
Determination of SeMet and Se(IV) in biofortified yeast by ion-pair reversed phase liquid chromatography-hydride generation-microwave induced nitrogen plasma atomic emission spectrometry (HPLC-HG-MP-AES)
Eunice Yañez
Barrientos
a,
Kazimierz
Wrobel
a,
Juan Carlos
Torres Guzman
b,
Alma Rosa
Corrales Escobosa
a and
Katarzyna
Wrobel
*a
aChemistry Department, University of Guanajuato, L. de Retana 5, 36000 Guanajuato, Mexico. E-mail: katarzyn@ugto.mx; Fax: +52 473 7326252; Tel: +52 473 7227555
bBiology Department, University of Guanajuato, L. de Retana 5, 36000 Guanajuato, Mexico
First published on 7th September 2015
Abstract
Selenium biofortified yeast is the most common dietary Se supplement in human nutrition and in farm animals. Therefore, the production and routine quality control of commercial products are highly demanded. In this work, a simple and cost-effective procedure is proposed for the determination of SeMet and Se(IV) in hydrolyzed yeast, consisting of ion-pair reversed phase separation, post-column hydride generation and Se quantification by atomic emission spectrometry with microwave plasma sustained by nitrogen (HPLC-HG-MP-AES). Freeze-dried biomass was hydrolyzed with methanesulfonic acid; chromatographic separation was performed with a mobile phase containing 0.08% v/v heptafluorobutyric and methanol (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) at a flow rate of 1 mL min−1; the column effluent was on-line mixed with an alkaline solution of potassium persulfate (K2S2O8 6% m/v, NaOH 3% m/v), passed through a reaction coil submerged in a water bath at 60 °C, and then 10 M hydrochloric acid was added prior to hydride generation in the MP-AES multimode sample introduction system (NaBH4 2% m/v, NaOH 0.3% m/v). The total chromatographic run was accomplished in 5 min and the evaluated on-column quantification limits were 59 ng Se mL−1 for Se(IV) and 0.52 μg mL−1 for SeMet. The procedure was tested using standardized Seleno Excell® high selenium yeast and then applied for the analysis of Saccharomyces cerevisiae biofortified under different fermentation and exposure conditions. The procedure was capable of detecting differences in selenium concentration among cultures and the results were consistent with those obtained while coupling HPLC separation directly to ICP-MS detection.
:8) at a flow rate of 1 mL min−1; the column effluent was on-line mixed with an alkaline solution of potassium persulfate (K2S2O8 6% m/v, NaOH 3% m/v), passed through a reaction coil submerged in a water bath at 60 °C, and then 10 M hydrochloric acid was added prior to hydride generation in the MP-AES multimode sample introduction system (NaBH4 2% m/v, NaOH 0.3% m/v). The total chromatographic run was accomplished in 5 min and the evaluated on-column quantification limits were 59 ng Se mL−1 for Se(IV) and 0.52 μg mL−1 for SeMet. The procedure was tested using standardized Seleno Excell® high selenium yeast and then applied for the analysis of Saccharomyces cerevisiae biofortified under different fermentation and exposure conditions. The procedure was capable of detecting differences in selenium concentration among cultures and the results were consistent with those obtained while coupling HPLC separation directly to ICP-MS detection.
1. Introduction
The importance of selenium in human health and nutrition has long been recognized. Different Se-containing dietary supplements are in use and among them biofortified yeast is the most common.1,2 Numerous experimental studies and clinical trials have proved the chemopreventive and anti-tumor activity of selenized yeast.3–5 Administration of Se-yeast in the form of different dietary supplements or its incorporation into specific foods has been widely approved, and it was concluded that a daily element intake of about 100 μg does not present a safety hazard; at the same time, the tolerable upper intake level was set at 300 μg per day.6,7 In addition to the human supplementation, Se-yeast has been accepted as an additive for animal feed, so the overall market demand is quite large.6 Commercial products are obtained from cultures of bakers's yeast (Saccharomyces cerevisiae) that are grown in the presence of sodium selenite (Se(IV)), which are then washed to remove residual inorganic selenium and other free minerals, and finally the biomass is pasteurized and dried to obtain a homogenous powder.6,8 Depending on the strain type, fermentation conditions (medium, pH, temperature, and aeration) and Se(IV) concentration, selenized yeast may contain up to 3 mg selenium per gram of the dry mass.9 Speciation studies have often been undertaken with the aim to understand the incorporation pathways and to fully characterize all selenium compounds in biofortified yeast.10–12 The reported species identity and their abundance have varied for different protocols applied in the fabrication and analysis of Se-yeast, indicating a need for the implementation of quality control schemes.2,13The primary organic, bioavailable and beneficial selenium compound in yeast is selenomethionine (SeMet) incorporated into proteins hence its reliable quantification has become the essential part of quality control; it should also be ensured that the residual inorganic selenium has been efficiently eliminated. Specifically, Se-yeast of good quality does not contain more than 1% of inorganic Se whereas SeMet analyzed in an enzymatic or acid digest accounts for 60–90% of the total selenium;9 these specifications dictate analytical requirements that should be met by any procedure employed in quality control.
Beyond any doubt, procedures based on the hyphenation of gas or liquid chromatography with species-specific or unspecific isotope dilution – inductively coupled plasma mass spectrometry – provide accurate and precise quantification results;14–19 however, they are still too demanding to be implemented in routine analysis. Simplification and miniaturization have been approached in several studies with an emphasis mainly on the sample pretreatment. In this regard, microwave assisted acid- or enzymatic digestion as well as ultrasonic enzymatic hydrolysis have been explored;18,20 enzymatic hydrolysis was also carried out on a miniaturized microtiter plate.21 On the other hand, less sophisticated and cost-effective techniques with respect to ICP-MS have been used as the detection systems for liquid chromatography.22 To compensate for lower detection power of these techniques and to avoid matrix interference, post-column hydride generation has been integrated while using atomic fluorescence, atomic emission or atomic absorption spectrometry.22–26 In such an approach, each selenium species eluting from the column has to be converted to Se(IV), because only this species forms hydrogen selenide in the reaction with sodium borohydride. For the oxidation of organic species, potassium persulfate in an alkaline medium or the mixture of hydrogen bromide/potassium bromate can be used, the resulting Se(VI) is usually reduced to Se(IV) by heating with hydrochloric acid 6–10 mol L−1; alternatively, UV irradiation has often been employed with addition of alkaline potassium iodide for the reduction of Se(VI) to Se(IV).24,26–29
Post-column hydride generation has proved to be a promising tool for efficient introduction of selenium species into an annular-shaped nitrogen microwave induced plasma formed in Okamoto cavity.30 More recently, Hammer has developed a magnetically excited microwave plasma emission source,31 which is now used in a commercially available instrument (microwave nitrogen plasma-atomic emission spectrometry, MP-AES). In this system, nitrogen is supplied via an air generator, which drastically lowers the operation cost as compared to any other atomic spectrometry technique. A robust diatomic gas (N2) plasma of toroidal shape is generated with a conventional torch and the introduction of sample aerosol into the core of the plasma occurs through the central channel, in a similar way to that in an ICP nebulizer and spray chamber configuration. The feasibility of HPLC-MP-AES coupling for speciation analysis has recently been demonstrated;32,33 however poor detection power is an important limitation of this hyphenated technique. In line with many earlier studies,28,34 post-column hydride generation might be helpful for enhanced transport efficiency of column effluent to the plasma and also for the elimination of the chemical matrix, which is especially troublesome at the most intense selenium emission line (196.026 nm).
Due to the high demand in the market, many manufacturers produce Se-yeast and need suitable analytical tools that would be helpful during optimization of the production process and could also be used for quality control purposes. Specifically, the determination of SeMet and Se(IV) by means of a simple and cost-effective procedure is required and for this purpose, ion-pair reversed phase liquid chromatography separation was coupled in this work with MP-AES detection via hydride generation for the first time (HPLC-HG-MP-AES). For comparative purposes, ICP-MS was used as an alternative element specific detection system, and the results obtained by the two procedures were in good agreement. The proposed procedure enabled for detecting differences in Se(IV) and SeMet concentrations among yeast biofortified under different fermentation conditions, which demonstrates its utility in the quality control schemes.
2. Experimental
2.1. Apparatus
An Agilent Series 1050 liquid chromatographic system controlled by using Chemstation (Agilent Technologies) was used with a Gemini C18 column (50 × 3 mm, 5 μm) from Phenomenex. The column effluent was transported via PEEK® tubing and typical FIA tubing (0.5 mm i.d.) to the multimode sample introduction system (MSIS) of an Agilent 4100 MP-AES nitrogen microwave plasma atomic emission spectrometer controlled by using Agilent MP Expert Software (actualized from MP-AES 4200). Two T-joints were incorporated between column exit and MSIS for on-line introduction of the oxidizing agent and hydrochloric acid, respectively. After mixing with an oxidizing agent, a reaction coil (100 cm) was integrated and it was submerged in a water bath (60 °C). The two reagents and the solution of sodium borohydride were pumped by using a MP-AES peristaltic pump, each of them at the flow rate of 1 mL min−1. Tygon peristaltic tubing (0.76 cm i.d.) was used. The above set-up is schematically presented in Fig. 1.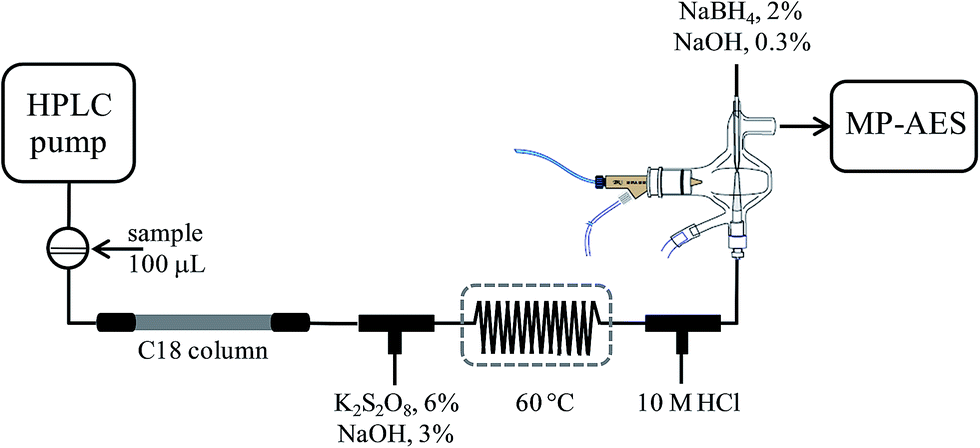 | ||
| Fig. 1 General scheme of HPLC-HG-MP-AES coupling. | ||
An Agilent Series 1050 system was coupled directly to inductively coupled plasma mass spectrometry detector, via a short-length Teflon tubing. A model 7500ce ICP-MS (Agilent Technologies, Tokyo, Japan) with a Meinhard nebulizer and Peltier-cooled spray chamber (2 °C) was used. The analytical column was a Polaris C18 column (100 × 3 mm, 3 μm) from Varian.
2.2. Reagents and samples
All chemicals were of analytical reagent grade. Deionized water (18.2 MΩ cm, Labconco, USA) and HPLC-grade methanol (Fisher Scientific, Pittsburgh, USA) were used throughout.Stock solutions of selenomethionine (SeMet) and sodium selenite (Se(IV)) were prepared by dissolving respective Sigma standards in deionized water. Other Sigma reagents used in this work were: potassium persulfate, sodium hydroxide, methanesulfonic acid, hydrochloric acid, nitric acid, heptafluorobutyric acid (HFBA), sodium borohydride, β-mercaptoethanol and hydride peroxide. An Agilent Technology mixed internal standard solution was also used (4 mg L−1 In, 20 mg L−1 Li, 4 mg L−1 Y, 4 mg L−1 Bi, 10 mg L−1 Sc, and 2 mg L−1 Rh).
Commercial high selenium yeast Seleno Excell®, containing 1255 μg g−1 Se, was from Cypress Systems, Inc.
Selenium biofortified yeast samples were obtained in the frame of an on-going study performed at the Department of Biology, University of Guanajuato (Juan Carlos Torres Guzman, unpublished data). In brief, different cultures of Saccharomyces cerevisiae were obtained under a series of fermentation and exposure conditions. In the first approach 2 × 107 cells mL−1 from S. cerevisiae strain (CC) were grown at 28 °C during 18 h with agitation, in an Erlenmeyer flask containing 50 mL of minimal medium (6.7 g L−1 yeast nitrogen base without amino acids, Difco; 40 g L−1 dextrose, BD Bioxon) in the presence of 0, 5, 10, 15, 20 and 30 mg L−1 Se added in form of sodium selenite (Sigma-Aldrich). The cells were recovered by centrifugation and washed with deionized water, these samples were encoded as C0, C5, C10, C20, C30, respectively. The second series of samples was obtained from S. cerevisiae strains CC, S, M and R (the last three isolated from strain CC by a directed evolution strategy) that were incubated under the same conditions in a minimal medium in the presence of 10 mg L−1 Se added in form of sodium selenite. The obtained yeast cultures were recovered by centrifugation and were washed with deionized water. These samples were denoted as CC10, S10, M10 and R10, respectively. Finally, all samples were lyophilized in a FreeZone 6 Freeze Dry System (Labconco) prior to analysis.
2.3. Determination of total selenium in yeast by ICP-MS
An aliquot (25 mg) of each biomass was placed in a polypropylene Eppendorf tube, 750 μL of internal standard mix and 250 μL of concentrated nitric acid were added, and the sample was heated at 110 °C during 1.5 h. After cooling to room temperature, 500 μL of hydrogen peroxide 30% m/m were added and the sample was kept at 110 °C during 1 h. Finally, the volume was adjusted to 1.5 mL, 25 μL were taken and diluted to 5 mL. The ICP-MS operating conditions were as follows: forward power 1500 W, plasma gas flow rate 15 L min−1, carrier gas flow rate 0.89 L min−1, make-up gas flow rate 0.15 L min−1, sampling depth 8 mm, platinum sampling and skimmer cones, dwell time 100 ms per isotope, and collision/reaction cell gas He, 4.5 mL min−1. The isotopes 78Se, 82Se were monitored (115In as IS). Five point external calibration was performed at element concentration levels 0–100 μg L−1 with the addition of the internal standard (4.0 μg L−1 In). The linear regression functions were obtained (r2 > 0.999) and the evaluated instrumental detection limit for 78Se was 25 ng L−1. For accuracy checking, Seleno Excell® was analyzed.2.4. Hydrolysis of yeast for speciation analysis
The procedure described previously was used.35 To a 100 mg aliquot of the biomass, 2 mL of methanesulfonic acid 4 mol L−1 were added and the mixture was heated at reflux during 16 h (120 °C). The obtained hydrolyzate was evaporated and the residue was re-suspended in 10 mL of HFBA 0.08% m/v for MP-AES detection and in 10 mL of HFBA 0.1% m/v for ICP-MS. For each sample, hydrolysis was carried out in triplicate. It is noteworthy that methanesulfonic acid hydrolysis was selected due to the efficient release of Se–methionine from proteins, as demonstrated elsewhere.35,362.5. Selenium speciation by HPLC-HG-MP-AES
Separation was achieved on a reversed phase column using heptafluorobutyric acid as an ion-pair reagent. Prior to hydride generation, the column effluent was mixed with an alkaline solution of potassium persulfate, heated in a water bath at 60 °C and then hydrochloric acid was added, as shown in Fig. 1. In the MP-AES multimode sample introduction system, the column effluent was introduced through the lower channel, sodium borohydride was pumped through the upper channel and nebulization gas was introduced perpendicularly. The chromatographic, hydride generation and MP-AES instrument operating conditions are given in Table 1. The chromatographic run and the detection system were started simultaneously; the raw data from continuous acquisition were saved as a Microsoft Excel file for further treatment using GRAMS 5.0 (Thermo Scientific). For each chromatogram, binominal filtration and extrapolation (32 points) were applied, followed by Fourier filtration 97%. For external calibration, eight solutions containing Se(IV) 0; 0.25; 0.5; 1.0; 2.0; 3.0; 4.0; 5.0 mg Se L−1and SeMet 0; 2.5; 5; 10; 20; 30; 40; 50 mg Se L−1 were used. The re-suspended yeast samples (10 mL) were filtered (0.22 μm Whatman filters) prior to their injection into the chromatographic system. For the recovery experiment, the Seleno Excell® digest was evaporated and re-suspended in 5 mL of HHBA 0.08% m/v. Two aliquots of 1 mL were taken, one was diluted to a 1:1 ratio with HFBA and the second was spiked with 500 μL of SeMet 40 mg Se L−1, 100 μL of Se(IV) 40 mg Se L−1 and brought to 2 mL with HFBA. Addition of two standards corresponded to 200 μg Se g−1 of Se(IV) and 1000 μg Se g−1 of SeMet in the yeast biomass.
| Chromatographic conditions | |
|---|---|
| a These solutions were carried via a MP-AES peristaltic pump. | |
| Column | Gemini C18 (50 × 3 mm, 5 μm), Phenomenex |
| Mobile phase | Heptafluorobutyric acid 0.08% m/v:methanol (92:8) |
| Flow rate | 1 mL min−1 |
| Injection volume | 100 μL |
|
|
| On-line treatment of the column effluent and hydride generation | |
| Oxidizing agenta | K2S2O8 6% m/v + NaOH 3% m/v, 1 mL min−1 |
| Reaction coil | 100 cm long, 0.4 mm i.d., 60 °C |
| Acidificationa | HCl 10 mol L−1, 1 mL min−1 |
| Hydride generationa | NaBH4 2% m/v + NaOH 0.3% m/v, 1 mL min−1 |
|
|
| MP-AES detection | |
| Nitrogen | 140 psi |
| Air injection flow rate | Low |
| Nebulizer/spray chamber | MiraMist Teflon®/multimode sample introduction system |
| Nebulizer pressure | 165 kPa |
| Viewing position | 12 |
| Acquisition mode | Continuum |
| Integration time | 5 s |
| Stop time/post time | 20 min/2 min |
| Detection wavelength | 196.026 nm |
| Background correction | Off-peak left + right |
| Number of pixels | 3 |
2.6. Selenium speciation by HPLC-ICP-MS
Separation was achieved on a reversed phase Polaris C18 column; isocratic elution with HFBA 0.1% m/v:methanol (95:5) was carried out at the flow rate of 1 mL min−1 and the injection volume was 20 μL. ICP-MS operating conditions were the same as those for total Se determination except for the carrier gas flow rate of 0.75 L min−1 and make-up gas flow rate of 0.10 L min−1. The isotopes 78Se and 82Se were monitored. For external calibration, a series of mixed Se(IV) and SeMet standard solutions was used, both species at the concentrations of 0, 10, 20, 50, 100, 250, and 500 μg Se L−1. Linear regression functions were obtained for peak area measurements, yielding good linearity (r2 > 0.999). The on-column detection limits (78Se) were 0.76 μg Se L−1 for Se(IV) and 0.38 μg Se L−1 for SeMet, respectively. The re-suspended yeast samples were 40 times diluted and filtered (0.22 μm Whatman filters) prior to their injection into the chromatographic system. For the recovery experiment, 250 μL aliquot of the re-suspended Selen Excell® digest was taken, 500 μL of Se(IV) 750 μg Se L−1 and 500 μL of SeMet 2.5 mg Se L−1 were added and the volume was brought to 10 mL with HFBA; by so doing the standard addition corresponded to 150 μg Se g−1 of Se(IV) and 500 μg Se g−1 of SeMet in the biomass. All results are presented as μg of selenium per gram of the freeze-dried biomass.
3. Results and discussion
3.1. Method development
Direct introduction of Se(IV) or SeMet standard solution into MP-AES was tested and by so doing, selenium at a concentration of 5 mg L−1 was not even detected, which confirmed the need for post-column hydride generation. Se-yeast contains a low concentration of Se(IV) which is efficiently converted to H2Se in the presence of sodium borohydride, so a desired sensitivity enhancement was expected for this species in the proposed system. On the other hand, SeMet is the most abundant Se compound and improving the detection power for this species was less critical. As briefly described in the Introduction, quantitative conversion of SeMet to Se(VI) and then to Se(IV) for hydrogen selenide generation is not straightforward and requires relatively harsh chemical conditions with prolonged heating, sonication and/or UV irradiation.24,26–29 To simplify this step, the direct oxidation of SeMet to Se(IV) was tested by using an alkaline solution of sodium persulfate,29 expecting incomplete but reproducible conversion.The conversion of SeMet to Se(IV) was examined using a set-up presented in Fig. 1, but without a chromatographic column. An aliquot (100 μL) of SeMet standard solution (5 mg Se L−1) was repeatedly injected into the flow system, while varying the following parameters: concentration of potassium persulfate (2–8% m/v), length of the reaction coil (50–150 cm), temperature (20–80 °C), concentration of hydrochloric acid (6–10 mol L−1) and concentration of sodium borohydride (1–4% m/v). For comparative purposes, the same conditions were also tested for Se(IV), 5 mg Se L−1. The selection criteria applied were: (1) the highest possible analytical signal and (2) the lowest possible standard deviation obtained for replicate injections; in these experiments the analytical signal was acquired as the peak area under manually selected MP-AES operating conditions listed in Table 1. As expected, the most important variables were those related to the oxidation step; in the absence of potassium persulfate, SeMet did not produce any signal whereas the highest and the most reproducible signal was obtained using 6% m/v persulfate. At this concentration, the Se(IV) signal was 20.7% lower as compared to that obtained without the addition of persulfate. It is noteworthy that when standard solution was introduced directly into MP-AES, Se(IV) 5 mg L−1 was not detected. Furthermore, the analytical signal of SeMet gradually augmented when temperature and the length of reaction coil were increased; however the signal improvement was accompanied by peak broadening and also by sensitivity loss for Se(IV) so the compromise conditions had to be adopted. It should be stressed that the addition of an oxidizing agent after the elution of Se(IV) was not considered in order to keep the flow system as simple as possible (persulfate, hydrochloric acid and borohydride solutions were delivered by using the MP-AES peristaltic pump). Other variables (hydrochloric acid and borohydride concentrations) were less critical and had a similar effect on the signals of two Se species. The finally selected parameters are given in Table 1 and in Fig. 2, typical records for three successive injections are presented for Se(IV) and SeMet. For Se(IV) 5 mg L−1, the average peak area was 58299 (RSD = 2.2%) and for this same concentration of SeMet, the average peak area was 8182 (RSD = 2.6%). For five between-day replicates the respective relative standard deviations were 2.6% and 3.1%. The signal of SeMet was about seven times lower than that of Se(IV) but it presented acceptable repeatability both in successive injections and for between-days measurements.
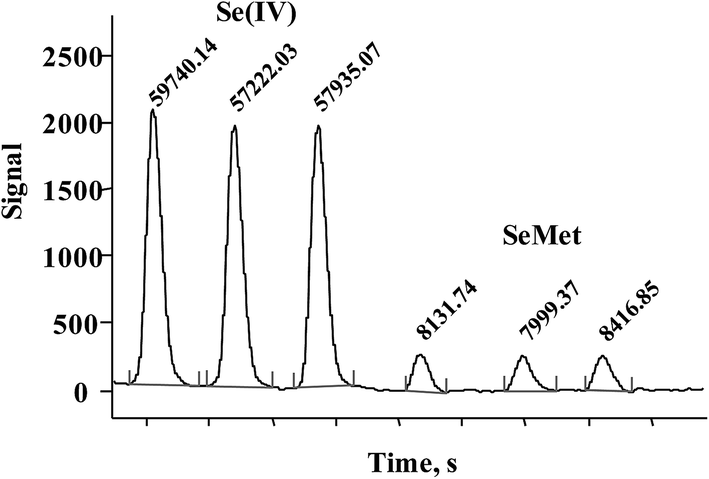 | ||
| Fig. 2 Successive FIA signals acquired for Se(IV) and SeMet (5 mg Se L−1 each) under hydride generation conditions listed in Table 1. | ||
Based on many earlier studies,12,37 anion exchange and ion-pair reversed phase separations were considered and in the first approach, the possible effect of the mobile phase composition on the analytical signal of SeMet and Se(IV) was examined in the FIA system. Phosphate buffer at pH 6.8 (25–100 mmol L−1), typically used in anion exchange separations, caused important depression of the SeMet signal even at the lowest concentration tested (about 30% decrease). On the other hand, heptafluorobutyric acid (0.05–0.2% m/v) did not affect the selenium signal for any species; therefore it was decided to use a reversed phase column. It was also verified that up to 15% v/v of methanol could be safely added to the mobile phase without deterioration of analytical signals. Isocratic separation was carried out on a Gemini C18 (50 × 3 mm, 5 μm) column under the conditions listed in Table 1; Se(IV) was eluted with a retention time of 1.16 ± 0.05 min and SeMet 2.97 ± 0.07 min; the total chromatographic run was 5 min.
3.2. Method validation
In Fig. 3, typical chromatograms obtained for the calibration solutions are presented. For six independent calibration processes, the linear regression functions were: ASe = 2862.8cSe(IV) − 238.6 and ASe = 451.78cSeMet + 644.18 with r2 > 0.998 (ASe – peak area; cSeMet/cSe(IV) – concentrations as mg Se L−1). The detection limits (DLs) and quantification limits (QLs) were evaluated based on the signal-to-noise ratio; the criterion of three and ten standard deviations was adopted using the Se signal obtained for the lowest calibration standard for each selenium species.38 The obtained calibration DLs were 18 μg Se L−1 for Se(IV) and 0.16 mg Se L−1 for SeMet; the respective QL values were 59 μg Se L−1 and 0.52 mg Se L−1. To evaluate method DLs and QLs, the baseline was acquired from the chromatogram of control biomass (C0) and this sample was analyzed after the addition of the lowest calibration standard of the two species. The obtained DL and QL for Se(IV) were 3.7 μg Se g−1 and 11.9 μg Se g−1, respectively, whereas for SeMet these values were 32 μg Se g−1 and 104 μg Se g−1. For five between-day replicates, the lowest RSD for analytical signals of both species was obtained in the concentration range of 10–40 mg Se L−1; <1.5% for SeMet and <2.9% for Se(IV). The above analytical parameters were suitable for the application of the proposed procedure in the analysis of Se-yeast digests.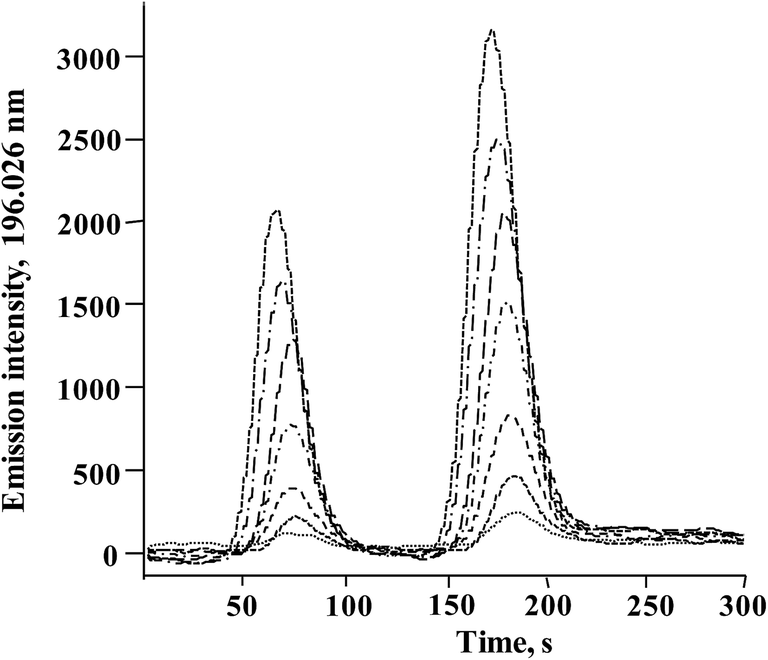 | ||
| Fig. 3 Typical HPLC-HG-MP-AES chromatograms obtained for calibration solutions (0; 0.25; 0.5; 1.0; 2.0; 3.0; 4.0; 5.0 mg Se L−1for Se(IV) and 2.5; 5; 10; 20; 30; 40; 50 mg Se L−1 for SeMet). | ||
The procedure was tested by analyzing standardized Seleno Excell® yeast, which had been evaluated and determined to be Generally Recognized As Safe (GRAS) in accordance with the Federal Food, Drug, and Cosmetic Act.7 This product was used by Clark et al. in the first clinical trial that demonstrated the potential of Se-yeast in cancer prevention.39 Total Se determined in HNO3/H2O2 digested biomass was 1289 ± 33 μg Se g−1 (n = 3), in agreement with the 1255 μg Se g−1 reported by the manufacturer. The concentration found in methanesulfonic acid hydrolyzate was 1293 ± 39 μg Se g−1, confirming efficient solubilization of all selenium forms in this sample. In Fig. 4a two chromatograms are presented that were obtained for Seleno Excell® and for the same sample after standard addition, and the respective quantification results are given in Table 2. Of note, a more complex composition of yeast hydrolyzate as compared to the calibration solutions caused a slight decrease of the retention times for both species (compare Fig. 3 and 4).
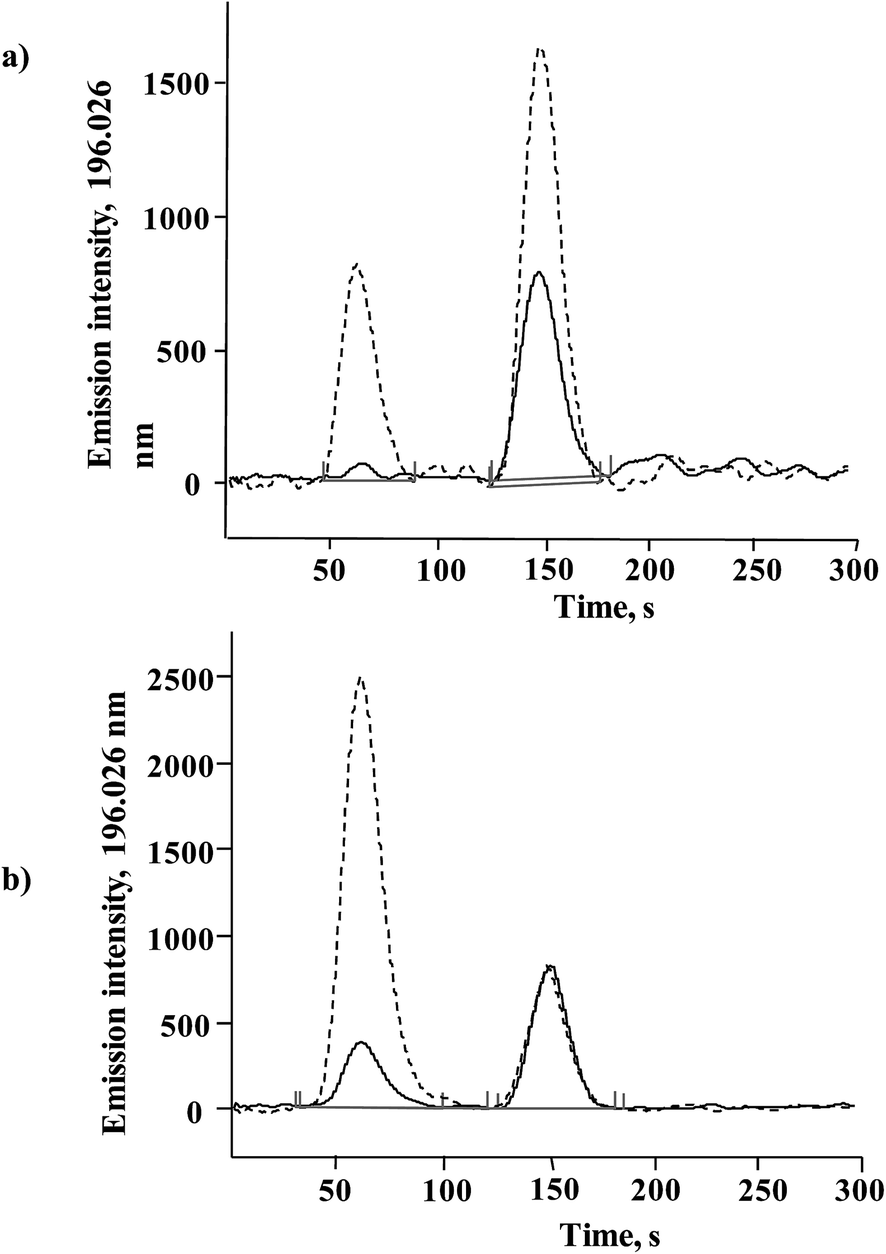 | ||
| Fig. 4 HPLC-HG-MP-AES chromatograms of methanesulfonic acid hydrolyzates of yeast: (a) (—) Seleno Excell and (----) the same sample after standard addition (details given in Section 2.5). (b) (—) Sample C10 and (----) sample C20. | ||
| Procedure | SeMet | Se(IV) | ||||
|---|---|---|---|---|---|---|
| Added, μg Se g−1 | Found ± SD, μg Se g−1 | Recovery, % | Added, μg Se g−1 | Found ± SD, μg Se g−1 | Recovery, % | |
| 1 | 0 | 1044 ± 42 | 0 | 16.5 ± 1.5 | ||
| 1000 | 2085 ± 49 | 104.1 | 200 | 221 ± 18 | 102.2 | |
| 2 | 0 | 1036 ± 37 | 0 | 15.5 ± 1.4 | ||
| 500 | 1529 ± 46 | 98.7 | 150 | 169 ± 15 | 102.6 | |
For further accuracy checking, methanesulfonic hydrolyzate of Seleno Excell® was also analyzed by the HPLC-ICP-MS procedure as described in Section 2.6. Two chromatograms corresponding to the spiked and non-spiked sample are shown in Fig. 5a, whereas the quantification results are included in Table 2. The elution of several minor species other than Se(IV) and SeMet is observed in Fig. 5a, in agreement with many earlier reports.9,10 Based on HPLC-ICP-MS results, the sum of SeMet and Se(IV) in the non-spiked sample accounted for 81.6% of total acid-digested selenium, whereas for HPLC-HG-MP-AES this percentage was 82.3%. Previously reported data for Seleno Excell indicated 84% SeMet and below 1% of Se(IV) as referred to total Se.7,9 Recovery results obtained for the two procedures after standard addition were in the range of 98.7–104.1% (Table 2); highly consistent speciation results obtained for the control sample by two different procedures demonstrates the suitability of the HPLC-HG-MP-AES system for the determination of SeMet and Se(IV) in Se-biofortified yeast.
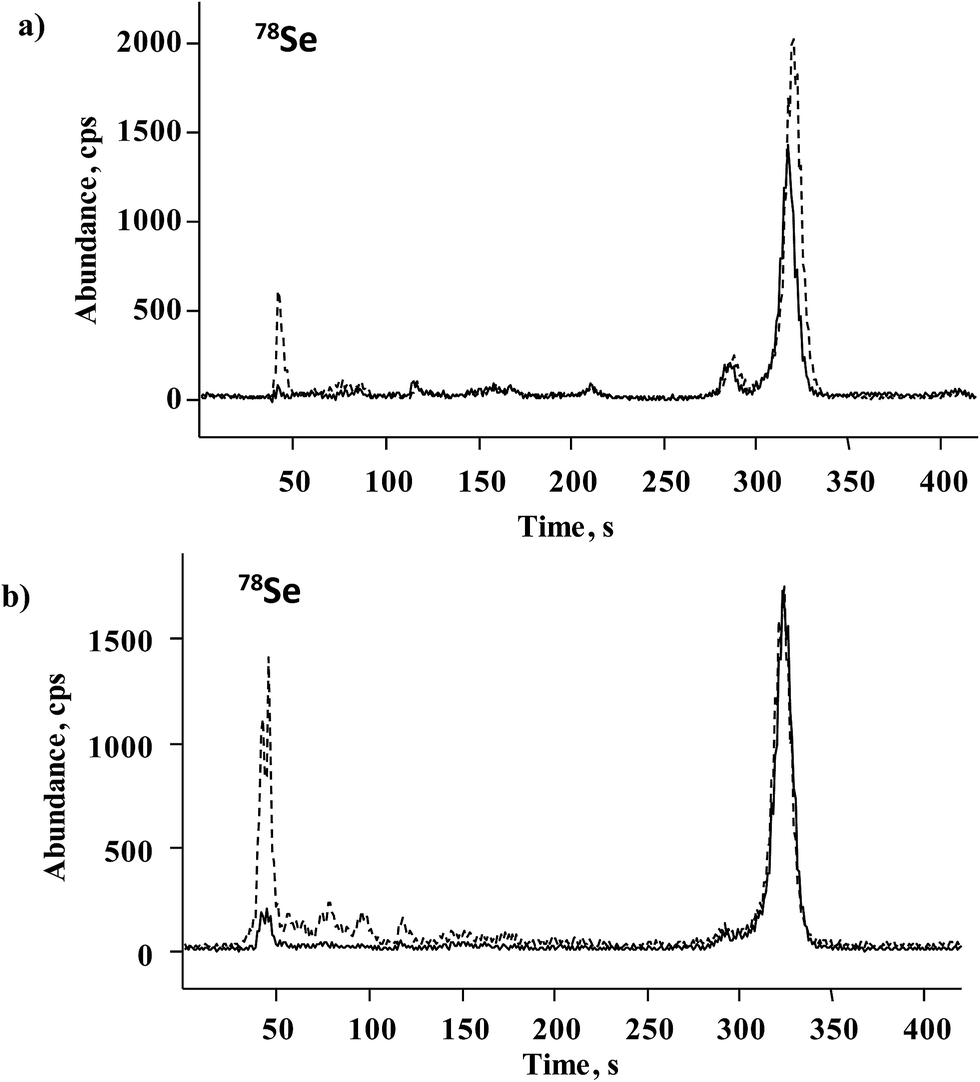 | ||
| Fig. 5 HPLC-ICP-MS chromatograms of yeast hydrolyzates (methanesulfonic acid): (a) (—) Seleno Excell and (----) the same sample after standard addition (details given in Section 2.5). (b) (—) Sample C10 and (----) sample C20. | ||
3.3. Application for the analysis of different yeast cultures
A series of S. cerevisiae cultures were obtained in the Biology Department of the University of Guanajuato as shortly described in Section 2.2. The results of Se determination in yeast digested with HNO3/H2O2 and hydrolyzed with methanesulfonic acid are shown in Fig. 6. For the first experiment (samples C0–C30), total acid-digested Se increased accordingly as the Se(IV) concentration added to the growth medium increased, whereas in methanesulfonic acid hydrolyzed samples this tendency was much less marked. It is noteworthy that starting from the Se(IV) concentration of 15 mg L−1 (C15), the yeast biomass was pink, likely revealing the presence of elemental selenium. Therefore, the difference observed between two treatments was indicative of the amount of Se0 in the sample and the Se concentration in methanesulfonic acid hydrolyzate corresponding to the total of other than Se0 element forms incorporated into the biomass. In the second experiment, varying growth conditions were used while keeping the same concentration of Se(IV) in the medium (10 mg L−1), none of these cultures presented pink color. It can be observed that for the series of samples R10, M10, N10 and CC10, no important differences in Se concentrations were found between two treatments and the results were similar to those obtained for Seleno Excell® (Fig. 6), which indicates efficient incorporation of the element in yeast. Speciation analysis in the yeast methanesulfonic acid hydrolyzates was carried out by HPLC-HG-MP-AES and by HPLC-ICP-MS; typical chromatograms obtained for two different cultures (C10 and C20) are presented in Fig. 4b and in Fig. 5b, respectively. The concentration-dependent chromatographic signals of Se(IV) and SeMet are clearly observed in both systems; however, a more sensitive HPLC-ICP-MS procedure allowed for the detection of several other, minor Se compounds (similarly as in Fig. 5a). Quantification results for SeMet and Se(IV) are summarized in Table 3, these data show good agreement between two different speciation systems which reaffirms the validity of the procedure proposed here. It should be stressed that the percentage amount of individual species and of their sum with respect to total HNO3/H2O2 digested Se were also consistent between the two procedures (Table 3). On the other hand, the concentrations of SeMet and Se(IV) varied, depending on the growing and exposure conditions applied. In the first series, SeMet was relatively stable within a range of 10–20 mg Se L−1 for Se(IV) in the medium; however, increasing concentrations of Se(IV) in the medium were accompanied by gradual buildup of this species in yeast. In the second series, CC10 was obtained under the same growth and exposure conditions as for C10 in the first series; total Se concentrations and speciation results were consistent for these two samples, indicating good repeatability of the microbiological experiment and also good repeatability of analytical results. The samples R10 and N10 corresponded to two different yeast strains, lower efficiency of Se incorporation was expected in these cultures and indeed the lowest total Se and SeMet were found (Table 3 and Fig. 6). Finally, the M10 sample contained the highest SeMet concentration, which was also expected because it was obtained from yeast grown by a directed evolution strategy in a minimal medium, deprived of amino acids. In this sample, the SeMet and total Se concentrations were slightly higher as compared to Seleno Excell®, yet respective percentages of SeMet in biomass were very similar in these two materials (81.5% versus 81.0% according to the proposed procedure and 81.9% versus 80.4% according to HPLC-ICP-MS; Table 3). Experimental data obtained in this work do not enable any biological interpretation; however the above discussion proves the feasibility of HPLC-HG-MP-AES in refining experimental conditions for the production of Se-yeast of good quality. In particular, this procedure was capable of detecting differences of SeMet and Se(IV) concentrations among samples obtained from different yeast cultures, hence it meets the requirements of sensitivity and detection power dictated by this analytical task.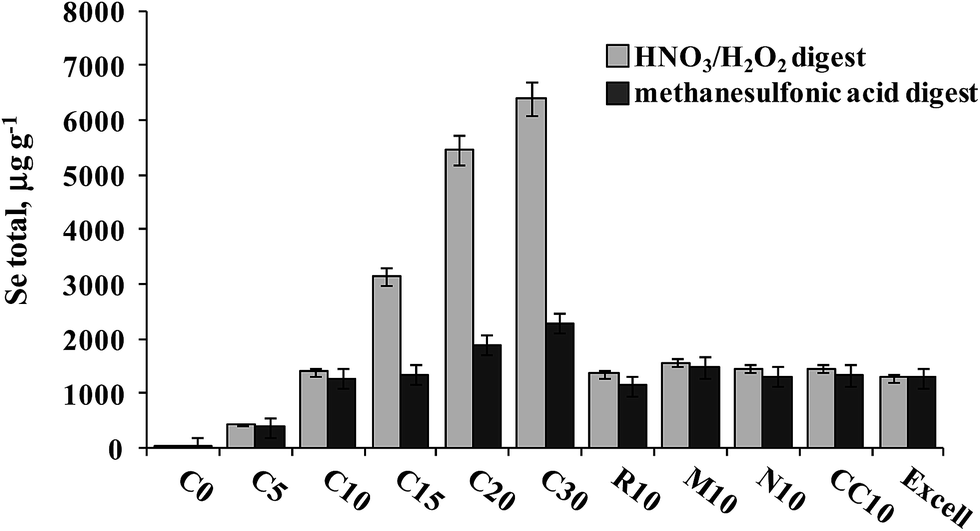 | ||
| Fig. 6 Total selenium concentrations found in HNO3/H2O2 digests and methanesulfonic acid hydrolyzates of different yeast cultures. | ||
| Yeast culture | SeMet | Se(IV) | SeMet + Se(IV) in hydrolyzate, % Se | ||
|---|---|---|---|---|---|
| Mean ± SD μg Se g−1 | % Se in biomass | Mean ± SD μg Se g−1 | % Se in biomass | ||
| HPLC-HG-MP-AES | |||||
| C0 | nd | — | nd | — | — |
| C5 | 322 ± 25 | 73.3 | 58.1 ± 2.7 | 13.2 | 99.5 |
| C10 | 1035 ± 41 | 73.5 | 98.1 ± 4.2 | 7.0 | 88.7 |
| C15 | 1044 ± 37 | 33.1 | 202 ± 5 | 6.4 | 92.2 |
| C20 | 1078 ± 42 | 81.0 | 675 ± 37 | 1.3 | 92.6 |
| C30 | 969 ± 23 | 15.1 | 989 ± 39 | 15.5 | 85.5 |
| R10 | 979 ± 21 | 71.7 | 108 ± 10 | 7.9 | 95.3 |
| M10 | 1275 ± 31 | 81.5 | 129 ± 13 | 8.2 | 94.8 |
| N10 | 791 ± 32 | 54.6 | 139 ± 18 | 9.6 | 70.7 |
| CC10 | 1041 ± 39 | 71.7 | 114 ± 10 | 7.9 | 86.5 |
| Excell | 1044 ± 42 | 81.0 | 16.5 ± 1.5 | 1.3 | 82.0 |
|
|||||
| HPLC-ICP-MS | |||||
| C0 | 7.3 ± 0.4 | 60.5 | nd | — | 62.6 |
| C5 | 318 ± 23 | 72.5 | 59.7 ± 2.8 | 13.7 | 98.9 |
| C10 | 1038 ± 34 | 73.7 | 96.7 ± 3.7 | 6.9 | 88.7 |
| C15 | 1056 ± 42 | 33.5 | 210 ± 5 | 6.7 | 93.6 |
| C20 | 1082 ± 33 | 19.8 | 663 ± 32 | 12.1 | 92.1 |
| C30 | 976 ± 21 | 15.3 | 973 ± 41 | 15.2 | 85.1 |
| R10 | 964 ± 24 | 70.6 | 105 ± 9 | 7.7 | 93.6 |
| M10 | 1281 ± 38 | 81.9 | 121 ± 11 | 7.7 | 94.7 |
| N10 | 802 ± 35 | 55.4 | 133 ± 16 | 9.2 | 71.1 |
| CC10 | 1050 ± 42 | 72.4 | 110 ± 8 | 7.6 | 86.9 |
| Excell | 1036 ± 37 | 80.4 | 15.5 ± 1.4 | 1.2 | 81.4 |
4. Conclusions
In this work, a HPLC-HG-MP-AES system was used for the first time for the speciation analysis in real-world samples. In particular, a procedure for the determination of SeMet and Se(IV) in biofortified yeast has been developed. Prior to the post-column hydride generation, relatively mild oxidation conditions (K2S2O8 6% m/v + NaOH 3% m/v, 100 cm reaction coil, 60 °C) were applied thus achieving H2Se formation from both species; HG was necessary to enhance the detection power and to avoid the introduction of a sample matrix into MP-AES that was tuned to the emission line highly susceptible to interference (196.026 nm). The compromise conditions applied allowed for the method quantification limits of 11.9 μg Se g−1 and 104 μg Se g−1 for Se(IV) and SeMet, respectively. Therefore, the proposed procedure is well suited for the determination of the two species in Se-yeast of good quality that should contain 60–90% SeMet and below 1% of Se(IV) with respect to the total Se content in biomass (1255 μg Se g−1 in Seleno Excell®). Furthermore, two series of S. cerevisiae cultures biofortified under different growth and exposure conditions were analyzed and the results were consistent with those obtained by using the HPLC-ICP-MS system. In summary, it was demonstrated that the simple and cost-effective HPLC-HG-MP-AES procedure proposed here can be helpful during optimization of the production process of selenized yeast and might be incorporated into the quality control scheme.Acknowledgements
The financial support from the National Council of Science and Technology, Mexico (CONACYT), projects 178553, 187749, 388394, is gratefully acknowledged. The authors also acknowledge the financial support from the University of Guanajuato projects 415/2014 and academic excellence 2014.References
- M. Kieliszek and S. Blazejak, Nutrition, 2013, 29, 713–718 CrossRef CAS PubMed.
- G. N. Schrauzer, Pure Appl. Chem., 2006, 78, 105–109 CrossRef CAS.
- M. P. Rayman, Proc. Nutr. Soc., 2005, 64, 527–542 CrossRef CAS.
- P. D. Whanger, Br. J. Nutr., 2004, 91, 11–28 CrossRef CAS.
- G. F. Combs, J. Nutr., 2005, 135, 343–347 CAS.
- F. Aguilar, H. Autrup, S. Barlow, L. Castle, R. Crebelli, W. Dekant, K. H. Engel, N. Gontard, D. Gott, S. Grilli, R. Gürtler, J. C. Larsen, C. Leclercq, J. C. Leblanc, F. X. Malcata, W. Mennes, M. R. Milana, I. Pratt, I. Rietjens, P. Tobback and F. Toldrá, EFSA J., 2008, 766, 1–42 Search PubMed.
- R. S. McQuate, R. C. Kraska, GRAS assessment of high selenium yeast, Food usage conditions for general recognition of safety for Cypress System, Inc., 2008.
- E. Rampler, S. Rose, D. Wieder, A. Ganner, I. Dohnal, T. Dalik, S. Hann and G. Koellensperger, Metallomics, 2012, 4, 1176–1184 RSC.
- M. P. Rayman, Br. J. Nutr., 2004, 92, 557–573 CrossRef CAS.
- E. Dumont, F. Vanhaecke and R. Cornelis, Anal. Bioanal. Chem., 2006, 385, 1304–1323 CrossRef CAS PubMed.
- K. Bierla, J. Szpunar, A. Yiannikouris and R. Lobinski, TrAC, Trends Anal. Chem., 2012, 41, 122–132 CrossRef CAS PubMed.
- K. Wrobel, K. Wrobel and J. A. Caruso, in The Determination of Chemical Elements in Food. Applications for Atomic and Mass Spectrometry, ed. S. Caroli, John Wiley & Sons, Hoboken, NJ, 2007, pp. 671–706 Search PubMed.
- H. Goenaga-Infante, R. Sturgeon, J. Turner, R. Hearn, M. Sargent, P. Maxwell, L. Yang, A. Barzev, Z. Pedrero, C. Cámara, V. Díaz Huerta, M. L. Fernández Sánchez, A. Sanz-Medel, K. Emese, P. Fodor, W. Wolf, R. Goldschmidt, V. Vacchina, J. Szpunar, L. Valiente, R. Huertas, G. Labarraque, C. Davis, R. Zeisler, G. Turk, E. Rizzio, L. G. Mackay, R. B. Myors, D. L. Saxby, S. Askew, W. Chao and W. Jun, Anal. Bioanal. Chem., 2008, 390, 629–642 CrossRef CAS PubMed.
- L. Hinojosa Reyes, J. M. Marchante-Gayon, J. I. Garcia Alonso and A. Sanz-Medel, J. Agric. Food Chem., 2006, 54, 1557–1563 CrossRef CAS PubMed.
- Z. Mester, S. Willie, L. Yang, R. Sturgeon, J. A. Caruso, M. L. Fernandez, P. Fodor, R. J. Goldschmidt, H. Goenaga-Infante, R. Lobinski, P. Maxwell, S. McSheehy, A. Polatajko, B. B. Sadi, A. Sanz-Medel, C. Scriver, J. Szpunar, R. Wahlen and W. Wolf, Anal. Bioanal. Chem., 2006, 385, 168–180 CrossRef CAS PubMed.
- L. Yang, Z. Mester and R. E. Sturgeon, Anal. Chem., 2004, 76, 5149–5156 CrossRef CAS PubMed.
- C. Swart, O. Rienitz and D. Schiel, Talanta, 2011, 83, 1544–1551 CrossRef CAS PubMed.
- L. Yang, P. Maxwell and Z. Mester, Anal. Methods, 2013, 5, 525–529 RSC.
- S. Sannac, F. Pannier, C. Oster, G. Labarraque, P. Fisicaro and M. Potin-Gautier, J. Anal. At. Spectrom., 2009, 24, 237–241 RSC.
- J. L. Capelo, P. Ximenez-Embun, Y. Madrid-Albarran and C. Camara, Anal. Chem., 2004, 76, 233–237 CrossRef CAS PubMed.
- M. Stilboller, M. Damm, A. M. Barbera, D. Kuehnelt, K. Francesconi and C. O. Kappe, Anal. Methods, 2011, 3, 738–741 RSC.
- J. L. Capelo, C. Fernandez, B. Pedras, P. Santos, P. Gonzalez and C. Vaz, Talanta, 2006, 68, 1442–1447 CrossRef CAS PubMed.
- D. Sanchez-Rodas, W. T. Corns, B. Chen and P. B. Stock, J. Anal. At. Spectrom., 2010, 25, 933–946 RSC.
- J. L. Gomez-Ariza, M. A. Caro de la Torre, I. Giraldez and E. Morales, Anal. Chim. Acta, 2004, 524, 305–314 CrossRef CAS PubMed.
- H. Li, Y. Luo, Z. Li, L. Yang and Q. Wang, Anal. Chem., 2012, 84, 2974–2981 CrossRef CAS PubMed.
- M. M. Gómez, T. Gasparic, M. A. Palacios and C. Camara, Anal. Chim. Acta, 1998, 374, 241–251 CrossRef.
- S. Simon, A. Barats, F. Pannier and M. Potin-Gautier, Anal. Bioanal. Chem., 2005, 583, 562–569 CrossRef PubMed.
- Y. Arslan, E. Yildirim, M. Gholami and S. Bakirdere, TrAC, Trends Anal. Chem., 2011, 30, 569–585 CrossRef CAS PubMed.
- J. Zhang, N. Moore and W. T. Frankenberger, Environ. Sci. Technol., 1999, 33, 1652–1656 CrossRef.
- A. Chatterjee, Y. Shibata and M. Morita, J. Anal. At. Spectrom., 2000, 15, 913–919 RSC.
- M. R. Hammer, Spectrochim. Acta, Part B, 2008, 63, 456–464 CrossRef PubMed.
- A. R. Corrales Escobosa, K. Wrobel, E. Yanez Barrientos, S. Jaramillo Ortiz, A. S. Ramirez Segovia and K. Wrobel, Anal. Bioanal. Chem., 2015, 407, 1149–1157 CrossRef CAS PubMed.
- K. Wrobel, A. R. Corrales Escobosa, A. A. Gonzalez Ibarra, M. Mendez Garcia, E. Yanez Barrientos and K. Wrobel, J. Hazard. Mater., 2015, 300, 144–152 CrossRef CAS PubMed.
- M. Slachcinski, Appl. Spectrosc. Rev., 2014, 49, 271–321 CrossRef CAS PubMed.
- K. Wrobel, S. S. Kannamkumarath and J. A. Caruso, Anal. Bioanal. Chem., 2003, 375, 133–138 CAS.
- L. Yang, R. E. Sturgeon, S. McSheehy and Z. Mester, J. Chromatogr. A, 2004, 1055, 177–184 CrossRef CAS PubMed.
- Z. Pedrero and Y. Madrid, Anal. Chim. Acta, 2009, 634, 135–152 CrossRef CAS PubMed.
- ICH Harmonized Tripartite Guideline, Validation of analytical procedures: text and methodology (Q2/R1), http://www.ish.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1_Guideline.pdf.
- L. C. Clark, B. W. Turnball, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glover, D. K. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith, J. R. Taylor, D. S. Alberts, R. J. Allison, J. C. Bradshaw, D. Curtus, D. R. Deal, M. Dellasega, J. D. Hendrix, J. H. Herlong, L. J. Hixon, J. Knight, J. Moore, J. S. Rice, A. I. Rogers, B. Schuman, E. H. Smith and J. C. Woodward, J. Am. Med. Assoc., 1996, 276, 1957–1963 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |