
Underwater measurements using laser induced breakdown spectroscopy
S. Michael
Angel
*,
Joseph
Bonvallet
,
Marion
Lawrence-Snyder
,
William F.
Pearman
and
Janna
Register
Department of Chemistry and Biochemistry, The University of South Carolina, Columbia, SC 29208, USA
First published on 17th November 2015
Abstract
New data is presented related to high-pressure underwater laser-induced breakdown spectroscopy measurements. LIBS calibration curves in high-pressure water are shown for elements that are relevant to hydrothermal vent fluid chemistry. The use of internal standards in high-pressure solutions is shown, and the use of hydrogen and oxygen, produced from decomposition of water by the LIBS plasma, as an internal standard for underwater LIBS measurements is demonstrated. The use of double-pulse LIBS for high pressure underwater measurements provides improved sensitivity but it is shown that the technique is limited by reduction in the size of the laser-induced vapor bubble with increased solution pressure.
Introduction
Laser-induced breakdown spectroscopy (LIBS) is a relatively simple spectroscopic technique that allows rapid multi-elemental analysis of solids, liquids and gases with little or no sample preparation, and was first reported by Brech and Cross in 1962.1 LIBS is well suited for in situ, non-contact and remote elemental analysis because, in principle, only optical access to the sample is required for analysis.2–20 Of particular interest to our research group is applying underwater LIBS to in situ elemental analysis of hydrothermal vent fluids in the deep ocean.21–24Hydrothermal vents occur at mid-ocean ridges where seawater circulates through the semipermeable ocean crust, exchanging ions along the way. Hot, mineral-laden water eventually is expelled at vent orifices at temperatures of 200–405 °C, and at ambient pressures from 8.1 × 106 to 3.6 × 107 Pa (8.1 to 360 Bar), corresponding to ocean depths of 800–3600 m.25 The fluid composition changes due to interactions with the host rock, phase separation, and possibly magma degassing. For example, alkali metals and transition metals are leached from the rock and concentrated in vent fluids, while Mg and SO4 are removed from the fluid. Cooling due to mixing with seawater also causes additional changes to the fluid chemistry.26 Conventional analytical measurements of vent fluids are difficult because of high temperatures and pressures at the vent sites, the corrosive nature of the vent fluids, the difficulty of taking samples at these depths, and irreversible changes in composition that occur when samples are removed to the surface. The relatively few in situ methods that have been described for deep-ocean hydrothermal vent measurements include voltammetry for dissolved species27–30 and mass spectrometry for volatile compounds.31,32 Thus, there is a need to build instrumentation and sensors that can measure in situ chemical changes in dynamic environments.28
LIBS is a promising technique for in situ measurements and has the potential for use in the deep ocean, but it has not yet been demonstrated under conditions like those found at deep-ocean hydrothermal vents. There are several reports of LIBS measurements of submerged solids,33–45 some at pressures relevant to hydrothermal vent applications.44 However, there have been few published reports of underwater LIBS of dissolved species since Cremer's first report in 1987,46–56 likely because water quenches the LIBS plasma, making sensitivity an issue for many important species.24 Only a handful of reports demonstrate analytical measurements of dissolved species in water at pressures that correspond to ocean depths where hydrothermal vents are found—100 Bar (10 MPa) in the case of Thornton, et al.,56 and up to 300 Bar (30 MPa) by our group.21–23 This paper includes new calibration results for relevant species at pressures up to 250 Bar in seawater, the use of internal standards for calibration, including the first demonstration of O and H as internal standards in underwater LIBS, and unpublished double-pulse (DP) LIBS results showing shadowgraphy images of plasma bubble growth and its effect on optimizing the laser interpulse delay time for high pressure solutions.
Experimental
Fig. 1 shows the general setup that was used for single-pulse (SP) and double-pulse (DP) LIBS measurements (A) and for time-resolved shadowgraphy (B). The high-pressure sample chamber used for SP and DP LIBS measurements, constructed by welding together stainless steel Swagelok fittings (Central Swagelok Company, Solon, OH), was described previously.21,22 The cell has 4, 25 mm diameter, 6 mm thick, sapphire optical windows which can be configured for collinear or orthogonal DP LIBS. For DP measurements, two Nd:YAG laser beams (Continuum Surelite III, 5 ns pulses, 1064 nm, 1 Hz) are used with the cell, aligned either collinear or orthogonal to one another, with the emission collinear with one or both lasers, or orthogonal to both lasers. The lasers were focused using five-cm focal length, spherical, fused silica lenses. For collinear light collection, a laser-focusing lens was also used to collect plasma emission, and another lens was used to focus the collected plasma emission onto a collection optical fiber (FO). For DP LIBS, the energies of the first and second laser pulses were low, typically 8 and 25 mJ at the laser, respectively. The e-folding scale (i.e., the distance over which the laser power attenuates by 1/e) for pure water at 1064 nm is about 2.2 cm,57 therefore at the laser focus (∼3 cm into the bulk solution) approximately 24% of the laser energy (including reflection losses) incident on the focusing lens reaches the laser focus region inside the sample chamber (e.g., ∼1.9 mJ for the first pulse). Assuming a laser spot size in solution of ∼100 microns, the fluence of the first pulse is ∼7 GW cm−2.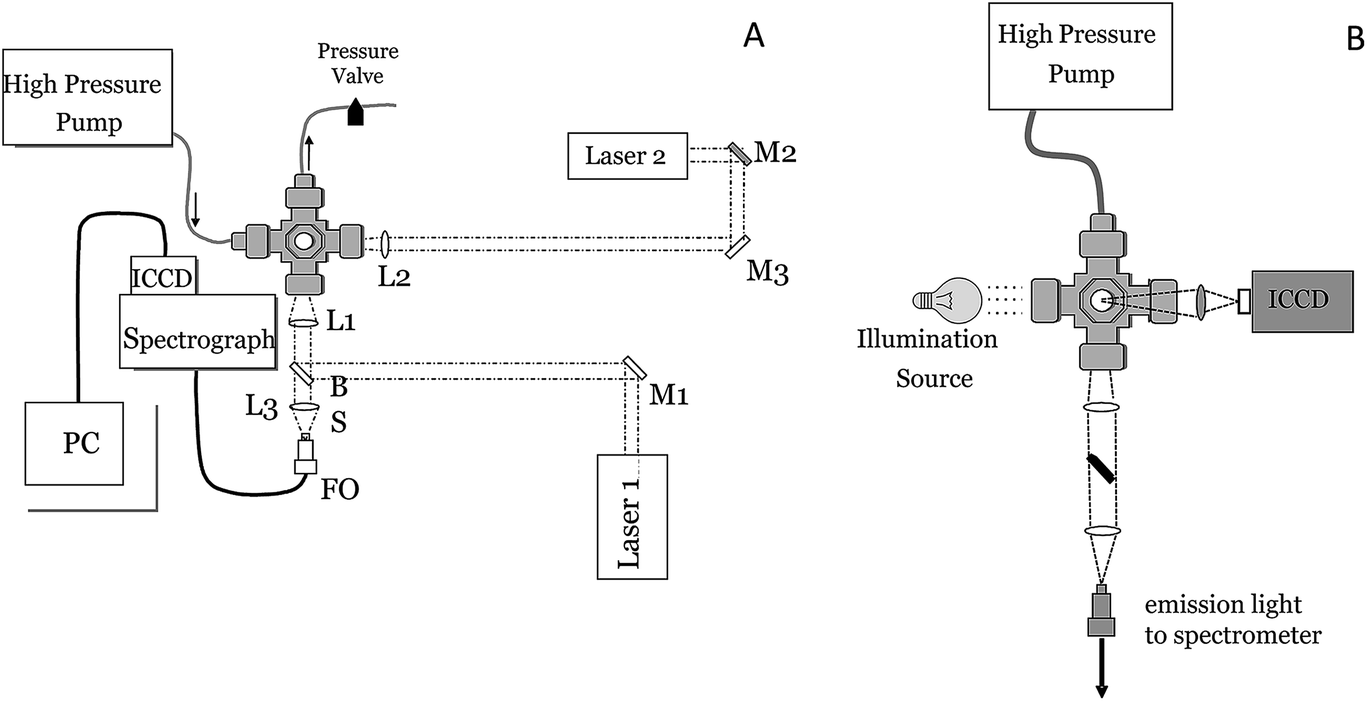 | ||
| Fig. 1 Schematic of the experimental setup used for SOP LIB and DP LIBS (left) and shadowgraphy (right). L = lens, M = mirror, FO = optical fiber. | ||
Spectral measurements were made using a Chromex spectrograph (Model 250IS/RF, 0.25-m, f/4) with a 1200 groove per mm grating blazed at 500 nm to provide 0.1 nm spectral resolution using a 25 μm-slit and an intensified CCD (ICCD) camera (Princeton Instruments I-Max 1024E). Detector gating was controlled using a pulse-timing generator, and a delay generator was used to synchronize laser timing for spectral measurements and for image acquisition. Shadowgraph images of the laser-induced plasma and bubble were obtained by shining a broadband light source through the cell, orthogonal to the incident laser pulse (see Fig. 1B). A camera lens (Nikon series E, 50 mm, f/1.8) was used to image the shadow of the plasma region directly onto an ICCD. The imaging lens was positioned approximately 6 and 9.5 cm from the plasma region and imaging ICCD, respectively, providing an image magnification of approximately 1.6 (corresponding to a field of view of ∼16 microns per CCD pixel) and a total imaged area of 9 × 6 mm around the plasma breakdown region. A neutral density filter (OD 2) was positioned in front of the imaging lens to reduce the amount of light incident on the detector. Imaging detector timing was controlled using a Princeton Instruments Model PG-200 pulse generator and Model ST-138 detector controller.
In the following discussion with regard to DP LIBS, td, tb, and Δt, refer to the interval between the second laser pulse and collection of the plasma emission (td), the ICCD gate width which determines the duration of the plasma emission integration (tb), and the inter-pulse delay time, which is the delay between the two sequential laser pulses (Δt), respectively. Unless otherwise stated, bubble and plasma images were acquired using a 1-μs integration time (the smallest window that provided significant image contrast between the laser-induced bubble and the background illumination) and a detector delay, td, of 200 ns (the shortest delay time after the laser pulse that emission could be easily recorded with the imaging ICCD). All plasma emission was recorded using a similar detector delay, td, of 200 ns and an integration time, tb, of 1 μs, to ensure that SP- and/or DP-LIBS plasma emission was collected over a significant duration of the plasma lifetime (plasma emission decreased to negligible levels within several μs).
Results and discussion
Most underwater SP LIBS measurements are made by collecting the emission light collinear with the excitation laser, in a backscatter geometry. Fig. 2 (top) shows SP LIBS spectra of 50 ppm Li in deionized water (DI) and in seawater (SW), top left. It was found that the constituents of seawater have little effect on SP LIBS emission spectra. The only significant difference in these spectra is a slightly higher background in seawater. This was also observed for several other species. Li spectra measured at 1 Bar and 250 Bar solution pressures are also shown in Fig. 2 (top right). There is no significant effect of solution pressure on the intensity or width of the 671 nm Li emission line, other than a small reduction of emission intensity at the higher pressure. Whether this is a real effect or an artifact of the response of the cell windows to pressure is not clear. This result has also been shown for other species in earlier published work.21 Calibration curves for Li are shown in the lower part of Fig. 2, for solutions that also contain 50 ppm K, at 1 Bar (left) and 250 Bar (right). On the left, the closed circles (solid fitted line) show plots of the emission intensity while the open circles (dashed fitted line) show plots of the Li/K ratio. The response is linear over the range shown, though there is an outlier in the emission intensity plot at 35 ppm, which is corrected using K as an internal standard. There also seems to be increased variability between measurements at the higher pressure, which is also reduced by measuring the ratio of Li to K, as shown by the open circle data points in Fig. 2, lower left. In separate unpublished studies the Li detection limit was found to be 0.125 ppm and 500 ppb, using 1064 nm and 532 nm excitation, respectively in solution at 280 Bar pressure. This is below the concentrations typically found in the regions around deep-ocean hydrothermal vents, suggesting that SP LIBS is suitable for measuring Li at these sites.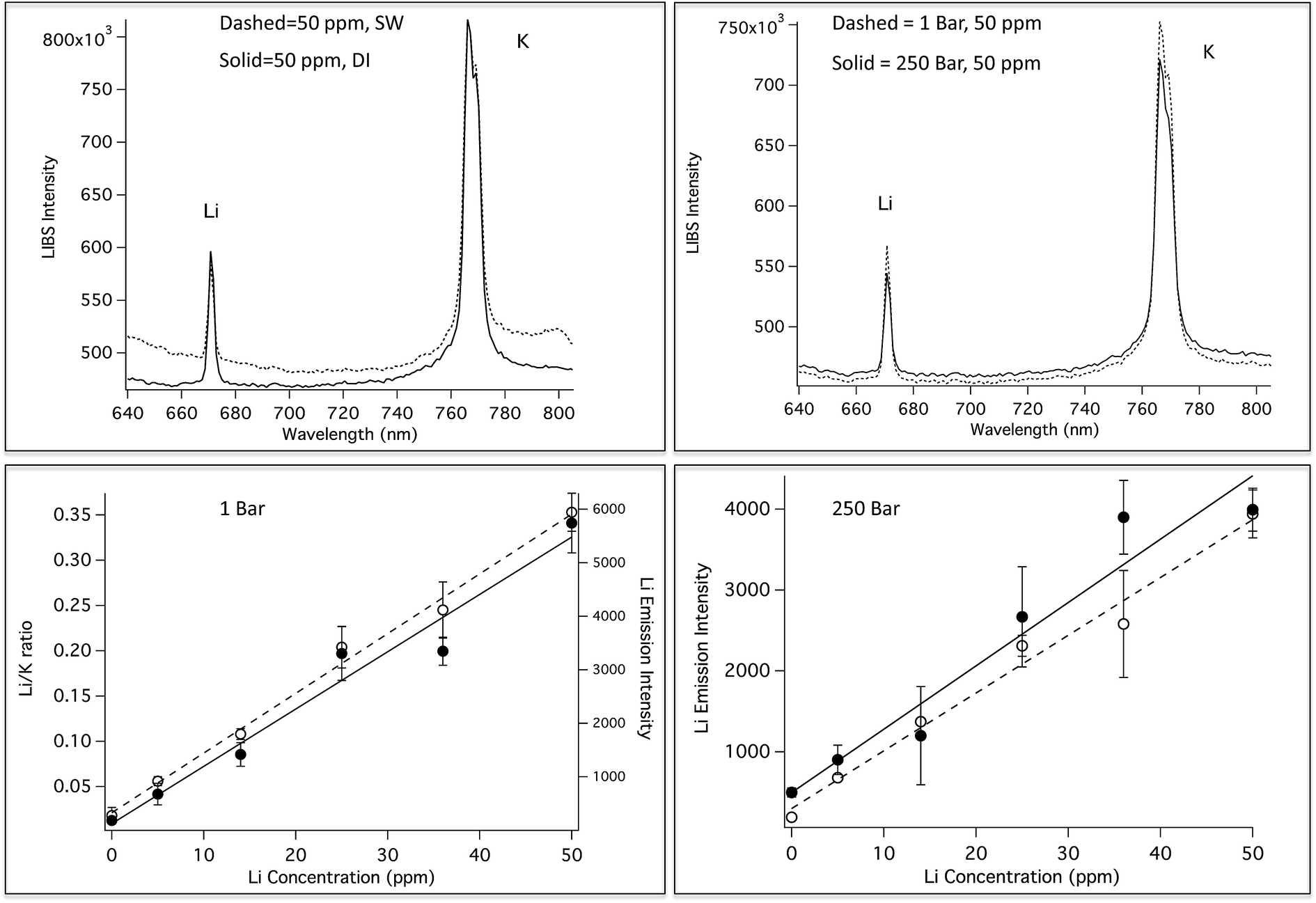 | ||
| Fig. 2 Top: SP LIBS spectra of a solution of 50 ppm Li and K in deionized (DI) water and seawater (SW, left), and for the solution at 1 Bar and 250 Bar (right). Lower Left: Li calibration curves at 1 Bar, plotted as Li emission intensity (solid circles, solid fitted line) and as the Li/K ratio (open circles, dashed fitted line). Lower Right: Li calibration curves at 250 Bar, plotted as Li emission intensity for SW (solid circles, solid fitted line) and DI (open circles, dashed fitted line). | ||
Previously published work shows that the emission intensity for Na and Mn decreases at high laser pulse energy.21,23Fig. 3 shows this quantitatively for Ca and Li, showing that there is an optimal laser pulse energy. Maximum emission for these elements was observed using 30–45 mJ, and 20–30 mJ per pulse, as shown in Fig. 3 for Ca (left) and Li (right), respectively. A similar power dependence of emission was observed up to 252 Bar as shown in this figure.
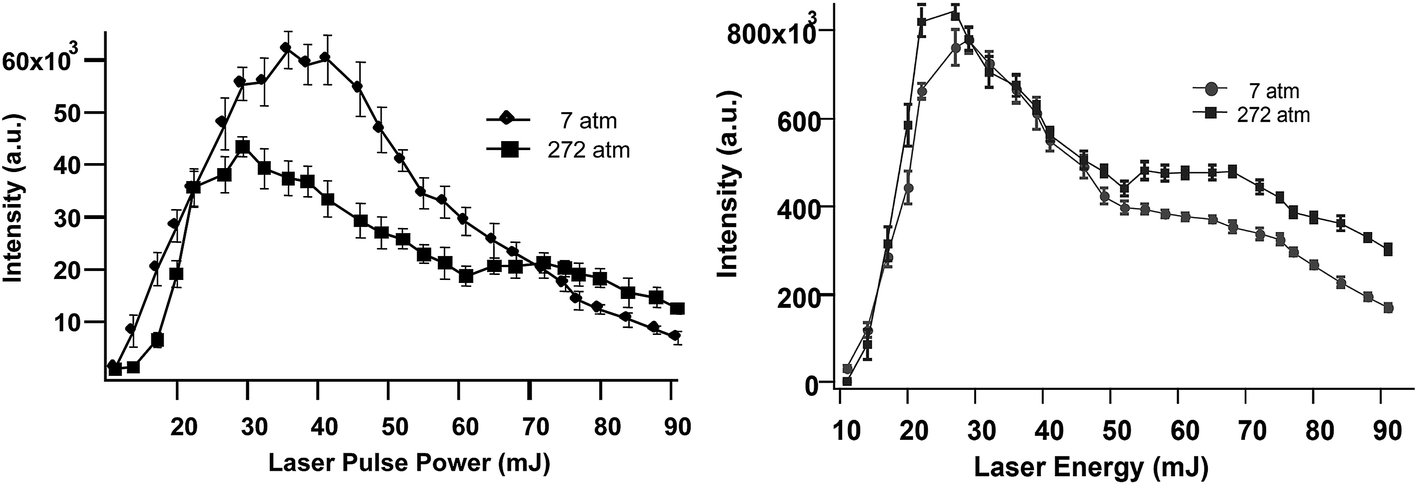 | ||
| Fig. 3 SP LIBS emission intensity of Ca (left) and Li (right) solutions versus laser pulse energy, at 7 Bar and 252 Bar solution pressure. | ||
Fig. 4 (top left) shows a comparison of Mn emission in DI and SW for a solution that contains 500 ppm Mn and 100 ppm Ca. The Mn emission intensity is the same in both solutions, however, as in the case of Li, the background is significantly higher for the SW solution. The Ca emission peak is higher in the SW solution because SW already contains high levels of Ca. Fig. 4 (top right) shows SP LIBS spectra of the same solution, measured at one and 250 Bar. As in the case of Li, no significant pressure effects are observed (the spectra are slightly offset for clarity). Previously published SP LIBS studies of Ca and K also showed no significant pressure effects up to 250 Bar.21,23 Mn calibration curves in DI and SW are shown in Fig. 4 (lower), for solutions that also contain 100 ppm Ca. As in the case of Li, measurement variability is reduced using Ca as an internal standard, as shown in Fig. 4 (lower right, open circles). In unpublished studies, the detection limit for Mn was found to be ∼50 ppb in solution at 280 Bar pressure using 1064 nm excitation, sufficiently low to be suitable for hydrothermal vent fluid measurements.
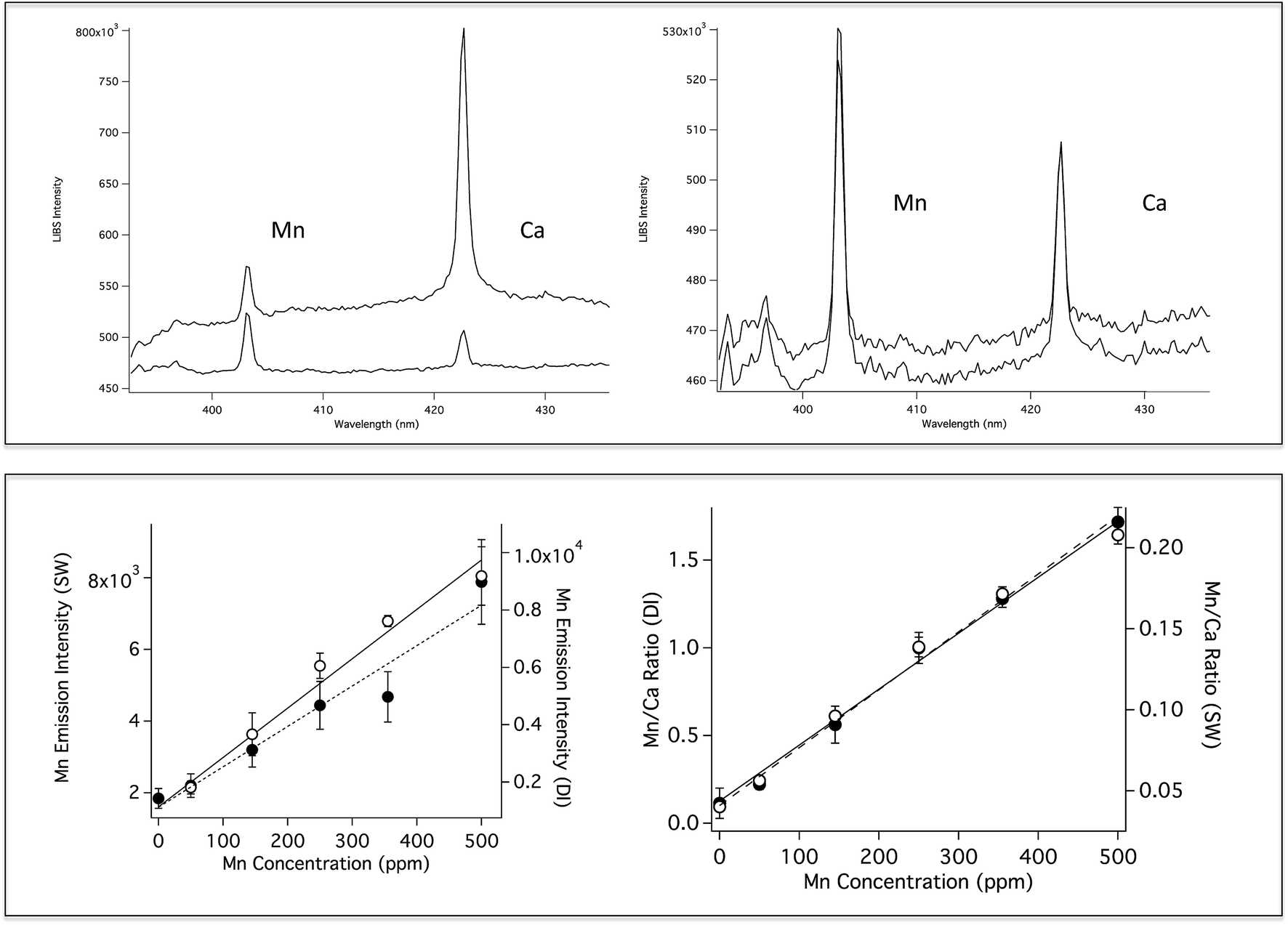 | ||
| Fig. 4 Top: (Left) SP LIBS spectra of a solution of 500 ppm Mn and 100 ppm Ca in deionized water (DI, lower) and seawater (SW, top), and (right) for the solution at 1 Bar (lower spectrum) and 250 Bar (top spectrum). Lower Left: Mn calibration curves at 1 Bar, plotted as Mn emission intensity in DI (open circles, solid fitted line) and in seawater (solid circles, dashed fitted line). Lower Right: Mn calibration curves at 1 Bar in DI water (open circles, solid fitted line) and in seawater (filled circles, dashed fitted line), plotted as Mn/Ca ratio. | ||
As shown by the calibration curves in Fig. 2 and 4, the use of an internal standard for SP LIBS improves measurement precision for bulk aqueous solution measurements. For oceanic LIBS measurements, H and O potentially provide built in internal standards. H and O are produced by breakdown of water in the LIBS plasma, so the emission of these species in a LIBS plasma should be relatively constant. The use of H and O as internal standards for surface water measurements has been reported.58,59 However, there have been no reports of the use of H or O as internal standards for underwater LIBS. It is important to demonstrate that O and H can be used as internal standards for underwater LIBS measurements because the formation of an underwater LIBS plasma is different from a surface measurement where the plasma is formed in air. Formation of the LIBS plasma in air is easier, requiring lower laser fluence than formation underwater, because of strong water quenching.
H and O emission in water is very short lived, and is only observed using short detector gate delays. Fortunately, the elements Li, K, Ca and Na can be measured over a relatively wide range of detector gate delay times, so it is possible to measure these species simultaneously with H and O at pressures up to 250 Bar. Fig. 5 shows SP LIBS spectra of a solution that contains Li and K, at 1 Bar (solid line) and 250 Bar (dashed line), using a detector gate delay of 135 ns. Using a short detector gate delay, Li and K emission is observed as well as strong emission from H and O. Although there is no effect on the Li intensity and little effect on the O emission, there is a relatively large pressure effect on the width of the H line, suggesting that H might not be as useful an internal standard as O at high pressures.
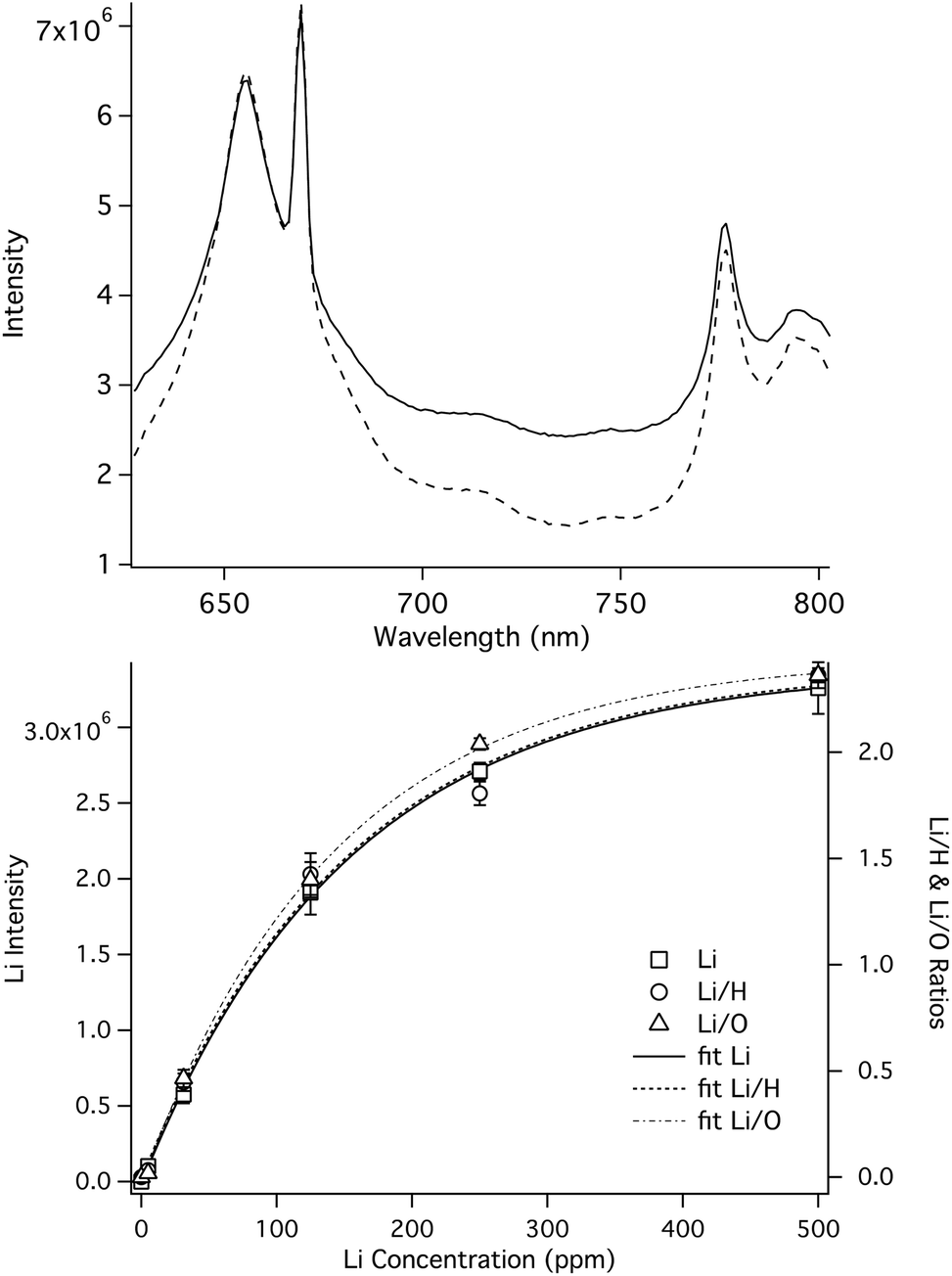 | ||
| Fig. 5 Top: SP LIBS spectra of a solution containing 50 ppm Li and K at 1 Bar (solid line) and 250 Bar (dashed line), showing Li, K, H and O emission lines measured simultaneously, using a 135 ns gate delay. The spectra were offset vertically so that the Li lines overlapped. Lower: Li calibration curves measured at 1 Bar. | ||
The effectiveness of H and O as internal standards at 1 Bar pressure was determined by measuring the relative standard deviation in the intensity of repeated measurements of Li and K solutions. The intensity of each line was determined by subtracting the baseline value. The baseline value was calculated by manually fitting a line to the baseline around the line of interest. In this study each solution was measured in triplicate by averaging 200 laser shots for each spectrum. For 25 ppm solutions, the relative standard deviation (RSD) of the intensity the Li and K emission lines was 21% and 16%, respectively. Using H as an internal standard, the RSD of the Li/H and K/H ratios was 8% and 16%, respectively, while the RSD of the Li/O and K/O ratios was 4% and 3%, respectively. Similar results were observed at other concentrations. Fig. 5 (lower) shows Li/H (left axis, open circles) and Li/O (right, open triangles) calibration plots over a wide range of concentrations that shows how the O ratio tracks the Li intensity with less variability than the Li/H ratio. A similar result was observed using K. High pressure calibration studies are ongoing and will be the subject of a future publication. However, based on high-pressure LIBS spectra like those shown in Fig. 5, the use of O, as an internal standard is expected to be as effective at 250 Bar as 1 Bar, and H might also be useful if pressure corrections are made.
SP LIBS works well for the measurement of dissolved Group I and II elements (e.g., alkali and alkaline earth metals).22,49 However, weak lines can be difficult to measure and many elements are not amenable to SP LIBS. For example, of the transition metals we have found that only Mn is easily measured using SP LIBS with 1064 or 532 nm excitation. Double pulse, or dual-pulse LIBS (DP LIBS) is being investigated to extend the range of elements that can be measured in water. In DP LIBS of liquids, two consecutive laser pulses are used.46,60–66 The first pulse breaks down water at the focus and the resulting high plasma temperature and pressure (6000–15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K and 20–60 kBar, respectively) causes thermal expansion of the plasma and consequent formation of vapor bubbles (i.e., thin layers of vapor and diffused gas) around the plasma volume.33,61 The initial laser-induced plasma quickly decays and cools (within <1 μs), and after tens of microseconds a relatively large vapor bubble forms. In DP LIBS, a second laser pulse is focused in the region of the vapor bubble at the point of maximum expansion, and excites another plasma, which rapidly fills the vapor bubble. It has been shown that the second laser plasma forms at the vapor bubble interface, then fills the vapor bubble.24 The second plasma is isolated from quenching by water and thus the resulting emission of the previously vaporized material is stronger than the emission from the plasma that is generated by the first laser pulse.24 Thus, DP LIBS can be used to measure a wider range of dissolved elements, and weaker lines than SP LIBS in bulk aqueous solution. Using orthogonal DP LIBS, Pearman reported a detection limit for Ca of <50 ppb and also reported the detection of Cr at ∼1 ppm and Zn at 17 ppm, the latter two species not detectable using SP LIBS.49
000 K and 20–60 kBar, respectively) causes thermal expansion of the plasma and consequent formation of vapor bubbles (i.e., thin layers of vapor and diffused gas) around the plasma volume.33,61 The initial laser-induced plasma quickly decays and cools (within <1 μs), and after tens of microseconds a relatively large vapor bubble forms. In DP LIBS, a second laser pulse is focused in the region of the vapor bubble at the point of maximum expansion, and excites another plasma, which rapidly fills the vapor bubble. It has been shown that the second laser plasma forms at the vapor bubble interface, then fills the vapor bubble.24 The second plasma is isolated from quenching by water and thus the resulting emission of the previously vaporized material is stronger than the emission from the plasma that is generated by the first laser pulse.24 Thus, DP LIBS can be used to measure a wider range of dissolved elements, and weaker lines than SP LIBS in bulk aqueous solution. Using orthogonal DP LIBS, Pearman reported a detection limit for Ca of <50 ppb and also reported the detection of Cr at ∼1 ppm and Zn at 17 ppm, the latter two species not detectable using SP LIBS.49
Fig. 6 shows two examples of DP LIBS for dissolved species in bulk aqueous solution. The spectrum on the left shows the weak Na doublet around 819 nm, using SP and DP LIBS in an all orthogonal configuration with an interpulse delay time of about 100 microseconds. Sodium emission is very strong in the DP LIBS spectrum (upper curve) while no emission line is observed in the SP LIBS spectrum (lower curve). Making the assumption that Na emission intensity in the SP spectrum is no larger than the noise, the Na intensity in the DP LIBS spectrum must be enhanced by several hundred. Fig. 6 (left) shows similar spectra for the 777 nm O emission line, also showing an enhancement factor of several hundred. The inset shows the O emission intensity as a function of the delay time between the laser pulses, showing that the O emission is strong over a wide range, hundreds of microseconds, of interpulse delay times.
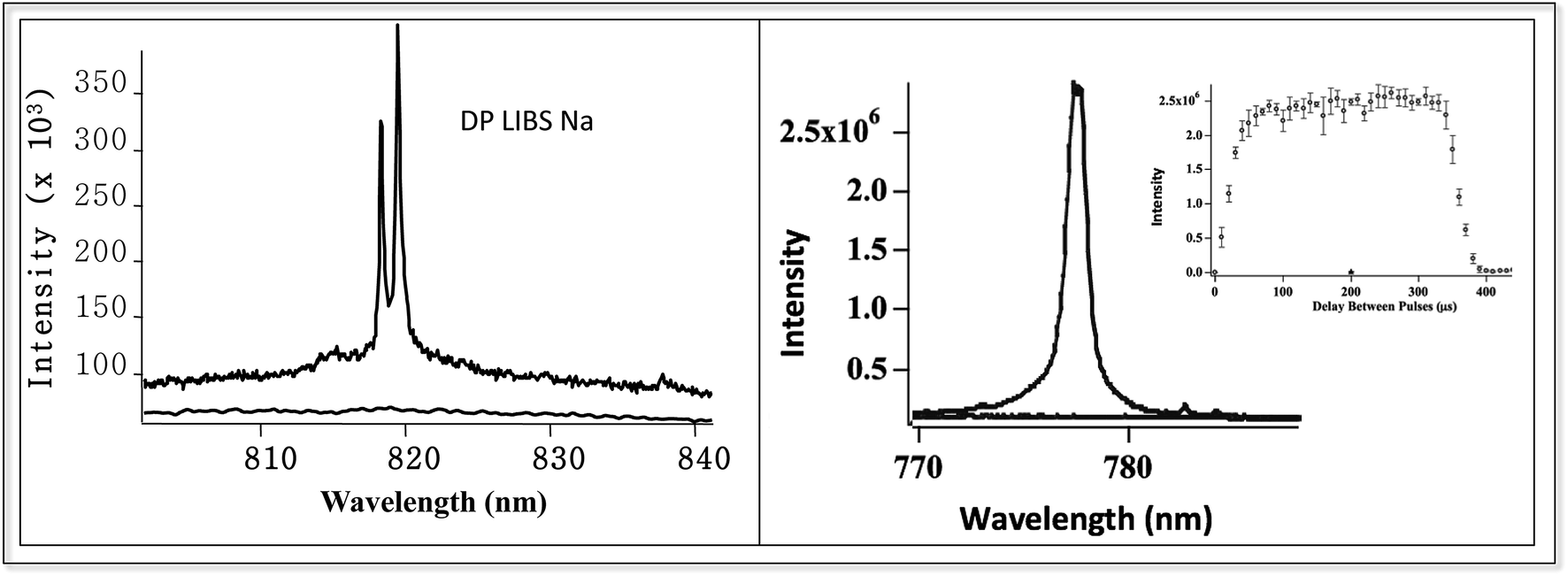 | ||
| Fig. 6 DP LIBS of weak 817 nm Na line and 777 nm O line, using orthogonal laser beam excitation and emission collection. Inset shows O emission intensity versus interpulse delay time. | ||
Although DP LIBS is well suited for oceanic measurements, the magnitude of the emission enhancement is strongly affected by solution pressure as shown in Fig. 7. Fig. 7 shows the 777 nm O(I) emission line measured using DP LIBS for pure water as a function of solution pressure, up to 50 Bar. This result shows that even moderate pressure leads to a large decrease in O(I) emission, and as shown by the top inset in Fig. 7, the emission intensity is inversely proportional to the solution pressure. This spectrum was measured using two orthogonal laser beams with emission collected collinear with the first laser beam. Recent time-resolved shadowgraphy imaging studies show that the emission intensity is proportional to the diameter of the vapor bubble that is formed by the first laser pulse, and the bubble diameter is inversely proportional to the solution pressure. Thus emission intensity is inversely proportional to the solution pressure.
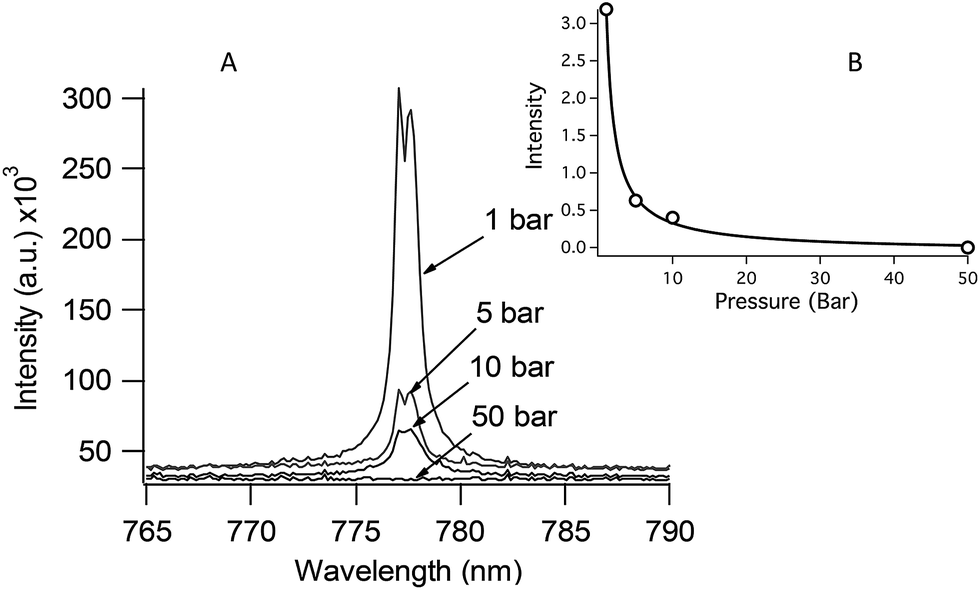 | ||
| Fig. 7 DP LIBS spectra showing 777 nm O emission line as a function of solution pressure. Inset: a plot of O emission intensity versus solution pressure. | ||
Time-resolved shadowgraphy images of the LIBS plasma region show clearly how DP excitation leads to enhanced LIBS emission. Fig. 8 (top left) shows a shadowgraphy image of the plasma that is produced by a single laser pulse. As shown in this image, the plasma spreads out from the laser focal point along the optical axis in the direction of the laser beam, quickly forming a stream of small vapor bubbles.24 Emission near the laser focal point that is collected collinear with the laser is attenuated by plasma shielding. The small vapor bubbles coalesce, over a period of ∼60 μs in this case, to form a large vapor bubble up to 2 mm in diameter (top center image). The top right image shows a second laser focused in the bubble region, 60 μs after the first laser pulse, initiating a second plasma that fills the vapor bubble. In DP LIBS, emission from dissolved species is measured from this second plasma.
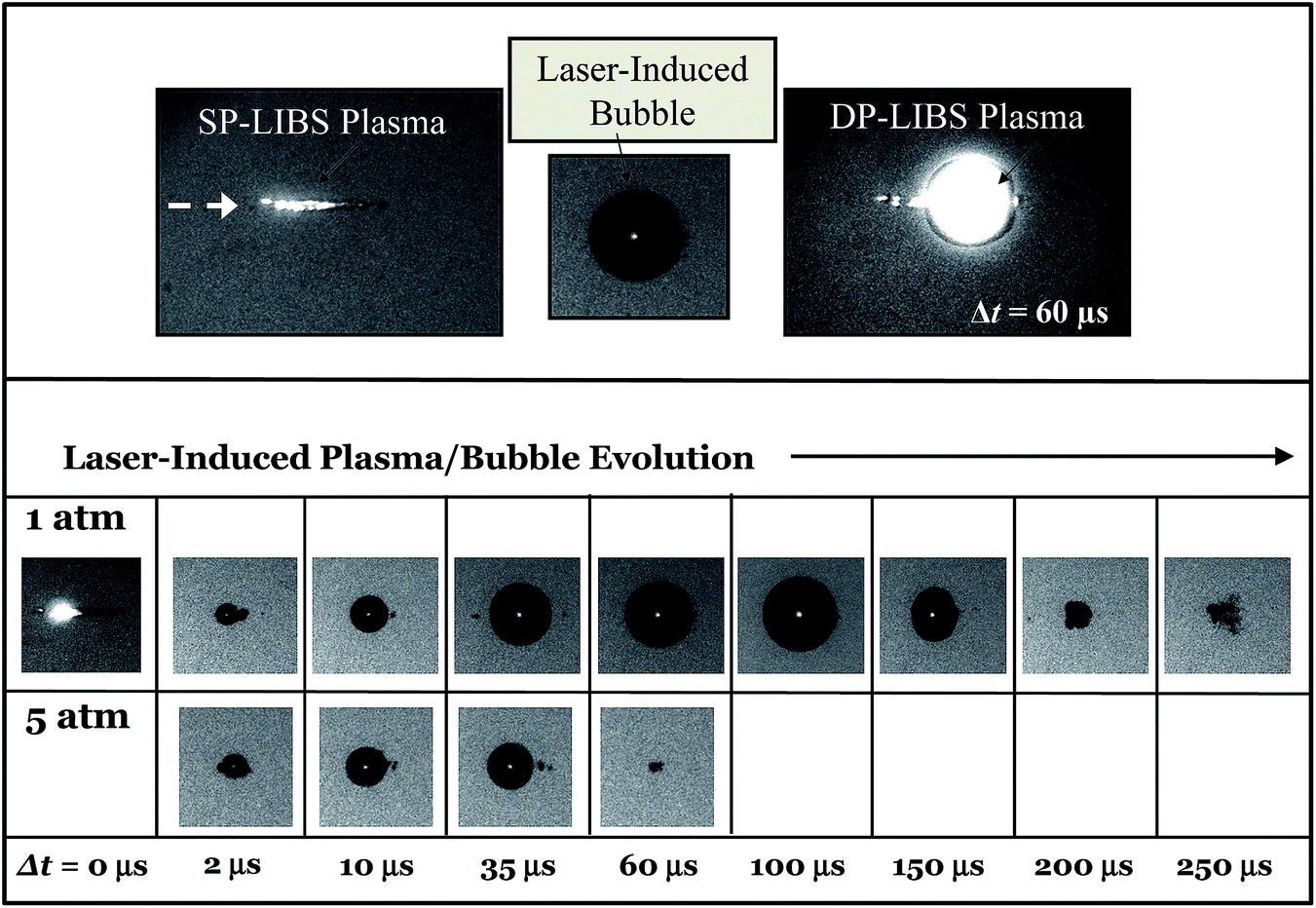 | ||
| Fig. 8 Top: (left) SP LIBS plasma measured immediately after firing laser. (Middle) Laser induced bubble, 60 μs after firing first laser pulse. (Right) DP-LIBS plasma formed by firing second laser pulse 60 μs after the first pulse. Lower: Sequence of time-resolved shadowgraphy images showing the growth of a laser induced bubble for 1 Bar and 5 Bar solutions. | ||
The sequence of shadowgraphy images in the lower part of Fig. 8, show how the vapor bubble evolves in time following the first laser pulse, for 1 Bar and 5 Bar solution pressures. At 5 Bar the vapor bubble expands at the same rate as the 1 Bar bubble, but the maximum size it reaches is much smaller. In the case of 1 Bar solution, the maximum bubble size was reached 100 μs after the first laser-induced plasma was formed, while at 5 Bar, the maximum bubble size occurred between 10 and 35 μs. Measuring the size of laser-induced bubbles in shadowgraphy images and comparing to the emission intensity shows that the maximum emission occurs at the time of maximum bubble size, and that the emission intensity is directly proportional to the bubble diameter. This is shown in Fig. 9, which shows bubble diameter and emission intensity for the 777 nm O line, versus interpulse delay time, at 1 Bar and 5 Bar solution pressures. The emission intensity measurements shown in Fig. 9 (lower curve) were made with both lasers collinear with the collection direction, and focused by the same lens. The inset shows a plot of emission intensity versus bubble diameter for all of the measured points. The fitted solid line shows clearly that the emission intensity is directly proportional to the bubble diameter.
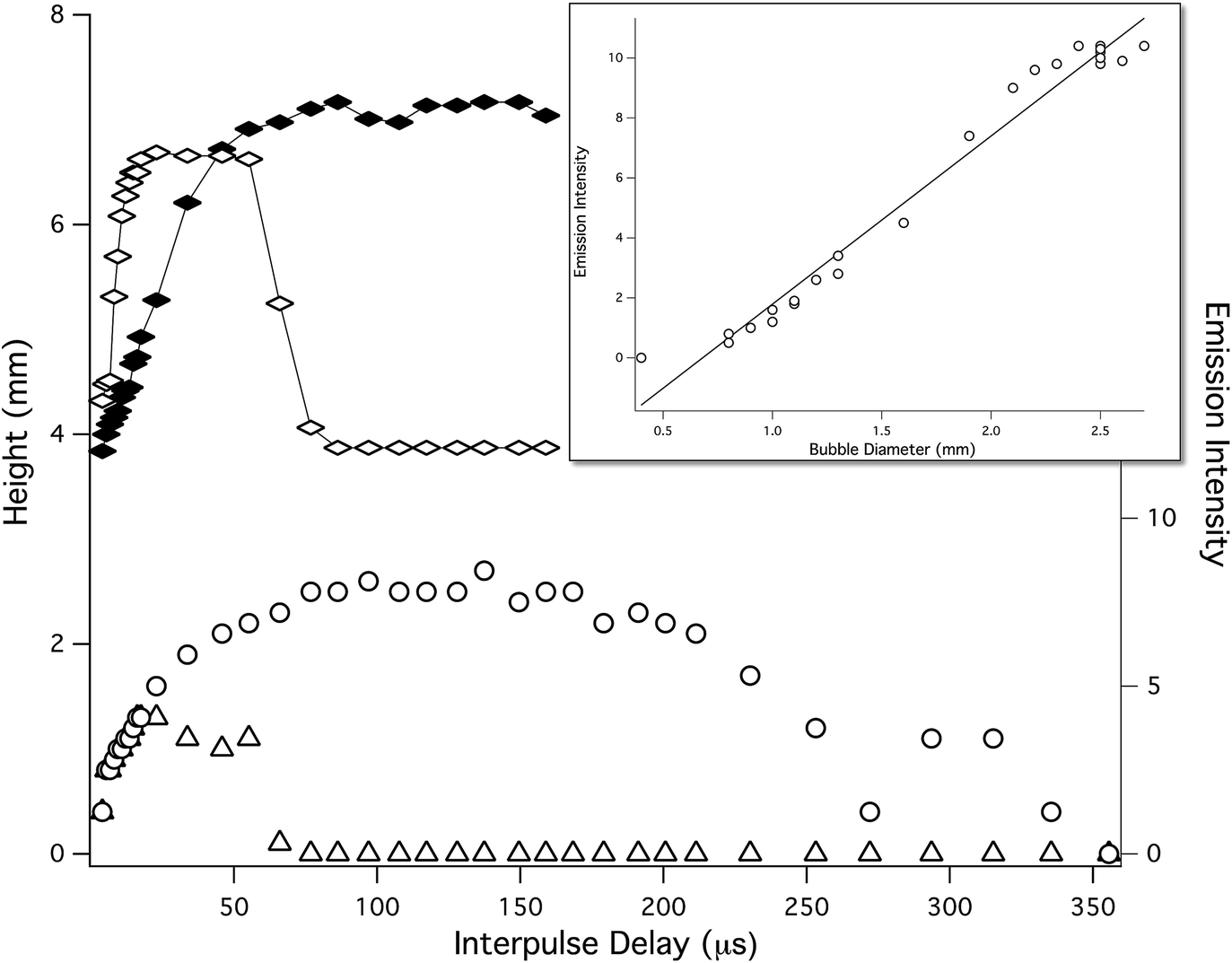 | ||
| Fig. 9 Bubble size (top) and DP LIBS, O emission intensity (lower) measurements, made as a function of delay time after the first laser pulse, at 1 Bar (filled diamonds, open circles) and 1.7 Bar (open diamonds, open triangles). Inset: a plot of DP LIBS O emission intensity versus bubble diameter for the date shown in the lower plots, at both pressures. | ||
The images in Fig. 8 suggest a way to increase the maximum pressure at which DP LIBS might provide enhanced emission. Plasma shielding (see Fig. 8, top left) reduces the amount of collected emission when a collinear collection geometry is used. Collection of light at an angle to the excitation beam should increase the amount of emission light collected. This is the approach used in previously reported DP LIBS studies in which a detection limit of ∼1 ppm Cr was achieved.49 In other previously reported DP LIBS shadowgraphy studies, it was shown that the second DP LIBS plasma was formed at the vapor bubble/water interface.24 Optimally focusing the second laser at the vapor bubble surface is difficult using a collinear geometry and is best achieved by changing the angle between the two lasers. This was also done in the previous paper,49 but this arrangement has not been extended to high pressure studies. In unpublished work we have also shown that the type and quality of lenses used can improve the size and reproducibility of bubble formation, which in turn should improve the overall sensitivity of DP LIBS. Optimization of the laser configurations, and the focusing and imaging optics should improve DP LIBS sensitivity and extend the usefulness of the technique to higher pressures.
Conclusions
SP LIBS is useful for bulk aqueous phase measurements of Li, K, Ca, and Mn, up to 280 Bar and no significant pressure effects were observed, with the exception of Na at very high concentrations. Internal standards are useful to improve measurement precision in SP LIBS of bulk aqueous solution and O has been shown to be a suitable internal standard for water measurements, at pressures up to at least 280 Bar. Hydrogen and oxygen emission can be measured simultaneously with Li and K emission using SP LIBS in bulk aqueous solution. Thus, SP LIBS as currently used is suitable for deep-water ocean measurements of hydrothermal vent fluids. DP LIBS is promising for increased sensitivity in underwater LIBS measurements at low pressures but sensitivity decreases rapidly with increasing solution pressure because of the reduced size of the laser-induced vapor bubble. Changing the laser configuration, and optimization of excitation and imaging optics might increase the maximum pressure attainable by DP LIBS.Acknowledgements
We would like to thank the National Science Foundation for support of this work under grant numbers OCE-0352242, OCE-0527927, OCE-0352278, OCE-0752664 and OCE-1154735.References
- F. Brech and L. Cross, Appl. Spectrosc., 1962, 16, 59 Search PubMed.
- D. A. Cremers, Appl. Spectrosc., 1987, 41, 57 CrossRef.
- C. M. Davies, H. H. Telle, D. J. Montgomery and R. E. Corbett, Spectrochim. Acta, Part B, 1995, 50, 1059 CrossRef.
- B. J. Marquardt, S. R. Goode and S. M. Angel, Anal. Chem., 1996, 68, 977 CrossRef CAS.
- B. J. Marquardt, D. N. Stratis, D. A. Cremers and S. M. Angel, Appl. Spectrosc., 1998, 52, 1148 CrossRef CAS.
- O. Samek, D. C. S. Beddows, J. Kaiser, S. V. Kukhlevsky, M. Liška, H. H. Telle and J. Young, Opt. Eng., 2000, 39, 2248 CrossRef CAS.
- H. H. Telle, D. C. S. Beddows, G. W. Morris and O. Samek, Spectrochim. Acta, Part B, 2001, 56, 947 CrossRef.
- S. Palanco, J. M. Baena and J. J. Laserna, Spectrochim. Acta, Part B, 2002, 57, 591 CrossRef.
- R. C. Wiens, S. K. Sharma, J. Thompson, A. Misra and P. G. Lucey, Spectrochim. Acta, Part A, 2005, 61, 2324 CrossRef PubMed.
- J. D. Blacic, D. R. Pettit, D. A. Cremers and N. Roessler, Laser-Induced Breakdown Spectroscopy for Remote Elemental Analysis of Planetary Surfaces, Proceedings of the International Symposium on Spectral Sensing Research, Maui, Hawaii, 15–20 November 1992, p. 302 Search PubMed.
- C. M. Davies, H. H. Telle and A. W. Williams, Fresenius' J. Anal. Chem., 1996, 355, 895 CAS.
- X. D. Hou and B. T. Jones, Microchem. J., 2000, 66, 115 CrossRef CAS.
- A. K. Knight, N. L. Scherbarth, D. A. Cremers and M. J. Ferris, Appl. Spectrosc., 2000, 54, 331 CrossRef CAS.
- J. Gruber, J. Heitz, H. Strasser, D. Bäuerle and N. Ramaseder, Spectrochim. Acta, Part B, 2001, 56, 685 CrossRef.
- A. I. Whitehouse, J. Young, I. M. Botheroyd, S. Lawson, C. P. Evans and J. Wright, Spectrochim. Acta, Part B, 2001, 56, 821 CrossRef.
- Z. A. Arp, D. A. Cremers, R. D. Harris, D. M. Oschwald, G. R. Parker Jr and D. M. Wayne, Spectrochim. Acta, Part B, 2004, 59, 987 CrossRef.
- C. López-Moreno, S. Palanco and J. J. Laserna, Spectrochim. Acta, Part B, 2005, 60, 1034 CrossRef.
- B. Sallé, J.-L. Lacour, P. Mauchien, P. Fichet, S. Maurice and G. Manhés, Spectrochim. Acta, Part B, 2006, 61, 301–313 CrossRef.
- S. Palanco, C. López-Moreno and J. J. Laserna, Spectrochim. Acta, Part B, 2006, 61, 88 CrossRef.
- C. López-Moreno, S. Palanco, J. J. Laserna, F. DeLucia Jr, A. W. Miziolek, J. Rose, R. A. Walters and A. I. Whitehouse, J. Anal. At. Spectrom., 2006, 21, 55 RSC.
- M. Lawrence-Snyder, J. Scaffidi, S. M. Angel, A. P. M. Michel and A. D. Chave, Appl. Spectrosc., 2006, 60, 786 CrossRef CAS PubMed.
- M. Lawrence-Snyder, J. Scaffidi, S. M. Angel, A. P. M. Michel and A. D. Chave, Appl. Spectrosc., 2007, 61, 171 CrossRef CAS PubMed.
- A. P. M. Michel, M. Lawrence-Snyder, S. M. Angel and A. D. Chave, Appl. Opt., 2007, 46, 2507 CrossRef CAS PubMed.
- M. Lawrence-Snyder, J. P. Scaffidi, W. F. Pearman, C. M. Gordon and S. Michael Angel, Spectrochim. Acta, Part B, 2014, 99, 172 CrossRef CAS.
- W. E. Seyfried Jr, D. R. Janecky and M. J. Mottl, Geochim. Cosmochim. Acta, 1984, 48, 557 CrossRef.
- C. van Dover, The ecology of deep-sea hydrothermal vents, Princeton, New Jersey, Princeton University Press, 2000 Search PubMed.
- D. J. MacDonald, A. J. Findlay, S. M. McAllister, J. M. Barnett, P. Hredzak-Showalter, S. T. Krepski, S. G. Cone, J. Scott, S. K. Bennett, C. S. Chan, D. Emerson and G. W. Luther III, Environ. Sci.: Processes Impacts, 2014, 16, 2117–2126, 10.1039/c4em00073k.
- D. B. Nuzzio, M. Taillefert, S. C. Cary, A. L. Reysenbach, and G. W. Luther, Environmental Electrochemistry, 2009, ACS Symposium Series, vol. 811, pp. 40–51 Search PubMed.
- G. W. Luther, C. E. Reimers, D. B. Nuzzio and D. Lovalvo, Environ. Sci. Technol., 1999, 33, 4352–4356 CrossRef CAS.
- C. P. Leonardo, Y. Mustafa, O. Dario, B. Jean-Pierre and L. B. Nadine, Deep Sea Research Part I, Oceanographic Research Papers, 2013, 80, 47–57, DOI:10.1016/j.dsr.2013.05.014.
- K. L. Frank, D. R. Rogers, H. C. Olins, C. Vidoudez and P. R. Girguis, ISME J., 2013, 1–11, DOI:10.1038/ismej.2013.17.
- H. C. Olins, D. R. Rogers, K. L. Frank, C. Vidoudez and P. R. Girguis, Geobiology, 2013, 11, 279–293, DOI:10.1111/gbi.12034.
- P. K. Kennedy, D. X. Hammer and B. A. Rockwell, Prog. Quantum Electron., 1997, 21, 155 CrossRef CAS.
- T. Bundschuh, R. Knopp and J. I. Kim, Colloids Surf., A, 2001, 177, 47 CrossRef CAS.
- C. Walther, C. Bitea, W. Hauser, J. I. Kim and F. J. Scherbaum, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 195, 374 CrossRef.
- A. de Giacomo, M. Dell'Aglio, F. Colao and R. Fantoni, Spectrochim. Acta, Part B, 2004, 59, 1431 CrossRef.
- V. Lazic, F. Colao, R. Fantoni and V. Spizzicchino, Spectrochim. Acta, Part B, 2005, 60, 1002 CrossRef.
- V. Lazic, F. Colao, R. Fantoni and V. Spizzichino, Spectrochim. Acta, Part B, 2005, 60, 1014 CrossRef.
- S. Guirado, F. J. Fortes, V. Lazic and J. J. Laserna, Spectrochim. Acta, Part B, 2005, 74–75, 137 Search PubMed.
- A. de Giacomo, M. Dell'Aglio, A. Casavola, G. Colonna, O. de Pascale and M. Capitelli, Anal. Bioanal. Chem., 2006, 385, 303 CrossRef CAS PubMed.
- T. Saeki, H. Oguchi, S. Masai, K. Hirata and Y. Ogata, Appl. Phys. Lett., 2006, 88, 061120 CrossRef.
- V. Lazic, F. Colao, R. Fantoni, V. Spizzichino and S. Jovićević, Spectrochim. Acta, Part B, 2007, 62, 30 CrossRef.
- A. de Giacomo, M. Dell'Aglio, O. de Pascale and M. Capitelli, Spectrochim. Acta, Part B, 2007, 62, 721 CrossRef.
- B. Thornton and T. Ura, Appl. Phys. Express, 2011, 4, 1 Search PubMed.
- V. Lazic, J. J. Laserna and S. Jovicevic, Spectrochim. Acta, Part B, 2013, 82, 42 CrossRef CAS.
- D. A. Cremers, L. J. Radziemski and T. R. Loree, Appl. Spectrosc., 1984, 38, 721 CrossRef CAS.
- R. Knopp, F. J. Scherbaum and J. I. Kim, J. Anal. Chem., 1996, 355, 16 CAS.
- O. Samek, D. C. S. Beddows, J. Kaiser, S. V. Kukhlevsky, M. Liska, H. H. Telle and J. Young, Opt. Eng., 2000, 39, 2248 CrossRef CAS.
- W. Pearman, J. Scaffidi and S. M. Angel, Appl. Opt., 2003, 42, 6085 CrossRef PubMed.
- S. Koch, W. Garen, M. Müller and W. Neu, Appl. Phys. A, 2004, 79, 1071 CrossRef CAS.
- A. de Giacomo, M. Dell'Aglio and O. de Pascale, Appl. Phys. A, 2004, 79, 1035 CrossRef CAS.
- L. St-Onge, E. Kwong, M. Sabsabi and E. Vadas, J. Pharm. Biomed. Anal., 2004, 36, 277 CrossRef CAS PubMed.
- V. Lazic, S. Jovicevic, R. Fantoni and F. Colao, Spectrochim. Acta, Part B, 2007, 62, 1433 CrossRef.
- A. P. M. Michel and A. D. Chave, Appl. Opt., 2008, 47, G131 CrossRef CAS PubMed.
- V. Lazic and S. Jovićević, Spectrochim. Acta, Part B, 2014, 101, 288 CrossRef CAS.
- B. Thornton, T. Takahashi, T. Sato, T. Sakka, A. Tamura, A. Matsumoto, T. Nozaki, T. Ohki and K. Ohki, Deep-Sea Res., 2015, I95, 30 Search PubMed.
- S. N. White, A. D. Chave and G. T. Reynolds, J. Geophys. Res., 2002, 108 DOI:10.1029/2000jb000015.
- O. Samek, D. C. S. Beddows, J. Kaiser, S. V. Kukhlevsky, M. Liska, H. H. Telle and J. Young, Opt. Eng., 2000, 39, 2248 CrossRef CAS.
- W. T. Y. Mohamed, Int. J. Pure Appl. Phys., 2006, 2, 11 Search PubMed.
- R. Nyga and W. Neu, Opt. Lett., 1993, 18, 747 CrossRef CAS PubMed.
- A. E. Pichahchy, D. A. Cremers and M. J. Ferris, Spectrochim. Acta, Part B, 1997, 52, 25 CrossRef.
- A. de Giacomo, M. Dell'Aglio, F. Colao and R. Fantoni, Spectrochim. Acta, Part B, 2004, 59, 1431 CrossRef.
- A. de Giacomo, M. Dell'Aglio, F. Colao, R. Fantoni and V. Lazic, Appl. Surf. Sci., 2005, 247, 157 CrossRef CAS.
- V. Lazic, F. Colao, R. Fantoni and V. Spizzicchino, Spectrochim. Acta, Part B, 2005, 60, 1002 CrossRef.
- A. Casavola, A. de Giacomo, M. Dell'Aglio, F. Taccogna, G. Colonna, O. de Pascale and S. Longo, Spectrochim. Acta, Part B, 2005, 60, 975 CrossRef.
- A. de Giacomo, M. Dell'Aglio, A. Casavola, G. Colonna, O. de Pascale and M. Capitelli, Anal. Bioanal. Chem., 2006, 385, 303 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |