
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsability of online-coupling ion exchange chromatography ICP-AES/-MS for the determination of trivalent metal complex species under acidic conditions
C.
Winter
and
A.
Seubert
*
Philipps-University Marburg, Faculty of Chemistry, Analytical Chemistry, Hans-Meerwein-Straße 4, D-35032 Marburg, Germany. E-mail: seubert@staff.uni-marburg.de
First published on 19th April 2016
Abstract
The determination of metal complex species in aqueous solutions by using chromatographic techniques is potentially affected or even impossible due to species decomposition during the separation. An often used technique for slower exchanging metal ions is ion exchange chromatography (IC), which is used for the separation and quantification of 1-1-complexes of trivalent metal ions. The test set consisted of chelating agents F−, Ox2−, NTA3− and EDTA4−, differing in their denticity, and the trivalent metals ions of Cr, Al, Fe, Ga, In and lanthanoids differing in their ligand exchange rate. It became apparent that the ligand exchange rate of the metal ion and the denticity of the chelator both play an important role. For slow exchanging metals and/or high denticity ligands, IC is a suitable tool for the determination of species distributions. A simple empirical equation is given to distinguish between inert, e.g. suitable for chromatographic separations, and labile 1-1-complexes of trivalent metal ions by using their complex formation constants and the aqua ligand exchange rate of the metal ions.
Introduction
Chelating agents are widely used in industrial processes, agriculture and as detergents. Although they are not toxic themselves, they are often poorly biologically degradable and capable of mobilizing metals from river sediments. The resulting metal complexes play an important role in several environmental and biological processes.1,2 The use of ion exchange chromatography for the determination of those complexes is obvious as the complexes are often charged.3 In the literature, two kinds of metal complexes are often analyzed by ion exchange chromatography, namely complexes with chelating agents of higher denticity or with kinetically inert metal ions. The most commonly used complexing agent is ethylenediamine tetraacetic acid (EDTA),4–12 the metal ions of aluminum and chromium are of special interest for IC determination. This is due to a variety of complexes with different ligands for aluminum, such as F−, Ac−, Ox2−, Cit3− and OH−,13–19 and the toxic behavior of chromium.20–22An important field of application of ion exchange chromatography for the determination of species distributions is biomaterials.23,24 These analyses are performed with samples of human serum,25 forest soil26 and plants,27 for example. In combination with an ICP-AES/-MS as a detector, a very selective and sensitive method of determination is provided, which is capable of monitoring the intake of aluminum by hydrangeas.28,29 Other di- and trivalent metal complexes were also analyzed regarding their occurrence in real samples.30–32
Retention models for ion exchange chromatography of metal complexes with multiple ionic eluents containing anions like CO32−, HCO3− and OH− have been developed using a chemical reaction named ion exchange. This has been done while taking into account the equilibria between the analyte and the eluent ions as well as the ion exchange equilibria.33–35
All the analyzed complexes in the fields mentioned before can be roughly summarized into complexes with chelating agents of higher denticity or with inert metal ions. This trend is also found in other analytical techniques related to ion exchange chromatography as explained in the following paragraphs.
The determination of metal ions in aqueous solutions is performed by ion exchange chromatography using their complexes. The eluent contains a chelating agent in excess regarding analyte ions, leading to complete complexation of metal ions. Here oxalate,36–38 NTA39 and EDTA40,41 are used, all being chelating agents of higher denticity.
A somewhat similar experimental setup with an opposing objective is used for the determination of amino polycarboxylic acids (APCAs) by means of IC-ICP-MS coupling via their indium and palladium complexes.42,43 The analyzed APCAs are possible ligands with at least three coordination sites.
High performance chelation ion chromatography (HPCIC) is a special type of cation exchange chromatography using immobilized chelating agents as exchange groups for the determination of metal ions. The most commonly used groups are related to oxalate ions due to the relatively high thermodynamic stability of the complexes and their medium lability compared to chelating agents with higher denticity. The use of immobilized tridentate ligands leads to slow mass transfer from the stationary to the mobile phase and poor separations.44,45
A combination of the two approaches stated in the paragraphs before is performed by using complexing agents as additives for the optimization of the separation in HPCIC. A prediction of the retention behavior of the metal ions can be done via the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β values of the metal complexes.46
β values of the metal complexes.46
Even in the field of the determination of complex stability constants with ion exchange chromatography, the kinetic lability of the metal ion complexes is of great importance.47–49 The analyzed complexes must be inert enough to be separated and ought not react or decompose on the separation column. This is fulfilled by multidentate chelating agents50,51 or metal ions with slow ligand exchange rates.52
A fundamental question concerning the use of IC for the determination of element species distributions and all the applications mentioned before arises, when we recall that complexation is an equilibrium reaction. Hence, in the state of thermodynamic equilibrium the forward and the reverse reactions are similarly fast. In the case of extraction of one reactant, the system tends to compensate this interference by recreating the missing reactant. While migrating through an ion exchange column this process also takes place. Metal ions, chelating agents and complexes normally differ in charge and are separated in the ion exchange process. The determination of the complex concentrations in the thermodynamic equilibrium is only possible if the reaction of the system due to the interference is much slower than the actual separation. For metals with slow ligand exchange rates this is apparently the case.53 Otherwise the measured species distribution and the state of the system before the separation do not match each other. The question whether a complex is inert enough to be analyzed by ion exchange chromatography ought to be answered before the chromatograms of samples are analyzed.
In order to answer the question of the applicability of IC for metal complexes, three factors need to be considered – the complex stability, the ligand exchange rate of the metal ion and the denticity of the chelating agent. The complex stability is quantified by the complex stability constant KStab, which provides information whether and to what extend a certain complex is formed for the given concentrations of the metal ion and the chelating agent. The KStab-value does not give information on how fast this equilibrium is reached and on the reaction rates in the equilibrium state of forward and reverse reactions. It is a purely thermodynamic description of the equilibrium state.
The ligand exchange rate of a metal ion is a kinetic property. For the comparison of the ligand exchange kinetics of metal ions of similar charge and related coordination geometry, the aqua ligand exchange rates kH2O can be used. As known from the literature, the kH2O-values differ from 10−10 s−1 for Ir3+ to 109 s−1 for lanthanoid ions.54 The underlying trend is transferable to other ligands, although the actual value may differ significantly. Nevertheless, it is a reference point for the reaction rates of the forward and reverse reactions in the equilibrium state, and therefore an indicator for the suitability of a metal complex for IC separations. For very fast exchanging metal ions, such as lanthanoids, a rapid complex decomposition is expected, leading to only the signal of the metal ion in the chromatogram, whereas for slow exchanging metal ions the chromatogram should correspond to the species distribution in the sample.
A characteristic contribution to both the thermodynamic and the kinetic sides of this question is the denticity of the chelating agent. Increasing denticity leads to higher KStab-values due to entropic effects on the thermodynamic side. On the kinetic side, it is apparent that the denticity is an important factor for the kinetic lability of a complex, because every coordinative bond between the metal ion and ligand has to break at the same time, so that complex decomposition can take place. The probability of that decreases exponentially with the number of coordination sites.
In light of these thoughts a thorough investigation of several trivalent metals and chelating agents with different denticities is still a task to be carried out. Based on the achieved data a general trend for the applicability of IC for the determination of 1-1-metal complexes is obtained.
Experimental
Selection criteria of the samples and calculation of species distribution
A maximum of the 1-1-complex with a minimum of hydroxides and higher complexes in the samples was aspired. For the elimination of hydroxides, a pH of 2.0 was selected and the metal-to-ligand-ratio was determined by thermodynamic calculation of the species distribution.55 For this purpose the stability constants of all complexes in the considered system need to be known. For most of the simple systems this requirement can be easily fulfilled. The chosen metal-to-ligand-ratios are reported in the Experimental procedure section. The elemental species distribution of the samples was calculated by Visual MINTEQ ver. 3.0 with the therein provided stability constants of the complexes.56 The temperature was set to 20 °C, ionic strength was calculated and pH was fixed at 2.0.Experimental procedure
364 nm (Ga), 230606 nm (In) and at m/z = 27 (Al), 53 (Cr), 57 (Fe), 139 (La), 140 (Ce), 141 (Pr), 146 (Nd), 147 (Sm), 153 (Eu), 157 (Gd), 159 (Tb), 163 (Dy), 165 (Ho), 166 (Er), 169 (Tm), 172 (Yb) and 175 (Lu). The flow rate of 1 mL min−1 of the chromatographic system is compatible with the Cross-Flow-Nebulizer (ICP-AES) and the Micro-Flow-Nebulizer (ICP-MS). The outlet of the IC column was directly connected to the nebulizer of the ICPs. A coupling of the software for the IC and both ICPs was not possible. The measurements were synchronized manually.
For some elements commercial calibration standards were employed: cerium 1000 mg L−1 ICP-standard (Fluka), dysprosium 1000 mg L−1 ICP-standard (Fluka), gadolinium 1000 mg L−1 ICP-standard and indium 1000 mg L−1 ICP-standard (Fluka). The remaining metals were obtained in the solid form and 1000 mg L−1 stock solutions in 0.7 mol L−1 nitric acid were self-prepared. Aluminumnitrate-nonahydrate (p.a., Merck), chromiumnitrate-nonahydrate (99%, ABCR), iron(III)nitrate-nonahydrate (p.a., Merck), erbiumchloride-hexahydrate (99.9%, Ventron), europiumchloride (99.9%, Ventron), holmiumchloride-hexahydrate (99.9%, Ventron), lanthanumnitrate-hexahydrate (99%, Riedel-de Haën), lutetiumchloride-hexahydrate (99.9%, Ventron), neodymium(III)oxide (99.9%, HEK), praseodymiumchloride-hexahydrate (99.9%, Ventron), samarium(III)oxide (99.9%, Ventron), terbiumchloride-hexahydrate (99.9%, Ventron), thuliumchloride-hexahydrate (99.9%, Ventron) and ytterbium(III)oxide (99.9%, Sigma Aldrich) were used. The stock solutions of the chelating agents were self-prepared out of the following salts by dilution in water: sodiumfluoride (p.a., Riedel-de Haën), oxalic acid-dihydrate (99.5%, Fluka), nitrilotriacetic acid (99%, Riedel-de Haën) and ethylenediamine tetraacetic acid disodium dihydrate (99%, Fluka).
The samples were prepared by diluting appropriate amounts of the stock solutions, so that a concentration of 10 mg L−1 (20 mg L−1 for Ga and In) of the metals and the aspired metal-to-ligand-ratio were achieved after dilution with the eluent. The pH of the samples was checked and adjusted if necessary. The chromium samples were heated for 24 hours at 60 °C after preparation to speed up the complex formation. For the metal–NTA and metal–EDTA solutions a molar ratio of 1:1, for the metal–fluoride solution 1:0.4, for chromium, aluminum and iron and 1:1 for the other metal ions and for the metal–oxalate solution 1:0.4 were chosen.
Results and discussion
Thermodynamic calculation of the species distribution
The thermodynamic determination of the species distribution of the M–L-species in the samples was done using Visual MINTEQ and the therein provided stability constants of the complexes.56 The stability constants of the chromium oxalate complexes were taken from the report of Ciavatta.58 In Table 1 the species distribution of the chromium, aluminum, iron, gallium, indium and lutetium samples is shown. The values are rounded to one decimal digit and ratios less than 0.1% are omitted.| M:F |
M3+ | [MF]2+ | [MF2]+ | [M(OH)]2+ | [M(OH)2]+ |
|---|---|---|---|---|---|
| Cr:F 1:0.4 |
78.1 | 20.7 | 0.2 | 1.1 | |
| Al:F 1:0.4 |
61.4 | 37.5 | 1.1 | ||
| Fe:F 1:0.4 |
22.2 | 57.9 | 6.6 | 13.1 | 0.2 |
| In:F 1:1 |
77.6 | 21.6 | 0.3 | 0.6 | |
| Ga:F 1:1 |
44.2 | 48.1 | 4.1 | 3.5 | |
| Lu:F 1:1 |
91.0 | 9.0 |
| M:Ox |
M3+ | [MOx]+ | [MOx2]− | [MHOx]2+ | [M(OH)]2+ |
|---|---|---|---|---|---|
| Cr:Ox 1:0.4 |
65.3 | 33.9 | 0.8 | ||
| Al:Ox 1:0.4 |
61.5 | 37.9 | 0.3 | 0.2 | |
| Fe:Ox 1:0.4 |
37.7 | 39.6 | 0.1 | 22.2 | |
| In:Ox 1:0.4 |
65.3 | 30.6 | 0.9 | 2.7 | 0.5 |
| Ga:Ox 1:0.4 |
58.5 | 35.2 | 1.6 | 4.6 | |
| Lu:Ox 1:0.4 |
72.6 | 27.1 | 0.2 |
| M:NTA |
M3+ | MNTA(aq) | [MHNTA]+ | [M(OH)NTA]− | [M(OH)]2+ |
|---|---|---|---|---|---|
| Cr:NTA 1:1 |
1.3 | 98.7 | 1.4 | ||
| Al:NTA 1:1 |
51.9 | 26.0 | 22.0 | ||
| Fe:NTA 1:1 |
0.6 | 90.3 | 8.3 | 0.3 | 0.4 |
| In:NTA 1:1 |
3.0 | 10.9 | 86.1 | ||
| Ga:NTA 1:1 |
7.0 | 92.1 | 0.3 | 0.6 | |
| Lu:NTA 1:1 |
57.6 | 42.4 |
| M:EDTA |
M3+ | [MEDTA]− | MHEDTA(aq) |
|---|---|---|---|
| Cr:EDTA 1:1 |
56.1 | 43.9 | |
| Al:EDTA 1:1 |
24.3 | 9.5 | 66.1 |
| Fe:EDTA 1:1 |
76.6 | 23.4 | |
| In:EDTA 1:1 |
99.1 | 0.9 | |
| Ga:EDTA 1:1 |
0.1 | 84.7 | 15.2 |
| Lu:EDTA 1:1 |
3.4 | 96.6 |
The amount of the main metal–ligand-species in the samples shown in Table 1 is far above the detection limits, so that they ought to be seen in the chromatograms of the samples when no or only minor species disintegration occurs. The species distributions of the other lanthanoid samples are similar to that of lutetium and the six metal ions in Table 1 are used as examples.
Ion exchange chromatography
Fig. 1 shows the overlays of chromatograms obtained for the chromium, aluminum, iron, gallium, indium and lutetium ion samples as mentioned in Table 1.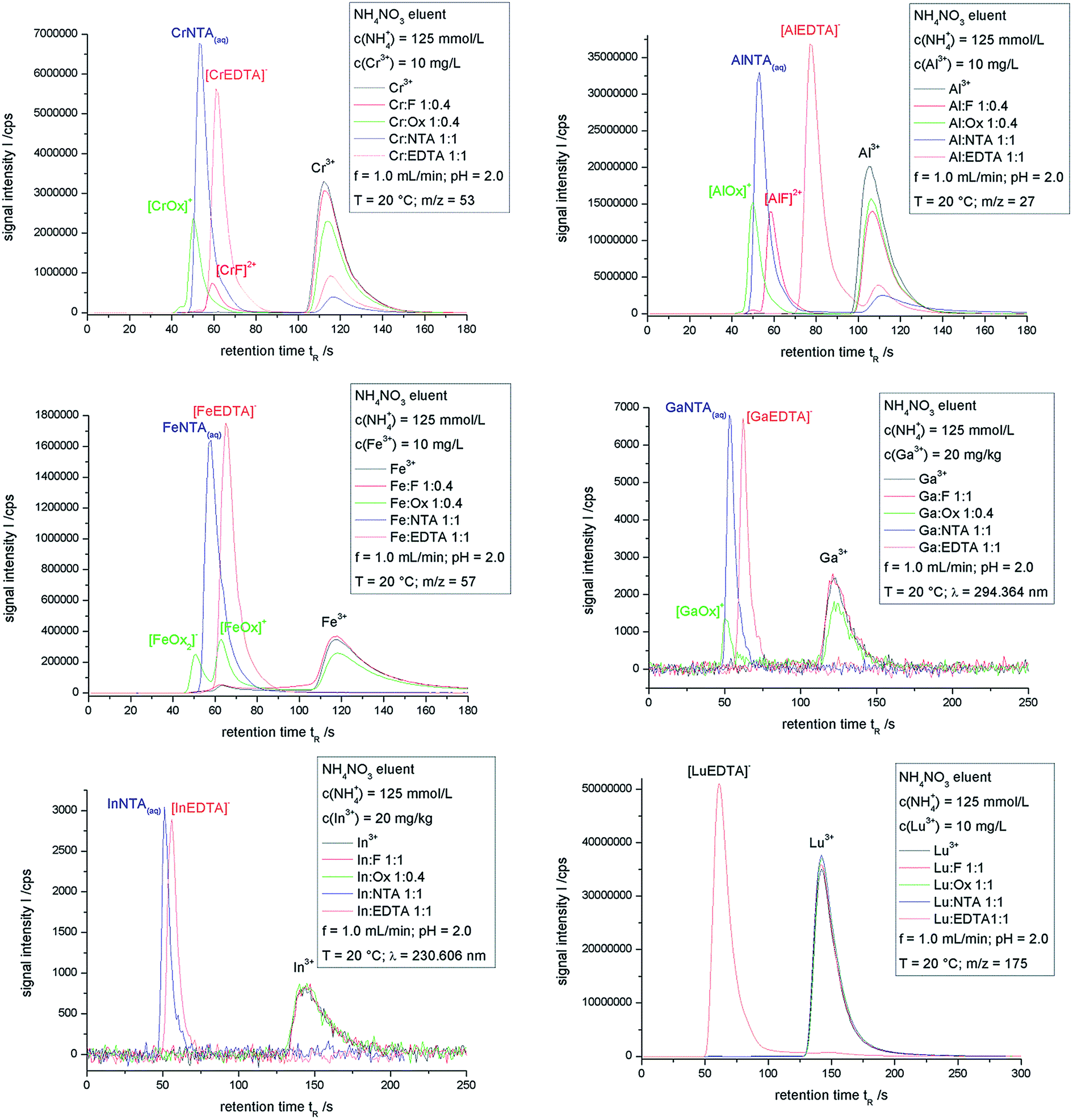 | ||
| Fig. 1 Overlaid chromatograms obtained for the samples described in Table 1. | ||
Signals of metal complexes cannot be obtained for the lutetium samples except for the [LuEDTA]− complex. All other ligand complexes of lutetium are kinetically not inert enough to be detected after IC separation. The complex decomposition is much faster than the measuring technique. Therefore, a complex with a very fast exchanging metal ion such as lutetium requires four coordinative bonds between the metal ion and the ligand to be inert on the time scale of ion exchange chromatography. This result is transferable to all other trivalent lanthanoid ions.
For the InF and InOx samples only the chromatographic signal of the In3+ ion can be seen, whereas for NTA and EDTA the corresponding complex signal is detected. In contrast to the lanthanoid ions, which exhibit a higher aqua-ligand-exchange-rate,50 the NTA-complex of indium is inert enough to persist the chromatographic run. So for indium ions, only three coordinative bonds to the chelating agent are needed to form an inert complex. One additional comment has to be made concerning the In–NTA-complexes. The thermodynamic calculation in Table 1 shows two different NTA-complexes, InNTA(aq)− and [InHNTA]+. These complexes undergo fast conversion resulting in one chromatographic peak containing both species. This is the case for all complexes with the possibility of the ligand to be protonated.
The chromatograms of the gallium samples also show the NTA and EDTA complexes and in addition a signal attributed to [GaOx]+ in the oxalate sample. The chromatogram of the gallium–fluoride-sample is identical to that of the gallium-sample containing no chelating agent. Therefore the species disintegration of GaF-complexes is much faster than the separation. Two further comments have to be made about the GaOx-sample: the area of the [GaOx]+-peak is smaller than expected after the thermodynamic calculation and the baseline in the sector between this signal and the Ga3+-peak is slightly lifted. Those two points indicate a species disintegration taking place on the same time scale as the chromatographic separation. Gallium complexes with bidentate chelating agents are at the boundary between inert and labile complexes, whereas complexes with higher numbers of coordinative bonds from one ligand to the metal ion are inert enough to be analyzed by IC.
The situation of the iron complexes is somewhat similar to that of the gallium complexes, but the analysis of the chromatograms is hindered by the distinct tendency of iron to form hydroxo complexes even at low values of pH. This can be seen by the lifted base line in the chromatogram between the retention times of 60 to 110 s. The chromatograms of the iron fluoride sample and the iron samples without a chelating agent are closely related to each other, which leads to the assumption that the iron fluoride complexes are labile and decomposing very fast. The NTA and EDTA complexes are inert and only the complex signal is obtained. The ion exchange chromatography of the iron oxalate sample leads to a chromatogram with peaks corresponding to [FeOx2]−, [FeOx]+ and Fe3+. The percentaged peak area of [FeOx2]− is higher than the thermodynamic calculation, whereas it is lower for [FeOx]+. The Fe3+ fraction is also higher than expected. The loss of [FeOx]+ and the gain of Fe3+ might suggest a species disintegration, whereas the [FeOx2]− results might be explained by faulty complex formation constants. The very similar aqua ligand exchange rates of iron and gallium, which exhibits species disintegration of the [GaOx]+ complex, also support the complex decomposition of [FeOx]+, but a definite classification is not possible. A complex decomposition on the column leads to analytes eluting in between the complex signal and the Fe3+ peak, resulting in the formation of a plateau. The iron hydroxo complexes are also eluted in the same time window, making a distinction between those species nearly impossible.
All the aluminum complexes analyzed here are kinetically inert. Even the chromatogram of the aluminum fluoride sample corresponds to the thermodynamic calculation of the species distribution. So for the slow exchanging Al3+ ion one coordinative bond to a fluorido ligand is enough to form an inert complex. The only real discrepancy between the calculation and the IC separation is seen for the Al–NTA sample. The percentaged peak area of AlNTA(aq) exceeds the calculated amount, which might also be explained by faulty complex formation constants.
The chromatographic separations of the chromium complexes are similar to those of the aluminum samples. All complexes are inert on the time scale of the measurement. This can be concluded to be due to the non-formation of a plateau in between the complex signal and the Cr3+ peak. Some discrepancies have to be marked between the IC separation and the thermodynamic calculation. The [CrEDTA]− and CrNTA(aq) complexes are found in lower amounts than expected. These deviations might be due to faulty complex formation constants. A major problem for the determination of KStab values for chromium complexes is the substitution rate of coordinated water by the ligand, which is extremely low. A waiting time for reaching thermodynamic equilibrium of more than a year at room temperature is required.53 This might lead to lower KStab values and minor amounts of complexes in the calculated species distribution.
The combination of those results leads to a summary of the inertness and lability of 1-1-complexes of trivalent metal ions as shown in Fig. 2.
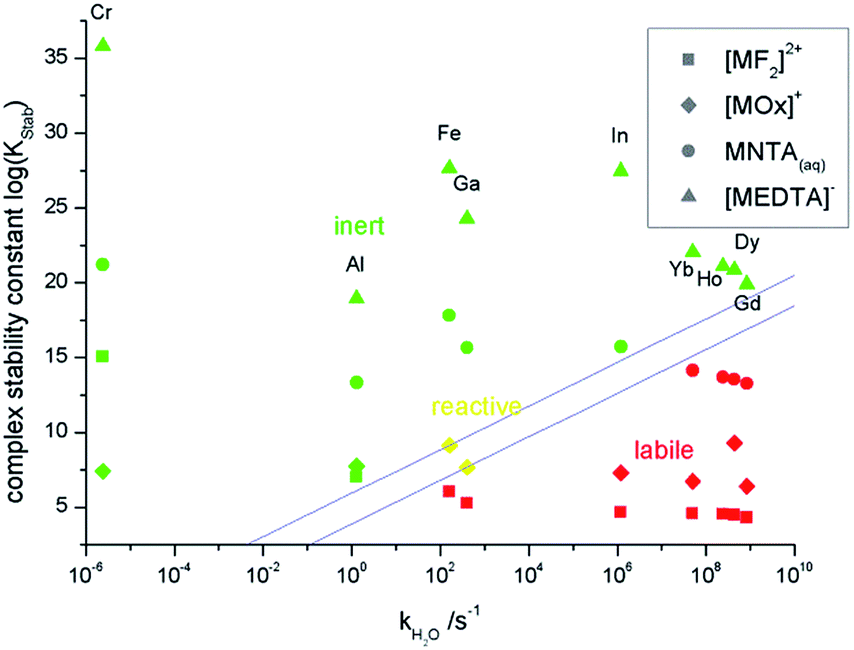 | ||
| Fig. 2 Summary of the kinetic results of the metal–ligand screening and their use in IC. | ||
Three regions can be identified in Fig. 2. In the upper part above the straight lines all inert metal complexes are found, whereas all labile complexes are located in the part below the straight lines. The figure gives a graphical answer to the question whether a thermodynamically stable 1-1-complex of the examined metal ions is analyzable by IC or not.
The EDTA-complexes are all kinetically inert enough to be analyzed via IC. For NTA this is only the case for metal ions with a slower aqua-ligand-exchange rate than the lanthanoids. This boundary between inertness and lability shifts for oxalate-complexes to the area of iron and gallium and for fluoride-complexes to the area between iron and aluminum. [GaOx]+ is neither inert nor labile on the IC-time scale as shown above. [FeOx]+ is expected to behave similarly, but analysis is hindered by the distinct tendency to form hydroxo-complexes.
Overall the inertness of a metal ion complex depends on the ligand-exchange kinetics of the metal ion, illustrated here by the aqua-ligand-exchange rate, on the thermodynamic stability of the complex and on the denticity of the ligand. The inert area increases to faster ligand exchanging metal ions with increasing denticity from monodentate (fluoride) to tetradentate (EDTA). In Fig. 2 an empirically derived frontier between the inert and the labile region is shown. Those two lines are described by the empirical i/l-value (inert/labile) defined as:
| i/l = log(KStab) − 1.45log(kH2O) | (1) |
This equation can be used to estimate the inertness or lability of a 1-1-complex of a trivalent metal ion by its complex formation constant and the aqua ligand exchange rate of the metal ion. For i/l < 3.9 the complex is labile, whereas for i/l > 6.0 it is inert. Values in between 3.9 and 6.0 indicate lability on the same time scale as the chromatographic separation. Eqn (1) ought to be usable for other trivalent metal ions and the chelating agent at pH = 2. The values in eqn (1) and the limits mentioned here are only applicable for the applied conditions. Similar equations with different actual values can be derived for other experimental setups.
Conclusions
The determination of the species distribution of trivalent metal ion samples by ion exchange chromatography is only suitable for complexes with slow ligand exchange rates. This exchange rate is dependent on the aqua-ligand-exchange rate of the metal ions and the denticity of the chelating agent. For complexes with ligand exchange rates being way too fast, only the chromatographic signal of the free metal ion can be detected and the information about the species distribution before the separation is lost. The gallium-oxalate sample gives a chromatogram that is an overlay of the chromatographic separation and the complex decomposition. The information about the species distribution is nevertheless in that chromatogram and ought to be gained from it by an appropriate mathematical analysis. The distinction between the inert and labile 1-1-complexes of trivalent metal ions can be achieved by the empirical i/l-value introduced here.The obtained results should be applicable to other ligands and their 1-1-complexes with trivalent metal ions depending on their denticity.
Notes and references
- L. A. Finney and T. V. O'Halloran, Science, 2003, 300, 931–936 CrossRef CAS PubMed.
- K. H. Thompson and C. Orvig, Science, 2003, 300, 936–939 CrossRef CAS PubMed.
- R. N. Collins, J. Chromatogr., A, 2004, 1059, 1–12 CrossRef CAS PubMed.
- R. N. Collins, B. C. Onisko, M. J. McLaughlin and G. Merrington, Environ. Sci. Technol., 2001, 12, 2589–2593 CrossRef.
- W. W. Bedsworth and D. L. Sedlak, J. Chromatogr., A, 2001, 905, 157–162 CrossRef CAS PubMed.
- R. Tófalvi, K. Horváth and P. Hajós, J. Chromatogr., A, 2013, 1272, 26–32 CrossRef PubMed.
- Z. Chen, Q. Sun, Y. Xi and G. Owens, J. Sep. Sci., 2008, 31, 3796–3802 CrossRef CAS PubMed.
- A. A. Ammann, J. Chromatogr., A, 2002, 947, 205–216 CrossRef CAS PubMed.
- Z. Chen, G. Owens, K.-R. Kim and R. Naidu, Anal. Chim. Acta, 2007, 599, 163–169 CrossRef CAS PubMed.
- M. C. Bruzzoniti, E. Mentasti and C. Sarzanini, Anal. Chim. Acta, 1999, 382, 291–299 CrossRef CAS.
- C. Sarzanini, G. Sacchero, E. Mentasti and P. Hajós, J. Chromatogr., A, 1995, 706, 141–147 CrossRef CAS.
- Z. Chen, G. Owens, M. Megharaj and R. Naidu, Rapid Commun. Mass Spectrom., 2009, 23, 419–424 CrossRef CAS PubMed.
- P. M. Bertsch and M. A. Anderson, Anal. Chem., 1989, 61, 535–539 CrossRef CAS PubMed.
- S. H. Sutheimer and S. E. Cabaniss, Anal. Chem., 1995, 67, 2342–2349 CrossRef CAS.
- B. Mitrovic, R. Milacic and B. Pihlar, Analyst, 1996, 121, 627–634 RSC.
- G. Borrmann and A. Seubert, Anal. Chim. Acta, 1996, 332, 233–239 CrossRef CAS.
- T. Bantan, R. Milacic and B. Pihlar, Talanta, 1998, 47, 929–941 CrossRef CAS PubMed.
- T. Bantan, R. Milacic and B. Pihlar, Talanta, 1998, 46, 227–235 CrossRef CAS PubMed.
- G. Borrmann and A. Seubert, Anal. Chim. Acta, 1999, 386, 77–88 CrossRef CAS.
- C. Ehrling, U. Schmidt and H. Liebscher, Fresenius' J. Anal. Chem., 1996, 354, 7–8 Search PubMed.
- C. Barnowski, N. Jakubowski, D. Stuewer and J. A. C. Broekaert, J. Anal. At. Spectrom., 1997, 12, 115–1161 RSC.
- P. P. Coetzee, J. L. Fischer and S. J. van Vuuren, S. Afr. J. Chem., 2004, 57, 8–14 CAS.
- J. Szpunar, TrAC, Trends Anal. Chem., 2000, 19, 127–137 CrossRef CAS.
- J. Szpunar, Analyst, 2000, 125, 963–988 RSC.
- T. Bantan, R. Milacic, B. Mitrovic and B. Pihlar, J. Anal. At. Spectrom., 1999, 14, 1743–1748 RSC.
- F. Michalas, V. Glavac and H. Parlar, Fresenius' J. Anal. Chem., 1992, 343, 308–312 CrossRef CAS.
- T. Bantan, R. Milacic, B. Mitrovic and B. Pihlar, Fresenius' J. Anal. Chem., 1999, 365, 545–552 CrossRef CAS.
- M. Busch, PhD thesis, Philipps-University, Marburg, 2001.
- O. Happel, PhD thesis, Philipps-University, Marburg, 2007.
- R. Svendsen and W. Lund, Analyst, 2000, 125, 1933–1937 RSC.
- A. A. Ammann, Anal. Bioanal. Chem., 2002, 372, 448–452 CrossRef CAS PubMed.
- E. Bakkaus, R. N. Collins, J.-L. Morel and B. Gouget, J. Chromatogr., A, 2006, 1129, 208–215 CrossRef CAS PubMed.
- P. Hajos and G. Revesz, J. Chromatogr., A, 1993, 640, 15–25 CrossRef CAS.
- P. Hajos, G. Revesz, O. Horvath, J. Peear and C. Sarzanini, J. Chromatogr. Sci., 1996, 34, 291–299 CAS.
- P. Hajos, O. Horvath and G. Revesz, Adv. Chromatogr., 1998, 39, 311–350 CAS.
- M. C. Bruzzoniti, S. Cavalli, A. Mangia, C. Mucchino, C. Sarzanini and E. Tarasco, J. Chromatogr., A, 2003, 997, 51–63 CrossRef CAS PubMed.
- P. Janos, J. Chromatogr., A, 1996, 719, 457–461 CrossRef CAS.
- R. M. Cassidy and L. Sun, J. Chromatogr., A, 1993, 654, 105–111 CrossRef CAS.
- R. S. Dybczynski, K. Kulisa, M. Pyszynska and A. Bojanowska-Czajka, J. Chromatogr., A, 2015, 1386, 74–80 CrossRef CAS PubMed.
- S. Motellier and H. Pitsch, J. Chromatogr., A, 1996, 739, 119–130 CrossRef CAS.
- A. M. Dolgonosov, React. Polym., 1992, 17, 95–99 CrossRef CAS.
- J. Knoell and A. Seubert, J. Chromatogr., A, 2012, 1270, 219–224 CrossRef CAS PubMed.
- D. Nette and A. Seubert, Anal. Chim. Acta, 2015, 884, 124–132 CrossRef CAS PubMed.
- P. Jones and P. N. Nesterenko, J. Chromatogr., A, 1997, 789, 413–435 CrossRef CAS.
- P. N. Nesterenko and P. Jones, J. Sep. Sci., 2007, 30, 1773–1793 CrossRef CAS PubMed.
- P. Jones and P. N. Nesterenko, J. Chromatogr., A, 2008, 1213, 45–49 CrossRef CAS PubMed.
- F. H.-J. Lin and C. Horváth, J. Chromatogr., A, 1992, 589, 185–195 CrossRef CAS.
- P. Janos, J. Chromatogr., A, 1995, 699, 1–10 CrossRef CAS.
- L. Xing and D. Beauchemin, J. Anal. At. Spectrom., 2009, 24, 336–339 RSC.
- P. Janos, J. Chromatogr., A, 1993, 641, 229–234 CrossRef CAS.
- C. Huang and D. Beauchemin, J. Anal. At. Spectrom., 2006, 21, 317–320 RSC.
- E. Sato, S. Miya, K. Saitoh, S. Saito and M. Shibukawa, J. Chromatogr., A, 2011, 1218, 922–928 CrossRef CAS PubMed.
- F. Helfferich, in Ion Exchange, Dover Publications, McGraw Hill, 1995 Search PubMed.
- L. Helm and A. E. Merbach, Coord. Chem. Rev., 1999, 187, 151–181 CrossRef CAS.
- J. E. Shaff, B. A. Schultz, E. J. Craft, R. T. Clark and L. V. Kochian, Plant Soil, 2010, 330, 207–214 CrossRef CAS.
- J. P. Gustafsson, Visual MINTEQ (Version 3.0), KTH, Sweden, 2010 Search PubMed.
- M. Raskop, A. Seubert and A. Grimm, Pat. EP 1 842 592 A1, 2007.
- L. Ciavatta, M. Iuliano and A. Vitiello, Ann. Chim., 2000, 90, 169–179 CAS.
| This journal is © The Royal Society of Chemistry 2016 |