
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium stable isotope measurements by double spike MC-ICPMS
Timo
Hopp
*,
Mario
Fischer-Gödde
and
Thorsten
Kleine
Institut für Planetologie, University of Münster, 48149 Münster, Germany. E-mail: timo.hopp@wwu.de
First published on 24th May 2016
Abstract
We developed a new technique for precise measurements of ruthenium (Ru) stable isotope compositions by multiple-collector inductively coupled plasma mass spectrometry (MC-ICPMS). Instrumental mass bias and potential isotope fractionation induced during the chemical purification of Ru were corrected using a 98Ru–101Ru double spike added prior to sample digestion. The separation and purification of Ru from natural samples is achieved by cation exchange chromatography followed by distillation of Ru as volatile oxides. A series of analytical tests demonstrates that this method results in very pure Ru cuts and accurate and reproducible Ru isotope measurements. The Ru isotope results are expressed relative to an Alfa Aesar™ Ru standard solution as the permil deviations of the 102Ru/99Ru ratio (δ102/99Ru). The external reproducibility (2 s.d.) of the entire analytical procedure (including sample digestion, chemical purification and isotope measurement) that has been determined using several doped terrestrial rock standards is 0.05‰ for δ102/99Ru. We obtained Ru stable isotope data for three different Ru standard solutions and five chromitite samples from two different localities in the Shetland Ophiolite Complex and from the Bushveld complex. All three standard solutions are isotopically distinct with δ102/99Ru values up to ca. 0.9‰. The different chromitite samples also show different Ru stable isotope compositions with δ102/99Ru values between ca. 0.2‰ and ca. 0.7‰. These natural Ru stable isotope variations can be readily resolved using the 98Ru–101Ru double spike method presented here. Overall, these data show that Ru stable isotopes hold promise as a tracer of a wide range of geochemical and cosmochemical processes.
1 Introduction
Stable isotope variations have proven to be powerful and versatile tools to investigate the nature and conditions of a wide range of important geological processes.1–3 Until now, stable isotope studies mainly utilized major elements (e.g., Fe, Si) or elements that are enriched in the silicate portion of the Earth (e.g., Ge, Zn, Ti).4–8 In contrast, only a few studies have investigated the stable isotope compositions of siderophile elements.9–11 This is not surprising, given that these elements are typically depleted in the accessible portion of the Earth, making precise isotope measurements of natural samples difficult. Nevertheless, the stable isotope composition of siderophile elements has great potential for a wide range of applications in geo- and cosmochemistry.Ruthenium (Ru) is a promising element to investigate for stable isotope variations. It is a highly siderophile element (HSE) with seven naturally occurring isotopes (96Ru, 98Ru, 99Ru, 100Ru, 101Ru, 102Ru, 104Ru) and belongs to the platinum group elements (PGEs). The highly siderophile character combined with the large range of oxidation states (−2 to +8)12—the widest range observed for any element—make significant stable isotope fractionations likely. Owing to its strong tendency to partition into metal, Ru is almost quantitatively incorporated into the metallic core of planetary bodies. As such Ru is one of the rarest metals in the silicate portion of the Earth and other planetary bodies.13 Ruthenium mostly occurs as a metal phase or as an alloy with other PGEs, but is also incorporated into sulfides (e.g., laurite, pentlandite) and in chromites.14 Although Ru can exhibit a wide range in oxidation states, natural Ru occurs as Ru0 (in metals) and Ru4+ (in sulfides).14 The main terrestrial deposits of Ru and other PGEs are related to large intrusive complexes (e.g., Bushveld complex) and are often associated with chromitite deposits.15 In these PGE deposits Ru is enriched with a factor of up to 5000 compared to its abundance in the Earth's mantle (∼7 ppb).16,17 Such extreme enrichments may have led to mass-dependent isotope fractionations, which when documented may help to assess the processes involved in the genesis of the ore deposits.
While Ru is strongly depleted in the Earth's mantle, it is abundant in chondrites (∼400 ppb to ∼1 ppm Ru) and iron meteorites (∼2 to ∼30 ppm Ru).18–20 Most iron meteorites are thought to have formed by fractional crystallization of metallic magma, a process that may have induced an isotopic fractionation between solid and liquid metal.21 As such, Ru stable isotopes may be a useful tool to investigate the process of core crystallization and cooling in planetary bodies. Likewise, in chondritic meteorites Ru predominantly is hosted in refractory inclusions and FeNi metal. At least some of these components are thought to have formed by condensation and evaporation within the solar nebula and so the Ru stable isotope composition of chondrite components may shed new light on the nature of these processes.18 Finally, although the Earth's mantle is strongly depleted in Ru (and other HSE), the abundances of these elements are still higher than expected on the basis of experimentally determined metal–silicate partition coefficients.22 This overabundance is generally thought to reflect the addition of a ‘late veneer’ of primitive meteoritic material to the mantle after cessation of core formation.23,24 Thus, the investigation of mass-dependent Ru isotope variations, especially when combined with nucleosynthetic Ru isotope studies, may also help to assess the nature and origin of the late veneer.25
Despite the wide range of possible applications, there have been to our knowledge no previous attempts to determine Ru stable isotope compositions. Instead, previous Ru isotope studies were exclusively aimed at identifying nucleosynthetic Ru isotope anomalies in meteorites. Such mass-independent isotope anomalies arise through the heterogeneous distribution of the distinct products of the s-, r- and p-processes of stellar nucleosynthesis in the early solar nebula.26,27 In addition, anomalies in 98Ru and 99Ru could potentially arise through the decay of now-extinct Tc.28–30 However, evidence for live Tc in the early solar system and corresponding radiogenic Ru isotope anomalies remains elusive. Note that in studies of nucleosynthetic and radiogenic Ru isotope anomalies the Ru isotope data are internally normalized to a fixed 99Ru/101Ru ratio to correct for instrumental and natural mass-dependent isotope variations. As such, these data provide no information on the potential presence of mass-dependent Ru stable isotope variations.
To utilize Ru stable isotopes as a new tracer in the Earth and planetary sciences, we have developed analytical methods for the chemical purification of Ru from natural samples and the precise Ru isotope measurements by double spike multi-collector inductively coupled plasma mass spectrometry (MC-ICPMS). Here we present the chemical purification procedure for Ru, followed by a detailed description and evaluation of the Ru double spike method. After evaluating the precision and accuracy of this new method, we obtained Ru stable isotopic data for different commercially available standard solutions and five natural chromitite samples, with the ultimate goal to assess the magnitude of naturally occurring Ru stable isotope variations.
2 Design and preparation of the ruthenium double spike
The double spike technique is the method of choice for the precise measurements of mass-dependent isotope variations.31 The approach of this technique was developed already in the 1960s,32 but only recently this method has been applied to a wide range of elements, mostly in combination with MC-ICPMS.8–11,33–36 The advantage of the double spike technique over the conventional standard-sample method is that, with a single isotope measurement, the natural mass fractionation (i.e., the quantity we are interested in) can be distinguished from any mass-dependent isotope fractionation induced during chemical purification and mass spectrometry. This technique, therefore, is less susceptible to potential analytical artefacts induced by different sample matrices and incomplete chemical yields during the purification of the element of interest. However, in the reduction of double spike data it is commonly assumed that any isotopic difference between the sample and the standard is purely mass-dependent. Thus, for samples having mass-independent isotope anomalies, a separate isotope measurement on an unspiked aliquot is necessary to ultimately determine the true mass-dependent isotope fractionation. For instance, nucleosynthetic Ru isotope anomalies exist in meteorites26,27 and these, therefore, will have to be taken into account when determining mass-dependent Ru isotope variations in meteorites using a double spike. Note, however, that all samples investigated in the present study are terrestrial samples for which no nucleosynthetic isotope variations exist. Another potential source of mass-independent Ru isotope variations are chemical exchange reactions, which may have occurred in nature or during the chemical purification of Ru prior to isotope measurements. Such chemically induced mass-independent Ru isotope fractionations were observed during solvent extraction experiments using DC18C6 crown ether and are probably related to the nuclear field shift effect leading to fractionation between Ru isotopes with odd and even masses.37,38 However, a prior study on nucleosynthetic Ru anomalies in meteorites, which applied the same Ru purification method as used in the present study, did not find any evidence for nuclear field shift effects on measured Ru isotope abundances.27 For Ru isotopic data internally normalized to a fixed 99Ru/101Ru ratio, the nuclear field shift effect would lead to large apparent anomalies in 104Ru/101Ru, but with one exception all meteorites and terrestrial chromitites analysed so far have indistinguishable 104Ru/101Ru ratios.26,27,39Ruthenium has seven stable isotopes (Fig. 1) and so there are many different options for the design of a double spike. The double spike approach requires the use of four different isotopes, two of which are enriched in the double spike, whereas the other two isotopes are used for the inversion of the measured isotopic data to determine α, the natural mass fractionation factor. To select the optimal double spike it is useful to minimise the (theoretical) error on α; using this approach, Rudge et al. (2009) created a list of double spike compositions and their associated theoretical uncertainties on the determined α for all elements having more than four isotopes.31 This list is based on the isotopic compositions of individual spikes available from the Oak Ridge National Laboratory, USA. On this basis, the optimal Ru double spike would be a 98Ru–102Ru double spike, where 96Ru, 98Ru, 102Ru and 104Ru would be used in the inversion. However, this error assessment does not take into account potential difficulties in the measurement of particular isotopes, as for instance the possible presence of isobaric interferences. In the specific case of Ru, using the isotopes 96Ru, 98Ru, 102Ru and 104Ru is not ideal, because all four isotopes are affected by isobaric interferences from Mo, Zr and Pd (Fig. 1). A further complicating factor is that the cup configuration of the Neptune Plus MC-ICPMS at Münster does not allow the simultaneous measurement of Zr, Mo and Pd interference monitors. We, therefore, selected another double spike composition, namely a 98Ru–101Ru double spike with 98Ru, 99Ru 101Ru and 102Ru used in the inversion. Of note, our double spike setup does not use 96Ru and, hence, we do not need to measure a Zr interference monitor. Moreover, we use two Ru isotopes, 99Ru and 101Ru, that are not affected by isobaric interferences, and so overall potential isobaric interferences need to be considered only for 98Ru and 102Ru. However, given the low abundance of 102Pd (1.02%), the effect of Pd interferences is expected to be very small (Fig. 1).
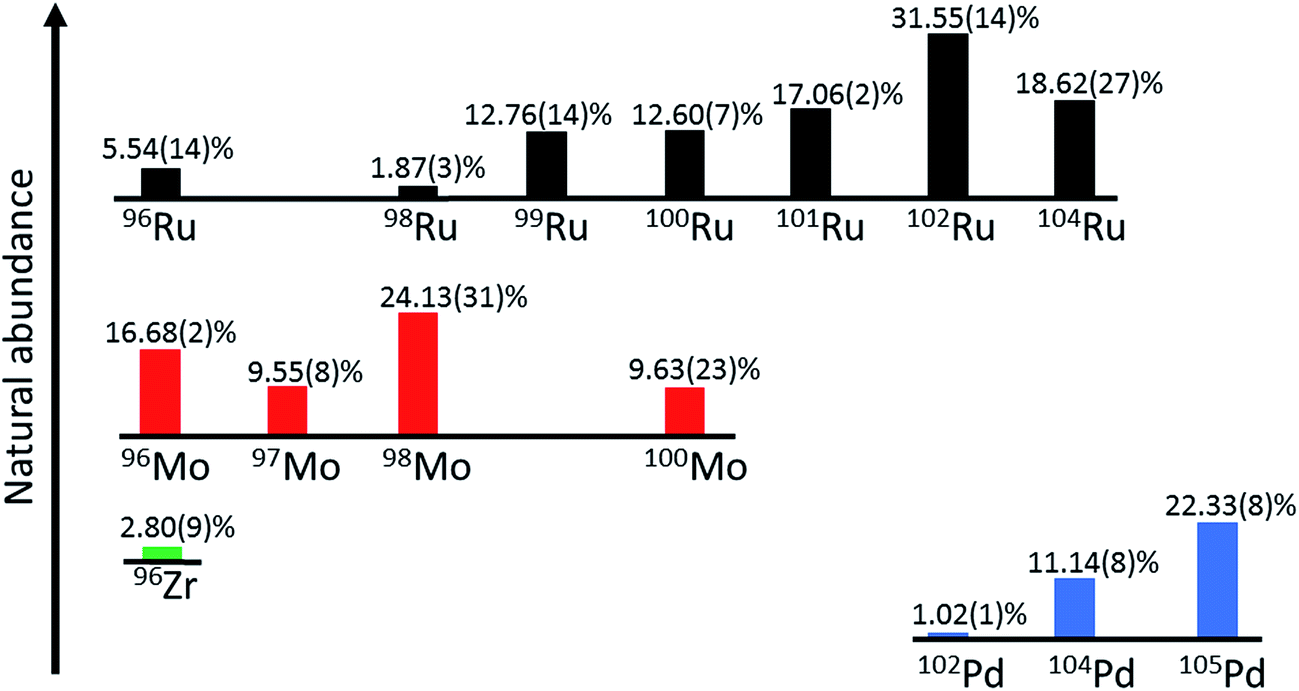 | ||
| Fig. 1 Schematic bar diagram of Ru stable isotopes and possible isobaric interferences. Ruthenium consists of seven stable isotopes (96Ru, 98Ru, 99Ru, 100Ru, 101Ru, 102Ru and 104Ru). The double spike method presented here uses only 98Ru, 99Ru, 101Ru and 102Ru and so only interferences on 98Ru and 102Ru may be significant. Isotopic abundances according to IUPAC.40 | ||
The 98Ru and 101Ru single spikes (batch no. 193101 and 193491) were obtained from the Oak Ridge National Laboratory, USA, as 10 mg each of fine powdered metal. The powders were carefully weighed (note that the actual weight of the spikes is not important because the concentrations of the individual spikes are determined later relative to Ru standard solutions of known concentrations) and transferred to Carius tubes for digestion. After addition of aqua regia (6 ml 37% HCl + 2 ml 69% HNO3), the spikes were digested inside the Carius tubes at 200 °C for 14 days. After complete digestion, the solutions were carefully transferred into 60 ml Savillex™ beakers and repeatedly evaporated to dryness using concentrated HCl, and finally re-dissolved in 6 M HCl. Aliquots of the two single spikes were then used to determine their Ru isotopic compositions and concentrations by isotope dilution using MC-ICPMS. After the calibration, portions of the single spikes were mixed together to yield a 98Ru–101Ru double spike containing the optimal proportions of the single spikes for minimizing the error propagation in the double spike inversion (47.87% 98Ru spike and 52.13% 101Ru spike; Fig. 2).31 The double spike was equilibrated for 48 h at 120 °C on a hot plate using 6 M HCl, dried down several times in concentrated HCl and finally re-dissolved in 6 M HCl. In the next step, small aliquot of the double spike were then used to determine its concentration (by isotope dilution) and isotopic composition. The calibration of the double spike is described in more detail below (Sect. 4.1).
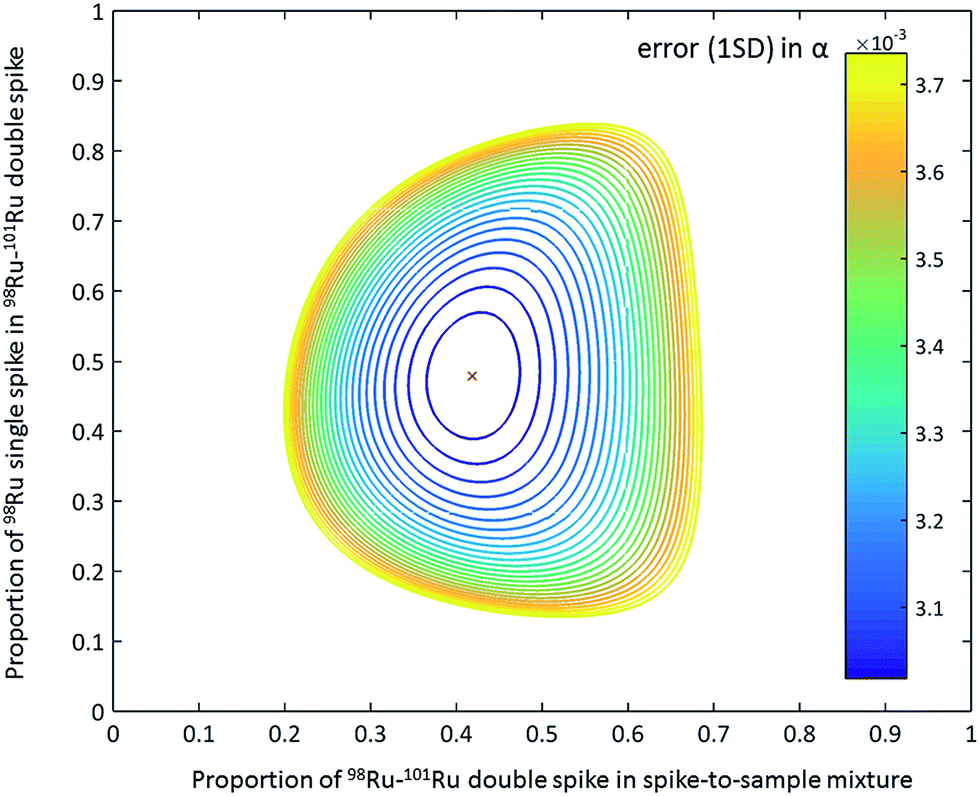 | ||
| Fig. 2 Contour plot of the error propagation as a function of proportion of 98Ru single spike in the 98Ru–101Ru double spike and the double spike-to-sample ratio.31 The ideal proportion of the 98Ru and 101Ru single spikes in the double spikes is 47.87% and 52.13%. The ideal double spike-to-sample ratio, using the measured isotopic composition of the 98Ru–101Ru double spike, is 0.43. The contour line express 1% increase of the error on the natural fractionation coefficient α relative to the minimum error.31 | ||
3 Materials and methods
3.1 Chemicals and standard solutions
All laboratory work was performed in a class-10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 clean room environment using class-10 laminar flow hoods in the Institut für Planetologie at the University of Münster. Pre-cleaned Savillex Teflon perfluoroalkoxy (PFA) vials and bottles were used for all samples and solutions processed in this study. Merck Millipore Emsure™ grade acids (69% HNO3, 37% HCl) were distilled twice using Savillex™ DST-1000 Acid Purification Systems and only Merck Suprapur™ H2O2 and an Alfa Aesar™ CrO3 powder (99.9% metal basis) were used as supplied without further purification. The chemicals were diluted with Merck Millipore Milli-Q™ water (18.2 MΩ cm) as necessary.
000 clean room environment using class-10 laminar flow hoods in the Institut für Planetologie at the University of Münster. Pre-cleaned Savillex Teflon perfluoroalkoxy (PFA) vials and bottles were used for all samples and solutions processed in this study. Merck Millipore Emsure™ grade acids (69% HNO3, 37% HCl) were distilled twice using Savillex™ DST-1000 Acid Purification Systems and only Merck Suprapur™ H2O2 and an Alfa Aesar™ CrO3 powder (99.9% metal basis) were used as supplied without further purification. The chemicals were diluted with Merck Millipore Milli-Q™ water (18.2 MΩ cm) as necessary.
There are no certified standard reference solutions for Ru available for purchase. We, therefore, used three different Ru standard solutions for this study: (1) an Alfa Aesar™ 1000 ppm Ru plasma standard in 20% HCl (Lot # 61300952 – 965 μg g−1 Ru), (2) a Merck Millipore Certipur™ 1000 ppm Ru ICP standard in 7% HCl (Lot # HC42234247 – 961 ± 8 μg g−1 Ru), and (3) an Inorganic Ventures™ 1000 ppm Ru standard solution in 10% HCl (Lot # G2-RU01069 – 976 ± 3 μg g−1 Ru).
3.2 Geological samples
To test the accuracy and reproducibility of the entire analytical method, different geochemical reference materials from the National Institute for Standards and Technology (NIST), USA, were used. These included the basalts BHVO-2 and BCR-2 as well as the peridotite UB-N. All three samples have very low Ru contents and to test our analytical method we, therefore, doped these samples with appropriate amounts of Ru from our standard solution prior to digestion of the samples.To investigate the magnitude and direction of natural Ru stable isotope variations, we applied our newly developed method to four chromitite samples (C-1, C-3, HG-1, HG-2) from the Shetland Ophiolite Complex (SOC).41 The SOC consists of serpentinised upper mantle material with a minimum age of 492 ± 3 Ma, which was emplaced during the closure of the Iapetus ocean at ∼470 Ma.42,43 Within the typical mantle lithologies (dunites, harzburgites, pyroxenites) discontinuous chromitite lenses of up to 2 m thickness occur.41 Such podiform chromitite deposits are common in ophiolite complexes and show anomalous relative platinum group element (PGE) abundances.44
We chose chromitite samples from two of the main deposits within the SOC (C-Cliff and HG-Harold's Grave).41 The PGE concentrations at Cliff show large variations in the total PGE concentrations (0.18 to 58 ppm) accompanied by enrichments in platinum (Pt), palladium (Pd) and rhodium (Rh).45 In contrast, the chromitites at Harold's Grave display smaller variations in the total PGE concentrations (4.77 to 14.7 ppm) and are relatively enriched in Ru and iridium (Ir) compared to Pt and Pd.45 The high enrichment and variability of the PGE abundances, in particular Ru, in these two localities could potentially have affected the Ru stable isotopic compositions of the samples, making these samples ideal to test our newly developed method and, at the same time, evaluate the magnitude of Ru isotope variations in natural samples.
In addition to these podiform PGE deposits we analysed the certified reference material SARM81, which is commercially available from MINTEK Corporation, South Africa. This reference material is a representative rock powder of the UG2 chromitite layer from the Bushveld complex. The Bushveld complex comprises the largest PGE and chromium (Cr) deposits in the world.46 In contrast to the small-scale, podiform chromitite deposits of the SOC, the Bushveld complex is a large-scale stratiform PGE deposit.46 These different formation processes could have potentially led to Ru isotopic variations between podiform and stratiform chromitite deposits.
3.3 Sample digestion and separation of ruthenium
All samples were weighed using Al foil and then transferred into Carius tubes (CT) using a burette funnel. An appropriate amount of the 98Ru–101Ru double spike was then weighed into a 7 ml Savillex PFA beaker and together with 7.5 ml reverse aqua regia (5 ml HNO3 + 2.5 ml HCl) also transferred into the CT. The burette funnel was carefully rinsed with the acid to ensure quantitative transfer of both the sample and spike into the Carius tube. The Carius tube was sealed using a gas flame and the sample-double spike mixture was then dissolved and equilibrated at 220 °C for 48 h.47 In most natural samples, Ru like other highly siderophile elements (HSE) is quantitatively incorporated into metals and sulphides, which are completely dissolved by the CT digestion. Thus, although silicates are not fully dissolved, the entire sample Ru is released by the CT digestion.After digestion the sample solutions were transferred from the Carius tubes into 50 ml centrifuge tubes and centrifuged for 20 min to remove silicate particles from the solution. The solutions were then pipetted into 60 ml Savillex™ Teflon beakers and evaporated at 100 °C on a hotplate. After drying, the samples were re-dissolved in 10 ml 6 M HCl and evaporated at 120 °C. This procedure was repeated two or three times to remove any residual HNO3. The samples were finally dissolved in 10 ml 0.2 M HCl and placed on a hot plate at 120 °C overnight. After cooling, the samples were loaded onto columns filled with 10 ml BioRad™ AG50W-X8 (100–200 mesh) cation exchange resin. The resin was batch cleaned in a PFA bottle using 6 M HCl, 6 M HCl-1 M HF and Milli-Q™ water. Prior loading of sample solutions, resin and columns were further cleaned using 40 ml 6 M HCl. The resin was then backwashed with Milli-Q™ water and finally conditioned with 20 ml 0.2 M HCl. On the cation exchange resin, Ru (and other elements like Re, Ir, Pt, Rh, Pd and Mo) is not absorbed because it forms negatively charged chloro-complexes, whereas most major elements (Fe, Mg, etc.) are retained by the resin.17,18,27 Thus, the cation exchange resin provides an effective means of separating HSE from the major sample matrix. The samples were loaded onto the column in two steps (2.5 ml and then 7.5 ml) and the columns were rinsed with an additional 6.5 ml of 0.2 M HCl. Ruthenium and other HSE were collected with the 7.5 ml loading and the 6.5 ml rinsing step. The total recovery of Ru from this column is >95%, as determined using a mixed HSE standard solution containing typical matrix elements. For natural samples the yield could in principle be somewhat lower, because Ru may occur in different oxidation states and, hence, may show different behaviour on the column. However, this effect is minimised by the repeated treatment of the samples with 6 M HCl enhancing the formation of Ru chloro-complexes. The Ru cuts from the cation exchange column were dried and, re-dissolved in one drop of concentrated HCl and two drops of concentrated HNO3 and dried down again at 80 °C.27 This step was necessary to destroy organic compounds from the cation resin and increase the Ru yields during the subsequent distillation.
The Ru fractions from the cation exchange column were further purified using a PFA distillation unit (Fig. 3), which consists of four 30 ml and four 20 ml Savillex™ Teflon beakers that are sealed with 33 mm impinge closures.27 A peristaltic pump is connected to the 30 ml beakers (oxidation beaker), which are placed on a hotplate and are connected to the 20 ml beakers (reduction beaker) with four PFA tubes. The Ru cuts from the cation column were re-dissolved in 8 ml of a 1:1 H2SO4–H2O mixture and transferred into the oxidation beakers, to which 3.6 ml of an aqueous CrO3 solution was also added.27,29 The oxidation beakers are placed onto a hot plate and heated to 70 °C, 80 °C and 90 °C in intervals of 2 h each. In these beakers, Ru is oxidised to volatile RuO4 and transferred by an air flow into the reduction beaker, which is filled with 10 ml of 3% H2O2, ultimately resulting in the reduction of the Ru to RuO2 (Fig. 3). The only other element that is oxidised alongside Ru is Os in the form of OsO4. However, the volatile Os is not reduced by H2O2, and so the resulting Ru cut trapped in the H2O2 is very pure.48 After distillation, the Ru cut is dried slowly at 80 °C and then re-dissolved in 2 ml of 0.28 M HNO3. To remove all residual H2O2, the sample solutions were transferred into 7 ml Savillex™ beakers and dried down again at 80 °C. The samples were then dissolved in 0.5 ml 0.28 M HNO3 for the Ru isotope measurements.
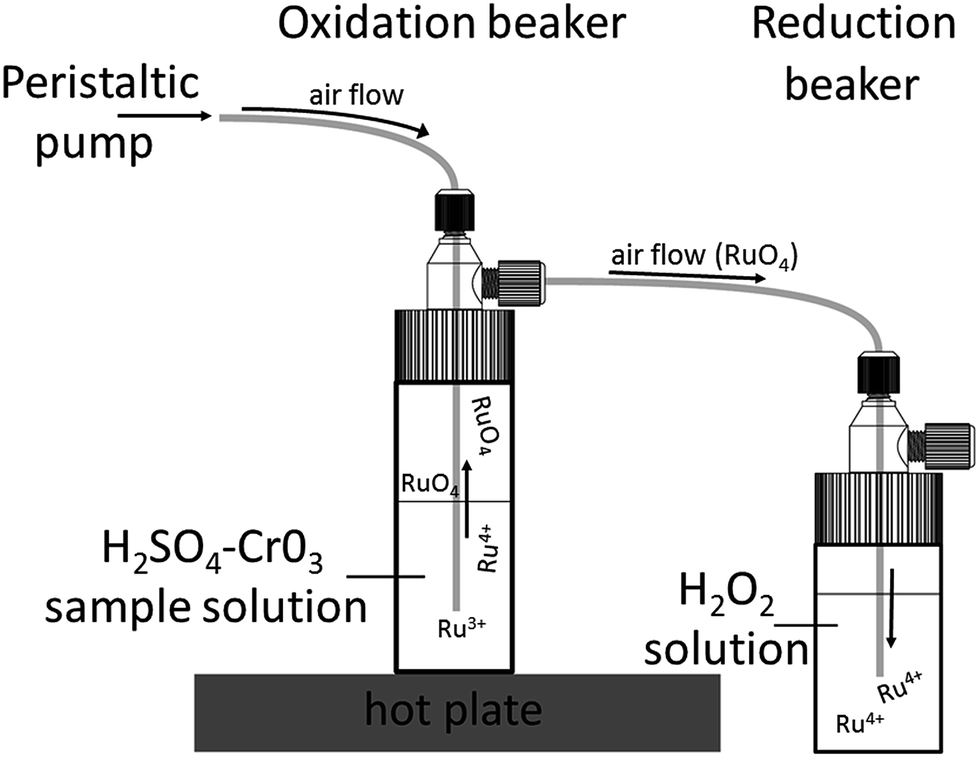 | ||
| Fig. 3 Schematic drawing of the PFA distillation unit. This unit consists of impinge vials that are connected via PFA tubing. The sample is placed inside the oxidation beaker with a solution of H2SO4–CrO3 and heated on a hot plate. The oxidised Ru forms volatile RuO4, which is transported via an airflow into the reduction beaker in which Ru is reduced again to RuO2. Drawing reproduced with permission from Savillex Corporation. | ||
The total procedural blank, including the digestion of the samples and the separation of Ru, is 41 ± 17 pg (1 s.d., n = 3) and, hence, insignificant given that more than 100 ng of Ru was analysed for each sample. The yield of the entire procedure is between 60 and 95%, as determined by distillation of pure Ru standard solutions and by processing doped terrestrial rock standards (Fig. 5). The lower yields most likely reflect Ru losses during the distillation or during evaporation of the samples in oxidising acids. The yields of the distillation may vary because different sample matrices may affect the efficiency with which Ru is oxidised; however, this effect is difficult to predict. Either way, because the double spike is added before digestion of the samples and spike–sample equilibration is achieved inside sealed Carius tubes, the loss of some Ru does not compromise the accuracy of the Ru isotopic data. This is evident from our tests using doped terrestrial standard rock materials, which, although they had variable yields, all gave the Ru isotope composition of the standard used for the doping (see Sect. 4.2 below).
3.4 Mass spectrometry and data reduction
The Ru stable isotope measurements were conducted using the ThermoScientific™ Neptune Plus MC-ICP-MS at the Institut für Planetologie at the University of Münster. The operating and measurement conditions of this instrument are summarised in Table 1. For the isotope measurements, the samples were dissolved in 0.28 M HNO3 and introduced into the mass spectrometer using an ESI Apex-IR™ desolvator coupled with a MicroMist™ glass nebuliser. The formation of oxides was minimised through the addition of N2 to the sample gases, resulting in oxide formation rates—measured as Ce/CeO—of <1%. Sample and standard solutions were typically measured at concentrations of 200 ppb Ru; the resulting total ion beam intensity was 2 × 10−10 A at an uptake rate of ∼100 μl min−1, corresponding to a sensitivity of ∼100 V ppm−1. During the Ru isotope measurements ion beams at 96Ru, 97Mo, 98Ru, 99Ru, 100Ru, 101Ru, 102Ru, 104Ru and 105Pd were simultaneously collected in a single cycle using Faraday cups connected to amplifiers with 1011 Ω feedback resistors for Ru isotopes and 1012 Ω feedback resistors for Mo and Pd isotopes. Each measurement comprised 50 × 4.2 s integrations of the ion beams and consumed ∼90 ng of Ru. Baselines were measured on peak for 40 × 4.2 s on a solution blank prior to each sample or standard analysis (Table 1). Between two successive analyses a total washout time of ∼30 min was necessary to clean the introduction system and reduce background levels to ≪1 mV for all Ru isotopes.| Instrument operating conditions | Neptune Plus (MS) |
|---|---|
| RF power | 1300 W |
| Plasma gas flow rate | 16 L min−1 |
| Interface cones – sampler | Nickel |
| Interface cones – skimmer | Nickel |
| Acceleration voltage | 10000 V |
| Instrumental resolution | Low resolution mode |
| Mass analyser pressure | 5–9 × 10−9 mbar |
| Detector | 9 Faraday detectors; all Pt |
| Sample introduction system | Apex IR |
| Sample uptake rate | ∼100 μl min−1 |
|
|
| Measurement parameters | |
| Solution concentration | 200 ppb |
| Typical sensitivity | 100 V ppm−1 |
| Sample measurement time | 50 × 4.2 s integrations |
| Washout time | 30 min |
| Background measurement time | 40 × 4.2 s integrations |
The measured raw datasets were processed off-line using the double spike toolbox of Rudge et al. (2009),31 which employs MATLAB's non-linear equation solving routine for the iteration. Interference corrections require the prior mass bias correction of the isotope ratios involved, but for spiked samples the magnitude of the instrumental mass fractionation factor β cannot be measured directly. We, therefore, first processed the raw isotope data using the double spike toolbox to obtain a first estimate of β; this value was then used for the mass bias correction of measured isotope ratios, followed by an interference correction of isotope ratios affected by isobaric interferences from Mo or Pd isotopes. The mass bias correction was then removed from the interference-corrected isotope ratios to obtain interference-corrected raw beams. These raw beams were then used in the double spike inversion, which returns a new estimate of β. This procedure is repeated until the β values converge, which typically is the case after two iterations. This method has been tested by the measurement of spiked Ru standard solutions doped with varying amounts of Pd and Mo (see Sect. 4.2 below).
The Ru stable isotope results are reported as the relative deviations from the composition of the Alfa Aesar standard solution as δ102/99Ru:
| δ102/99Ru = (102/99Rusample/102/99Rustandard − 1) × 1000. |
4 Results and discussion
4.1 Calibration of the ruthenium double spike
For the preparation and calibration of the 98Ru–101Ru double spike, the concentrations and isotopic compositions of the single 98Ru and 101Ru spikes as well as the final 98Ru–101Ru double spike were determined. All spike-standard mixtures used in the calibration were equilibrated in closed Savillex beakers with 6 M HCl on a hot plate at 120 °C. The use of 6 M HCl suppressed the formation of volatile Ru oxides, which could have been lost before spike-standard equilibration was achieved. The good agreement of the δ102/99Ru values determined for different standard-double-spike mixtures (see below and Fig. 4) demonstrates that complete spike-standard equilibration was obtained prior to any potential loss of volatile Ru oxides from the mixtures. | ||
| Fig. 4 (a) Measured δ102/99Ru as a function of different sample–spike mixtures. The proportion of the mixtures is expressed as deviation from the optimal amount of spike. Each point is the mean of four measurements with given error (2 s.d.). The measured δ102/99Ru is insensitive to the sample–spike ratio and even largely underspiked or overspiked samples provide accurate results. The grey horizontal bar indicates the mean δ102/99Ru of all measured sample–spike mixtures with its associated uncertainty of ±0.04‰ (2 s.d.). (b) Long-term reproducibility of the δ102/99Ru measurements of the spiked Alfa Aesar standard solution over a period of one year. Measurements are from seven different sessions (Nov. 2014–Nov. 2015). The grey area represents the long-term reproducibility of spiked solution standards of ±0.04‰ (2 s.d.). | ||
The Ru concentrations of the single and the double spikes were determined by isotope dilution using three different standard solutions (Alfa Aesar, Merck Millipore Certipur, Inorganic Ventures; see Sect. 3.1). The determined concentrations of the 98Ru and 101Ru single spikes using three different sample–spike ratios for each of the three standard solutions agree within <1% (2 s.d.), indicating that the given Ru concentrations of the standard solutions are accurate at the ∼1% level. The concentration of the double spike was determined using three different mixtures of the double spike and the Alfa Aesar standard solution. Additionally, one standard-spike mixture each of the other two standard solutions were also prepared. The Ru concentration of the 98Ru–101Ru double spike stock solution obtained from each of the five spike-standard mixtures agree with each other and average at 68.53 ± 0.40 ppm (2 s.d.).
The Ru isotopic compositions measured for the Alfa Aesar™ Ru standard solution, the individual spikes and the 98Ru–101Ru double spike are summarised in Table 2. In this table, published or recommended isotopic compositions for these materials are also given for comparison. In the absence of a certified standard solution for Ru, we chose the Alfa Aesar™ standard solution as the reference composition for the double spike measurements, which was used for the determination of the isotopic compositions of the 98Ru and 101Ru single spikes and the 98Ru–101Ru double spike (Table 2). The 101Ru/99Ru ratio measured for the standard before and after each spike analysis was used for the correction of instrumental mass fractionation of the measured isotope ratios of the spike.
| 96Ru | 98Ru | 99Ru | 100Ru | 101Ru | 102Ru | 104Ru | Atomic wt [amu] | |
|---|---|---|---|---|---|---|---|---|
| a Uncertainties are given in parentheses and refer to last significant digits. | ||||||||
| Natural Ru40 | 5.54 (14) | 1.87 (3) | 12.76 (14) | 12.60 (7) | 17.06 (2) | 31.55 (14) | 18.62 (27) | 101.07 (2) |
| Alfa Aesar Ru [this study] | 5.5529 (9) | 1.8797 (12) | 12.7646 (7) | 12.6002 (2) | 17.0661 (9) | 31.5390 (17) | 18.5977 (23) | 101.06314 (19) |
| 98Ru spike31 | 0.56 | 89.34 | 3.61 | 1.7 | 1.71 | 2.17 | 0.9 | 98.14650 |
| 98Ru spike [this study] | 0.5350 (11) | 89.4393 (4) | 3.6880 (7) | 1.6801 (12) | 1.6515 (14) | 2.1410 (5) | 0.8649 (9) | 98.15215 (7) |
| 101Ru spike31 | 0.07 | 0.07 | 0.15 | 0.32 | 97.82 | 1.35 | 0.21 | 100.9035 |
| 101Ru spike [this study] | 0.0309 (21) | 0.0171 (24) | 0.1582 (9) | 0.3243 (7) | 97.7937 (2) | 1.44966 (7) | 0.2260 (11) | 100.91836 (12) |
| Opt. 98Ru–101Ru DS31 | 0.30 | 42.78 | 1.81 | 0.98 | 51.85 | 1.74 | 0.54 | 99.59463 |
| 98Ru–101Ru DS [this study] | 0.2750 (2) | 43.3635 (16) | 1.8722 (3) | 0.9844 (4) | 51.1766 (14) | 1.7905 (9) | 0.5379 (5) | 99.57753 (5) |
To test the accuracy of the double spike calibration we analysed nine Alfa Aesar standard-double spike mixtures covering a wide range of spike-to-sample ratios of between 0.2 and 0.7. The optimal spike-to-sample ratio for minimizing the error propagation in the double spike inversion is ∼0.43 (Fig. 2).31 The δ102/99Ru values obtained for the different spike-standard mixtures all agree within 0.04‰ (2 s.d.) (Fig. 4a). Of note there is no dependence of the measured δ102/99Ru from the spike-to-sample ratio, as would be expected if the isotopic composition measured for the double spike would deviate from its true value. The constant δ102/99Ru values obtained over a wide range of spike-to-sample ratios, therefore, indicates that the calibration of the double spike is accurate. The smallest scatter on measured δ102/99Ru was obtained for spike-to-sample ratios between 0.35 and 0.55 and all samples analysed in the present study were, therefore, spiked such as to yield spike-to-sample ratios between 0.40 and 0.45 (Fig. 4a).
4.2 Reproducibility of ruthenium double spike measurements
The internal precision (2 s.e.) of each individual Ru double-spike measurement of standards or samples is ca. ±0.02‰ for δ102/99Ru. However, the δ102/99Ru values obtained for the spiked Alfa Aesar standard slightly vary between different sessions and not always scatter around zero. These small offsets (<0.08 δ102/99Ru) are probably caused by slightly different tuning settings of the MC-ICPMS or non-exponential mass fractionation effects not accounted for by the double spike correction. To account for these small offsets between individual sessions, we employed a standard-sample bracketing technique, in which the average composition measured for the Alfa Aesar standard during one session, αMeanAlfa Aesar, is subtracted from each sample analysis in the same session:| δ102/99Rusample = −1000 × (αsample − αMeanAlfa Aesar) × ln(m102/m99), |
The long-term reproducibility of the optimally spiked Ru standard solution does not account for potential effects induced by the chemical purification of Ru and by the presence of matrix elements in the analysed Ru fraction. To evaluate such effects and, hence, to arrive at a more appropriate estimate of the external reproducibility of the Ru double spike method, we processed the rock standards UB-N, BHVO-2 and BCR-2, which represent typical rock matrices, through the entire analytical procedure (Carius tube digestion, cation exchange chromatography, distillation). These three samples have very low Ru concentrations of ca. 6.4 ppb (UB-N), 0.14 ppb (BHVO-2) and 0.03 ppb (BCR-2).17,49 Even for UB-N these concentrations are too low for precise isotope measurements using the sample masses of ∼0.5 g typically processed with the method described in the present study. Prior to digestion we, therefore, added ∼500 ng Ru from the Alfa Aesar™ standard solution to ∼0.5 g of the rock powders, together with an appropriate amount of double spike. Note that even in the case of UB-N—the sample with the highest Ru content of the three investigated rock powders—only ∼3 ng of Ru would derive from the sample, compared to ∼500 ng Ru from the standard solution. Thus, UB-N itself contributes less than 1% of the total Ru analysed, meaning that only if the δ102/99Ru of UB-N would be more than 4‰ different from that of the Alfa Aesar standard, would we be able to measure a resolvable Ru isotope difference outside the uncertainty of the δ102/99Ru analyses. That UB-N is characterised by such a large Ru isotope fractionation is highly unlikely, however, and we, therefore, expect to measure the Ru isotopic composition of the added standard solution. The same holds for the other two doped terrestrial rock standards (BHVO-2, BCR-2), given their much lower Ru concentrations compared to UB-N. As a further test of our analytical protocol, we processed an aliquot of the spiked Alfa Aesar solution standard alongside the terrestrial rock powders through the entire procedure. Taken together, these test make it possible to evaluate potential effects of sample processing and sample matrix on measured Ru isotope compositions; if such effects are present they would manifest themselves in deviations from δ102/99Ru = 0. Moreover, these test also provide a robust estimate of the external reproducibility of the Ru isotope measurements.
The δ102/99Ru values measured for the doped terrestrial samples and the processed standard solution are all zero within uncertainty (Fig. 5; Table 3) and the mean δ102/99Ru of all the measurements of the four samples is 0.00 ± 0.05‰ (2 s.d.; n = 20). Each sample was analysed five times and the reproducibility of these five measurements was better than 0.05‰ (2 s.d.) for each sample and is thus similar to the reproducibility obtained for the measurement of unprocessed standard solutions (see Fig. 4b and 5). A reproducibility of 0.05‰ (2 s.d.), therefore, provides a good approximation of the external error of our analytical protocol. Additionally, the varying yields of the four processed samples (65 to 98%) show that even losses of up to 35% Ru do not compromise the accuracy of the Ru isotopic data (Fig. 5, Table 3).
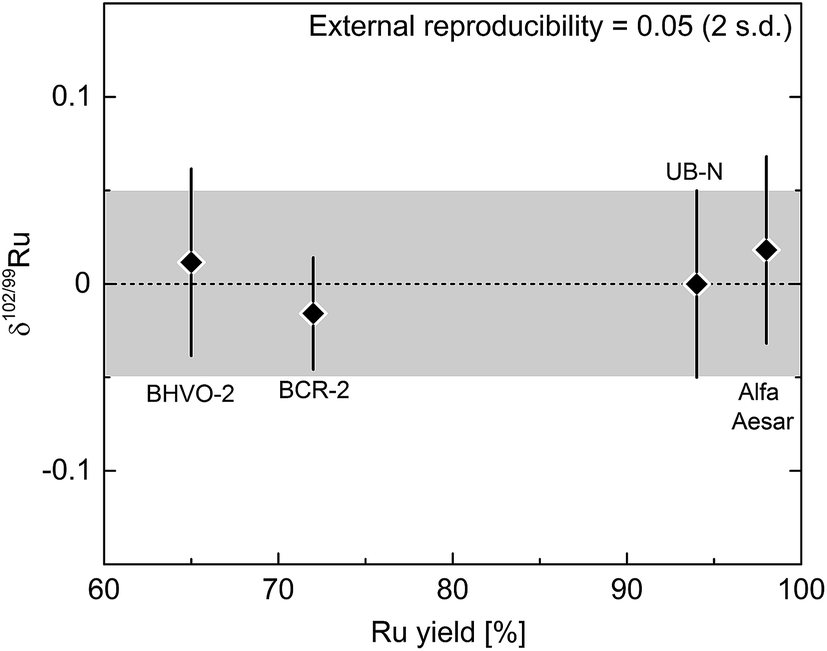 | ||
| Fig. 5 Ruthenium isotopic composition of processed reference materials and standard solutions plotted against the Ru yield of the chemistry. Each point is the mean of five measurements with given error (2 s.d.). Note that the varying Ru yields (65 to 98%) do not affect the measurements. The external error of the entire procedure (digestion, purification, measurement) is given by the error of the results for all processed materials (2 s.d.) and is 0.05‰ for δ102/99Ru and represented by the grey area. | ||
| Sample | N | δ 102/99Ru | Rumeasured [ppm] | Ruliterature [ppm] |
|---|---|---|---|---|
| Processed standard and reference materials | ||||
| Alfa Aesar solution | ||||
| #1 | 0.04 | |||
| #2 | 0.01 | |||
| #3 | 0.01 | |||
| #4 | −0.02 | |||
| #5 | 0.05 | |||
| Mean (±2 s.d.) | 5 | 0.02 ± 0.05 | ||
| Doped BCR-2 | ||||
| #1 | 0.00 | |||
| #2 | −0.03 | |||
| #3 | 0.00 | |||
| #4 | −0.02 | |||
| #5 | −0.03 | |||
| Mean (±2 s.d.) | 5 | −0.02 ± 0.03 | ||
| Doped BHVO-2 | ||||
| #1 | 0.04 | |||
| #2 | −0.01 | |||
| #3 | 0.02 | |||
| #4 | −0.03 | |||
| #5 | 0.00 | |||
| Mean (±2 s.d.) | 5 | 0.01 ± 0.05 | ||
| Doped UB-N | ||||
| #1 | 0.02 | |||
| #2 | 0.02 | |||
| #3 | 0.00 | |||
| #4 | −0.03 | |||
| #5 | −0.02 | |||
| Mean (±2 s.d.) | 5 | 0.00 ± 0.05 | ||
|
||||
| Standard solutions | ||||
| Merck Millipore | ||||
| #1 (Sep. 2015) | 0.11 | |||
| #2 (Sep. 2015) | 0.12 | |||
| #3 (Sep. 2015) | 0.13 | |||
| #4 (Nov. 2015) | 0.11 | |||
| #5 (Nov. 2015) | 0.11 | |||
| Mean (±2 s.d.) | 5 | 0.11 ± 0.02 | ||
| Inorganic Ventures | ||||
| #1 (Sep. 2015) | 0.90 | |||
| #2 (Sep. 2015) | 0.91 | |||
| #3 (Sep. 2015) | 0.91 | |||
| #4 (Nov. 2015) | 0.91 | |||
| #5 (Nov. 2015) | 0.90 | |||
| Mean (±2 s.d.) | 5 | 0.91 ± 0.01 | ||
|
||||
| Chromitites | ||||
| Shetland Ophiolite | ||||
| C-3 | ||||
| #1 (Sep. 2015) | 0.20 | |||
| #2 (Sep. 2015) | 0.20 | |||
| #3 (Sep. 2015) | 0.21 | |||
| #4 (Nov. 2015) | 0.19 | |||
| #5 (Nov. 2015) | 0.21 | |||
| Mean (±2 s.d.) | 5 | 0.20 ± 0.02 | 18.5 | 20.0 (ref. 41) |
| C-1 | ||||
| #1 | 0.25 | |||
| #2 | 0.23 | |||
| #3 | 0.24 | |||
| Mean (±2 s.d.) | 3 | 0.24 ± 0.05 | 17.0 | 16.7 (ref. 41) |
| HG-1 | 0.67 | 1.94 | 2.36 (ref. 41) | |
| HG-2 | 0.66 | 2.91 | 3.44 (ref. 41) | |
| Mean (±2 s.d.) | 2 | 0.67 ± 0.05 | ||
| Bushveld complex | ||||
| UG2 | ||||
| #1 | 0.42 | |||
| #2 | 0.43 | |||
| Mean (±2 s.d.) | 2 | 0.43 ± 0.05 | 0.67 | 0.64 − 0.79SARM81 |
4.3 Effects of interferences on double spike measurements
The accuracy and precision of the Ru isotope measurements can be compromised by the presence of isobaric interferences of 96Mo, 98Mo, 100Mo, 102Pd and 104Pd on Ru isotopes (Fig. 1). Of these, interferences from 96Mo, 100Mo and 104Pd as well as potential interferences from 96Zr are inconsequential for the Ru isotope measurements, because they do not directly interfere with the Ru isotopes used in the double spike inversion (i.e., 98Ru, 99Ru 101Ru and 102Ru). However, given the high natural abundance of 98Mo combined with the low abundance of 98Ru, the presence of Mo in the analysed Ru fractions in particular may have a significant effect on measured δ102/99Ru. Thus, to test which amounts of Mo (and Pd) in the analysed Ru fractions are tolerable and can reliably be corrected without compromising the accuracy and precision of measured δ102/99Ru values, we performed a series of doping test, in which varying amounts of Mo and Pd were added to the spiked Alfa Aesar Ru standard solution.The Ru fractions obtained after cation exchange chromatography and distillation typically have Mo/Ru and Pd/Ru of ≪0.0005. We, therefore, doped the spiked Alfa Aesar standard to yield Mo/Ru and Pd/Ru ratios between 0.00001 and 0.02; note that the lower of these ratios are typical ratios obtained for processed samples. Fig. 6 summarises the results of the doping tests and illustrates that the presence of Mo and Pd does not induce measureable offsets in δ102/99Ru for Mo/Ru <0.0001 and Pd/Ru <0.002 (Fig. 6). For larger amounts of Mo or Pd measured δ102/99Ru values deviate from the true value. These offsets can readily be corrected for Mo/Ru ratios up to ∼0.0006 and for Pd/Ru ratios up to ∼0.02, but for higher Mo/Ru and Pd/Ru ratios the interference correction becomes inaccurate (Fig. 6). Possible reasons for these deviations include an improper correction for instrumental mass bias and deviations in the isotopic composition of Mo and Pd used in the interference correction. Note, however, that our chemical purification procedure results in Mo/Ru and Pd/Ru ratios ≪0.0005, which is well within the range of ratios for which the interference corrections are accurate and precise. Thus, these interference corrections are inconsequential for the precision and accuracy of the Ru isotope measurements.
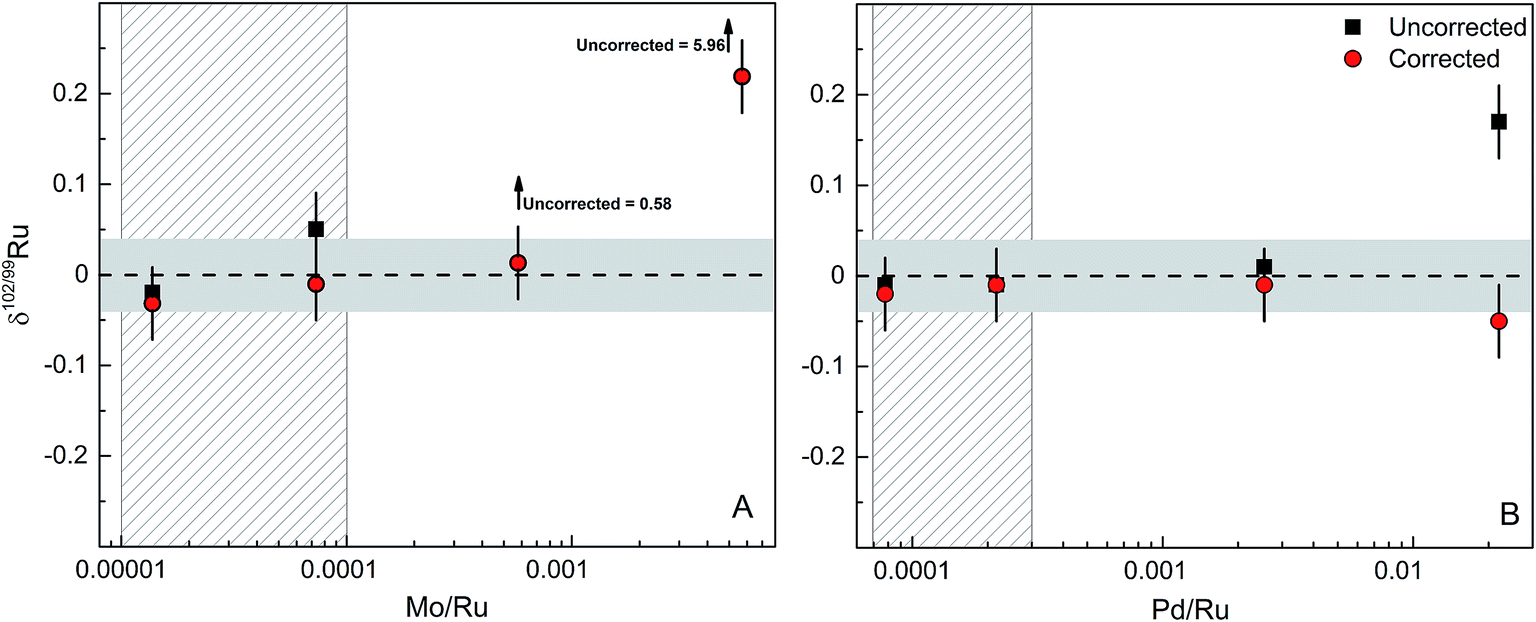 | ||
| Fig. 6 Effects of isobaric Mo and Pd interferences on the δ102/99Ru. (A) Plot of δ102/99Ru versus Mo/Ru. (B) Plot of δ102/99Ru versus Pd/Ru. Horizontal grey areas represent long-term reproducibility of spiked solution standards of 0.04‰. The vertical patterned grey areas represent the typical range of Mo/Ru and Pd/Ru in processed reference materials, standard solution and chromitite samples used in this study. | ||
Other potential interferences that may affect the quality of the Ru isotope measurements include Ar oxides (e.g., 40Ar2O+), Ru hydrides, doubly charged Pt, and polyatomic Cr species (e.g., 50Cr52Cr+). However, several observations and tests indicate that these potential interferences have no measurable effect on the Ru isotopic data. First, oxide rates (i.e., CeO/Ce) were always <1%, minimising the formation of potential interferences of Ar oxides on Ru masses. Note that the major Ar oxide in the Ru mass range is 40Ar216O at mass 96, but that 96Ru is not used in the double spike inversion. Second, there is no detectable signal on mass 105 (i.e., 104RuH+) during the measurement of a pure Ru standard solution, indicating that there is no significant Ru hydride formation beyond baseline levels. Third, our chemical purification procedure efficiently separates Ru from all other HSE, so that none of the analysed samples contains Pt and that, therefore, no doubly charged Pt could have formed. Finally, we assessed the effects of Cr contamination on measured Ru isotope composition by measuring spike-standard mixtures admixed with varying amounts of Cr. A prior study occasionally observed a contamination of the final Ru cut with Cr from the aqueous CrO3 solution used for the distillation.27 We note, however, that this contamination has only been observed when purifying Ru by micro-distillation and seems to be absent when using the larger PFA distillation unit employed in the present study. Either way, our tests show that the possible presence of Cr in the analysed Ru solutions has no effect on the measured δ102/99Ru values, even for Cr/Ru ratios as high as ∼1 (Fig. 7).
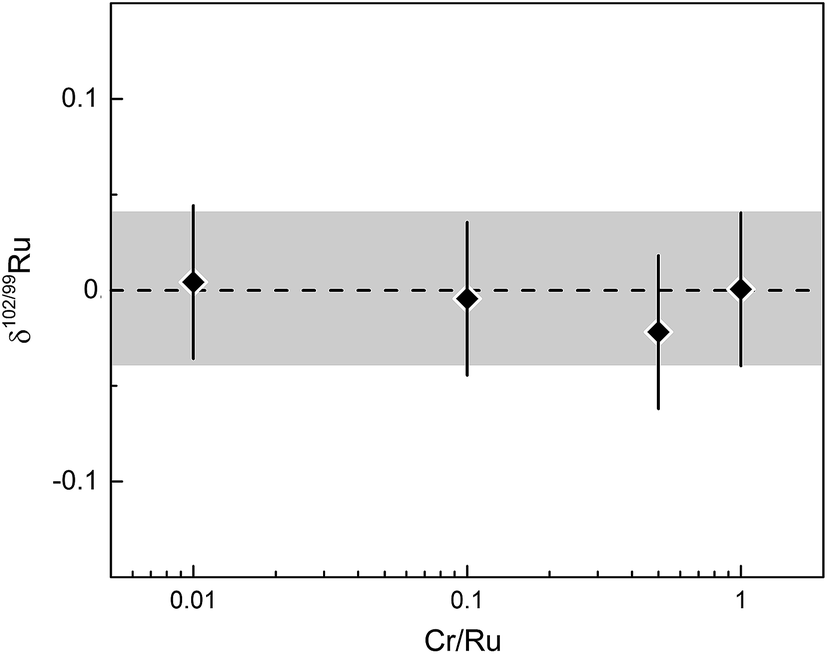 | ||
| Fig. 7 δ 102/99Ru versus Cr/Ru ratio of analysed spike-standard mixture. Error bars and grey area represent long-term reproducibility of unprocessed spiked solution standards of 0.04‰. | ||
4.4 Natural ruthenium stable isotope variations
To assess the magnitude of Ru stable isotope variations, we analysed two commercially available Ru standard solutions from Merck Millipore™ and Inorganic Ventures™ (referred to as M-1 and I-1), four chromitite samples (C-1, C-3, HG-1, HG-2) from the Shetland Ophiolite Complex and one certified reference material (SARM81) from the UG2 chromitite layer of the Bushveld complex, South Africa.41,44 The Ru standard solutions were mixed with an appropriate amount of the double spike, equilibrated on a hot plate at 120 °C using 6 M HCl and directly measured without further processing. This equilibration procedure is the same as used for the preparation of standard-double spike mixtures for the calibration of the double spike; this method produced reproducible results (see above and Fig. 4), indicating that efficient spike-standard equilibration was achieved. For the chromitites, about 50 to 500 mg of each powder were spiked and digested inside Carius tubes and the Ru was purified by cation exchange chromatography and distillation (see above).The results for the standard solutions and chromitites are summarized in Table 3 and shown in Fig. 8. The Ru concentrations obtained for the SOC chromitites vary substantially from published values for the same samples, with deviations of up to ∼20% (Table 3). Note that we obtained both higher and lower values compared to previous studies and so this offset cannot reflect improper spike calibration. Instead, the variable Ru concentrations determined on different splits of the same samples most likely reflect the heterogeneous distribution of PGE phases at the sampling scale, consistent with conclusions of prior studies.41
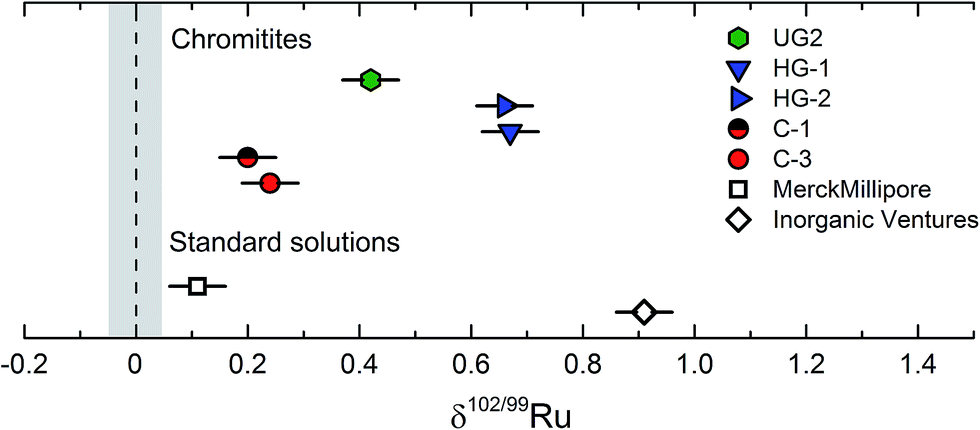 | ||
| Fig. 8 Measured δ102/99Ru of two standard solution (Merck, Inorganic Ventures) and five chromitite samples from the Shetland Ophiolite Complex and the Bushveld complex.37,42 Error bars are given as the external error of the method of ±0.05‰ (2 s.d.). The grey vertical bar represents the uncertainty on the δ102/99Ru of the Alfa Aesar Ru standard solution. | ||
The chromitite samples as well as the different standard solutions exhibit variable Ru stable isotopic compositions, and all have higher δ102/99Ru values (up to ∼0.9‰) and, hence, are isotopically heavier than the Alfa Aesar™ standard solution. As such these data demonstrate that naturally occurring Ru stable isotope fractionation are on the order of at least ca. 1‰; these variations can readily be resolved using the analytical techniques developed in the present study, given an external error of only ±0.05‰ (2 s.d.). The different Ru stable isotope compositions observed for the three investigated standard solutions reflect either isotope fractionation induced by the chemical processing necessary for the purification and enrichment of Ru during production of the standard solutions, or, alternatively, natural Ru isotope variations inherited from different starting materials. Although the current data do not allow to distinguish between these two possibilities, the latter option seems more likely, given that the investigated chromitites show a similar range in Ru stable isotope compositions (Fig. 8).
The four investigated SOC chromitites (C-1, C-3, HG-1, HG-2) are all from podiform chromitite deposits of the Shetland Ophiolite Complex. While the chromitites from one location exhibit indistinguishable Ru stable isotopic signatures, the samples from the Cliff (δ102/99Ru ≈ 0.22‰) and Harold's Grave (δ102/99Ru ≈ 0.66‰) locations are different (Table 3; Fig. 8). Thus, samples formed within the same large geological context (the Shetland Ophiolite Complex) can have different Ru stable isotopic compositions. The Cliff and Harold's Grave chromitites probably derive from different magmas, as is evident from their different PGE abundances, major element compositions and petrology.41,45 Moreover, the high variability of Ru concentrations in the Cliff chromitites is attributed to the remobilization of the PGEs during hydrothermal alteration by an As-rich fluid.45,50 Thus, the different Ru stable isotopic compositions of the Cliff and Harold's Grave chromitites may reflect differences in composition of the source magmas, different magnitudes of Ru enrichments and differences in the enrichment processes (magmatic vs. magmatic and hydrothermal). Likewise, the UG2 sample derived from a stratiform chromitite deposit of the Bushveld complex (UG2) has another a Ru isotopic composition in between those of the two analysed podiform SOC chromitites, which again probably reflects either different formation processes or differences in magma compositions.46
5 Conclusion
We have developed a method for the precise measurement of mass-dependent Ru isotope variations by MC-ICPMS combined with a 98Ru–101Ru double spike. This spike provides accurate results over a wide range of spike-to-sample ratios and is insensitive to the incomplete recovery of Ru from the analysed samples. Our chemical purification procedure, which includes cation exchange chromatography and distillation, results in sufficiently clean Ru fractions with only very minor amounts of Mo and Pd remaining in the final Ru cut. Isobaric interferences from Mo and Pd are, therefore, small and, if present, can be reliably corrected for. Repeated measurements of different Ru standard solutions and terrestrial rock standards show that an external precision of ±0.05‰ for δ102/99Ru can routinely be obtained using the analytical method presented in this study.The analyses of different commercially available Ru standard solutions and chromitite samples from three different localities show that natural Ru stable isotope variations of up to at least ca. 1‰ (for δ102/99Ru) exist. These variations can readily be resolved using the analytical method presented here. Thus, Ru stable isotopes hold promise as a tracer for a wide range of processes, including the condensation of metal in the solar nebula, the segregation and crystallization of metal cores in asteroids and the terrestrial planets as well as the formation of terrestrial PGE deposits.
Acknowledgements
We are thankful to Brian O'Driscoll and Katherine Bermingham for providing the samples of the chromitites from the Shetland Ophiolite Complex. Further we thank Savillex for permission to reproduce the drawing of the distillation unit. Comments of two anonymous reviewers helped to improve the manuscript and are gratefully acknowledged. This research received funding to Thorsten Kleine from the European Research Council under the European Community's Seventh Framework Program (FP7/2007–2013 Grant Agreement 616564 ‘ISOCORE’).References
- P. Fritz and J. C. Fontes, Handbook of Environmental Isotope Geochemistry. Vol. 1. The Terrestrial Environment, A, 1980 Search PubMed.
- D. J. Depaolo, Earth Planet. Sci. Lett., 1981, 53, 189–202 CrossRef CAS.
- A. D. Anbar and O. Rouxel, Metal stable isotopes in paleoceanography, Annu. Rev. Earth Planet. Sci., 2007, 35, 717–746 CrossRef CAS.
- H. M. Williams, A. Markowski, G. Quitte, A. N. Halliday, N. Teutsch and S. Levasseur, Earth Planet. Sci. Lett., 2006, 250, 486–500 CrossRef CAS.
- R. C. Hin, C. Fitoussi, M. W. Schmidt and B. Bourdon, Earth Planet. Sci. Lett., 2014, 387, 55–66 CrossRef CAS.
- F. Moynier, J. Blichert-Toft, P. Telouk, J.-M. Luck and F. Albarède, Geochim. Cosmochim. Acta, 2007, 71, 4365–4379 CrossRef CAS.
- B. Luais, Chem. Geol., 2012, 334, 295–311 CrossRef CAS.
- M. Millet and N. Dauphas, J. Anal. At. Spectrom., 2014, 29, 1444–1458 RSC.
- J. B. Creech, J. Baker, M. Handler, M. Schiller and M. Bizzarro, J. Anal. At. Spectrom., 2013, 28, 853–865 RSC.
- R. C. Hin, C. Burkhardt, M. W. Schmidt, B. Bourdon and T. Kleine, Earth Planet. Sci. Lett., 2013, 379, 38–48 CrossRef CAS.
- C. Burkhardt, R. C. Hin, T. Kleine and B. Bourdon, Earth Planet. Sci. Lett., 2014, 391, 201–211 CrossRef CAS.
- E. A. Seddon and K. R. Seddon, The Chemistry of Ruthenium, Elsevier Science Publishers, Amsterdam, 1984 Search PubMed.
- E. Medard, M. Schmidt, M. Wälle, N. S. Keller and D. Günther, Geochim. Cosmochim. Acta, 2015, 162, 183–201 CrossRef CAS.
- M. Locmelis, N. J. Pearson, S. J. Barnes and M. L. Fiorentini, Geochim. Cosmochim. Acta, 2011, 75, 3645–3661 CrossRef CAS.
- H. Renner, G. Schlamp, I. Kleinwächter, E. Drost, H. M. Lüschow, P. Tews, P. Panster, M. Diehl, J. Lang, T. Kreuzer, A. Knödler, K. A. Starz, K. Dermann, J. Rothaut, R. Drieselmann, C. Peter and R. Schiele, Platinum Group Metals and Compounds. Ullmann's Encyclopedia of Industrial Chemistry, 2001 Search PubMed.
- H. Becker, M. F. Horan, R. J. Walker, S. Gao, J.-P. Lorand and R. L. Rudnick, Geochim. Cosmochim. Acta, 2006, 70, 4528–4550 CrossRef CAS.
- M. Fischer-Gödde, H. Becker and F. Wombacher, Geochim. Cosmochim. Acta, 2011, 280, 365–383 Search PubMed.
- M. Fischer-Gödde, H. Becker and F. Wombacher, Geochim. Cosmochim. Acta, 2010, 74, 356–379 CrossRef.
- R. J. Walker, W. F. McDonough, J. Honesto, N. L. Chabot, T. J. McCoy, R. D. Ash and J. J. Belluci, Geochim. Cosmochim. Acta, 2008, 72, 2198–2216 CrossRef CAS.
- T. J. McCoy, R. J. Walker, J. I. Goldstein, J. Yang, W. F. McDonough, D. Rumble, N. L. Chabot, R. D. Ash, C. M. Corrigan, J. R. Michael and P. G. Kotula, Geochim. Cosmochim. Acta, 2011, 75, 6821–6843 CrossRef CAS.
- E. R. D. Scott, Geochim. Cosmochim. Acta, 1972, 36, 1205–1236 CrossRef CAS.
- R. J. Walker, Geochemistry, 2009, 69, 101–125 CAS.
- K. Kimura, R. Lewis and E. Anders, Geochim. Cosmochim. Acta, 1974, 38, 683–701 CrossRef CAS.
- C. Chou, in Proceedings of the 9th Lunar and Planetary Science, 1978, vol. 1, pp. 219–230 Search PubMed.
- R. J. Walker, K. Bermingham, J. Liu, I. S. Puchtel, M. Touboul and E. A. Worsham, Chem. Geol., 2015, 411, 125–142 CrossRef CAS.
- J. H. Chen, D. A. Papanastassiou and G. J. Wasserburg, Geochim. Cosmochim. Acta, 2010, 74, 3851–3862 CrossRef CAS.
- M. Fischer-Gödde, C. Burkhardt, T. S. Kruijer and T. Kleine, Geochim. Cosmochim. Acta, 2015, 168, 45–62 CrossRef.
- I. D. Hutcheon, J. T. Armstrong and G. J. Wasserburg, Geochim. Cosmochim. Acta, 1987, 51, 3175–3192 CrossRef CAS.
- H. Becker, C. Dalpe and R. J. Walker, Analyst, 2002, 127, 775–780 RSC.
- H. Becker and R. J. Walker, Chem. Geol., 2003, 196, 43–56 CrossRef CAS.
- J. F. Rudge, B. C. Reynolds and B. Bourdon, Chem. Geol., 2009, 265, 420–431 CrossRef CAS.
- M. Dodson, Geochim. Cosmochim. Acta, 1970, 34, 1241–1255 CrossRef CAS.
- C. Siebert, A. Ross and J. McManus, Geochim. Cosmochim. Acta, 2006, 70, 3986–3995 CrossRef CAS.
- P. Bonnand, I. J. Parkinson, R. H. James, A.-M. Karjalainen and M. A. Fehr, J. Anal. At. Spectrom., 2011, 26, 528–535 RSC.
- M. Millet, J. A. Baker and C. E. Payne, Chem. Geol., 2012, 304, 18–25 CrossRef.
- J. B. Creech, J. A. Baker, M. R. Handler and M. Bizzarro, Chem. Geol., 2014, 363, 293–300 CrossRef CAS PubMed.
- K. R. Bermingham, R. J. Walker and E. A. Worsham, Int. J. Mass Spectrom., 2016 DOI:10.1016/j.ijms.2016.02.003 , in press.
- T. Fuji, F. Moynier and F. Albarede, Chem. Geol., 2009, 139–156 CrossRef.
- T. Fuji, F. Moynier, P. Telouk and F. Albarede, Astrophys. J., 2006, 1506–1516 CrossRef.
- M. Berglund and M. E. Wieser, Pure Appl. Chem., 2011, 83, 397–410 CrossRef CAS.
- B. O'Driscoll, J. M. D. Day, R. J. Walker, J. S. Daly, W. F. McDonough and P. M. Piccoli, Earth Planet. Sci. Lett., 2012, 333, 226–237 CrossRef.
- J. G. Spray and G. R. Dunning, Geol. Mag., 1991, 128, 667–671 CrossRef CAS.
- D. M. Chew, J. S. Daly, T. Magna, L. M. Page, C. L. Kirkland, M. J. Whitehouse and R. Lam, Geol. Soc. Am. Bull., 2010, 122, 1787–1799 CrossRef CAS.
- H. M. Prichard and C. P. Brough, New developments in magmatic Ni–Cu and PGE deposits, 2009, pp. 277–290 Search PubMed.
- C. P. Brough, H. M. Prichard, C. R. Neary, P. C. Fisher and I. McDonald, Econ. Geol., 2015, 110, 187–208 CrossRef.
- R. G. Cawthorn, Platinum Met. Rev., 2010, 54, 205–215 CrossRef.
- S. Shirey and R. Walker, Anal. Chem., 1995, 67, 2136–2141 CrossRef CAS.
- A. Westland and F. Beamish, Anal. Chem., 1954, 26, 739–741 CrossRef CAS.
- T. Meisel and J. Moser, Geostand. Geoanal. Res., 2004, 28, 233–250 CrossRef CAS.
- R. A. Lord, H. M. Prichard and C. R. Neary, Trans. - Inst. Min. Metall., 1994, B179–B193 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |