
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of ultra-low level 129I in vegetation using pyrolysis for iodine separation and accelerator mass spectrometry measurements
Xiaolin
Hou
*ab and
Yanyun
Wang
ac
aXi'an AMS Center, SKLLQG, Shaanxi Key Laboratory of Accelerator Mass Spectrometry Technology and Application, Institute of Earth Environment, CAS, Xi'an, 710061, China. E-mail: houxl@ieecas.cn
bTechnical University of Denmark, Center for Nuclear Technologies, Risø, 4000 Roskilde, Denmark. E-mail: xiho@dtu.dk
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 19th April 2016
Abstract
Radioactive isotopes of iodine are the most common radiological toxins from nuclear accidents due to their high release and high enrichment in human thyroid. Among the radioactive isotopes, long-lived 129I can not only be used for the estimation of the radioactive risk of short-lived radioactive isotopes of iodine to humans and ecosystems, but also for the investigation of the biogeochemical cycle and environmental behavior of iodine. Accurate determination of 129I in various environmental and biological samples is the key issue for these purposes. Due to its beta decay, low specific activity and ultra-low concentration in normal environmental and biological samples, it is important to efficiently separate iodine from the sample matrix and sensitively measure 129I. However, the complicated chemical properties of iodine and high organic content in biological samples make efficient separation of iodine very difficult using conventional acid digestion and alkaline ashing methods. By optimizing the key parameters related to the separation of iodine by pyrolysis using a tube furnace, including carbonization temperature, heating protocol, combustion assisting gas, iodine volatilization process and iodine trapping, a safe, robust and reliable pyrolysis method was established for the separation of trace amounts of iodine from biological samples with a recovery of more than 80%. By further separation of iodine, preparation of sample targets, and measurement of 129I using AMS, a highly efficient and sensitive method for the determination of ultra-low level 129I in biological samples was developed. With this method, a detection limit of 6 × 10−17 g g−1 (or 0.4 nBq g−1) for 129I was obtained. Compared with conventional methods, this method is easy to operate, provides highly efficient recovery of iodine, and has simple processing and less cross contamination. 5 different species of vegetation were analyzed using both the developed method and the conventional alkaline ashing method for sample decomposition, and the results agree very well with each other. The method has been successfully used for the determination of 129I in a large number of vegetation samples.
Introduction
Iodine-129 is a long-lived fission product of 238U and 239Pu. Although it is a naturally occurring radionuclide, human nuclear activities have released a large amount of 129I into the environment, increasing the 129I/127I atomic ratio to >10−10 from the pre-nuclear level of 10−12. Anthropogenic 129I is now the dominant source of 129I in the environment.1,2 Due to its high solubility in water and volatility, anthropogenic 129I is widely spread in the environment through ocean circulation and atmospheric dispersion.2,3 It is therefore used as a tracer to investigate environmental processes, reconstruct radiation levels of short-lived radioiodines (e.g.131I) and monitor nuclear environmental safety.4–15 Iodine is a biophilic element; it is concentrated in plants and animals, and accumulated in human thyroid through the food chain.16 Radioactive isotopes of iodine, especially short-lived radioiodines such as 131I, are highly radiologically toxic during nuclear accidents due to their high release. Due to the same production model, the same chemical properties and environmental and biological behaviors, 129I can be used to reconstruct the level and distribution of short-lived radioiodines released from a nuclear accident after the short-lived radioiodines disappear by radioactive decay.15,16 For these purposes, determination of 129I in various environmental samples is required, and the accurate determination of ultra-low level 129I in vegetation samples plays a key role for investigating radioiodine transfer in different ecosystem compartments and assessing their radiation risk in nuclear emergency.129I is a beta decay radionuclide with the emission of low energy gamma rays of low intensity. Although radiometric methods such as gamma spectrometry and beta counting can be used for its measurement, the low specific activity and ultra-low concentration of 129I in the environment make the determination of 129I in normal environmental samples using these techniques impossible.2 Although ICP-MS has also been proposed for the measurement of 129I, the low ionization efficiency of iodine and serious isobaric interference of 129Xe make it only possible to measure 129I in nuclear waste or contaminated environmental samples with 129I/127I ratios higher than 10−8 and 129I concentrations higher than 10−10 g g−1.17 Accelerator mass spectrometry (AMS) and neutron activation analysis (NAA) are the techniques for its determination in present environmental samples, and AMS is the only technique for the determination of 129I in samples with 129I/127I ratios lower than 10−10 and 129I concentrations lower than 10−14 g g−1.2,18 Using these methods, pre-concentration of iodine from the sample matrix and purification of iodine from other elements have to be carried out to improve the detection limit of 129I and remove interferences.19–21 Meanwhile, the separated iodine has to be prepared in suitable form for 129I measurements by using different techniques such as inorganic iodine species in aqueous solution or I2 in gas form for ICP-MS,17 dried inorganic iodine compounds (MgI2) or adsorbed in active charcoal for NAA,18–20 and AgI or AgI–AgCl form for AMS measurements of 129I.21,22 Due to the high volatility, active chemical properties and complicated behavior of iodine, iodine can easily be lost during chemical separation and sample preparation. Effective separation of iodine from a big sample is a key issue for the accurate determination of low level 129I in environmental samples. Environmental studies using 129I mainly focused on the analysis of water, soil and sediment, and biological samples with high iodine concentrations such as animal thyroid and seaweed or vegetation samples collected from surrounding nuclear facilities contaminated by 129I releases, while the analysis of vegetation samples collected in a background area with low 129I concentration is limited; this is mainly attributed to the difficulties in the decomposition and separation of volatile iodine from organic enriched biological samples, as well as the low iodine concentration and ultra-low level of 129I in the normal environmental vegetation samples.
In biological samples, iodine is often bound to organic compounds; alkaline leaching cannot release iodine from the sample to the solution. The sample has to be decomposed to convert iodine to inorganic form and release it into solution for further separation and purification. Three methods have been used to decompose biological samples for releasing iodine to solution, i.e. alkaline ashing following by water leaching,16,19,20 acid digestion23–25 and pyrolysis (combustion).26–31 Based on the lower volatility of iodine in alkaline media, biological samples are first mixed with alkaline solution (NaOH/KOH or Na2CO3) and evaporated to dryness, which is then ashed at 550–650 °C to decompose organic substance, and iodine converted to inorganic form during ashing is then leached using water or sulfite solution.19,20 Alkaline ashing is easy to operate, but very tedious, the whole procedure takes a few days. In addition, a high loss of iodine and varied recovery of iodine might occur, because high alkaline content prevents complete decomposition of organic substances and due to the high adsorption of released inorganic iodine to the carbonized sample. Meanwhile, the higher temperature can cause loss of iodine gas. The further separation of iodine in a large and complicated leachate is also time consuming.20 Decomposition of biological samples using acid digestion for the separation of iodine has to be carried out in a closed system, iodine is released from organic substances during decomposition, and evaporated as I2 under acidic conditions, and needs to be collected in an alkaline trap solution. The decomposition of biological samples using acids is therefore time consuming, especially for big size samples (>5 g), which cannot be applied for the simultaneous treatment of many samples, and is consequentially difficult to be used for the analysis of a large number of samples. Meanwhile, high oxidative acids such as HClO4, HF and H2SO4 are dangerous and difficult to handle. Microwave assisted acid digestion has been proposed for shortening the decomposition time for biological samples,24 but this method cannot be used to treat big size samples (>0.5 g). In addition, adsorption of I2 produced during acid digestion on the Teflon material of the container will result in the loss of iodine and cross contamination between samples.
Pyrolysis is an effective method for releasing iodine from solid samples, and has been widely used for the separation of iodine from soil, sediment and rocks.15,17–19,23 This method is based on the volatility of iodine: the solid sample is combusted in an oxygen flow to convert all species of iodine to gaseous forms (I2 and HI), which are then trapped by an alkaline solution (NaOH or NaOH–NaHSO3). With this method, up to 50 g soil samples can be treated.17,19,30 However, the pyrolysis is not well applied for the separation of iodine from biological samples for 129I determination. This is mainly attributed to the high risk of explosion due to the violent ignition of organic substances and outflow of uncombusted organic compounds which enter the trapping solution, making the separation of iodine a failure. A combustion system with two tube furnaces17,19,27 and a tube furnace with a Bunsen burner18 have been proposed for analyses of samples with high organic contents. The sample is weighed in a quartz tube and is combusted using a tube furnace. The first furnace or Bunsen burner is slowly moved to gradually ignite and combust the sample, and the second furnace is used to combust the particles or uncombusted organic compounds. However, this modification cannot avoid the problem of explosion and incomplete combustion of biological samples. By employing catalysts such as V2O5 and CuO in the combustion system and optimizing the O2 flow rate and volatilization temperature, the pyrolysis has been applied to separate iodine from biological samples.26,27 However, the problems of explosion and incomplete combustion during the analysis of biological samples are still not well solved. This work aims to investigate the ignition and combustion processes of biological samples, in order to establish a safe, robust and effective method for the separation of iodine from biological samples using the pyrolysis technique.
During pyrolysis, gaseous iodine released from the sample during combustion is often trapped in active charcoal18 and/or alkaline solution (TMAH or NaOH–NaHSO3).17,19,28,29,30 The trapped iodine needs to be further separated by a second combustion using active charcoal,18 or by solvent extraction,17,19,28–30 and converted to suitable forms for measurements, e.g. AgI precipitate for AMS and water soluble solid (LiOH or MgI2) for NAA.27 However, the trapping process is still not well understood; the separation procedure is time consuming and organic waste is produced. In this work, the trapping process was investigated and the separation procedure was improved in order to establish a rapid, reliable and sensitive analytical method for the determination of 129I in environmental vegetation samples by using AMS for 129I measurements.
Experimental
Instruments and chemical reagents
A pyrolysis system (Pyrolyser-4 Trio™, Raddec Inc. UK) was applied for the combustion of vegetation samples. The system (Fig. 1) consists of 4 quartz tubes which are placed in a tube furnace. The furnace is equipped with two heating zones which can be controlled and programmed, respectively.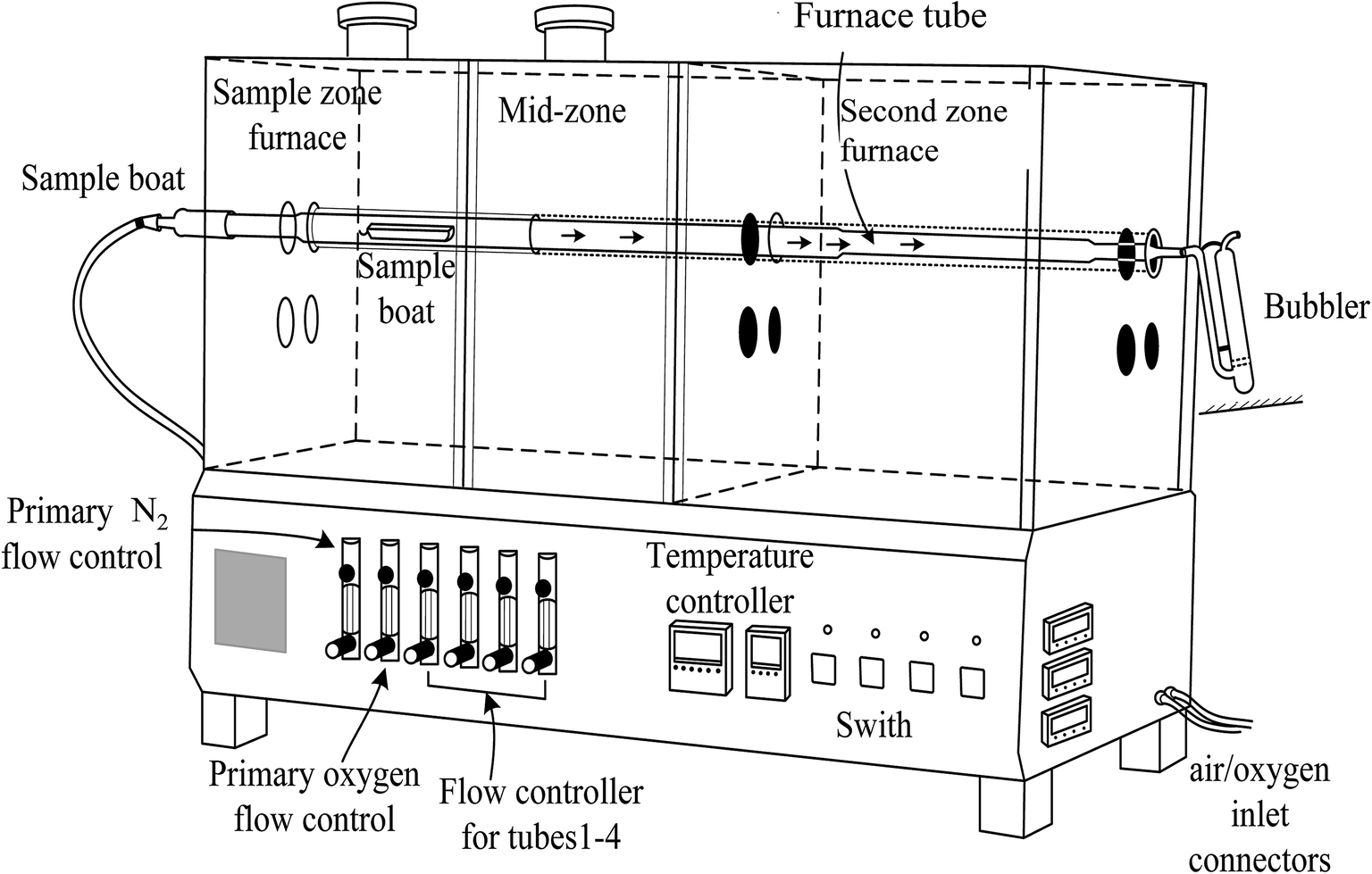 | ||
| Fig. 1 Schematic diagram of a Pyrolyser-4 Trio™ tube furnace with two separated heating zones, the first sample zone and second combustion zone. | ||
A muffle oven (Carbolite Gero Limited, UK) was applied for the alkaline ashing of vegetation samples, the oven can be heated up to 1100 °C. A NaI gamma detector (Xi'an Nuclear instrument Company, China) was used to measure 125I for chemical yield monitoring.
129I standard solution (NIST-SRM-4949c) was purchased from the National Institute Standard and Technology (Gaithersburg, MD), 127I carrier solution with a low 129I level (129I/127I atomic ratio < 2 × 10−14) was prepared by the dissolution of iodine crystals (Woodward iodine, Woodward Iodine Corporation, Oklahoma, U.S.A.) into 0.40 mol L−1 NaOH–0.05 mol L−1 NaHSO3 solution. All chemical reagents used were of analytical grade, and all solutions were prepared using deionized water (18.2 MΩ cm). A 129I/127I ratio standard of 1.03 × 10−10 (atom/atom) was prepared by the dilution of the 129I standard solution with the 127I carrier; AgNO3 solution was then added to this solution, and the AgI precipitate was separated by centrifugation and dried at 60 °C.
Sampling and sample pretreatment
Pine needle and grass (Green Bristle grass herb) were collected in Xi'an city (34.22°N, 109.00°E), and spinach and lichens were collected in an agricultural area in Baoji (34.15°N, 107.94°E), both sites are located in Shaanxi province, China. Seaweed (Laminaria japonica) was collected in Fujian province, China, and purchased from the market. The samples were first air dried, and then dried at 60 °C in an oven until constant weight. The dried samples were pulverized to powder (<50 mesh).Separation of iodine from vegetation by pyrolysis
The vegetation powder sample (1–10 g) was weighed and placed in a quartz boat, 125I tracer solution (100 Bq, as Na125I) was spiked on the sample. The sample boat was placed in the furnace tube in the pyrolysis system in the first heating zone. A U-type bubbler containing 35 mL of trapping solution (NaOH, NaOH–NaHSO3 solution or H2O) was attached to the outlet of the combustion tube with ground glass joints. The flow rate of input gases (N2 + O2 before 400 °C and O2 after 400 °C) was adjusted to 100–500 mL min−1. The second heating zone was directly heated to 900 °C in 30 min. The ramp procedure of the first heating zone was programmed according to the requirement.After combustion completed or at different times (to investigate releases of iodine with combustion time), the trap solution was transferred to a centrifuge tube, and the bubbler was washed 3 times with 6 mL water, and combined with the trap solution. 3.0 mL trap solution was taken to a counting tube for 125I measurements using a gamma detector for monitoring the recovery of iodine. This 3 mL solution was recombined with the original solution after gamma measurements. 1.0 mL of trap solution was transferred to a 14 mL centrifuge tube for 127I measurements using ICP-MS. The remaining solution was used for further separation of iodine for 129I measurements.
Separation of iodine from vegetation samples using alkaline ashing
Vegetation samples (2–3 g) were weighed into a nickel crucible. 125I tracer solution (100 Bq, as Na125I form) and 2.0 mol L−1 NaOH solution (sample![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH solution = 1 g:5 mL) were added to the samples and the samples were mixed. The sample in the crucible was evaporated to dryness on a hotplate, and then placed in a muffle oven and ashed using a ramp protocol shown in Fig. 2. After drying at 100 °C for 30 min, the temperature was increased to 350 °C and maintained for 1 h to carbonize the vegetation sample. The carbonized samples were finally ashed at 650 °C for 3 h. After cooling to room temperature, the sample crucible was taken out from the oven and 30 mL deionized water was added to the sample. The crucible with the sample and leaching water was heated at 70–80 °C on a hot plate for 30 min to leach iodine from the sample ash. The leachate was separated by filtration through a membrane of 0.45 μm pore size. The residue was transferred back to the crucible and 30 mL deionized water was added again to leach the remaining iodine. This leaching procedure was repeated 3 times, and all leachates were combined. 3.0 mL of the leachate was taken to measure 125I using a gamma detector to monitor the chemical yield of iodine. This solution was re-combined with the original solution after gamma measurements. 1.0 mL trap solution was transferred to a 14 mL centrifuge tube for 127I measurements using ICP-MS. The remaining solution was used for further separation of iodine for 129I measurements.
:NaOH solution = 1 g:5 mL) were added to the samples and the samples were mixed. The sample in the crucible was evaporated to dryness on a hotplate, and then placed in a muffle oven and ashed using a ramp protocol shown in Fig. 2. After drying at 100 °C for 30 min, the temperature was increased to 350 °C and maintained for 1 h to carbonize the vegetation sample. The carbonized samples were finally ashed at 650 °C for 3 h. After cooling to room temperature, the sample crucible was taken out from the oven and 30 mL deionized water was added to the sample. The crucible with the sample and leaching water was heated at 70–80 °C on a hot plate for 30 min to leach iodine from the sample ash. The leachate was separated by filtration through a membrane of 0.45 μm pore size. The residue was transferred back to the crucible and 30 mL deionized water was added again to leach the remaining iodine. This leaching procedure was repeated 3 times, and all leachates were combined. 3.0 mL of the leachate was taken to measure 125I using a gamma detector to monitor the chemical yield of iodine. This solution was re-combined with the original solution after gamma measurements. 1.0 mL trap solution was transferred to a 14 mL centrifuge tube for 127I measurements using ICP-MS. The remaining solution was used for further separation of iodine for 129I measurements.
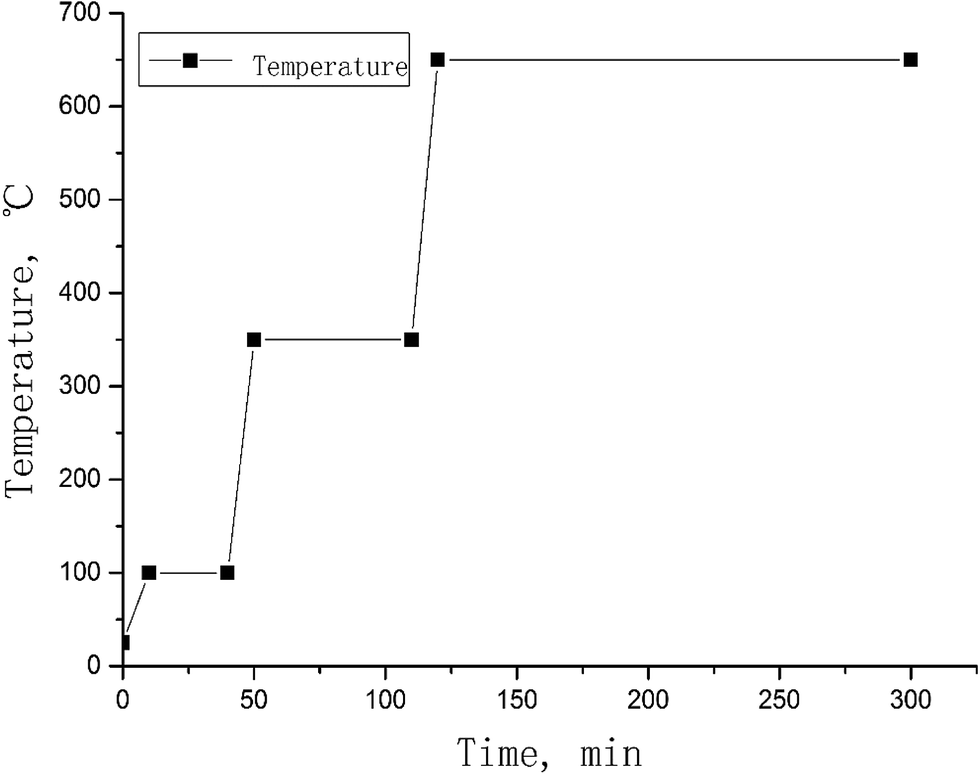 | ||
| Fig. 2 Protocol of alkaline ashing for vegetation samples. | ||
Procedure blanks were prepared using the same procedure (pyrolysis and alkaline ashing) as the samples, which were used to correct the analytical results of 129I and 127I in samples.
Separation of iodine from the trap solution and leachate for target preparation
Iodine in the trap solution was further separated by direct precipitation of AgI. 1.0 mg of 127I carrier and 1.0 mL of 1.0 mol L−1 NaHSO3 solution were first added to the trap solution, 3.0 mol L−1 HNO3 solution was then added to adjust to pH 1–2, iodate was reduced to iodide by sulphite under acidic conditions. 0.5 mL of 1.0 mol L−1 AgNO3 solution was then added to the sample solution, the formed AgI precipitate was separated by centrifugation at 3000 rpm for 5 min. 3.0 mL of the supernatant was taken to measure 125I using a gamma detector to monitor the recovery of iodine in this step. 5–8 mL of NH3 solution (7.5–25%) was added to the centrifuge tube and well mixed with the precipitate to wash the precipitate. After centrifuging at 3000 rpm for 5 min to separate the precipitate, 3.0 mL of supernatant was taken to measure 125I. The precipitate was transferred to a 1.5 mL centrifuge tube using water, and then separated by centrifugation. The precipitate (AgI) was finally dried at 60 °C for 129I measurements.Iodine in the leachate of alkaline ashing was separated using solvent extraction, and then precipitated as AgI. 1.0 mg of 127I carrier and 1.0 mL of 1.0 mol L−1 NaHSO3 solution were added to the leachate, 3.0 mol L−1 of HNO3 was then added to adjust to pH 1–2 to reduce all iodine to iodide. HNO3 solution was added slowly under stirring to avoid the violent reaction of HNO3 with carbonate which causes the release of a large amount of gas (CO2). After the gas in the solution was removed, the solution was transferred to a separatory funnel. 30 mL of CHCl3 was added and then 2 mL of 1.0 mol L−1 NaNO2 was added to oxidize iodide to I2, which was extracted into the CHCl3 phase by shaking. The organic phase was collected in a beaker, and 20 mL of CHCl3 was added to the separatory funnel again to extract the remaining iodine. The organic phases were combined and transferred to a separatory funnel, 10 mL H2O and 0.2 mL of 0.1 mol L−1 NaHSO3 solution were added. Iodine was back extracted into the aqueous phase by reducing I2 to iodide. This extraction and back extraction procedure was repeated. The aqueous phase was transferred to a centrifuge tube, and 0.5 mL of 1.0 mol L−1 AgNO3 was added into the tube to precipitate iodide as AgI. The AgI precipitate was separated by centrifugation, and then transferred to a 1.5 mL centrifuge tube using water, and separated again by centrifugation. The separated AgI in the 1.5 mL centrifuge tube was dried at 60 °C.
Measurement of 129I by AMS and 127I by ICP-MS
The dried AgI precipitates of samples, standard and procedure blanks were ground to fine powder and mixed with five times by mass of niobium powder (325-mesh, Alfa Aesar, Ward Hill, MA), which was finally pressed into a copper target holder using a pneumatic press (Zhenjiang Aode Presser Instruments Ltd.). 129I/127I atomic ratios in the prepared targets were measured by AMS using a 3 MV Tandem AMS system (HVEE) in the Xi'an AMS center. I5+ ions were chosen for the measurement, where 127I5+ was measured as charges (current) using a Faraday cup and 129I5+ was measured using a gas ionization detector. All samples were measured for 6 cycles and 5 min per sample in each cycle. The 129I/127I atomic ratios in the samples were normalized against the 129I/127I ratio in the standard. A detailed description of the AMS system and the measurement of 129I has been reported elsewhere.22129I/127I atomic ratios in procedure blanks were measured to be (1–2) × 10−13, which is two orders of magnitude lower than that in the samples (>1 × 10−11). The 129I/127I ratios in all samples were corrected for procedure blanks.1.0 mL of the trap solution was transferred to a vial, and deionized water was added to dilute the trap solution. For terrestrial vegetation (grass, pine needles, spinach and lichen), the trap solution was diluted 10 times, while for seaweed samples, the trap solution was diluted 500 times. The Cs+ solution (CsNO3) was spiked to the diluted trap solution at a Cs concentration of 2 ng mL−1. 127I in the prepared samples (0.0004 mol L−1 NaOH for seaweed and 0.02 mol L−1 NaOH media for other vegetation samples) was measured using ICP-MS (X series II, Thermo Scientific, USA). 1% NH3 solution was used as a rinsing solution between samples. The detection limit of 0.02 ng mL−1 for 127I was obtained. The iodine concentration in the sample was calculated by correction for chemical yield in the pyrolysis separation.
Results and discussion
In normal terrestrial vegetation samples, the concentration of iodine (<1 μg g−1 dry mass) and 129I levels (129I/127I atomic ratios in the range of 10−10 to 10−8 in the environmental sample without direct exposure to nuclear contamination) are very low. Although AMS is very sensitive for the measurement of 129I, the separation of a sufficient amount of iodine is still a key issue for the accurate determination of ultra-low level 129I in vegetation samples. Pyrolysis is a simple, rapid and effective method for the separation of iodine from solid samples, but the combustion process has to be well controlled, especially for vegetation samples of more than 0.5 g. This is very important for preventing possible explosion because of violent ignition and outflow of uncombusted particles and organic compounds into the trap solution because of rapid ignition, which will cause a failure of separation of iodine from samples. Our preliminary experiment has shown that the composition and flow rate of combustion assisting gases, temperature ramp procedure, temperature and time of ignition are critical parameters for the safe combustion of vegetation samples and reliable separation of iodine. For obtaining a sufficient amount of iodine from a vegetation sample for reliable AMS measurements of 129I, the recovery of iodine during combustion is another important issue besides using a large mass of sample. Combustion temperature and time are important issues to ensure a high volatilization efficiency of iodine from vegetation samples. In addition, the composition of the trap solution is also critical for ensuring effective trapping of iodine in the combustion process, as well as for the sensitive and reliable measurements of 127I using ICP-MS. In AMS measurements, only a small amount of iodine (<10 μg AgI per min) can be sputtered out from the target to produce the iodine ion beam for 129I measurements. The total mass and purity of the final precipitate of iodine is therefore important for measurement efficiency and the detection limit of 129I by AMS. All these parameters were investigated in this work in order to obtain optimal conditions for the separation of iodine from vegetation by pyrolysis and the reliable and accurate determination of low level 129I by AMS.Reliable ignition of vegetation in the pyrolysis process
Due to the high organic content, huge amounts of gases (CO2, CO, H2O, NOx, etc.) are created during the combustion of the sample, which cause a dramatically increased pressure in the furnace tube in a short time when a large amount of high organic content sample (>0.5 g) is ignited because of the limited volume of the tube furnace (<500 cm3). The connection of the outlet of the furnace tube to a bubbler filled with the trapping solution further exacerbates the buildup of the high pressure in the furnace tube, and raises the risk of explosion or damage, which has been often observed in the analysis of high organic content samples.18 In our preliminary experiment, it has been observed in a few cases that the trap solution was blown out of the bubbler during ignition. A few small explosions have happened, which broke the furnace tube. It was often reported that quartz wool was inserted into the second part of the furnace tube to improve the combustion efficiency of high organic content samples by trapping organic particles that escaped from the first part of the furnace and combusting these materials in the second part of the furnace (second zone of the furnace).17–19,29 This can prevent the uncombusted particles from entering the trap solution and creating a yellow and oil-like solution, which prevents further separation of iodine in the trapping solution using solvent extraction and/or AgI precipitation. However, we have observed in a few cases in our preliminary experiment that the sample with the quartz boat was flushed out from the inlet of the furnace tube and the quartz wool was pushed to another end of the furnace tube when quartz wool was inserted into the second part of the furnace tube for the pyrolysis of bigger size vegetation samples (3 g), meanwhile the trap solution was sucked into the high temperature furnace tube (900°) breaking the furnace tube. This is attributed to the further buildup of the pressure in the furnace tube by inserting quartz wool. The rapidly created gases during the ignition of the organic substance pushed the loose quartz wool in the second part of the furnace tube towards the narrow outlet of the furnace tube, which pressed and piled up the quartz wool, meanwhile the uncombusted particles were trapped on the quartz wool, causing the outlet of the working tube to be blocked. This sequentially prohibited the flowing out of created gases from the outlet of the furnace tube, causing the buildup of pressure, detaching the inlet cap from the furnace tube and flushing the sample, with a quartz boat, out of the tube from the inlet side. The sudden decrease in pressure in the furnace sucked the trap solution into the furnace tube which has been heated to 900 °C, breaking the furnace tube. In addition, the application of pure oxygen as the combustion assisting gas instead of air (or N2 + O2) did not improve the ignition process, and made the combustion difficult to control.All these incidents happened at the beginning of the ignition of the vegetation samples and occurred at different temperatures for different species of vegetation, indicating that the control of the ignition process is most critical for the successful pyrolysis of high organic content samples. The ignition temperature and the ramp speed at the ignition point are the key points for the ignition process control, and the composition of the combustion assistant gases is also important for the ignition process.
The pyrolysis experiments using different species of powdered vegetation samples (spinach, pine needles, Bristle grass, lichen, Laminaria japonica) were carried out to investigate the parameters influencing the ignition process of the vegetation samples by observing the bubbles in the trap solution. Dramatically increased bubbles (flow rates) were observed at about 250 °C (230–270 °C) when pure O2 was used as the combustion assisting gas, which causes a high risk of outflow of the trap solution from the bubbler, indicating a violent ignition. When mixed gases of O2 and N2 were applied, flow rates (bubblers in the trap solution) were still significantly increased at about 250 °C, but not as violently as with pure O2. It was observed that the outflow of the trap solution or oil-like yellow trap solution (with black particles) occurred when O2 or mixed gases with a volume ratio of O2:N2 less than 1 were applied, even for the combustion of 3 g of vegetation (spinach and grass) samples. For the application of mixed gases with O2:N2 volume ratios of less than 1:2, the ignition process is much less violent, meaning that the ignition process can be better controlled. During ignition and combustion, O2 is necessary for decomposing organic substances to gases (CO2, H2O, CO, and NOx), but excessive amounts of O2 can stimulate and accelerate the ignition process to create a large amount of gases in a short time, causing combustion failure. It is therefore important to control the ratio of O2:N2 in the combustion assisting gases, and better to use mixed gases of O2 and N2 in volume ratios of 1:2–1:3.
The flow rate of the combustion assisting gas is another important parameter for the ignition process of vegetation. It is better to adjust the flow rate of input gases below 200 mL min−1 to prevent the outflow of the trap solution from the bubbler and to obtain a sufficient contact time of the flowing gases containing iodine with the trap solution. The flow rate of the input gases should not be lower than 50 mL min−1 to maintain constant bubbles in the trap solution and to prevent the back flow (sucking) of the trap solution to the furnace tube. The optimal flow rates are observed to be 100–200 mL min−1.
The ramp speed of the temperature is the most critical parameter for controlling the ignition process, especially at the ignition point of the sample. By observing the variation of the flow rate in the trap solution, the ignition points of vegetation samples can be estimated, which vary from 230 °C to 270 °C depending on the species of vegetation. The rapid ramp up of temperature in the first zone of the furnace (where the sample was loaded) could result in violent ignition and dramatic creation of combustion gases, which caused the problems observed in our preliminary experiment. When the temperature ramp was controlled to less than 1.5 °C min−1, the ignition process was relatively smooth, although visibly increased flow rate occurred in the trap solution at the ignition point. The bubbling rate (flow rate of the outflow gases) was reduced to the level before the ignition point when the temperature ramped over 300 °C for most of the vegetation samples, and 350 °C for all types of high organic content samples. This indicates that in the ignition (carbonization) process, most of the vegetation samples were mineralized (burned) creating a large amount of gases (CO2, CO, and NOx). Afterwards, the combustion of the remaining carbon to CO2 (or CO) is relatively smooth.
Both species of vegetation and sample mass loaded in the furnace tube significantly influence the ignition process. For most species of vegetation (spinach, lichen and grass), the ignition process is quite smooth for up to 10 g samples, the trap solution is clear after pyrolysis, and a high recovery of iodine (>80%) was obtained. However, a problem occurred for pine needle samples when more than 5 g of sample was used; the trap solution was blown out from the bubbler when the temperature was increased to about 250 °C. This might be attributed to the high oil component in the pine needle samples, which accelerates the ignition speed when the temperature reaches the ignition point of the pine needle, resulting in the creation of a large amount of gases in a short time, causing elevated pressure in the furnace tube, and therefore an enhanced flow rate of gases, which blows out the trap solution from the bubbler. When the speed of the temperature ramp was decreased to 1 °C min−1 while all other parameters remained the same, the ignition process became smooth and the trap solution remained clear even after using up to 10 g of pine needle samples.
Based on the experimental results and above discussion, the optimal conditions for the ignition of vegetation samples are selected as follows: slow ramp of temperature of the furnace in the first zone at 220–300 °C at 1 °C min−1; applying mixed gases of O2:N2 with volume ratios of 1:2–1:3 as combustion assisting gases; a flow rate of assisting gases of 100–200 mL min−1.
When the temperature increases to 400 °C, all organic substances in the vegetation are carbonized, and the decomposition of the remaining carbon and mineralized components becomes slow. In this case, enhancing the O2 component of the assisting gases and flow rate does not significantly increase the evolved gases, but can accelerate the combustion process of the remaining samples and improve the volatilization of iodine from the sample as a gaseous form. Therefore, the assisting gas was changed to pure O2 and the flow rate was increased to 200 mL min−1.
For monitoring the chemical yield of iodine, 125I solution (Na125I) was spiked into the powdered vegetation samples before pyrolysis. In addition, a small amount of water might also exist in the vegetation sample even if it has been dried during sample preparation. It is important to remove all water from the sample before raising the temperature to the ignition point, to avoid splashing of the sample from the quartz boat when raising the temperature to more than 100°. Therefore, after placing the sample boat in the first zone of the furnace tube, the temperature is gradually increased to 100 °C and maintained for 20–30 min. Fig. 3 shows the optimal combustion protocol for vegetation samples, which proves to be safe and robust for the analysis of different species of vegetation, and has been successfully used for the analysis of more than 20 species of vegetation collected in China.
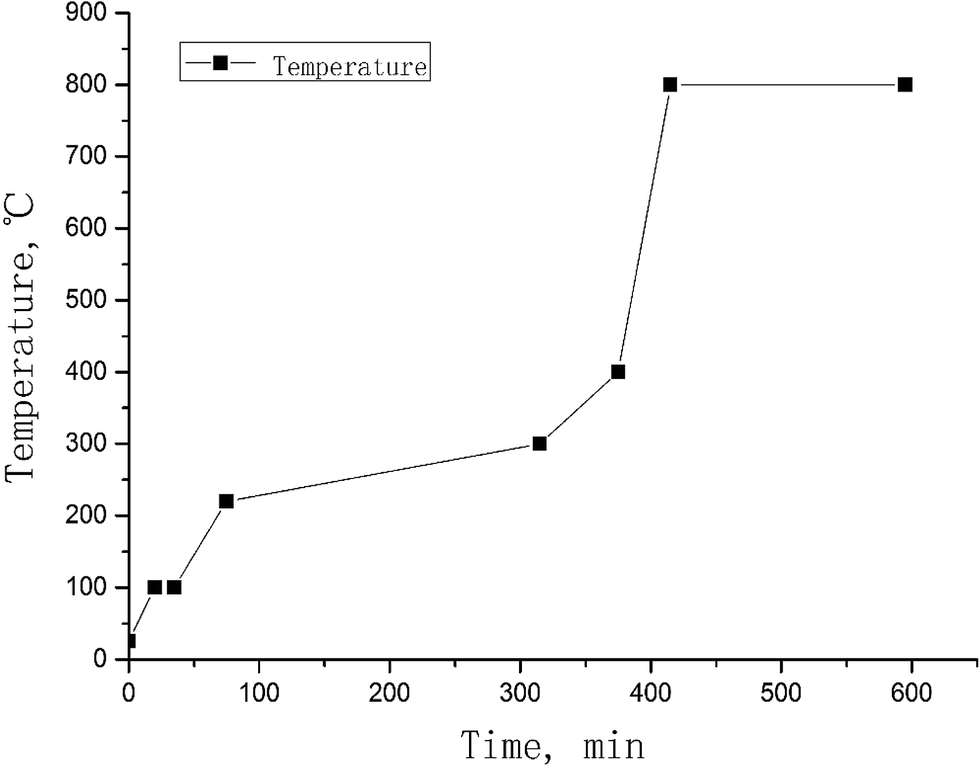 | ||
| Fig. 3 Pyrolysis protocol recommended for the separation of iodine from vegetation. | ||
The second zone of the furnace was directly ramped up to 900 °C after the pyrolysis process was started. This can combust any uncombusted particles escaping from the sample (first zone), and prevent them from entering the trap solution, which can lower the recovery of iodine. The high temperature in the second zone of the furnace can also reduce condensation and adsorption of iodine onto the tube wall at the outlet of the furnace tube. In addition, the length of the outlet end of the tube was adjusted to be less than 3 cm at the inlet of the bubbler, to ensure that the temperature of the outlet end of the furnace tube was higher than 300 °C, and therefore prevent the deposition of iodine on the inner wall of the furnace tube. A significant amount of iodine has been observed in the outlet end of the furnace tube when a long outlet end was applied.31 An extra heating wire wrapped around the outlet part of the furnace tube has been proposed to solve this problem.26,29 The approach of shortening the outlet end of the furnace tube and maintaining a high temperature in the second zone of the furnace from the beginning of the combustion until the end is proposed in this work, which is simple and easy to operate.
Effective vaporization of iodine in the pyrolysis of vegetation samples
During the carbonization process of vegetation at 230–300°, iodine could not be released in a gaseous form from the samples. Our 125I tracer experiment has shown that only a small fraction of iodine (<2%) was vaporized from the samples and collected in the trap solution below 400 °C. A combustion temperature range of 800–1000 °C has been used for the vaporization of iodine from solid samples (soil, sediment, and vegetation).17–19,22,26–31 It has been reported that the remarkable releases of iodine from solid samples do not occur before 700 °C, and a temperature of more than 800 °C does not further increase the releases of iodine from solid samples.32 Practically, 800 °C was often applied for the combustion of samples and vaporization of iodine.22,26,29,30 Our previous experiment for the separation of iodine from soil and sediment samples using pyrolysis has shown that combustion at 800 °C for 1 hour can completely vaporize iodine with a recovery of 93–98%.22 However, our preliminary experiments on the pyrolysis of vegetation samples have indicated that only about 50% of iodine was collected in the trap solution after 1 hour combustion at 800 °C. It has also been reported that iodine release is decreased with increased organic content in the samples.29The combustion experiment using 5 g of grass and the optimal protocol for ignition (Fig. 3) was conducted to investigate the influence of the combustion temperature and time. The results (Fig. 4) indicate that <10% and <16% of iodine was vaporized from vegetation samples before the temperature reached to 700 °C and 800 °C, respectively. Only about 40% of iodine was vaporized from the vegetation samples at 700 °C after 7 hours of combustion. The recovery of iodine was significantly improved when the temperature was increased to 800 °C, but more than 3 hours is needed to get a recovery of iodine higher than 80% (Fig. 4). It was also observed that the recovery of iodine increases rapidly in the first 2 hours of the combustion at 800 °C, afterward small amount of iodine was released and almost no more iodine was released after 4 hours of combustion. There is a slight variation among different species of vegetation, but the same trend was observed for all species of vegetation samples, the recoveries of iodine for the investigated vegetation samples vary from 83% to 92%. Increasing the combustion temperature to 900 °C can accelerate the vaporization of iodine from the carbonized samples. After one hour combustion at 900 °C, most of iodine can be vaporized from the vegetation sample (Fig. 4). The above experiments indicate that the release of iodine from high organic content vegetation during combustion is more difficult compared to soil and sediment which contain less organic substances. This might be attributed to relative alkalinity in the burned residue of the vegetation sample during combustion, which prevents iodine remaining in the sample to be released during combustion.
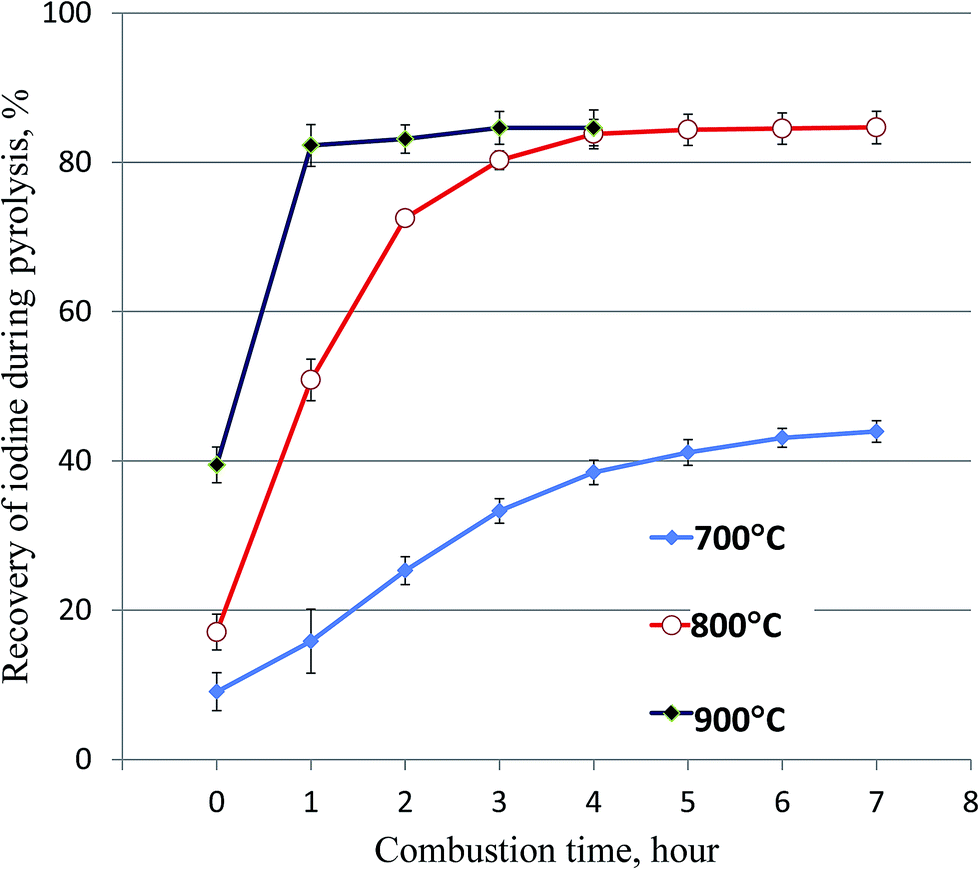 | ||
| Fig. 4 Influence of the combustion temperature and duration on the recovery of iodine in the pyrolysis of vegetation samples. The error bars represent one standard division of the results of 3 parallel experiments. | ||
A high recovery of iodine (about 85%), but not quantitative separation of iodine (up to 15% not collected in the trap solution) was obtained in the pyrolysis of vegetation samples. This might result from the incomplete release of iodine from vegetation and/or insufficient absorption of the released iodine in the trap solution. The experimental results (Fig. 4) show that only 40% of iodine was released at 700 °C and about 85% of iodine at 800 °C and 900 °C was released after 4–5 h combustion, and no more iodine was released when extending the combustion time. Examination of the outlet of the furnace tube by washing it using dilute NaOH (0.01 mol L−1) confirmed that no iodine was absorbed on the inner surface of the furnace tube. The incomplete recovery of iodine in the combustion process might relate to the formation of some species of iodine during the combustion of a high organic content sample, which is much less volatile at high temperatures up to 700–800 °C. The measurement of the residue sample for the 125I tracer after combustion showed that some iodine still remained in the residue after combustion at 700 °C, but almost no iodine (<1.0%) remained in the residue after combustion at 800 °C and 900 °C for more than 4 hours. The lost iodine has to be attributed to the insufficient trapping of the released iodine in the trap solution.
The tightness of the connection between the furnace tube and the bubbler through ground glass joints is an important issue for the collection of the vaporized iodine, which was always checked before combustion and during combustion to prevent any loss of iodine through this way. The similar recoveries of iodine in the 4 parallel experiments also confirm the tightness of the connection.
The flow rate of the input gases and the combustion time are also important for preventing the loss of the absorbed iodine in the trap solution. A long time gas flow through the trap solution (5–10 h) during combustion might cause loss of some trap solution from the bubbler through evaporation or nebulized droplets created and carried by the hot blowing gas. A tracer experiment has been conducted by spiking 125I into the trap solution and conducting the combustion experiment using vegetation samples and the same combustion protocol. The results showed that although the volume of the trap solution reduced by a factor of 5–10% after 3 hour combustion at 800 °C and a 5 h ignition process from room temperature to 800 °C under a gas flow rate of 150 mL min−1, 125I in the trap solution was not lost (<0.3%). An enhanced flow rate of input gases increases the loss of trap solution, but do not influence the 125I content in the trap solution.
To further investigate the trapping efficiency, a double trapping system was applied by two sequentially connected bubblers. The results showed that no iodine (<0.2%) was trapped in the second bubbler. This confirmed that negligible iodine was lost from the trap solution although gas flowed for a long time (7–10 h) through the trap solution during combustion. This might imply that the lost iodine escaped from the trap solution. It is well known that alkaline solution is efficient for trapping inorganic iodine, but not for organic gaseous iodine. The lost iodine that escaped from the trap solution might be some species of gaseous iodine, which is more common in high organic content samples, such as vegetation analyzed in this work. It should be mentioned that this investigation is based on the measurement of 125I spiked to the sample as NaI, which is not the same form as in the vegetation samples, in which most of iodine might be organic associated iodine. However, the previous investigation using 125I labeled vegetation26 demonstrated that this difference in the iodine species might not significantly change the results obtained in this work.
Based on the results and the discussion above, the optimal condition for the decomposition of vegetation to vaporize iodine is combustion at 800° for 3 hours or 900 °C for 1 hour. The optimal procedure for the pyrolysis of vegetation samples for the separation of iodine is shown in Fig. 3.
Trapping iodine released in the pyrolysis of vegetation
Iodine released in the pyrolysis of solid samples is often trapped in alkaline solution (e.g. NaOH, KOH, or tetramethyl ammonium hydroxide), which is further purified and converted to a suitable target for the measurement of 129I.17–19,26–31 Reductive reagents such as NaHSO3 or Na2SO3 at different concentrations (0.02–0.06 M) are often added to the trap solution to better absorb the released iodine in the solution.17–19,26,29,30 Trap solution without a reductant has also been used.27,28 Our experiment (Table 1) shows that the addition of 0.01–0.06 M NaHSO3 did not improve the recovery of iodine during the pyrolysis of vegetation samples (5 g of grass). In principle, the sulfite reductant can convert volatile I2 to stable iodide. It therefore can improve the trapping efficiency of I2 in the alkaline solution and the recovery of iodine during pyrolysis. The luck of influence of the addition of sulfite in the pyrolysis of vegetation might be attributed to the formation of reductive sulfite in the trap solution during the combustion of vegetation. Sulfur is often present in vegetation, which can be converted to SO2 during combustion with O2, and then to sulfite in NaOH solution. The formed sulfite in the trap solution may improve the trap efficiency of iodine, and prevent the loss of the trapped iodine.The influence of the NaOH concentration on the trap efficiency of iodine was also investigated. The results (Fig. 5) show that 60% of iodine was trapped when H2O was used as the trap solution, indicating a significant trapping function of H2O. This might be attributed to the fact that the composition of the trap solution was changed by the absorption of some released substances during combustion. Besides the absorption of the released SO2 and conversion to reductant NaHSO3, it was also observed that the pH of the H2O trap solution (pH 6.5) in the bubbler was elevated to >8 after the combustion of 5 g of the vegetation sample. This might be attributed to the releases of alkaline gases which were trapped in H2O, resulting in an elevated pH of the trap solution. The trap efficiency increased with the increased NaOH concentration from 0 to 0.2 mol L−1, while the recoveries of iodine remain constant (80–85%) for NaOH concentrations of 0.2–1.0 mol L−1 (Fig. 5). Besides the determination of 129I, stable 127I has to be determined to obtain the 129I/127I ratio, which is more useful in the evaluation of the 129I level in the environment compared to only 129I concentration. 127I in the trap solution is often measured using ICP-MS; the high salt content in the trap solution will reduce the measurement efficiency of iodine by ICP-MS, and deteriorate the detection limit of iodine. Therefore, 0.20 M NaOH was selected as the trap solution for iodine in the pyrolysis of vegetation samples.
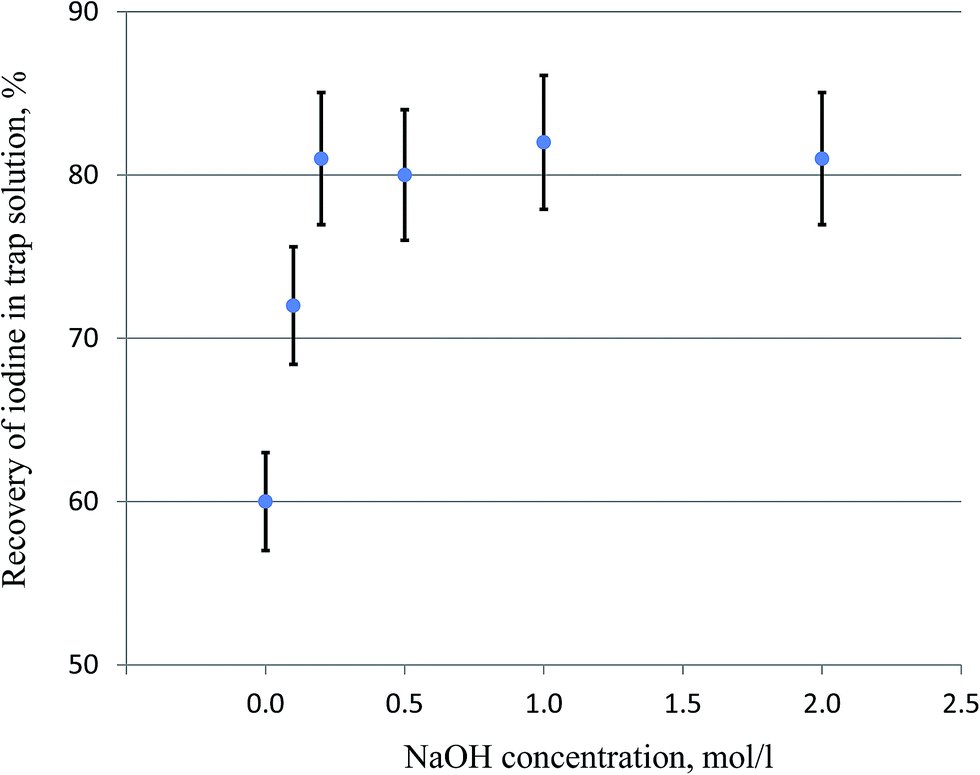 | ||
| Fig. 5 Influence of the NaOH concentration on the trapping efficiency of iodine released during pyrolysis using the protocol shown in Fig. 3 (except that for the last combustion step). The error bars represent one standard division of the results of three parallel experiments. | ||
The exact species of iodine released from the sample during combustion at high temperature is not well known, but is probably I2, HI, or HIO. But it is clear that all gaseous iodine released from soil samples can be collected in an alkaline trap solution because of the almost quantitative recovery of iodine (98–100%) in the trap solution (0.40 mol L−1 NaOH–0.04 mol L−1 NaHSO3) during the combustion of soil and sediment samples at 800 °C for 1 hour. However, organic gaseous iodine might be created in the combustion of vegetation samples, although the second zone of the furnace is heated to 900 °C from the beginning of pyrolysis.
Target preparation for the AMS measurement of 129I
In the AMS measurement of 129I, the separated iodine needs to be prepared in a solid form, often as AgI, which is mixed with thermally and electrically conductive materials (e.g. Ag and Nb powder) and pressed into a cathode holder. Because only a small amount of material (<0.5 mg) can be introduced into the AMS system by sputtering the target materials using a Cs+ ion source in <20 min and extracting I− to the beam line for 129I measurements,33 therefore other elements precipitated with AgI will increase the total mass of the target, decrease the amount of AgI pressed into the target holder because only totally 2–3 mg mixed powder (precipitate plus Nb powder) can be pressed into the target holder, and consequently deteriorate the detection limit of 129I, and worsen the analytical uncertainty. In addition, a complicated matrix in the target holder will also introduce additional interferences into the 129I measurement and deteriorate the analytical accuracy.Solvent extraction using CHCl3 or CCl4 has been widely applied to purify inorganic iodine in aqueous solution, and the purified iodide is then precipitated as AgI for AMS measurements. However, toxic organic wastes (CHCl3 and CCl4) are produced in the step. Meanwhile the multi-step separation also introduces more risk of 129I contamination during the experiment.
A direct precipitation of AgI was proposed to separate iodine in the trap solution in this work. However, the preliminary experiment showed a large amount of precipitation (>50 mg) after the addition of AgNO3 into the trap solution. This might result from the high chlorine and bromine contents in the trap solution because of a white precipitate (AgCl) instead of a yellow one (AgI). Chlorine is an essential nutrient in plants with a concentration of 2–20 mg g−1 (dry mass).33 Bromine is also widely present in vegetation at a level of more than 0.05 mg g−1 (dry mass). Chlorine and bromine can be also vaporized from the vegetation during the pyrolysis and be absorbed in the trap solution. The trapped chlorine and bromine can be reduced to Cl− and Br− when NaHSO3 is added and the pH is adjusted to <2 to reduce iodate to iodide. When AgNO3 was added to precipitate iodide as AgI, Cl− and Br− were also precipitated as AgCl and AgBr, causing a large amount of white precipitate. For 5 g of vegetation samples, >10 mg Cl− in the trap solution causes >40 mg of AgCl precipitate, which is much larger than the formed AgI (about 2 mg for 1.0 mg iodine carrier). Therefore, iodine in the precipitation is significantly diluted, which reduces the iodine amount pressed into the target holder. Based on the high solubility of AgCl and AgBr in NH3 solution, while AgI is insoluble, AgCl and AgBr can be selectively removed from the precipitate. However, the experiments (Table 2) show that a significant amount of iodine (>6%) was lost when a concentrated NH3 solution (25%) was directly used to remove the huge amount of AgCl. This might be attributed to the fact that AgI was wrapped in a large amount of AgCl and AgBr precipitates; when concentrated NH3 was added, AgCl and AgBr was quickly dissolved, and the small AgI particles wrapped in the AgCl and AgBr precipitates were released and suspended in the sample solution, which could not enter the precipitate during centrifugation. It was observed that the application of a low concentration of NH3 (7.5%) can remarkably prevent the loss of iodine in this step. When washing the AgI–AgCl–AgBr precipitate with 7.5% NH3 2 times, less than 1% of iodine was lost in this step (Table 2), meanwhile the entire weight of the precipitate was reduced to about 2 mg and the recovery of iodine monitored using 125I in this step is more than 97%, indicating a quantitative removal of AgCl and AgBr. This might be explained that AgCl and AgBr can be slowly dissolved in dilute NH3 solution and therefore repeated addition of NH3 is needed to sufficiently remove AgCl and AgBr from the precipitate. In this case, AgI particles are slowly released from the AgCl–AgBr precipitate. The released AgI particles were then aggregated to large particles and were separated by centrifugation. Based on these results, a simple direct precipitation method followed by washing the precipitate with 7.5% NH3 to remove AgCl and AgBr was applied for the preparation of the AgI precipitate suitable for the AMS measurement of 129I.
| NH3 concentration | 25.0% | 12.5% | 7.5a% |
|---|---|---|---|
| a Washing twice; the results are presented as the average and 1 SD of three replicate experiments. | |||
| Loss of iodine during NH3 washing (%) | 6.2 ± 1.0 | 4.6 ± 2.9 | 0.9 ± 0.1 |
| Loss of iodine in H2O washing (%) | 1.9 ± 1.8 | 2.6 ± 1.3 | 1.2 ± 0.4 |
| Total loss of iodine (%) | 8.0 ± 0.8 | 7.2 ± 1.6 | 2.1 ± 0.4 |
Analytical quality and detection limit
Due to the lack of suitable standard reference materials of vegetation for 129I, the vegetation samples were decomposed using both the traditional alkaline ashing and the developed pyrolysis, and 129I in the separated iodine was measured by AMS to evaluate the analytical quality of the developed method. Four species of terrestrial vegetation samples collected in Shaanxi and one seaweed sample, collected from Fujian, China, have been analyzed for 129I and 127I using the two methods. Meanwhile, blank samples for each batch of samples were also prepared and measured using the same procedure as for the samples. The analytical results (Table 3) show that the 129I/127I ratios in the procedure blanks for the developed method and the traditional alkaline ashing method are 2.56 × 10−13 and 2.44 × 10−13, respectively. They are two orders of magnitude lower than the measured values for the samples. The results shown in Table 3 are blank corrected values. The 129I concentrations in all samples measured by the developed method are in good agreement with that by the traditional alkaline ashing method (p < 0.05), indicating that the established pyrolysis method for the decomposition of vegetation samples and the direct precipitation method for preparing the AgI target are reliable for the determination of 129I and 127I in the vegetation samples.| Sample species | Sampling region | 127I concentration (μg g−1, dry) | 129I concentration (×106, atoms per g) | 129I/127I (×10−9) | Recovery of iodine (%) | |||
|---|---|---|---|---|---|---|---|---|
| Pyrolysis | Alkaline ash | Pyrolysis | Alkaline ash | Pyrolysis | Alkaline ash | |||
| a The uncertainties presented here are estimated in consideration of measurements and other steps in all analytical procedures, and extended uncertainties using a coverage factor of k = 1. | ||||||||
| Lichen | Baoji | 6.06 ± 0.28 | 262 ± 27.1 | 258 ± 23.3 | 9.14 ± 0.94 | 8.98 ± 0.81 | 83.72 ± 0.51 | 71.52 ± 9.78 |
| Spinach | Baoji | 1.73 ± 0.08 | 61.3 ± 6.30 | 56.5 ± 5.31 | 7.44 ± 0.76 | 6.86 ± 0.64 | 86.71 ± 4.14 | 77.03 ± 2.15 |
| Pine needles | Xi'an | 0.49 ± 0.02 | 11.9 ± 2.42 | 9.30 ± 0.94 | 4.77 ± 0.71 | 4.01 ± 0.40 | 78.53 ± 3.07 | 45.83 ± 2.91 |
| Grass | Xi'an | 0.76 ± 0.04 | 8.23 ± 1.12 | 8.64 ± 0.63 | 2.29 ± 0.31 | 2.41 ± 0.22 | 70.66 ± 4.00 | 81.75 ± 7.42 |
| Laminaria japonica | Fijian | 3978 ± 196 | 1840 ± 130 | 1740 ± 113 | 0.098 ± 0.008 | 0.087 ± 0.007 | 73.52 ± 4.01 | 91.38 ± 4.02 |
The detection limit of the presented method is estimated as 3 times the blank value, i.e. 7.5 × 10−13 for 129I/127I ratio. By using 1.0 mg of 127I carrier and 10 g of samples for analysis, a detection limit of 3.5 × 105 atoms per g (or 0.5 nBq g−1, or 7.5 × 10−17 g g−1) is calculated. Because the reported 129I/127I at the pre-nuclear level is 1.5 × 10−12, the developed method can be used for the analysis of all types of environmental vegetation samples, even pre-nuclear samples.
Determination of 129I and 127I in vegetation samples in China
The analytical results of 129I in vegetation from Shaanxi, China, (Table 3) show that even for the same sampling region, the 129I concentrations vary remarkably. The 129I concentration in lichen (260 × 106 at g−1) is 4 times higher than that in spinach (60 × 106 at g−1) and 25 times higher than that in grass and pine needles ((8–12) × 106 at g−1). However the differences of 129I/127I atomic ratios in these samples are much small. This is attributed to their significantly different concentrations of iodine (127I). Therefore, 129I/127I ratios, instead of the 129I concentration, are a more suitable indicator to evaluate the 129I level in the environment. In addition, the concentration of 129I in seaweed up to 1800 × 106 at g−1 is 10–100 times higher than that in the terrestrial samples in Shaanxi, China. However, the 129I/127I atomic ratio in the seaweed is 20–90 times lower than that in terrestrial samples. This is attributed to the strong enrichment of iodine in seaweed, especially in brown seaweed like Laminaria japonica,34 and the higher iodine concentration in seawater (50–60 ng g−1) compared to fresh water and soil solution (0.5–2 ng g−1).The 129I/127I ratios in pine needles and grass collected in Xi'an are more than 2 times lower than those in pine needles and lichen collected in Baoji, Shaanxi, China, showing a regional difference (Table 3). This might be attributed to the sampling sites. The sampling site in Baoji was in the rural area with less human and industrial activity, and iodine isotopes in this site are mainly at natural background levels. By contrast the samples from Xi'an were collected in the urban district, where the utilization of fossil fuels (natural gas, coal, and gasoline) in industry and for civil purposes releases a large amount of “old iodine” (with 129I/127I ratio lower than 1 × 10−12 in fossil fuel) into the atmosphere, which dilutes the 129I/127I ratio in the atmosphere, as well as the soil due to long term iodine releases, resulting in a low 129I/127I ratio in the vegetation from the urban area compared to the rural area in the same region (the similar 129I sources). The 129I/127I ratio in pine needles is 2 times higher than that in grass collected in the same site in Xi'an, China (Table 3), meanwhile, the 129I/127I ratio in lichen is significantly higher than that in spinach collected in the same site in Baoji, China. This might result from the different sources of iodine isotopes in different vegetation. Lichen and pine needle mainly take up iodine from air, while spinach and grass take iodine from both air and soil through root. Besides the recent deposition of atmospheric iodine (dry and wet deposition), iodine in the soil originates also from the long time atmospheric deposition and weathering of bed rocks which contain iodine of low 129I/127I ratio. Iodine deposited on the soil before human nuclear activity also has a low 129I/127I ratio, this results in a lower 129I/127I ratio in the soil compared to the present atmosphere due to the continuously increased 129I level in the atmosphere in the past 50 years.30,35 This result indicates that the species of plant and growing circumstance have a strong influence on the 129I level and 129I/127I ratio is the reliable and suitable parameter for the evaluation of the 129I level in the environment and its sources. However, the 129I concentration is more useful for the evaluation of radiation dose through ingestion.
Table 4 compares the 129I concentrations and 129I/127I ratios measured in this work with the reported values in the corresponding species of plants collected from different sites and dates. The 129I/127I ratios measured in the plants from Shaanxi, China, in this work are comparable to the values measured in the same region in 2009, indicating no significant variation of the 129I level in the past 5 years. Meanwhile, it also indicates that the measured value is reliable. Compared with the value in pine needles and grass collected in Beijing in 2000, the 129I/127I ratios in Xi'an in 2015 are lower by a factor of 2, while the 129I/127I ratios in the lichen and spinach collected in Baoji, China, in 2015 are similar to those observed in grass and needles in Beijing, China. This is attributed to the fact that the samples from Baoji and Beijing were collected in rural areas, with less dilution influence of 127I emitted mainly by burning fossil fuels in the urban area. This also indicates that the 129I level in Xi'an is similar to that in Beijing, China.
| Sample species | Sampling region | Sampling date | 127I concentration (μg g−1 dry) | 129I concentration (×106 atoms per g) | 129I/127I atomic ratio (×10−9) | Separation method | Measurement method | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pine needle | Xi'an | 2015 | 0.49 | 9.30 | 4.77 | Pyrolysis | AMS | This work |
| Beijing | 2000 | 0.40 | 8.11 | Alkaline ash | AMS | 37 | ||
| Lichen | Baoji | 2015 | 6.06 | 262 | 9.1 | Pyrolysis | AMS | This work |
| Sweden | 1987–1998 | 3160–14200 |
Acid digestion | AMS | 38 | |||
| Sweden | 1961–1975 | 950–5640 | Acid digestion | AMS | 38 | |||
| Chernobyl region | 1990 | 2.5–5.8 | 4330–56000 |
190–3900 | Pyrolysis | AMS | 38 | |
| Grass | Xi'an | 2015 | 0.76 | 8.23 | 2.29 | Pyrolysis | AMS | This work |
| Shaanxi | 2009 | 0.19–0.37 | 3.71–4.63 | 2.7–4.3 | Alkaline ash | AMS | 30 | |
| Beijing | 2000 | 2.08 | 5.97 | Alkaline ash | AMS | 37 | ||
| Denmark | 1998 | 0.46–1.0 | 607–659 | 135–277 | Alkaline ash | NAA | 20 | |
| Laminaria japonica | Fujian | 2015 | 3978 | 1840 | 0.087 | Pyrolysis | AMS | This work |
| Tianjin | 1998 | 3750 | 3453 | 0.19 | Alkaline ash | NAA | 37 | |
| Xiamen | 1996 | 2980 | 2473 | 0.17 | Alkaline ash | NAA | 37 | |
| Weihai | 1997 | 2850 | 1530 | 0.11 | Alkaline ash | NAA | 37 | |
| Seaweed | Shenzhen | 2012 | 254–535 | 250–730 | 0.16–0.28 | Pyrolysis | AMS | 38 |
| Slovenia | 2005–2006 | 234–506 | 1200–5100 | 0.53–3.1 | Alkaline ash | NAA | 39 | |
| Denmark | 1986–1999 | 99–594 | 22600–502000 |
34–370 | Alkaline ash | NAA | 40 | |
| Norway | 1980–1995 | 102–535 | 7250–151000 |
19–185 | Alkaline ash | NAA | 40 | |
| Greenland | 1997 | 212–368 | 990–1940 | 0.70–1.52 | Alkaline ash | NAA | 40 |
The measured 127I concentration in Laminaria japonica is comparable to the value in the same species of seaweed collected in other locations and dates. The 129I/127I atomic ratio in the seaweed collected in 2015 in Fujian, China, measured in this work is similar, but slightly lower than that in the seaweed collected in other sites along the China coast. This might be attributed to the fact that the sampling site in Fujian is located in the southeast coast of China at a lower latitude (24°N), which received a lower fallout of 129I and other radionuclides. A trend of decreasing 129I/127I ratios in the seaweed from north to south has been reported in the China coast.36 This also agrees with the global fallout mode of nuclear weapon testing, a high fallout level was observed in the middle latitude in the north hemisphere, and declined to the low latitude with the lowest level observed in the equatorial area, and Antarctica.
The 129I level in plants in Shaanxi, China, is much lower than that in Europe, the 129I concentration in lichen from Shaanxi, China, is 15–50 times lower than that collected in Sweden in 1987, and 4–20 times lower than that collected before 1987.38 The measured 129I/127I ratio in grass in Shaanxi, China, is 50–120 times lower than that collected in Denmark.20 This should be attributed to the different sources of 129I in Europe and China. Three large nuclear fuel reprocessing plants at Sellafield (UK), La Hague (France) and Marcoule (France) in Europe operating since the 1950s and 1960s have released a large amount of 129I both to air and to the seas, accounting for more than 90% inventory of 129I in the present environment. This has significantly enhanced the 129I level in Europe compared to other locations due to the high local deposition of the released 129I to the environment.2 The 129I level in the plants measured in this work is also 20–240 times lower than that in the lichen samples collected in the Chernobyl region; this is attributed to the high contamination of the local area of Chernobyl by the nuclear accident that happened in Chernobyl NPP in 1986, which released 1.6–6 kg of 129I into the atmosphere.2
The concentrations of iodine in different species of seaweed vary by a few orders of magnitude, high iodine concentrations have been observed in brown seaweed (1000–5000 mg kg−1), and the highest concentration of up to 3000–5000 mg kg−1 was measured in Laminaria japonica.34 Therefore, 129I/127I has to be applied to evaluate the environmental level of 129I in the marine environment. The 129I/127I ratio in seaweed collected in Fujian is lower compared to that reported in the seaweed in Shenzhen, China in 2012.39 The seaweed in Shenzhen, China was collected in Daya bay, where some rivers flow into the bay, 129I deposited on the catchment of these rivers was transported to the Bay, which increased the 129I concentration and the 129I/127I ratio in the seawater in Daya Bay, consequently increasing the 129I/127I ratios in the seaweed collected in this region. In addition, a nuclear power plant operating since the 1990s is located nearby the sampling site in the Daya Bay, so a small discharge of 129I from this nuclear power plant to the seawater and to the air followed by deposition on the local sea cannot be excluded. The 129I/127I ratio measured in the seaweed in this work is more than 10 times lower than that in the seaweed collected in the coast of Slovenia and Greenland, and 100–2000 times lower than that collected in the coast of Norway and Denmark.40,41 This should be attributed to the releases of European reprocessing plants to both air and the marine system. The marine discharged 129I from the European reprocessing plants has been transported to the Greenland coast through the North Sea, the Norwegian coast and the Arctic. The atmospheric releases of 129I from the European reprocessing plants were deposited, which highly enhanced the level of 129I in the environment of both the terrestrial and marine systems in Europe.
Conclusion
(1) A reliable pyrolysis method is developed for the safe and effective separation of iodine from vegetation samples by using a tube furnace. A recovery of more than 80% can be obtained, and up to 10 g vegetation samples can be analyzed. A detection limit of 6 × 10−17 g g−1 is obtained. The analysis of samples using both traditional alkaline ashing and the developed pyrolysis for sample decomposition showed that the results by two methods agree with each other, confirming the reliability of the developed method.(2) The temperature ramp speed during the carbonization of vegetation, especially around the ignition point temperature of the vegetation, is the most critical point for the safe and reliable decomposition of vegetation samples using pyrolysis. The ignition points vary with the species of plants, ranging from 230 °C to 300 °C.
(3) Combustion at more than 800 °C is needed to release most of the iodine from vegetation samples and more than 3 h at 800 °C or 1 h at 900 °C is needed to obtain a high recovery of iodine, which is much longer than that for soil and sediment samples.
(4) A direct and simple AgI precipitation method is presented to separate iodine from the trap solution from the pyrolysis. The excessive amount of AgCl and AgBr present in the precipitate due to the high Cl and Br contents in vegetation samples has to be removed, which can be easily completed by washing with NH3. The application of diluted NH3 is helpful to reduce the loss of AgI during the removal of AgCl.
(5) The 129I/127I atomic ratio is the suitable parameter to evaluate the 129I level in the environment because of different enrichment factors of iodine in different species of plants and less isotope fractionation of iodine in plants during uptake from the environment, while the 129I concentration has to be used when evaluating the intake of 129I and its radiation dose.
(6) A constant low 129I level was observed in Shaanxi, China compared to that in Europe. This is mainly attributed to the fact that the contribution of other sources is limited compared to the global fallout.
Acknowledgements
This work is supported by the Ministry of Science and Technology of China (Grant No. 2012IM030200 and 2015FY110800). Y. Y. Wang appreciates Dr Q. Liu for the AMS measurement of 129I and Dr Y. K. Fan and other staff in the Xi'an AMS center for their help in the experiment.References
- J. Fabryka-Martin, H. Bentley, D. Elmore and P. L. Airey, Geochim. Cosmochim. Acta, 1985, 49, 337–347 CrossRef CAS.
- X. L. Hou, V. Hansen, A. Aldahan, G. Possnert, O. C. Lind and G. Lujaniene, Anal. Chim. Acta, 2009, 632, 181–196 CrossRef CAS PubMed.
- G. Snyder, A. Aldahan and G. Possnert, Geochem., Geophys., Geosyst., 2010, 11, Q04010, DOI:10.1029/2009GC002910.
- X. L. Hou, A. Aldahan, S. P. Nielsen, G. Possnert, H. Nies and J. Hedfors, Environ. Sci. Technol., 2007, 41, 5993–5999 CrossRef CAS PubMed.
- X. L. Hou, H. Dahlgaard and S. P. Nielsen, Mar. Chem., 2001, 74, 145–155 CrossRef CAS.
- G. M. Raisbeck, F. Yiou, Z. Q. Zhou and L. R. Kilius, J. Mar. Syst., 1995, 6, 561–570 CrossRef.
- S. Xing, X. L. Hou, A. Aldahan, G. Possnert, K. L. Shi, P. Yi and W. J. Zhou, Environ. Sci. Technol., 2015, 49, 6691–6700 CrossRef CAS PubMed.
- P. Yi, A. Aldahan, V. Hansen, G. Possnert and X. L. Hou, Environ. Sci. Technol., 2011, 45, 903–909 CrossRef CAS PubMed.
- H. Reithmeier, V. Lazarev, W. Ruhm and E. Nolte, Sci. Total Environ., 2010, 408, 5052–5064 CrossRef CAS PubMed.
- M. Karcher, J. N. Smith, F. Kauker, R. Gerdes and W. M. Smethie Jr, J. Geophys. Res., 2012, 117, 72–82 CrossRef.
- X. L. Hou, P. P. Povinec, L. Y. Zhang, K. L. Shi, D. Biddulph, C. C. Chang, Y. K. Fan, R. Golser, Y. K. Hou, M. Jeskovsky, A. J. T. Jull, Q. Liu, M. Y. Luo, P. Steier and W. J. Zhou, Environ. Sci. Technol., 2013, 47, 3091–3098 CAS.
- C. H. He, X. L. Hou, Y. L. Zhao, Z. W. Wang, H. B. Li, N. Chen, Q. Liu, L. Y. Zhang, M. Y. Luo, W. G. Liang, Y. K. Fan and X. L. Zhao, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 632, 152–156 CrossRef CAS.
- P. He, X. L. Hou, A. Aldahan, G. Possnert and P. Yi, Sci. Rep., 2013, 3, 2045–2322 Search PubMed.
- X. L. Hou, A. Aldahan, S. P. Nielsen and G. Possnert, Environ. Sci. Technol., 2009, 43, 6522–6528 CrossRef CAS PubMed.
- R. Michel, A. Daraoui, M. Gorny, D. Jakob, R. Sachse, L. D. Romantschuk, V. Alfimov and H. A. Synal, J. Environ. Radioact., 2015, 150, 20–35 CrossRef CAS PubMed.
- X. L. Hou, A. F. Malencheko, J. Kucera, H. Dahlgaard and S. P. Nielsen, Sci. Total Environ., 2003, 302, 63–73 CrossRef CAS PubMed.
- T. Ohno, Y. Muramatsu, Y. Shikamori, C. Toyama, N. Okabe and H. Matsuzaki, J. Anal. At. Spectrom., 2013, 28, 1283–1287 RSC.
- A. Schmidt, C. Schnabel, J. Handl, D. Jakob, R. Michel, H.-A. Synal, J. Lopez and M. Suter, Sci. Total Environ., 1998, 223, 131–156 CrossRef CAS PubMed.
- Y. Muramatsu, Y. Ohmomo and M. Sumiya, J. Radioanal. Nucl. Ch. Ar., 1988, 123, 181–189 CrossRef CAS.
- X. L. Hou, H. Dahlgaard, B. Rietz, U. Jacobsen, S. P. Nielsen and A. Aarkrog, Analyst, 1999, 124, 1109–1114 RSC.
- N. Buraglio, A. Aldahan and G. Possnert, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 161, 240–244 CrossRef.
- X. L. Hou, W. J. Zhou, N. Chen, L. Y. Zhang, Q. Liu, M. Y. Luo, Y. K. Fan, W. G. Liang and Y. C. Fu, Anal. Chem., 2010, 82, 7713–7721 CrossRef CAS PubMed.
- R. J. Cox, C. J. Pickford and M. Thompson, J. Anal. At. Spectrom., 1992, 7, 635–640 RSC.
- J. M. Gómez-Guzmán, S. M. Enamorado-Báez, A. R. Pinto-Gómez and J. M. Abril-Hernández, Int. J. Mass Spectrom., 2011, 303, 103–108 CrossRef.
- J. E. Moran, U. Fehn and R. T. D. Teng, Chem. Geol., 1998, 152, 193–203 CrossRef CAS.
- M. N. Herod, R. J. Cornett, I. D. Clark, W. E. Kieser and G. St Jean, J. Environ. Radioact., 2014, 138, 323–330 CrossRef CAS PubMed.
- I. Krausova, J. Kucera and I. Svetlik, J. Radioanal. Nucl. Chem., 2013, 295, 2043–2048 CrossRef CAS.
- A. Shimada, M. Ozawa, Y. Kameo, T. Yasumatsu, K. Nebashi, T. Niiyama, S. Seki, M. Kajio and K. Takahashi, Nuclear Back-end and Transmutation Technology for Waste Disposal, ed. K. Nakajima, Springer, Japan, 2015, pp. 311–317 Search PubMed.
- E. Englund, A. Aldahan, G. Possnert and V. Alfimov, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 259, 365–369 CrossRef CAS.
- L. Y. Zhang, W. J. Zhou, X. L. Hou, N. Chen, Q. Liu, C. H. He, Y. K. Fan, M. Y. Luo, Z. W. Wang and Y. C. Fu, Sci. Total Environ., 2011, 409, 3780–3788 CrossRef CAS PubMed.
- K. I. Burns and M. R. Ryan, J. Radioanal. Nucl. Ch. Ar., 1995, 194, 15–23 CrossRef CAS.
- Y. Muramatsu, Y. Takada, H. Matsuzaki and S. Yoshida, Quaternary Geochronology, 2008, 3, 291–297 CrossRef.
- W. R. Chen, Z. L. L. He, X. E. Yang, S. Mishra and P. J. Stoffella, J. Plant Nutr., 2010, 33, 943–952 CrossRef CAS.
- X. L. Hou and X. J. Yan, Sci. Total Environ., 1998, 222, 141–156 CrossRef CAS.
- Y. K. Fan, X. L. Hou, W. J. Zhou and G. S. Liu, J. Environ. Radioact., 2016, 154, 15 CrossRef CAS PubMed.
- X. L. Hou, H. Dahlgaard, S. P. Nielsen and W. J. Ding, Sci. Total Environ., 2000, 246, 285–291 CrossRef CAS PubMed.
- B. Li, P. Q. Zhang, C. Y. Chen, M. He and Z. F. Chai, Chin. J. Anal. Chem., 2005, 33, 904–908 CAS.
- J. M. Gomez-Guzman, J. M. Lopez-Gutierrez, E. Holm and A. R. Pinto-Gomez, J. Environ. Radioact., 2011, 102, 200–205 CrossRef CAS PubMed.
- R. B. Zhang, H. Zhang, X. L. Hou, Z. F. Chai, Y. F. Chen and Y. K. Fan, J. Radioanal. Nucl. Chem., 2014, 301, 57–63 CrossRef CAS.
- A. Osterc and V. Stibilj, J. Environ. Radioact., 2008, 99, 757–765 CrossRef CAS PubMed.
- X. L. Hou, H. Dahlgaard and S. P. Nielsen, Estuarine, Coastal Shelf Sci., 2000, 51, 571–584 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |