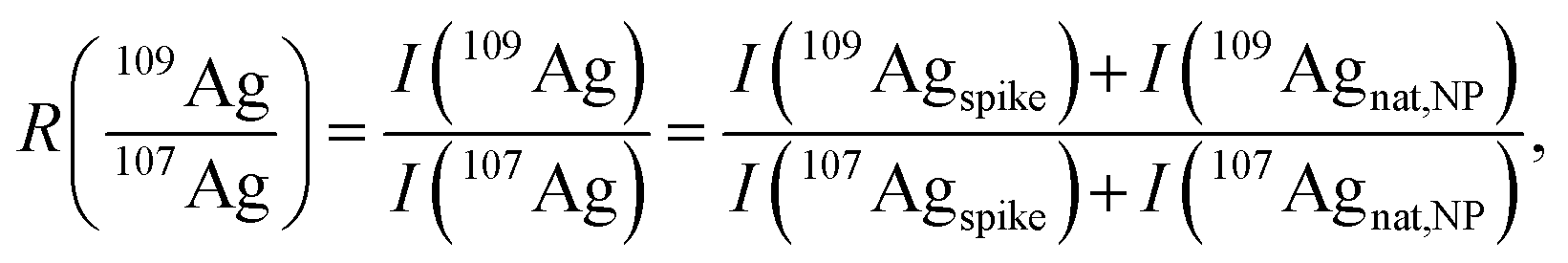 | (3) |
Provided that the signal intensity of 109Agspike is much higher than that of 109Agnat,NP (I(109Agspike) ≫ I(109Agnat,NP)) eqn (3) can be simplified:
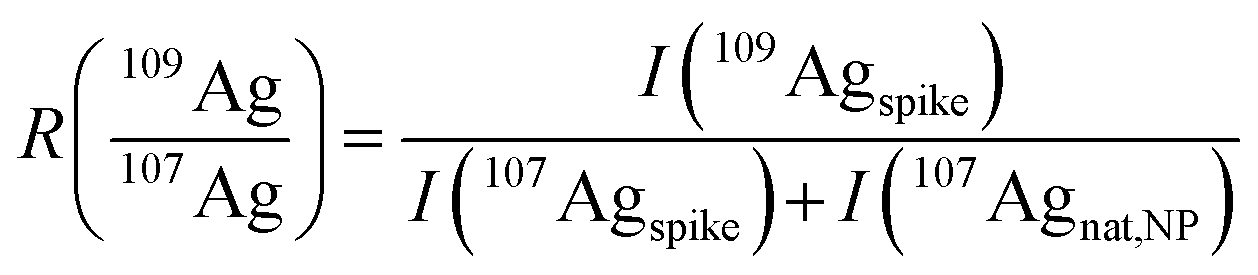 | (4) |
Using this approximation, the determination of 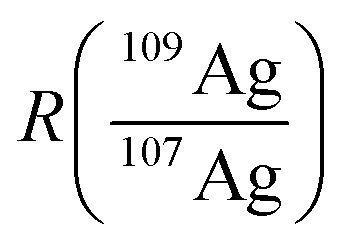 with a QMS is possible, provided that the signal intensity of 109Ag is relatively constant from one measurement cycle to the next. Finally, the mass flow of Ag in the sample (Mfs) can be calculated by applying the known mass flow equation (eqn (5)) and considering the transport efficiency of the spike solution (ηn).
with a QMS is possible, provided that the signal intensity of 109Ag is relatively constant from one measurement cycle to the next. Finally, the mass flow of Ag in the sample (Mfs) can be calculated by applying the known mass flow equation (eqn (5)) and considering the transport efficiency of the spike solution (ηn).
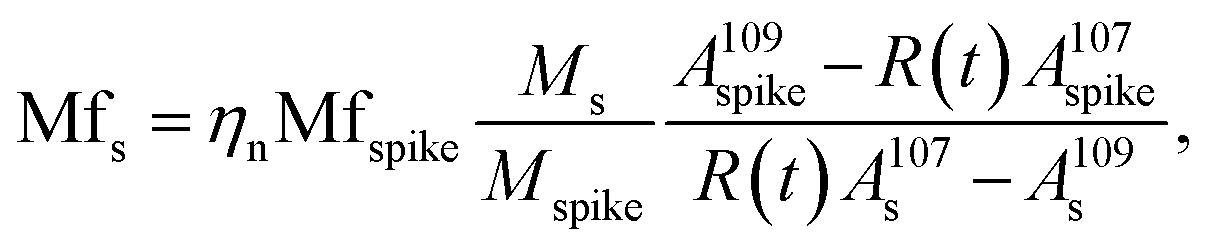 | (5) |
IDA–spICP-MS
Based on the theoretical considerations mentioned above, diluted suspensions of Ag NPs of different sizes in the presence of the 109Ag spike were introduced into the plasma. Fig. 1a shows a typical time-resolved scan of the two isotopes under the chosen experimental conditions (Table 1) with a measurement time of 10 min. The enriched 109Ag signal showed intensities of around 320000 cps. In this trace, the NP events that occur cannot be distinguished from the signal noise. The depleted isotope 107Ag (Fig. 1b) revealed a relatively constant background signal of approximately 8000 cps corresponding to its low abundance in the isotopically enriched 109Ag standard (1.77%). The periodic particle events can be monitored as usual in spICP-MS. Consequently, all information on the NPs is stored in the trace of the depleted isotope, in this case 107Ag. Due to the measurement of two isotopes and the required mass jumps, a maximum of 45% NP events can be detected in the depleted isotope.† As a consequence, relatively long acquisition times (10 min) were applied in this study in order to monitor minimum 1000 events. The histogram of the signals obtained by monitoring 107Ag (Fig. 1c) demonstrates that the NPs can be easily distinguished from the background signals. The obtained signal distribution is equivalent to the results of the conventional spICP-MS for the analysis of a NP suspension containing dissolved species.
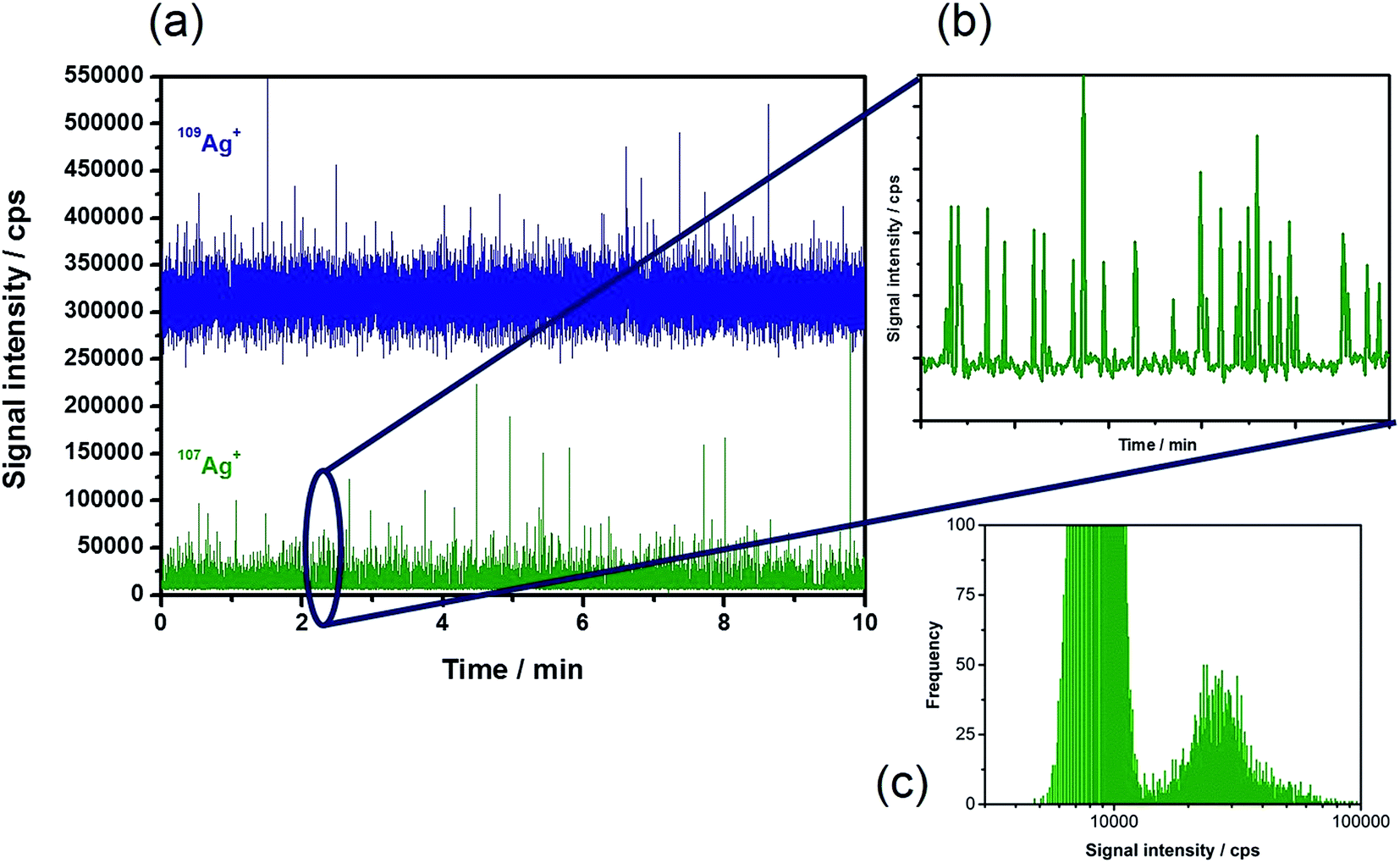 | ||
| Fig. 1 Typical time-resolved scan of IDA–spICP-MS analyzing 20 ng L−1 40 nm Ag NPs with 1 μg L−1 109Ag+ solution for (a) both isotopes, (b) isotope 107Ag in detail, and (c) the corresponding histogram for 107Ag. | ||
The next step was to convert the obtained Ag signal intensities into Ag particle masses. A modified version of the single particle calculation tool developed by Peters et al.22 was used for the particle size calculations using isotopic dilution. Based on the existing calculation tool, the cut-off between the background and the particle was selected visually from the signal distribution of the raw data. As a result, the signals were sorted into either a background or a particle signal. Note that any mass flow of the background different from the blank could be assigned to the concentration of the dissolved Ag+ species. The mass flow of the background was then subtracted from the mass flow of the particles (eqn (5)). As some background corrected particle mass flow signals might show a negative value due to fluctuations of the spike, further calculations were carried out using the positive signals only.
Fig. 2 depicts a 15 s long section of a time-resolved scan and its conversion into the corresponding mass flow, based on the analysis of 60 nm Ag NPs (total Ag concentration: 20 ng L−1) with the addition of a 1 μg L−1 109Ag solution. In Fig. 2a, it can repeatedly be observed that the particle events appeared in the depleted 107Ag trace. The application of eqn (5), with the restrictions explained above, results in the corresponding mass flow presented in Fig. 2b. A comparison of Fig. 2a and b demonstrates that the number of NPs detected in the mass spectrum exactly coincided with the number of NPs observed in the mass flow. This finding supports the hypothesis that the selection of the particle events as described above included uniquely ‘positive’ events. By multiplying the obtained mass flow of the particles with the chosen integration time, the particle mass of each event was obtained.
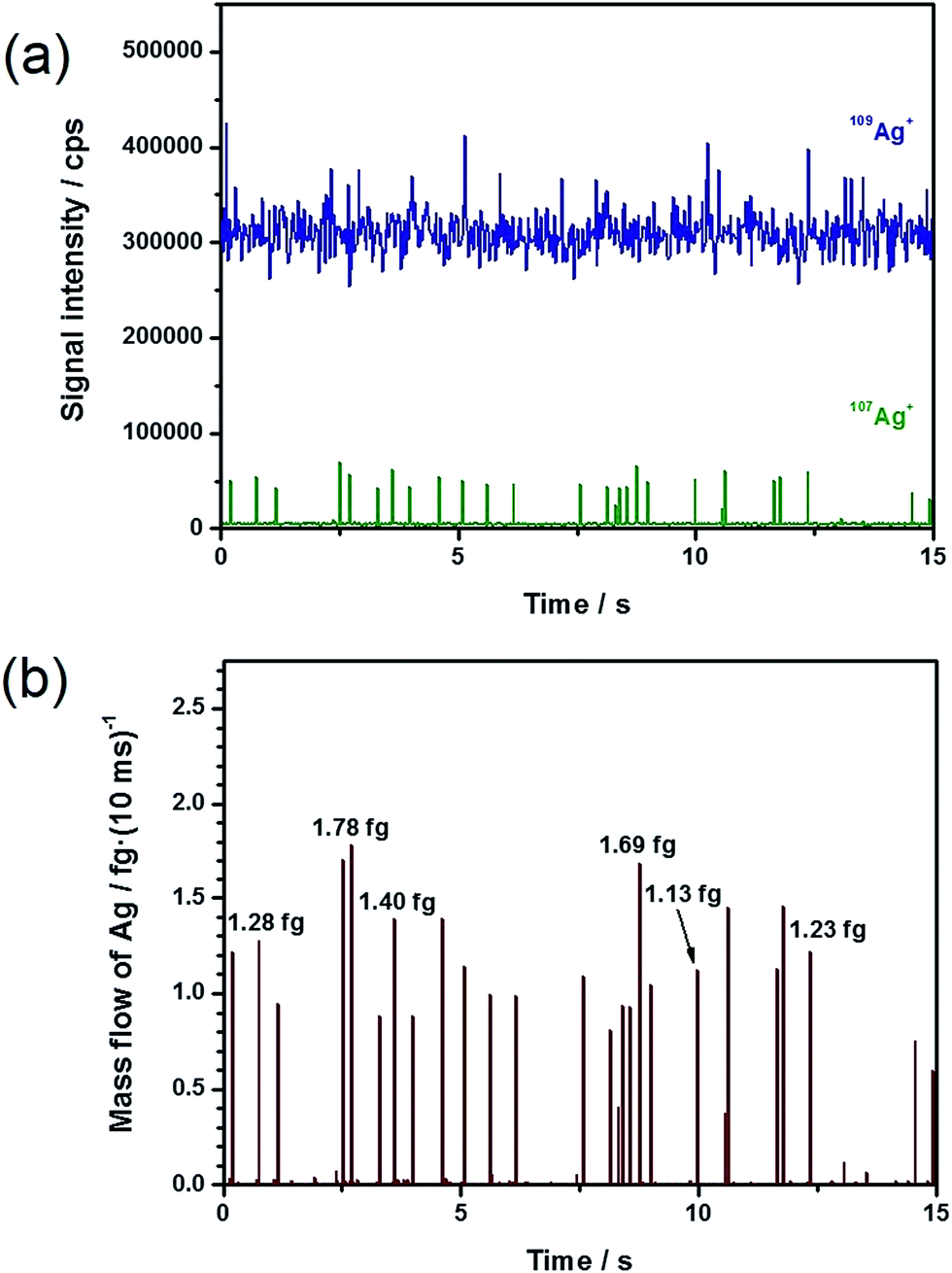 | ||
| Fig. 2 From intensity to mass flow. (a) Time-resolved scan of 20 ng L−1 60 nm Ag NPs with 1 μg L−1 109Ag+ solution, and (b) resulting mass flow. | ||
Through integration of the signals, the Ag particle masses were directly accessible ranging from approximately 0.81 to 1.78 fg and corresponding to particle sizes between 53 and 70 nm based on the spherical particle assumption. This represents an enormous advantage over the conventional spICP-MS experiments, in which the signal intensities need to be converted into masses by an external calibration. Exemplarily, the obtained masses for individual Ag NPs are marked in Fig. 2b to show the feasibility of the suggested approach. Under the assumption of spherical particles, the particle diameters d were calculated using eqn (6).
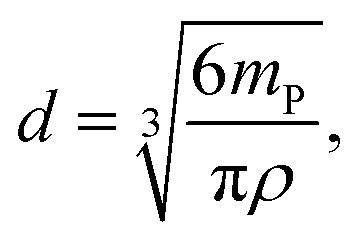 | (6) |
The average size was determined to be 62 nm (>1000 particle events).
The next step was to evaluate the quality of the results in comparison to conventional spICP-MS. For this purpose, Ag NPs with different nominal sizes between 30 and 80 nm were analyzed applying the experimental conditions given in Table 1. During each experiment, minimum 1000 events were recorded. For the IDA analyses, different concentrations of 109Ag+ solution (between 0.1 and 4.0 μg L−1) were added to the Ag NP suspensions. Table 2 summarizes the obtained results. The averages and corresponding standard deviations for the NPs analyzed by IDA with different spike concentrations were as follows: 28.0 nm ± 2.8 nm for NC30, 44.7 nm ± 1.5 nm for NC40, 63.0 ± 4.7 nm for NC60, and 83.2 nm ± 7.8 nm for NC80. It is assumed based on the present data in Table 2 that the determined particle sizes did not depend on the added spike concentration. Further experiments are necessary to fully verify this finding. However, our result hints at the spike concentration being almost independent of the particle sizes present in the sample. Thus, no preliminary information about the size of the NPs would be needed. Secondly, the results obtained with the suggested approach using IDA–spICP-MS were in good agreement with those of the conventional approach. Both the average particle sizes (Table 2) and the histograms (results not shown here) did not show significant differences.
| Parameter | NC30 [nm] | NC40 [nm] | NC60 [nm] | NC80 [nm] |
|---|---|---|---|---|
| Particle size (spICP-MS) | 24.0 | 39.9 | 55.4 | 77.0 |
| Particle size (IDA–spICP-MS): 0.1 μg L−1 109Ag+ | 30.5 | — | — | — |
| Particle size (IDA–spICP-MS): 0.5 μg L−1 109Ag+ | 27.6 | 46.3 | 68.2 | — |
| Particle size (IDA–spICP-MS): 1.0 μg L−1 109Ag+ | 26.0 | 44.2 | 61.7 | 88.9 |
| Particle size (IDA–spICP-MS): 2.0 μg L−1 109Ag+ | — | 43.5 | 59.0 | 74.4 |
| Particle size (IDA–spICP-MS): 4.0 μg L−1 109Ag+ | — | — | — | 86.4 |
In addition to the particle sizes, the particle number concentrations were determined (Table 3). For this, the number of events obtained for the isotope 107Ag was ascertained for the measuring period and with the knowledge of the sample uptake rate converted into the particle number concentration.23 In the case of IDA–spICP-MS, the needed time for an entire measurement cycle (22 ms) simply had to be related to the dwell time for the isotope 107Ag (10 ms) in which the particle events were observed. As demonstrated in Table 3, the resulting values were in good agreement with those for the conventional spICP-MS approach.
| Parameter | NC40 | NC60 | NC80 |
|---|---|---|---|
| a Particle number concentrations (particles per L) of the original suspensions. For the determination of the particle number concentrations, the suspensions were diluted to particle number concentrations of approximately 107 L−1. | |||
| Particle number concentration (spICP-MS) | 2.6 × 1013 | 1.2 × 1013 | 7.1 × 1012 |
| Particle number concentration (IDA–spICP-MS): 0.5 μg L−1 109Ag+ | 2.9 × 1013 | 1.4 × 1013 | — |
| Particle number concentration (IDA–spICP-MS): 1.0 μg L−1 109Ag+ | 2.6 × 1013 | 1.4 × 1013 | 7.3 × 1012 |
| Particle number concentration (IDA–spICP-MS): 2.0 μg L−1 109Ag+ | 2.9 × 1013 | 1.4 × 1013 | 7.3 × 1012 |
| Particle number concentration (IDA–spICP-MS): 4.0 μg L−1 109Ag+ | — | — | 6.2 × 1012 |
In summary, the chosen experimental conditions permitted the analysis of Ag NPs with equivalent results obtainable by conventional spICP-MS. The IDA approach used here comprised the advantage of direct access to the particle mass (fraction) of differently sized Ag NPs.
Matrix influence in spICP-MS
A general benefit of IDA is that the matrix effects resulting in a change of detection sensitivity can be successfully eliminated.36,37 Besides that, the instrumental drifts during the day are effectively compensated. These observations might also affect quantitative results during the use of spICP-MS with external calibration.22 Therefore, the strong matrix effect of 30 mmol L−1 NaCl (corresponding roughly to the NaCl concentration in 20-fold diluted sea water) was used as an example to explore its influence on the detection and the results of IDA–spICP-MS. Fig. 3 demonstrates the signal suppression of a Ag NP suspension containing the NaCl solution (Fig. 3b) in comparison to that of Milli-Q water (Fig. 3a). A decrease of nearly 70% of the 109Ag+ signal intensity was observed. Thanks to the concept of IDA, the matrix had the same impact on the depleted isotope including the particle events. The average intensity for the Ag NP events declined from approximately 60000 to 20000 cps. As an example, Fig. 4 shows the particle size histograms for NC60 NPs obtained under the chosen conditions. Although the histograms for IDA–spICP-MS were similar in terms of size maximum and distribution, the matrix showed strong influence on the results obtained by spICP-MS with external calibration. A significant shift from about 55 to 40 nm was observed considering the most abundant particle size. This tendency was confirmed by the analysis of further NP suspensions (Table 4). The data for spICP-MS with external calibration clearly showed a significant decrease (between 18 and 32%) in particle size after the addition of NaCl. These observations reflected the importance and influence of matrix effects in spICP-MS for obtaining reliable results.22
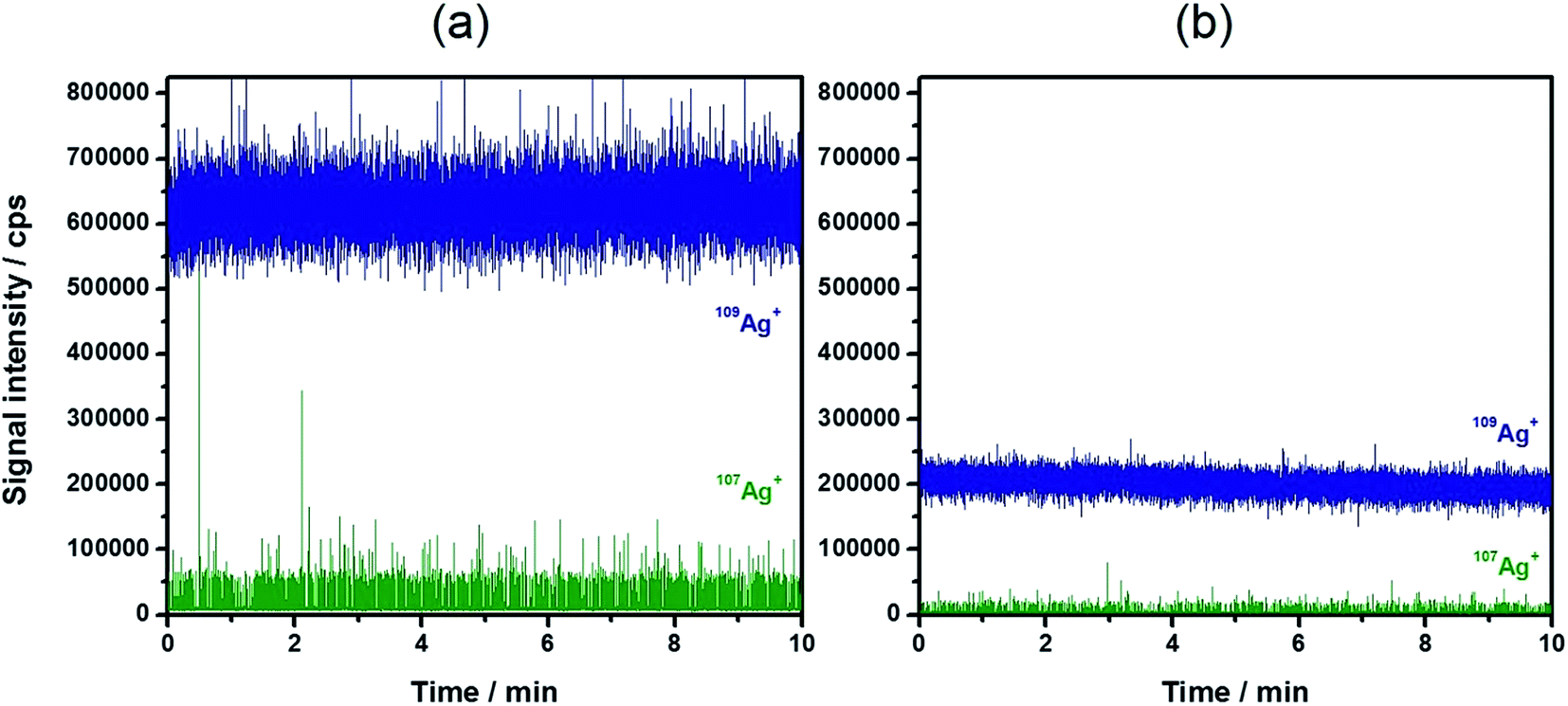 | ||
| Fig. 3 Typical time-resolved scan of 20 ng L−1 60 nm Ag NPs with 2 μg L−1 109Ag+ solution (a) in water and (b) in 30 mmol L−1 NaCl. | ||
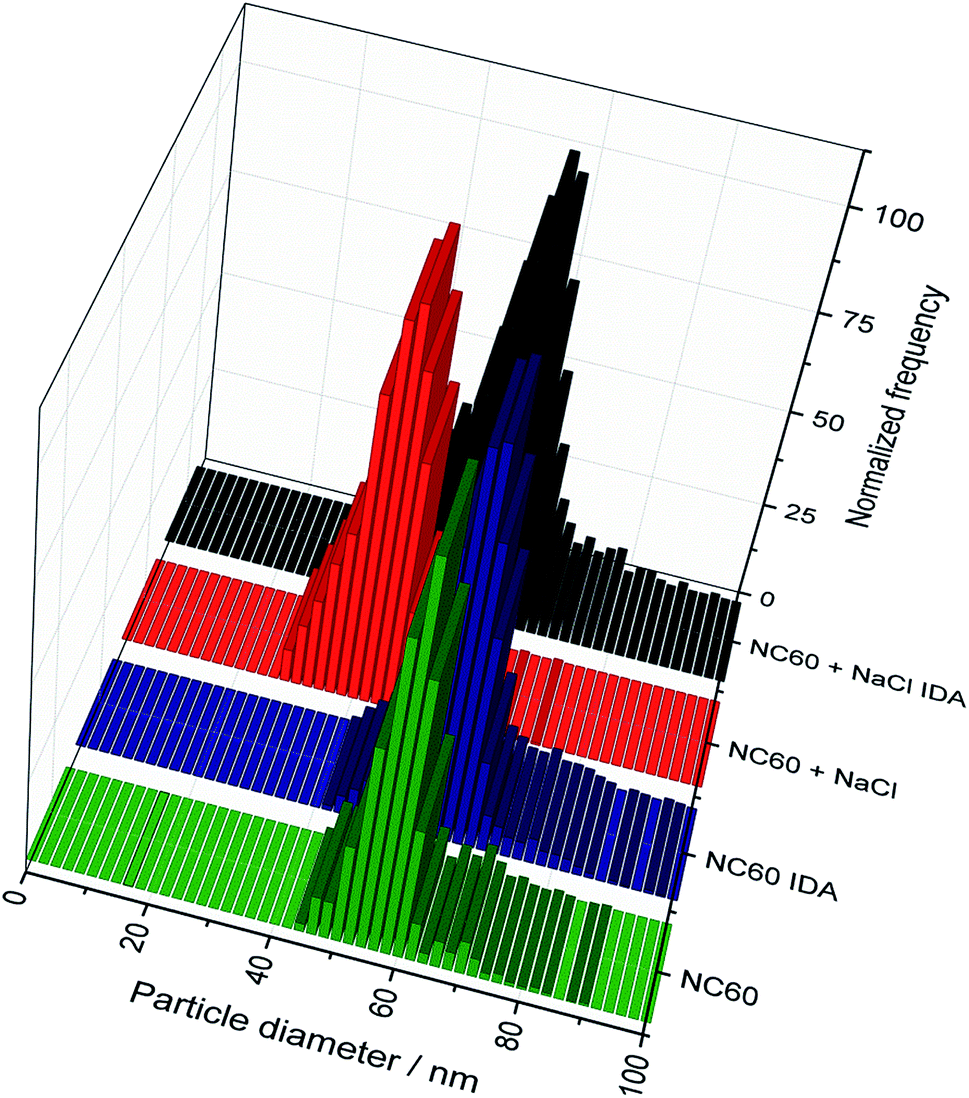 | ||
| Fig. 4 Comparison of the histograms of the 60 nm Ag NPs with and without the addition of NaCl for the conventional and the IDA–spICP-MS approaches. | ||
| NC40 [nm] | NC60 [nm] | NC80 [nm] | |
|---|---|---|---|
| a Average particle size (median particle size). | |||
| spICP-MS in water | 39.9 (39.9)a | 55.4 (54.7) | 77.0 (75.2) |
| spICP-MS in 30 mmol L−1 NaCl | 32.5 (32.6) | 40.4 (40.3) | 52.1 (51.7) |
| IDA–spICP-MS in water | 44.2 (43.1) | 59.0 (58.3) | 86.4 (82.9) |
| IDA–spICP-MS in 30 mmol L−1 NaCl | 40.4 (38.3) | 54.8 (54.6) | 78.1 (76.3) |
In contrast, the values obtained with IDA–spICP-MS were not affected by the signal suppressions induced by the matrix. However, the slight decrease (about 10%) of the obtained particle sizes could be attributed to the following fact: as isotope dilution analysis can effectively compensate for any signal suppressions, the observed tendency might be related to a matrix-induced change of the transport efficiency of the solutions. The determined rates with and without matrix were 6.1 and 7.3%, respectively. The latter value is higher than the previously determined transport efficiency of 6.3% due to a replacement of the nebulizer and tubing. Additionally, it was noticed that the intensity of the NP is more influenced by the signal depression by the matrix than the intensity of the spike. The latter results in lower diameters. For the NC60 NPs the average spike intensities of 628478 cps without and 201610 cps with the matrix, resulting in a ratio of 3.1, were found. The NPs showed the intensities of 55576 cps without and 16901 cps with the matrix at the peak maximum giving a ratio of 3.3. This is in good agreement with the NC40 and NC80 NPs for which the ratios of 2.2 (NC40, spike), 2.5 (NC40, NP), 3.5 (NC80, spike) and 3.9 (NC80, NP) were detected. The same transport and atomization efficiencies for both the NP and the spike are fundamental for this and the conventional spICP-MS approach. However, our results suggest that this might not always be the case. These two factors were already thoroughly investigated for slurry sampling.38 O'Hanlon et al. showed for example that the particles in the slurry with a size of 3–4 μm can have a smaller transport efficiency than a solution of the same element.39 Therefore, parallels between single particle and slurry sampling analyses could be conceivable and should be further investigated. Unfortunately, IDA–spICP-MS cannot compensate for this noticed effect, but it shows again the importance of taking into account any matrix effects in spICP-MS. Further studies including the signal depression and transport efficiencies of the spikes and the NPs are necessary to comprehend the finding. These could for example include the investigation of the ionization regions of aqueous standard solutions and NPs in the plasma.
Conclusions
In this work, we present the combination of spICP-MS and IDA for the analysis of Ag NPs using an ICP-QMS. This method is based on the addition of an isotopically enriched 109Ag+ to the NP suspensions. With the application of a modified version of the post-column isotope dilution equation, equivalent results were obtained for the determination of particle sizes of Ag NPs between 30 and 80 nm in comparison to the conventional spICP-MS approach. The methodology presented herein was superior in terms of robustness for the analysis of samples containing a NaCl matrix as signal suppressions could be efficiently compensated. This finding is of great advantage when analyzing samples in unknown matrices – hence, where a matrix-matched external calibration is impossible. In addition to the work of Telgmann et al.,21 results on the particle size distribution and the particle number concentration were presented, again, being in good agreement with the conventional spICP-MS approach.In future, it is expected that IDA–spICP-MS will have an important impact on the analysis of metallic NPs and will be a valuable tool for routine analysis.
In particular in combination with ICP-MS instruments that allow the simultaneous detection of various isotopes, like ICP-ToF-MS,33 the possibilities include the simultaneous detection of NPs containing different elements without the necessity of external calibration. Challenges nowadays, however, include the instrument sensitivity as the signal intensities of particles with a nominal diameter of 30 nm or lower suspended in a NaCl matrix are close to the detection limit of the used ICP-QMS.
This combination is thought to become a very promising tool in complex matrices such as environmental or biological samples, which can potentially suppress the instrument sensitivity.
Acknowledgements
J. Bettmer kindly acknowledges the financial support of the Spanish MICINN (Spanish Ministry for Science and Innovation, grant number CTQ2011-23038) and a fellowship provided by BAM, Berlin, Germany.References
- European Commission, Commission Recommendation of 18 October 2011 on the definition of nanomaterial (2011/696/EU).
- P. Krystek, A. Ulrich, C. C. García, S. Manohar and R. Ritsema, J. Anal. At. Spectrom., 2011, 26, 1701–1721 RSC.
- J. F. Liu, J. B. Chao, R. Liu, Z. Q. Tan, Y. G. Yin, Y. Wu and G. B. Jiang, Anal. Chem., 2009, 81, 6496–6502 CrossRef CAS.
- R. Allabashi, W. Stach, A. de la Escosura-Muniz, L. Liste-Calleja and A. Merkoci, J. Nanopart. Res., 2009, 11, 2003–2011 CrossRef CAS.
- A. Helfrich, W. Brüchert and J. Bettmer, J. Anal. At. Spectrom., 2006, 21, 431–434 RSC.
- A. Helfrich and J. Bettmer, Int. J. Mass Spectrom., 2011, 307, 92–98 CrossRef CAS.
- B. Franze and C. Engelhard, Anal. Chem., 2014, 86, 5713–5720 CrossRef CAS PubMed.
- K. Tiede, A. B. A. Boxall, D. Tiede, S. P. Tear, H. Davide and H. Lewis, J. Anal. At. Spectrom., 2009, 24, 964–972 RSC.
- K. Tiede, A. B. A. Boxall, X. Wang, D. Gore, D. Tiede, M. Baxter, H. David, S. P. Tear and J. Lewis, J. Anal. At. Spectrom., 2010, 25, 1149–1154 RSC.
- J. Soto-Alvaredo, M. Montes-Bayón and J. Bettmer, Anal. Chem., 2013, 85, 1316–1321 CrossRef CAS PubMed.
- N. Fernández-Iglesias and J. Bettmer, Nanoscale, 2014, 6, 716–721 RSC.
- E. K. Lesher, J. F. Ranville and B. D. Honeyman, Environ. Sci. Technol., 2009, 43, 5403–5409 CrossRef CAS PubMed.
- E. Bolea, J. Jiménez-Lamana, F. Laborda and J. R. Castillo, Anal. Bioanal. Chem., 2011, 401, 2723–2732 CrossRef CAS PubMed.
- B. Schmidt, K. Loeschner, N. Hadrup, A. Mortensen, J. J. Sloth, C. Bender Koch and E. H. Larsen, Anal. Chem., 2011, 83, 2461–2468 CrossRef CAS PubMed.
- M. M. Pornwilard and A. Siripinyanond, J. Anal. At. Spectrom., 2014, 29, 1739–1752 RSC.
- B. Meermann, A.-L. Fabricius, L. Duester, F. Vanhaecke and T. Ternes, J. Anal. At. Spectrom., 2014, 29, 287–296 RSC.
- C. A. Sötebier, F. S. Bierkandt, S. Rades, N. Jakubowski, U. Panne and S. Weidner, J. Anal. At. Spectrom., 2015, 30, 2214–2222 RSC.
- B. Meermann, K. Wichmann, F. Lauer, F. Vanhaecke and T. A. Ternes, J. Anal. At. Spectrom., 2016, 31, 890–901 RSC.
- C. Degueldre and P.-Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2013, 28, 1220–1232 RSC.
- L. Telgmann, C. D. Metcalfe and H. Hintelmann, J. Anal. At. Spectrom., 2014, 29, 1265–1272 RSC.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, J. Anal. At. Spectrom., 2015, 30, 1274–1285 RSC.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- B. Franze, I. Strenge and C. Engelhard, J. Anal. At. Spectrom., 2012, 27, 1074–1083 RSC.
- G. Cornelis and M. Hassellöv, J. Anal. At. Spectrom., 2014, 29, 134–144 RSC.
- J. Tuoriniemi, G. Cornelis and M. Hassellöv, J. Anal. At. Spectrom., 2014, 29, 743–752 RSC.
- B. Ramkorun-Schmidt, S. A. Pergantis, D. Esteban-Fernández, N. Jakubowski and D. Günther, Anal. Chem., 2015, 87, 8687–8694 CrossRef CAS PubMed.
- J. Bettmer and J. Meija, Presentation on European Winter Conference on Plasma Spectrochemistry, Krakow, 2013, vol. 2013, pp. 10–15, Book of Abstracts, ISBN 978-83-934442-3-6 Search PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155 RSC.
- M. D. Montaño, H. R. Badiei, S. Bazargan and J. F. Ranville, Environ. Sci.: Nano, 2014, 1, 338–346 RSC.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- O. Borovinskaya, S. Gschwind, B. Hattendorf, M. Tanner and D. Günther, Anal. Chem., 2014, 86, 8142–8148 CrossRef CAS PubMed.
- L. Rottmann and K. G. Heumann, Fresenius. J. Anal. Chem., 1994, 350, 221–227 CrossRef CAS.
- L. Rottmann and K. G. Heumann, Anal. Chem., 1994, 66, 3709–3715 CrossRef CAS.
- P. Rodríguez-González, J. M. Marchante-Gayón, J. I. García-Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef.
- K. G. Heumann, Anal. Bioanal. Chem., 2004, 378, 318–329 CrossRef CAS PubMed.
- P. Goodall, M. E. Foulkes and L. Ebdon, Spectrochim. Acta, Part B, 1993, 48, 1563–1577 CrossRef.
- K. O’Hanlon, L. Ebdon and M. Foulkes, J. Anal. At. Spectrom., 1997, 12, 329–331 RSC.
Footnote |
† One entire measurement cycle was 22 ms (∼2 ms for the required mass jumps and 20 ms for monitoring the two isotopes 107Ag and 109Ag, 10 ms each). Taking into account that the particle events could be only detected in the depleted isotope (107Ag), the number of detectable NPs was equal to 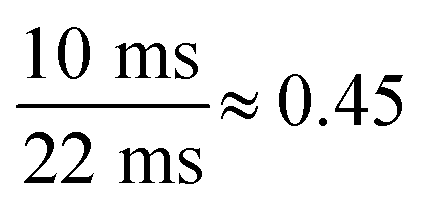 or ∼45%. or ∼45%. |
| This journal is © The Royal Society of Chemistry 2016 |