
Assessment of the natural variability of B, Cd, Cu, Fe, Pb, Sr, Tl and Zn concentrations and isotopic compositions in leaves, needles and mushrooms using single sample digestion and two-column matrix separation†
Ilia
Rodushkin
*ab,
Nicola
Pallavicini
 ab,
Emma
Engström
ab,
Dieke
Sörlin
b,
Björn
Öhlander
a,
Johan
Ingri
a and
Douglas C.
Baxter
b
ab,
Emma
Engström
ab,
Dieke
Sörlin
b,
Björn
Öhlander
a,
Johan
Ingri
a and
Douglas C.
Baxter
b
aDivision of Geosciences, Luleå University of Technology, S-971 87 Luleå, Sweden
bALS Laboratory Group, ALS Scandinavia AB, Aurorum 10, S-977 75 Luleå, Sweden. E-mail: Ilia.Rodushkin@alsglobal.com
First published on 9th September 2015
Abstract
An analytical procedure allowing multi-elemental analyses and isotope ratio measurements of eight of these (B, Cd, Cu, Fe, Pb, Sr, Tl and Zn) in matrices relevant for bio-monitoring using a single high-pressure acid digestion was developed. Method blanks, separation efficiency of matrix elements, repeatability and reproducibility were evaluated using sets of preparation blanks, certified reference materials and duplicate samples prepared and analyzed over a period of several months. The method was used to assess the natural variability of concentrations and isotopic compositions in bio-indicators (tree leaves, needles and mushrooms, over 240 samples) collected mainly from a confined area in North-East Sweden. Ranges found from leaves and needles were compared with data obtained for limited numbers of samples collected in Spain, Italy, France, United Kingdom and Iceland.
1. Introduction
Isotopic information can be used to aid a wide range of scientific disciplines, including environmental geochemistry and plant sciences. Such uses include tracing metal contamination sources/pathways, studying biological processes (nutrient and anthropogenic uptakes/cycles, within plant transport mechanisms),1 remediation and geographical provenance.2–5 Variability in the isotopic composition of radiogenic elements, e.g. lead (Pb), strontium (Sr) and osmium (Os), has been frequently utilized in environmental studies6–12 and application niches continue to grow. Mass-dependent fractionation of boron (B) and other light elements (carbon, oxygen and nitrogen) has now been used for provenance studies, tracing pollution sources and water mixing for decades.2,13–15Enhancements in measurement precision due to developments in analytical instrumentation, e.g. the advent of multiple collector inductively coupled plasma mass spectrometry (MC-ICP-MS), as well as continual refining of preparation/separation and pre-concentration methods1 have allowed inclusion of heavier stable elements in the ‘isotope toolbox’ and the number of published stable isotope studies has grown exponentially in the last decade.16 For example, in environmental studies involving bio-indicators, Pb11,17 and Sr18 isotopes have been relied upon for at least the past three decades, whereas Os,19–22 Cd23 and Tl have been relatively new additions contributing to the expanding array of investigations.
Another relatively new development is the application of multi-tracer studies,2,24–28 as source tracing in two or three-dimensional space potentially allows distinguishing samples having overlapping isotope signatures for a single element.16 In an exemplary study, Sherman et al.24 used Pb, Sr and Hg isotopes in precipitation to identify the signature of coal combustion. δ11B coupled with 87Sr/86Sr ratios has also proved to be effective in determining the origin of coal combustion residuals.25
Apart from anthropogenic assessment, a great deal of attention in recent studies has been given to the study of the biological processes responsible for variations in isotopic compositions in plants and animals.27,29–35 In the plant sciences, the major focus has been on elements essential for plant growth. With the help of isotopic data, Rosner et al.32 concluded that B assimilation by plants is directly influenced by the local conditions (both natural and anthropogenic). This has been further confirmed in another recent multi-isotope study where the coupling of Sr and B isotope ratios has been used to trace the geographic origins of coffee beans.2 Jouvin et al.31 described two models for fractionation of the micro-nutrients Cu and Zn during uptake by plants, proposing different fractionation patterns for different uptake strategies. Uptake mechanisms for Cu, Fe and Zn by plants have been the focus of a number of studies.29–31,36,37
Wider applications of isotope signatures in environmental geochemistry and plant sciences are often hampered by the high cost of instrumentation, the need for often tedious, elaborative and time-consuming sample preparation and analyte separation schemes,38,39 as well as challenges to verify the accuracy of analytical methods.16,40–42 As a result, many studies are still based on very limited numbers of samples and often consider only a single isotope system, which may affect the transferability of any conclusion drawn. Clearly, a massive amount of isotopic information would need to be acquired for a meaningful assessment of the natural variability in various eco-systems and to identify the factors responsible for such variability. In approaching an investigation, special importance must also be given to the geographical scale of the study or else important local details may resist detection.11 Thus the possibility to obtain isotope data for many elements coupled to concentration information in a reasonable time and without the need to use several types of instruments is of extreme value.
The aim of this work is to provide a detailed description of an analytical procedure allowing for MC-ICP-MS isotope ratio measurements of at least eight elements from a single digestion of a limited amount (approximately 0.5 g) of plant material. The analytical procedure was applied to a substantial number of leaves, needles and mushrooms (frequently used bio-indicators11,43,44) collected from urban environments in several European countries to assess the extent of the natural variability of B, Cd, Cu, Fe, Pb, Sr, Tl and Zn isotope compositions as well as seasonal variations.
2. Experimental
2.1. Instrumentation
All isotope ratio measurements except for B in those samples low in the analyte were performed using a NEPTUNE PLUS (Thermo Scientific, Bremen, Germany) MC-ICP-MS instrument operated with variously configured introduction systems, including Aridus II (Teledyne CETAC Technologies, Omaha, NE, USA) and Apex (Elemental Scientific, Omaha, NE, USA) desolvating nebulizers. Cup configurations used, operating conditions and measurement parameters are given in Table 1.| Elementa | Configuration of the introduction system | Resolution mode | Integration time (s) | Sample uptake rate (L min−1) | Cup configuration | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L4 | L3 | L2 | L1 | C | H1 | H2 | H3 | H4 | |||||
| a One element is used as the internal standard for the second element, except for Zr which is only used as the internal standard. b RF power: 1400–1450 W. Coolant gas flow: 15 L min−1. Auxiliary gas flow: 1.4 L min−1. Sample gas flow: 0.9–1.25 L min−1. Additional gas flow (N2, Aridus and Apex) 0.01–0.02 L min−1. Ion lens settings: adjusted daily to obtain maximum sensitivity and signal stability. Zoom optic settings: adjusted daily to obtain maximum resolution. Number of blocks: 9. Number of cycles per block: 5. Number of integrations: 3–5. Amplifier rotation: left. | |||||||||||||
| Cd/Ag | Aridus/Apex desolvating systems, self-aspirating micro-concentric PFA nebulizer, X-type skimmer cone | Low | 0.524 | 0.04–0.06 | 107Ag | 108Cd (108Pd) | 109Ag | 110Cd (110Pd) | 111Cd | 112Cd (112Sn) | 114Cd (114Sn) | 116Cd (116Sn) | 117Sn |
| Zn/Cu | Pumped Micromist nebulizer, double spray chamber, H-type skimmer cone | Medium | 0.262 | 0.20–0.25 | — | 63Cu | 64Zn (64Ni) | 65Cu | 66Zn | 67Zn | 68Zn | 70Zn (70Ge) | — |
| Fe/Ni | Pumped Micromist nebulizer, double spray chamber, H-type skimmer cone | Medium | 0.262 | 0.20–0.25 | 54Fe (54Cr) | — | 56Fe | 57Fe | — | 60Ni | 61Ni | 62Ni | — |
| Sr/Zr | Pumped Micromist nebulizer, double spray chamber, H-type skimmer cone | Low | 0.262 | 0.20–0.25 | 82Kr | 83Kr | 84Sr (84Kr) | 85Rb | 86Sr (86Kr) | 87Sr (87Rb) | 88Sr | 90Zr | 91Zr |
| B | Pumped Micromist nebulizer, mini cyclonic spray chamber, X-type skimmer cone | Low | 0.524 | 0.40–0.50 | — | — | 10B | — | — | — | 11B | — | — |
| Pb/Tl | Aridus/Apex desolvating systems, self-aspirating micro-concentric PFA nebulizer, X-type skimmer cone | Low | 0.524 | 0.04–0.06 | — | 202Hg | 203Tl | 204Pb (204Hg) | 205Tl | 206Pb | 207Pb | 208Pb | — |
B isotope ratio measurements in some samples and all measurements of elemental concentrations were performed by double-focusing sector field ICP-MS (ICP-SFMS; ELEMENT XR, Thermo Scientific).23 Methane addition to the plasma was used to decrease formation of oxide-based spectral interferences, improve sensitivity for elements with high first ionization potentials, and to minimize matrix effects.45 Operating conditions and measurement parameters for concentration measurements were as described in a previous study.46
A laboratory UltraCLAVE single reaction chamber microwave digestion system (Milestone, Sovisole, Italy) was used for sample digestions.
2.2. Chemicals and reagents
Nitric acid (HNO3) and hydrochloric acid (HCl), both from Sigma-Aldrich Chemie GmbH (Munich, Germany) and hydrogen fluoride (HF, 48%, Merck, Darmstadt, Germany) used in this work were all of analytical grade. Water used in all experimental procedures was de-ionized Milli-Q water (Millipore, Bedford, MA, USA) purified by reverse osmosis followed by ion-exchange cartridges. For dilution of sample digestion aliquots intended for B isotope ratio measurements, water was further purified by sub-boiling distillation in Teflon stills (Savillex, Minnetonka, MN, USA). AG MP-1M ion-exchange resin (macroporous, 100–200 dry mesh size, 75–150 μm wet bead size, Bio-Rad Laboratories AB, Solna, Sweden) was cleaned by soaking in 0.7 M HNO3 followed by rinsing with Milli-Q water and loading as slurry into 2 mL columns. Pre-packed 2 mL columns with Sr-Spec resin (Eichrom Technologies, IL, USA) were used as supplied.The following chemicals were used as ‘δ-zero’ standards: NIST SRM 3108 Cd solution Lot 130116, NIST SRM 976 Cu standard solution, NIST SRM 951a – boric acid, NIST SRM 981 common lead, and NIST SRM 987 (NBS-987) – strontium carbonate (all from the National Institute of Standards and Technology, Gaithersburg, MD, USA); IRMM 3702 zinc solution and IRMM-014 Fe metal (both from the Institute for Reference Materials and Measurements, Geel, Belgium). For Ag and Tl isotopic analyses, commercial standards (1000 mg L−1 mono-elemental solutions supplied by Ultra Scientific, North Kingstown, RI, USA; Ag: Lot M00474; and Tl: Lot L00709) were used as ‘δ-zero’ standards.
2.3. Samples
A large part of the samples was collected in the city and suburbs of Luleå (northern Sweden), a medium-sized town (population approximately 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) located in the province of Norrbotten. The study area lies almost entirely on a 1.9 Ga granitic bedrock with a minor metasedimentary constituent.47 Clay and silt loam are the main soil constituents48 even though it is important to note that some of the soil components in the urban areas can be non-native. The area surrounding the town of Luleå is heavily industrialized, with steelworks as the dominant local industry.
000) located in the province of Norrbotten. The study area lies almost entirely on a 1.9 Ga granitic bedrock with a minor metasedimentary constituent.47 Clay and silt loam are the main soil constituents48 even though it is important to note that some of the soil components in the urban areas can be non-native. The area surrounding the town of Luleå is heavily industrialized, with steelworks as the dominant local industry.
The sets of biological samples collected during 2013–2015 from approximately 50 individual locations included common birch (Betula pubescens) leaves, Norway spruce (Picea abies) needles and fruit bodies of edible mushrooms (Boletus edulis, Leccinum scabrum, Leccinum versipelle, Leccinum aurantiacum and Suillus variegatus). At a few locations leaves of oak (Quercus), aspen (Populus tremula) and rowan (Sorbus aucuparia) were also sampled. The amounts of material from each sampling location (corresponding to approximately 0.5–1.5 g dry weight per matrix) consisted of 10–50 leaves (depending on the growth stage) collected from different branches/trees, needles from the last year grown on parts of lower branches and mushrooms collected under sampled trees (where available). Sampling was performed either at the beginning of the growing season (May-early June, birch leaves only) or just before senescence (early September), though in the majority of locations samples were taken from different trees in spring and autumn. All samples were collected wearing powder-free laboratory gloves into zip-lock plastic bags marked with geographic coordinates, the type of sample and collection date. Sampling locations were chosen on the base of a sampling grid of 1 km2 mesh size, covering a total area of ca. 200 km2. Sampling height was limited to roughly 2.5 m from the ground for all leaf and needle samples.
Samples from other geographic locations – Genoa (Italy), suburbs of Barcelona (Spain), the city of Reykjavik and location Þórsmörk (Iceland), the city of Paris (France) and suburbs of Birmingham (United Kingdom) – were collected during autumn 2014 and spring 2015 though in significantly less numbers. When no birch or spruce trees were found (Italy and Spain), leaves and needles of other tree species, oak (Quercus), olive (Olea europaea), and pine (Pinus sylvestris), were collected.
For verifying the method, a set of certified reference materials (CRMs) has been included and processed in parallel to “natural samples” throughout the procedure (digestion, separation and analysis): ERM BB186 pig kidney (Institute for Reference Materials and Measurements), TORT-1 lobster hepatopancreas and NASS-4 open ocean water (National Research Council of Canada, Ottawa, Canada), NIST SRM 1547 peach leaves (National Institute of Standards and Technology) and NJV 94-5 wood fuel (Swedish University of Agricultural Sciences, Sweden), providing representative variability in the concentrations of analytes and ranges of isotope compositions (Table 2). Note that none of the materials mentioned above has a certified isotopic composition.
| Element | LOD (μg g−1) | Total procedural blank (ng) | Minimum concentration for isotope measurements (μg g−1) | NIST 1547 (n = 16) | IRMM-BB186 (n = 8) | NJV 94-5 (n = 10) | NRCC-TORT-1 (n = 8) |
|---|---|---|---|---|---|---|---|
| a The uncertainty given in parentheses is either the reproducibility expressed as twice the standard deviation (SD) for n replicates performed by at least two different operators or the confidence interval for the certified value. Data for CRMs are presented in the order: experimental mean values (2 SD) certified value (95% confidence interval). | |||||||
| B | 0.07 | 40 | 2 | 29.1(1.8) 29(2) | 0.58(0.12) – | 5.32(0.20) – | 5.21(0.27) – |
| Cd | 0.001 | 0.15 | 0.05 | 0.0254(0.0011) 0.026(0.003) | 1.04(0.04) 1.09(0.05) | 0.269(0.013) 0.27(0.028) | 26.2(1.0) 26.3(8.0) |
| Cu | 0.01 | 8 | 2 | 3.45(0.16) 3.7(0.4) | 34.4(1.8) 36.5(1.8) | 2.02(0.15) 2.2(0.30) | 421(22) 439(5.0) |
| Fe | 0.05 | 450 | 20 | 209(13) 218(14) | 244(15) 255(13) | 71.8(3.7) 70(16) | 196(9) 186(5.9) |
| Pb | 0.002 | 0.25 | 0.025 | 0.795(0.066) 0.87(0.03) | 0.036(0.004) 0.040(0.005) | 0.652(0.040) 0.68(0.025) | 9.32(0.42) 10.4(1.9) |
| Sr | 0.02 | 20 | 2 | 53.5(2.5) 53(4) | 0.204(0.034) – | 11.6(0.4) – | 108(6) 113(4.4) |
| Tl | 0.0001 | 0.08 | 0.01 | 0.021(0.002) | 0.015(0.001) – | 0.166(0.006) – | 0.005(0.001) – |
| Zn | 0.1 | 90 | 5 | 17.2(0.8) 17.9(0.4) | 128(7) 134(5) | 37.7(1.9) 38(8.5) | 168(8) 177(5.6) |
| Ag | 0.0004 | 0.2 | 0.025 | 0.0016(0.0004) | 0.0009(0.0003) – | 0.020(0.002) – | 6.91(0.41) – |
| Hg | 0.001 | 0.05 | 0.5 | 0.034(0.003) 0.031(0.007) | 0.017(0.002) 0.023(0.011) | 0.021(0.002) – | 0.309(0.013) 0.330(0.006) |
| Isotopes | Mean instrumental repeatability at the 2 SD level for n > 240 sample duplicates (‰) | Reproducibility at the 2 SD level for n > 10 sample duplicates (‰) | NIST 1547 (n = 16) | IRMM-BB186 (n = 8) | NJV 94-5 (n = 10) | NRCC-TORT-1 (n = 8) |
|---|---|---|---|---|---|---|
| δ 11B | 1.8 | 2.5 | 40.3(2.6) | −9.1(7.2) | 13.8(4.4) | 26.1(4.1) |
| δ 114Cd | 0.047 | 0.158 | 0.068(0.148) | 0.455(0.065) | −0.052(0.084) | −0.143(0.053) |
| δ 65Cu | 0.045 | 0.130 | 0.433(0.205) | 2.669(0.245) | −0.395(0.265) | −0.083(0.097) |
| δ 56Fe | 0.038 | 0.072 | −0.304(0.054) | −2.066(0.144) | −0.160(0.077) | −0.200(0.094) |
| 208Pb/206Pb | 0.079 | 0.330 | 2.482(0.010) | 2.478(0.048) | 2.424(0.011) | 2.464(0.005) |
| 206Pb/207Pb | 0.094 | 0.360 | 1.213(0.006) | 1.191(0.034) | 1.159(0.007) | 1.192(0.004) |
| 87Sr/86Sr | 0.036 | 0.210 | 0.71339(0.00009) | 0.71003(0.00109) | 0.73151(0.00013) | 0.70925(0.00009) |
| δ 205Tl | 0.054 | 0.115 | −0.269(0.153) | NA | −0.244(0.123) | NA |
| δ 66Zn | 0.032 | 0.084 | −0.337(0.144) | −0.608(0.105) | 0.075(0.101) | 0.616(0.099) |
| δ 109Ag | 0.048 | NA | NA | NA | NA | NA |
2.4. Sample preparation
All sample manipulations were performed in clean laboratory areas (Class 10000) by personnel wearing clean room gear and following all general precautions to reduce contaminations.49 All laboratory ware coming into contact with samples/sample digests was soaked in 0.7 M HNO3 (>24 h at room temperature) and rinsed with MQ water prior to use.Mushrooms were mechanically cleaned from external exogenous material and divided into approximately 1 cm3 pieces using a ceramic knife on a Teflon plate. Samples were then dried at 50 °C to constant weight, homogenized by crushing in plastic bags and stored air-tight packed at room temperature.
:1 v/v). The chamber was pressurized with compressed argon and the pre-programmed digestion cycle (30 min ramp to 220 °C followed by 20 min holding time at that temperature) was initiated. The total processing time, including cooling and subsequent transfer and dilution of sample digests to a final volume of 10 mL into storage polypropylene tubes, was approximately three hours per digestion batch.
In some samples, minor quantities of white precipitates of siliceous material were formed. Rapid dissolution of the precipitate was achieved after addition of 30 μL of 16 M HF and manual agitation for a few minutes. Sets of method blanks and CRMs were prepared with each batch of samples.
As for any of the wide variety of methods used for the preparation of biological matrices for subsequent ICP-SFMS analysis in the laboratory, e.g. ashing, hot-block and microwave digestions, and high pressure ashing,10,50,51 digestion using the UltraCLAVE has its merits and limitations. The former include complete oxidation of carbonaceous material thus ensuring negligible effects of undigested organics on the subsequent separation procedure, applicability to all matrixes tested in this study, ease of sample handling/loading (limited material manipulation and thus lowered risk of contamination), and relatively high throughput. The major limiting factor is the amount of material that can be digested in a 12 mL vessel (approximately 0.5 g dried material), which may require processing parallel digestions for samples low in some analytes, though this approach was not required in the present work.
Aliquots of digests were diluted 50-fold with 1.4 M HNO3, providing a total digestion factor of approximately 1000 v/m, and analyzed by ICP-SFMS using a combination of internal standardization and external calibration.50 Portions of diluted digests remaining after this analysis (approximately 6 mL) were used for B isotope ratio measurements either directly or after additional dilution. The rest of the original sample digest was evaporated to dryness in a 25 mL Teflon beaker at 95 °C on a ceramic-top hot-plate, followed by dissolution in 4 mL of 9.6 M HCl, thus being ready for subsequent purification.
All separated analyte fractions except those for Sr and Hg were evaporated to dryness and dissolved in 2–10 mL of 0.3 M HNO3. (An aliquot of 14 M HNO3 was pipetted directly onto the solid residue as a first step, allowed to react for 15–25 min, and then diluted appropriately by addition of MQ water.) 0.1 mL aliquots of separates were diluted 50-fold with 1.4 M HNO3 and analyzed by ICP-SFMS (same approach as for sample digests). This provides: (I) information on analyte contents needed to prepare concentration- and acid strength-matched solutions for isotope ratio measurements; (II) direct assessment of analyte recovery; (III) control over separation efficiency from matrix elements; and (IV) a test for the presence of potentially spectrally interfering elements either from the sample matrix or from handling contamination.
The absence of artificially introduced fractionation during separation/evaporation and analysis stages was tested by separating a mixture of ‘δ-zero’ standards with concentrations typical for birch leaves.
For B, MC-ICP-MS measurements were performed on unseparated digests which can be diluted to provide at least 20 μg L−1 concentrations in measurement solution yielding >1 V intensity for 11B. Profound B memory effects in the introduction system were minimised by using a low volume spray chamber, diluting all samples, standards and blanks in 1.4 M HNO3, employing higher sample uptake rates and increasing the washing time between samples and standards to 200 s. This ensures that the instrumental 11B blank is below 0.02 V before measurement of the next solution is started.
For the remaining elements, separated fractions were diluted to equal concentration levels; because of limited analyte content in some biological samples, several fixed measurement concentrations were used with minimum requirements listed in Table 2. A set of bracketing standards matching samples in terms of analyte and internal standard concentrations as well as acid strength was prepared for each isotope system. High degrees of concentration matching between samples and bracketing standards (better than ±10%) are needed for accurate measurements of Cd, Ag, Cu and Zn. Measuring samples against standards with halved or doubled analyte concentrations will result in up to 0.3‰ errors. The matching tolerance for Sr, Pb, Tl and Fe isotope ratio measurements is significantly broader, where up to ±30% concentration difference between samples and standards will not affect results notably.
The best signal stability and in-run precision in isotope ratios is obtained with an introduction system consisting of a self-aspirating PFA nebulizer with approximately 0.05 mL min−1 sample uptake, cyclonic/Scott double spray chamber arrangement and H-skimmer cone. Sample throughput can be increased by almost 40% by increasing the sample uptake four-fold using a peristaltic pump (due to much shorter solution-in, signal stabilization and wash-out times) with less pronounced matrix effects as an extra benefit. However this option requires replacing PFA with a Micromist (Glass Expansion Ltd, West Melbourne, Australia) nebulizer. For ultra-trace elements, the intensity provided by such a configuration is insufficient and the use of desolvating nebulizers and the X-skimmer cone is mandatory for Cd, Ag, Pb and Tl in the majority of samples, providing, depending on the analyte, seven- to 25-fold intensity gains. Intensity can be increased even further (by a factor of 3–4) by increasing the sample uptake of desolvating nebulizers to 0.15–0.20 mL min−1, though this results in increasingly unstable and ‘spiky’ signals. Even with 0.05 mL min−1 sample uptake typical in-run precision is almost three times poorer than with the standard introduction system configuration.
The use of Aridus was found to be unsuitable for Hg isotope ratio measurements because Hg vapour is lost from the system while passing the desolvating membrane. As it was impossible to pre-concentrate Hg by evaporation of the purified fraction (again because of analyte losses) and the need to further dilute 7 M HNO3 matrix of this fraction prior to analysis, even the use of the APEX did not allow reliable Hg isotope ratio measurements in samples collected for this study. Though other means of Hg introduction to MC-ICP-MS have been suggested (e.g. cold vapour and purge-and-trap52–54), these were not tested here and plans to measure Hg isotopes were abandoned.
For samples with B concentrations <20 μg g−1, isotope ratio measurements were performed by ICP-SFMS using the same configuration of the introduction system as for MC-ICP-MS and paying special attention to avoid tailing from Ar4+ spectral interference appearing on the low-mass side of the 10B isotope in the 5% acquisition window. Though the in-run precision of single collector instrumentation is inferior to that of MC-ICP-MS by a factor of 5–10, it nevertheless allows isotope ratio measurements at much lower B concentrations in measurement solutions. B isotope ratio measurements in NIST 1547 and NASS-4 CRMs were performed during every measurement sequence, with results from both techniques agreeing to within measurement uncertainty. Additionally, Sr and Pb isotope ratio measurements in samples low in analytes (<10 μg g−1 and <0.5 μg g−1, respectively) were repeated by ICP-SFMS using solution remaining from MC-ICP-MS measurement sessions and an introduction system consisting of a self-aspirating PFA nebulizer, a cyclonic/Scott double spray chamber arrangement and an H-skimmer cone.
Three sample solutions were analyzed between two standards. Two consecutive measurements were performed for each solution in the sequence. The MC-ICP-MS software option of excluding pass, run and block outliers was deactivated as it was found that the presence of some outliers actually improves the correlation between instrumental mass bias levels for analyte and internal standard isotope ratios.
Data evaluation, including correction for blanks, spectral interferences and instrumental mass bias, was performed off-line using commercially available spreadsheet software. Instrumental mass bias was corrected in a two-step procedure using first exponential correction by internal standardization using an algorithm proposed by Baxter et al.55 followed by standard–sample bracketing (SSB). Mean ratios from two consecutive measurements of the first and the third samples in each analytical block (standard 1–sample 1–sample 2–sample 3–standard 2) were calculated against ratios for standards 1 and 2, respectively. For the second sample, mean ratios from the bracketing standards were used assuming linear changes in instrumental mass bias persisting after internal standard correction. Results from two consecutive measurements of each sample allow calculation of mean δ-values and respective standard deviations for all isotope ratios that are less affected by variations caused by imperfect amplifier gain calibration.
For Ag, B, Cd, Cu, Fe and Zn δ-values were calculated using the general formula widely adopted in isotopic studies:

Further details on isotope ratio measurement, data processing and corrections can be found in previous studies.23,40,56,57
3. Results and discussion
3.1. Performance of the separation procedure
Wider use of isotope ratio measurements by MC-ICP-MS, especially in multi-tracer studies, has resulted in the development of numerous matrix separation/analyte pre-concentration schemes evaluated for various elements and sample types. The complexity of the schemes varies from the very simple, i.e. a single pass through a commercially available pre-packed column containing a specific ion-exchanger (e.g. Sr,58 U, Th59,60), to the very time-consuming and elaborate, consisting of several different purification steps (e.g. Cd, Mo38,39,61). As the number of steps increases, so do the risks associated with potential sample contamination and/or artificially induced isotope fractionation. Application of published procedures to new types of samples often requires re-validation because the sample matrix, the concentrations of analyte and interfering elements, as well as the particulars of the sample digestion approach may severely affect separation performance.The analyte purification scheme used in this study is an amalgamation of several published separation procedures merged to maximize separation efficiency and reduce procedural time, while ensuring high analyte recoveries and low contamination levels. Based on published isolation procedures for Cu, Fe and Zn,62–64 Cd,7,23 as well as Sr and Pb,5,65 introduction of a few prudent modifications extended the number of elements isolated from the single sample digest. Volumes needed at each step were obtained through replicate calibration of columns by collecting the sample load, matrix wash and all elution fractions with 1 mL resolution before analysis by ICP-SFMS to obtain detailed elution profiles for all elements present in these samples. A flow chart depicting all steps of the purification procedure is shown in Fig. 1.
 | ||
| Fig. 1 Flowchart of the purification procedure. | ||
Average method blanks for the entire procedure, assessed by applying all preparation and separation steps to a set of reagent blanks handled as samples, are listed in Table 2. In spite of extensive sample handling, method blanks, with few exceptions, correspond to <2% contribution to concentrations found in samples containing the minimum analyte content required for isotope ratio measurements (Table 3) and therefore have a negligible effect on measured ratios. Analyte recovery was above 95% from the majority of samples and CRMs separated during this study with the sole exception of Pb (above 85%). Though the lower recovery of Pb implies a risk for artificially introduced mass-dependent fractionation, the bias introduced can be tolerated given the range of radiogenic Pb ratios found. Samples with recoveries below these thresholds were either re-prepared and re-analyzed (when the amount of sample collected was sufficient) or results for affected analytes were excluded from the following evaluations.
| Parameter | Birch leaves Luleå, May–June (n = 54) | Birch leaves Luleå, September (n = 58) | Spruce needles Luleå, September (n = 31) | Mushrooms Luleå, September (n = 30) | Leaves Genoa, May–June (n = 10) | Leaves Paris, June (n = 9) | Needles Paris, June (n = 4) | Birch leaves Iceland, August (n = 8) | Leaves Barcelona, October (n = 7) | Birch leaves Birmingham, June (n = 3) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Results are presented in the format: average (standard deviation) median, minimum ÷ maximum; note that n is the number of different samples of the specified material collected and averaged in each dataset. | ||||||||||
| B (μg g−1) | 13(6)11 | 29(15)25 | 14(6)14 | 1.7(1.6)1.0 | 48(26)43 | 32(10)29 | 30(20)23 | 50(16)44 | 62(53)45 | 17(8)19 |
| 7 ÷ 40 | 7 ÷ 76 | 3 ÷ 31 | 0.2 ÷ 6 | 15 ÷ 88 | 18 ÷ 46 | 13 ÷ 58 | 36 ÷ 75 | 13 ÷ 160 | 8 ÷ 25 | |
| δ 11B (‰) | 7.7(6)9.4 | 7.9(8)8.0 | 18(9)20 | −1(10)–4 | 21(15)18 | 6.4(5.5)6.0 | 25(8.6)22 | 13(10)16 | 22(9)18 | 16(5)16 |
| −4 ÷ 21 | −7 ÷ 25 | −2 ÷ 37 | −12 ÷ 29 | 4 ÷ 40 | −1 ÷ 16 | 18 ÷ 37 | −6 ÷ 26 | 14 ÷ 36 | 12 ÷ 22 | |
| Cd (ng g−1) | 380(150)330 | 280(130)240 | 57(43)40 | 1600(1500)1100 | 490(470)420 | 560(550)330 | 60(80)30 | 150(70)120 | 61(59)28 | 43(15)34 |
| 190 ÷ 820 | 74 ÷ 630 | 13 ÷ 160 | 50 ÷ 5100 | 1 ÷ 1100 | 30 ÷ 1600 | 5 ÷ 180 | 6 ÷ 180 | 6 ÷ 180 | 31 ÷ 65 | |
| δ 114Cd (‰) | 0.42(0.21)0.44 | 0.50(0.15)0.49 | 0.10(0.25)0.11 | 0.16(0.22)0.23 | 0.13(0.13)0.10 | 0.09 (0.11)0.07 | −0.15 (0.06)–0.15 | 0.51(0.29)0.52 | −0.05 (0.14)–0.01 | 0.40 (0.33)0.25 |
| −0.09 ÷ 0.73 | 0.17 ÷ 0.95 | −0.42 ÷ 0.56 | −0.52 ÷ 0.43 | 0.01 ÷ 0.49 | −0.14 ÷ 0.21 | −0.21 ÷ −0.09 | 0.11 ÷ 0.96 | −0.23 ÷ 0.09 | 0.18 ÷ 0.79 | |
| Cu (μg g−1) | 10(3.4)10 | 4.7(0.8)4.6 | 3.2(0.9)3.1 | 46(27)42 | 16(15)13 | 11(7.1)10 | 13(9.1)12 | 5.7(1.3)5.1 | 11(9.0)6.4 | 7.0(3.3)6.9 |
| 4.0 ÷ 17 | 3.1 ÷ 6.8 | 2.1 ÷ 5.2 | 10 ÷ 120 | 4 ÷ 58 | 4.5 ÷ 28 | 3.3 ÷ 24 | 4.6 ÷ 7.9 | 3.3 ÷ 24 | 3.3 ÷ 11 | |
| δ 65Cu (‰) | −0.44(0.24)–0.42 | −0.55(0.27)–0.50 | −1.17(0.43)–1.16 | −1.17(0.71)–1.26 | −0.50(0.52)–0.59 | −0.26(0.22)–0.34 | −0.24(0.34)–0.33 | −0.30(0.41)–0.26 | −0.14(0.50)–0.14 | −0.28(0.27)–0.19 |
| −1.0 ÷ −0.05 | −1.3 ÷ −0.09 | −2.0 ÷ −0.41 | −2.3 ÷ −0.15 | −1.3 ÷ −0.28 | −0.55 ÷ 0.07 | −0.51 ÷ 0.10 | −0.82 ÷ 0.19 | −1.1 ÷ 0.27 | −0.58 ÷ −0.06 | |
| Fe (μg g−1) | 150(60)140 | 310(210)240 | 150(110)120 | 260(550)50 | 250(210)210 | 94(31)95 | 440(310)420 | 760(440)680 | 240(230)160 | 120(50)110 |
| 70 ÷ 360 | 110 ÷ 1500 | 40 ÷ 440 | 20 ÷ 3500 | 110 ÷ 1500 | 50 ÷ 150 | 150 ÷ 750 | 290 ÷ 1300 | 55 ÷ 750 | 77 ÷ 180 | |
| δ 56Fe (‰) | −0.30(0.14)–0.28 | −0.21(0.18)–0.20 | −0.35(0.34)–0.27 | −0.35(0.35)–0.35 | −0.10(0.14)–0.13 | −0.33(0.13)–0.30 | 0.12(0.15)0.12 | −0.09(0.10)–0.06 | −0.04(0.10)–0.04 | −0.32(0.23)–0.26 |
| −0.58 ÷ −0.03 | −0.84 ÷ 0.09 | −1.32 ÷ 0.05 | −1.11 ÷ 0.25 | −0.31 ÷ 0.09 | −0.53 ÷ −0.12 | −0.05 ÷ 0.26 | −0.31 ÷ 0.01 | −0.17 ÷ −0.09 | −0.57 ÷ −0.12 | |
| Pb (ng g−1) | 170(130)110 | 540(330)480 | 160(110)130 | 190(150)130 | 250(150)210 | 570(300)520 | 800(1500)580 | 73(70)55 | 2800(5400)750 | 290(150)260 |
| 60 ÷ 590 | 120 ÷ 2100 | 47 ÷ 540 | 15 ÷ 510 | 40 ÷ 480 | 340 ÷ 1400 | 470 ÷ 2200 | 25 ÷ 240 | 220 ÷ 15000 |
130 ÷ 470 | |
| 208Pb/206Pb | 2.44(0.02)2.44 | 2.43(0.02)2.42 | 2.42(0.03)2.42 | 2.44(0.02)2.43 | 2.46(0.02)2.47 | 2.45(0.01)2.44 | 2.44(0.02)2.45 | 2.45(0.02)2.46 | 2.44(0.01)2.44 | 2.43(0.01)2.42 |
| 2.386 ÷ 2.477 | 2.396 ÷ 2.464 | 2.344 ÷ 2.472 | 2.405 ÷ 2.480 | 2.431 ÷ 2.489 | 2.435 ÷ 2.459 | 2.417 ÷ 2.466 | 2.423 ÷ 2.477 | 2.433 ÷ 2.466 | 2.417 ÷ 2.435 | |
| 206Pb/207Pb | 1.19(0.03)1.19 | 1.17(0.02)1.17 | 1.18(0.02)1.18 | 1.18(0.02)1.18 | 1.18(0.02)1.18 | 1.18(0.01)1.18 | 1.16(0.01)1.16 | 1.18(0.02)1.18 | 1.17(0.02)1.17 | 1.16(0.01)1.17 |
| 1.134 ÷ 1.252 | 1.126 ÷ 1.220 | 1.145 ÷ 1.259 | 1.155 ÷ 1.240 | 1.163 ÷ 1.210 | 1.164 ÷ 1.199 | 1.154 ÷ 1.167 | 1.149 ÷ 1.223 | 1.158 ÷ 1.218 | 1.154 ÷ 1.170 | |
| Sr (μg g−1) | 29(14)26 | 36(12)35 | 18(16)15 | 0.53(0.38)0.37 | 73(38)64 | 150(110)150 | 110(90)90 | 71(23)69 | 56(60)23 | 21(20)12 |
| 12 ÷ 70 | 17 ÷ 70 | 2 ÷ 63 | 0.1 ÷ 1.8 | 29 ÷ 150 | 14 ÷ 380 | 25 ÷ 230 | 49 ÷ 110 | 11 ÷ 160 | 6 ÷ 46 | |
| 87Sr/86Sr | 0.729(0.003)0.729 | 0.734(0.005)0.733 | 0.735(0.007)0.733 | 0.731(0.009)0.730 | 0.710(0.002)0.710 | 0.708(0.001)0.708 | 0.708(0.001)0.708 | 0.707(0.001)0.707 | 0.710(0.001)0.710 | 0.710(0.001)0.710 |
| 0.723 ÷ 0.734 | 0.722 ÷ 0.748 | 0.724 ÷ 0.753 | 0.714 ÷ 0.763 | 0.708 ÷ 0.711 | 0.708 ÷ 0.711 | 0.708 ÷ 0.709 | 0.706 ÷ 0.709 | 0.709 ÷ 0.712 | 0.710 ÷ 0.711 | |
| Tl (ng g−1) | 4.9(5.0)2.6 | 7.6(6.8)6.0 | 15(18)10 | 17(17)11 | 4.7(3.5)6.4 | 3.9(2.8)3.2 | 7.9(4.9)7.4 | 1.6(1.8)0.9 | 5.7(4.1)4.4 | 1.5(0.9)1.0 |
| 1 ÷ 18 | 2 ÷ 48 | 0.8 ÷ 90 | 2 ÷ 70 | 0.2 ÷ 9 | 0.6 ÷ 8 | 3 ÷ 15 | 0.6 ÷ 5.7 | 2 ÷ 14 | 0.9 ÷ 2.6 | |
| δ 205Tl (‰) | NA | NA | −0.40(0.17)–0.38 | −0.39(0.13)–0.40 | NA | NA | −0.29(0.17)–0.27 | NA | NA | NA |
| −0.62 ÷ −0.18 | −0.62 ÷ −0.14 | −0.49 ÷ −0.11 | ||||||||
| Zn (μg g−1) | 140(70)120 | 320(150)280 | 52(20)51 | 120(50)100 | 120(130)60 | 160(80)160 | 64(40)50 | 360(150)340 | 50(40)30 | 91(50)82 |
| 35 ÷ 380 | 75 ÷ 820 | 20 ÷ 96 | 40 ÷ 240 | 10 ÷ 360 | 60 ÷ 320 | 38 ÷ 120 | 190 ÷ 570 | 12 ÷ 120 | 38 ÷ 150 | |
| δ 66Zn (‰) | −0.15(0.22)–0.12 | −0.09(0.15)–0.12 | −0.07(0.24)–0.07 | 0.53(0.35)0.55 | 0.05(0.33)0.11 | −0.23(0.20)–0.18 | −0.11(0.04)–0.10 | 0.20(0.16)0.22 | 0.13(0.46)–0.22 | −0.15(0.06)–0.12 |
| −0.94 ÷ 0.13 | −0.62 ÷ 0.24 | −0.95 ÷ 0.47 | 0.02 ÷ 1.21 | −0.61 ÷ 0.47 | −0.59 ÷ −0.01 | −0.18 ÷ −0.07 | 0.03 ÷ −0.46 | −0.59 ÷ −0.54 | −0.21 ÷ −0.10 | |
| Ag (ng g−1) | 23(15)20 | 18(36)9 | 21(10)19 | 3000(2300)2500 | 5.5(3.8)4.6 | 13(3.8)12 | 11(3.7)11 | 3.8(1.9)3.1 | 18(22)13 | 8.5(0.7)8.3 |
| 3 ÷ 54 | 2 ÷ 220 | 5 ÷ 39 | 280 ÷ 9300 | 0.7 ÷ 11 | 6 ÷ 18 | 8 ÷ 15 | 2 ÷ 7 | 3 ÷ 67 | 7.9 ÷ 9.4 | |
| δ 109Ag (‰) | NA | NA | NA | −0.21(0.13)–0.20 | NA | NA | NA | NA | NA | NA |
| −0.31 ÷ −0.07 | ||||||||||
| Hg (ng g−1) | 3.4(1.6)3.0 | 12(2.8)11 | 7.1(1.7)7.1 | 510(460)260 | 17(7.8)13 | 17(8.4)16 | 52(33)47 | 8.7(1.0)8.8 | 38(31)23 | 9.4(6.1)5.9 |
| 0.5 ÷ 5.1 | 5 ÷ 20 | 4 ÷ 11 | 18 ÷ 1500 | 9 ÷ 31 | 6 ÷ 36 | 17 ÷ 96 | 7 ÷ 10 | 5 ÷ 95 | 5–17 | |
3.2. Precision
Instrumental repeatability was estimated as twice the standard deviation (SD) of duplicate consecutive measurements of a single sample preparation. The mean instrumental repeatability for isotopic measurements, averaged over all samples (n > 240), was as a rule <0.05‰ (Table 2). The slightly poorer repeatability for Pb ratios is due to the high proportion of samples in the datasets containing too little Pb for optimum MC-ICP-MS measurement. B repeatability represents both MC-ICP-MS (approximately 2/3 of all results) and ICP-SFMS data. These figures are by a factor of 2–2.5 times better than instrumental, between-block SDs of individual measurements due to the fact that the contribution from imperfect amplifier gain calibration has been cancelled out.For accurate assessment of the overall reproducibility of the entire method, duplicate digestions and separations were performed for 14 samples analyzed during different analytical sessions conducted by various operators. Data for the four CRMs that were a part of each analytical batch can also be used for this purpose. Results are summarized in Table 2 and demonstrate generally that reproducibility values are two- to five-fold poorer than those of repeatability, reflecting cumulative effects arising from minor differences in the efficiency of separation, blanks, spectral interferences and mass-bias corrections, instrumental mass calibration stability and the quality of instrument optimization. This reproducibility provides a more realistic assessment of the developed method's ability to detect minor variations in isotope compositions than repeatability. It should be stressed though that reproducibility figures presented in Table 3 are valid for sample types analyzed in this study and may not be applicable to other matrices with higher (or lower) concentrations of analytes and interfering elements.
One observation made during the precision assessment for Cd isotope ratio measurements deserves special note. It was found that agreement between duplicates analyzed using Aridus was significantly inferior to that obtained with the standard sample introduction system. In extreme cases δ114Cd between-run variations exceeded 0.3‰, even when the same separated digest fractions were re-analyzed. In order to investigate the reason for poor precision, a comprehensive set of experiments was performed by measuring Cd isotopes in standard solutions: (I) with variable acid strength; (II) at different plasma sampling depths; (III) with variable sample flow rate; and (IV) with variable dry gas (Ar) flow through the Aridus desolvating membrane. It was found that changes in instrumental mass bias for Cd caused by the aforementioned variations were adequately corrected using Ag for internal standardization in tests (I), (II) and (III), while even minor changes in Ar flow through the desolvating membrane resulted in severe uncorrected effects, pointing to decoupling of the mass-bias correlation between Cd and Ag. Most probably, this is due to partial Cd losses through the membrane that could be explained by the evaporation/sublimation of Cd from cadmium nitrates Cd(NO3)2·XH2O in the Aridus because the desolvating module reaches temperatures above the boiling point of Cd(NO3)2·XH2O.66 The effect would result in preferential losses of lighter Cd isotopomers that have higher degrees of volatilization and diffusion rates through the membrane. A combination of these effects would be expected to lead to an isotope shift towards higher apparent δ114Cd as the flow of drying gas through the membrane increases, which was indeed observed in the test. To avoid this undesirable effect, the Apex desolvating nebulizer was used for all subsequent Cd isotope ratio measurements, with desolvation occurring by passing through a cooled condenser rather than a heated membrane.
3.3. Accuracy
The accuracy of concentration determinations by ICP-SFMS was verified by analyses of various CRMs (Table 2). For the majority of analytes, recovery is within the 90–110% range.Though a set of in-house isotope standards (seawater CRM for B, isotope standard solutions, commercial mono-element standards or dissolved pure salts/metals with isotope composition different from the respective δ-zero standards for the rest of analytes) was analyzed in every measurement session, such quality controls only apply to the instrumental stage and are thus unsuitable for assessment of the overall accuracy of the method. An absence of measurable fractionation in δ-zero standards passed through the separation procedure assures sufficiently good analyte recoveries, but as these standards are both matrix- and interfering element-free, they are not the perfect solution to quality assurance either. As to the best of our knowledge no matrix-matched CRMs with certified isotopic compositions for the elements encompassed by this study are available, the only means to evaluate method accuracy is to compare data obtained in our study (Table 2 and 3) with those previously published for similar matrices, where such data exist. Though only two of four CRMs used were of plant origin, the inclusion of IRMM-BB186 and NRCC TORT-1 provided heavy isotope extremes for Cu and Zn, respectively.
Roux et al.67 have reported δ11B of (40.12 ± 0.21)‰ in NIST 1547 using purification of B by cation exchange chromatography and micro-sublimation followed by MC-ICP-MS measurements, identical within uncertainties to our result. Isotopically heavy Cu in animal kidney has been explained by fractionation during the breakdown of ascorbate into oxalate.68 Maréchal et al.69 have reported δ66Zn of +0.51‰ in NRCC TORT-2 (lobster hepatopancreas) and Balter et al.70 lighter Zn isotope composition in sheep kidney compared to other organs. Cd and Zn isotope data for IRMM-BB186 and NRCC TORT-1 agree well with our previously published results23 confirming if not high accuracy at least good reproducibility for datasets generated in large analytical campaigns two years apart. Measured δ-values for different Cd, Fe and Zn isotope ratios normalized by respective mass differences agree well (correlation coefficient >0.98) for samples with analyte contents above the minimum required concentrations.
For some of the samples, B, Pb and Sr isotope ratios were measured by both MC-ICP-MS and ICP-SFMS. Sr and Pb datasets obtained by these techniques are compared in Fig. 2 demonstrating very satisfactory agreement.
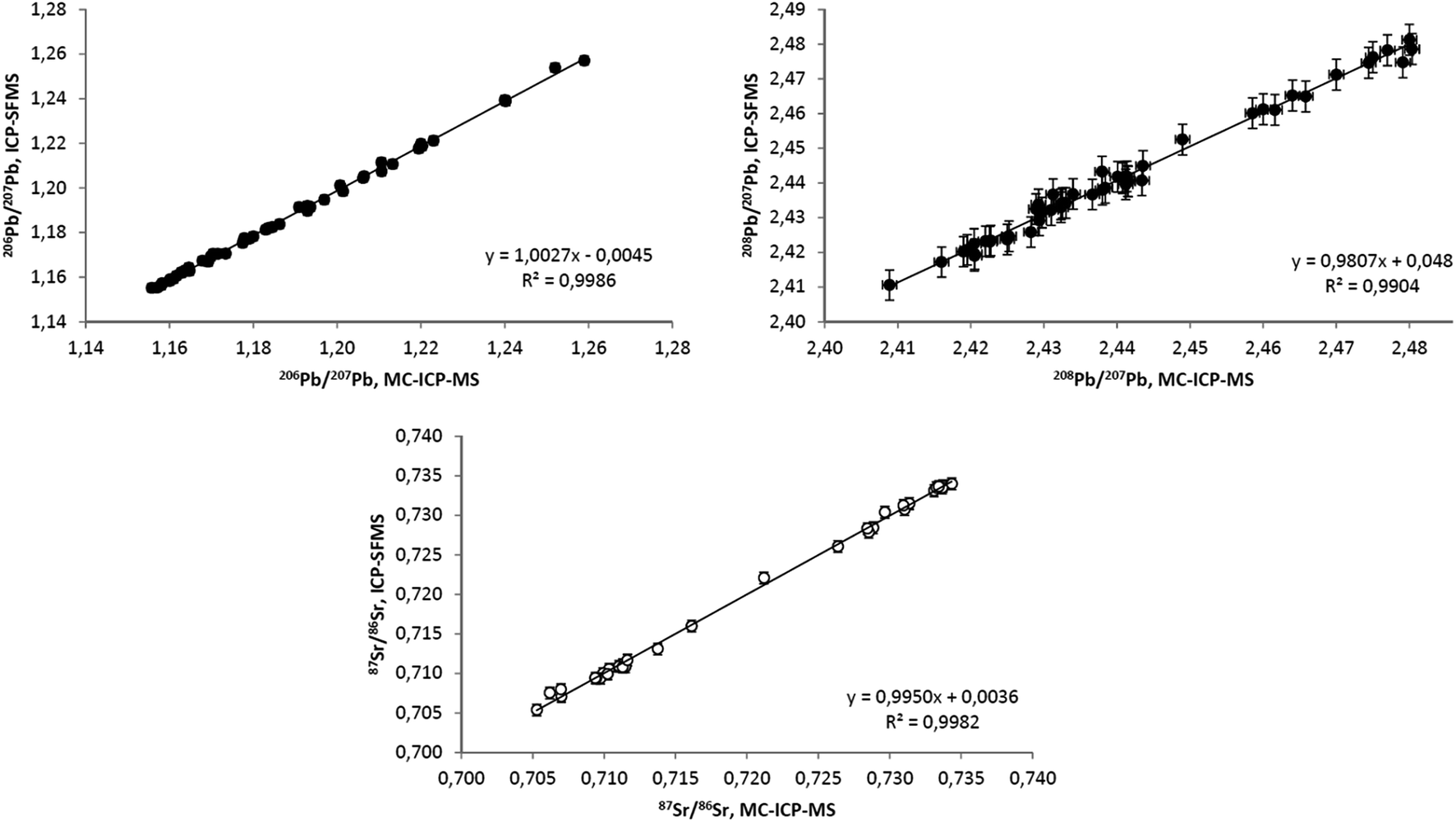 | ||
| Fig. 2 Comparison of Pb and Sr isotope ratios obtained in bio-indicators by ICP-SFMS and MC-ICP-MS. The ordinary linear regression equations are: (top left) y = (−0.0045 ± 0.0175) + (1.0027 ± 0.0148)x; (top right) y = (0.0480 ± 0.0913) + (0.9807 ± 0.0374)x; and (bottom) y = (0.0036 ± 0.0155) + (0.9950 ± 0.0215)x where the uncertainties correspond to 99% confidence intervals. | ||
3.4. Throughput
Given a batch size of 36 samples plus two preparation blanks and two CRMs (as limited by the maximum of 40 digestion vessel positions in the UltraCLAVE), unrestricted availability of hot plate(s) for evaporation of digests and purified fractions (performed mostly overnight), and simultaneous access to ICP-SFMS and MC-ICP-MS, the entire procedure from sample weighing to data evaluation can be done by two chemists in approximately two weeks. This is approximately three-fold more time-consuming than complete isotopic analysis of a single element (e.g. Cd), but because many operations in the procedure can be performed in parallel, this is significantly more time-effective (and less sample consuming) compared to using individual preparation/separation schemes for all eight elements.3.5. Variations in concentrations and isotopic compositions in leaves, needles and mushrooms
Element content and isotope composition in leaves can (and does) reflect different accumulation pathways from a variety of natural and anthropogenic sources. Variations in the same depend upon the type of tree (different accumulation mechanisms, nutrition supply strategies, leaf morphology), age/size, tree location (on continental as well as local scales; soil type and composition, sub-soil geology, proximity to local contamination sources, etc.), location of leaves on the tree (height, orientation of branches), sampling period, weather preceding sampling occasion, etc.23,44,71 The ability to detect such variability will entirely depend on the ‘resolution’ (overall reproducibility) of the analytical procedure used and data in Table 3 demonstrate the extent of such effects.A statistical summary of concentrations and isotope data for the majority of samples analyzed in the course of this study is presented in Table 3. Data for oak, aspen and rowan leaves, as well as for pine needles collected in Luleå are omitted as there were less than five samples in each category, and because the results for these samples fall into ranges found for birch leaves or spruce needles from the same collection period.
In the ESI† data for four similarly sized birch trees sampled annually for three years are discussed in more detail. The key finding from the latter dataset is that the individual trees, growing within a radius of 100 m, exhibit significant spatial and temporal variations in concentrations and isotopic compositions. This strongly suggests that the elemental and isotopic signatures of deciduous plants are as unique as the trees themselves and bear little connection to events on a regional scale.
Although the extensive variations observed for 208Pb/206Pb and 206Pb/207Pb ratios (from 2.344 to 2.489 or 5.9% and from 1.126 to 1.259 or 11.3%, respectively) encompass the majority of all ratios reported for biological samples from Europe,11,12,76 ranges for each individual matrix, location and sampling occasion are significantly narrower. The RSD for mean 208Pb/206Pb and 206Pb/207Pb ratios for all sample groups is 0.48% and 0.74%, respectively. In birch leaves from the same location (Luleå) the range of measured Pb isotope ratios decreases considerably from spring to fall sampling.
There are minor differences in mean 87Sr/86Sr ratios between different matrices from the same location, becoming more radiogenic in the order: leaves from Iceland (0.707), leaves and needles from Paris (0.708), leaves from Genoa, Barcelona and Birmingham (0.710) and all samples collected in Luleå (0.729–0.735). The ratios reflect the ages of the underlying bedrock at each location, with the oldest being found in the Luleå area ranging from 1.8 to 2.8 Ga.77 Except for the latter location, ranges of 87Sr/86Sr ratios observed elsewhere overlap significantly, potentially complicating the use of this isotope system for confirmation of the geographical origin of the plant material.3,4,18 Highly radiogenic mean 87Sr/86Sr ratios found in Luleå samples agree well with figures published by Åberg et al.78 for plant samples collected from the central part of Sweden. Except for the Luleå location where wide ranges of observed 87Sr/86Sr ratios can be caused by heterogeneity of the 87Sr/86Sr in the granitic bedrocks,79 variations in Sr ratios in plant samples from the same group are seldom >0.3% RSD.
As the use of Zr for mass-bias correction80 allows for assessment of mass-dependent fractionation of the 88Sr/86Sr ratio, it can be stated that none of the samples shows fractionation outside the −0.04‰ to +0.04‰ range. Therefore, 88Sr/86Sr ratios can safely be used for mass bias correction of 87Sr/86Sr ratios in plant samples, providing identical results to Zr corrected data (Fig. 3) with considerably less effort.
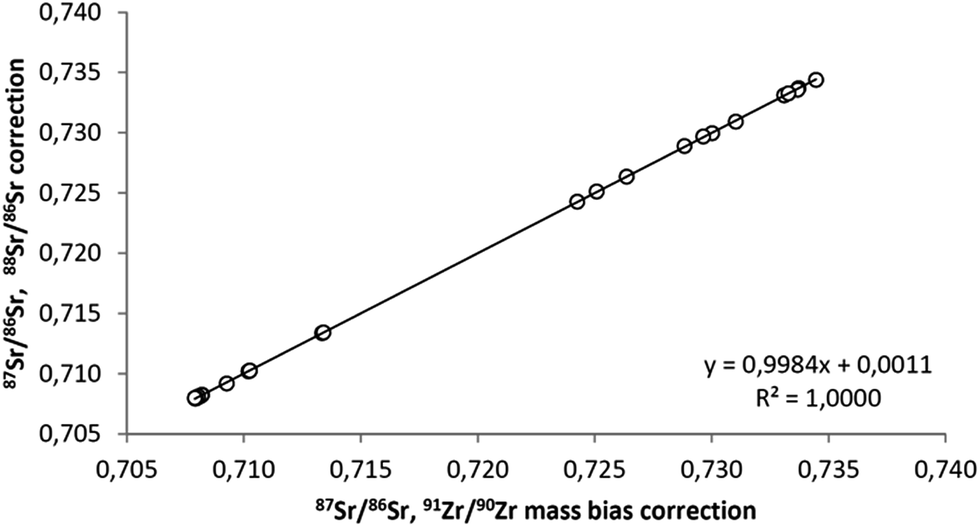 | ||
| Fig. 3 Comparison between the two approaches for MC-ICP-MS Sr mass bias correction. The ordinary linear regression equation is y = (0.0011 ± 0.0015) + (0.9984 ± 0.0021)x where the uncertainties correspond to 99% confidence intervals. | ||
4. Conclusions
The analytical protocol tested in this study has proved to be suitable for isotope ratio measurements of at least eight elements in leaf, needle and mushroom samples. Digestion using the UltraCLAVE device provides complete oxidation of the organic material, while two-column separation ensures low blank levels, efficient separation of matrix elements, sufficiently high analyte recoveries and relatively high sample throughput.It was shown that losses of Cd may occur in membrane desolvation systems, which may result in poor precision. Therefore, utilization of an Apex desolvation system can be a better alternative than the frequently used Aridus82,83 for Cd isotope ratio measurements.
Regular use of duplicate sample preparations and analyses performed in different batches or measurement sessions is a must for overall reproducibility assessment. The use of synthetic, matrix-free isotope standards or replicate analyses within the same measurement session results in artificially optimistic estimates of precision.
The absence of commercially available CRMs with certified isotopic compositions continues to hamper straightforward accuracy assessment. In contrast to the geological field, there are still only a very few published datasets containing information on the isotopic composition of CRMs of biological origin. There is thus a growing need for inter-laboratory exercises, preferably using commercially available CRMs representing various matrices, to fill this gap and ensure data transferability until appropriate CRMs become available.
In general the proposed method represents a starting point for further development in the direction of multi-tracer studies. With some relatively modest modifications, the list of separated elements can be extended to include Ca, Mg and Ga. Vapor-phase introduction of Hg can overcome the insufficient sensitivity of the current system. The possibility to obtain precious information on several isotope systems from a single sample will aid the interpretation of natural processes and enable more reliable pollution source attribution.
The majority of results obtained for eight isotope systems in more than 240 samples agree well with previously published data where they exist. For some bio-indicators (e.g. needles and mushrooms), our data represent the first combined characterization of B, Cd, Cu, Fe, Pb, Sr, Tl and Zn isotopic compositions. Even after removing some known variation sources such as the type of bio-indicator, sampling height and sampling period, very broad ranges in isotopic compositions of many elements were found in samples collected from relatively confined geographical areas (Table 3) or even from within 100 m (see ESI Table S1†), significantly exceeding method reproducibility. The observed degree of isotopic variability, irrespective if caused by natural or anthropogenic factors, may complicate such isotope applications as source tracing, geographical origin authentication, studying plant metabolism, etc., and should be carefully considered for each given study object.
Better understanding of regularities in observed isotope variability in leaves and needles would require acquiring isotope profiling of different soil compartments and soil solution as a function of soil depth – work which is currently underway.
Acknowledgements
ALS Scandinavia AB is gratefully acknowledged for technical support. MetTrans Initial Training Network (funded by the European Union under the Seventh Framework Programme) is acknowledged for financial support. We wish to thank Katerina Rodiouchkina for field assistance and help with sample preparation. We would also like to show our gratitude to Enzo Stranieri, Maria Grazia and Sergio Pallavicini for field assistance. The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7 2007–2013, under the REA grant agreement no. 290336. The views expressed in this article are those of the authors and may not necessarily reflect those of the European Union.
References
- T. D. Bullen, in Handbook of Environmental Isotope Geochemistry, ed. M. Baskaran, Springer, Berlin, Heidelberg, 2012, pp. 177–203 Search PubMed.
- H.-C. Liu, C. F. You, C. Y. Chen, Y. C. Liu and M. T. Chung, Food Chem., 2014, 142, 439–445 CrossRef CAS PubMed.
- M. V. Baroni, N. S. Podio, R. G. Badini, M. Inga, H. A. Ostera, M. Cagnoni, E. A. Gautier, P. P. García, J. Hoogewerff and D. A. Wunderlin, J. Agric. Food Chem., 2015, 63, 4638–4645 CrossRef CAS PubMed.
- P. Degryse, A. Shortland, D. de Muynck, L. van Heghe, R. Scott, B. Neyt and F. Vanhaecke, J. Archaeol. Sci., 2010, 37, 3129–3135 CrossRef.
- I. Rodushkin, D. C. Baxter, E. Engström, J. Hoogewerff, P. Horn, W. Papesch, J. Watling, C. Latkoczy, G. van der Peijl, S. Berends-Montero, J. Ehleringer and V. Zdanowicz, J. Food Compos. Anal., 2011, 24, 70–78 CrossRef CAS.
- A. E. Shiel, D. Weis, D. Cossa and K. J. Orians, Geochim. Cosmochim. Acta, 2013, 121, 155–167 CrossRef CAS.
- C. Cloquet, J. Carignan, G. Libourel, T. Sterckeman and E. Perdrix, Environ. Sci. Technol., 2006, 40, 2525–2530 CrossRef CAS PubMed.
- G.-X. Sun, X.-J. Wang and Q.-H. Hu, Environ. Pollut., 2011, 159, 3406–3410 CrossRef CAS PubMed.
- M. Rehkämper, F. Wombacher, T. J. Horner, and Z. Xue, in Handbook of Environmental Isotope Geochemistry, ed. M. Baskaran, Springer, Berlin, Heidelberg, 2012, pp. 125–154 Search PubMed.
- I. Rodushkin, T. Bergman, G. Douglas, E. Engström, D. Sörlin and D. C. Baxter, Anal. Chim. Acta, 2007, 583, 310–318 CrossRef CAS PubMed.
- J. Sucharová, I. Suchara, C. Reimann, R. Boyd, P. Filzmoser and P. Englmaier, Appl. Geochem., 2011, 26, 1205–1214 CrossRef.
- M. J. M. Notten, N. Walraven, C. J. Beets, P. Vroon, J. Rozema and R. Aerts, Appl. Geochem., 2008, 23, 1581–1593 CrossRef CAS.
- M. A. Smith and S. Pell, J. Archaeol. Sci., 1997, 24, 773–778 CrossRef.
- M. Perini, F. Camin, L. Bontempo, A. Rossmann and E. Piasentier, Rapid Commun. Mass Spectrom., 2009, 23, 2573–2585 CrossRef CAS PubMed.
- D. Xue, B. de Baets, O. van Cleemput, C. Hennessy, M. Berglund and P. Boeckx, Environ. Pollut., 2012, 161, 43–49 CrossRef CAS PubMed.
- J. G. Wiederhold, Environ. Sci. Technol., 2015, 49, 2606–2624 CrossRef CAS PubMed.
- J. Klaminder, R. Bindler and I. Renberg, Appl. Geochem., 2008, 23, 2922–2931 CrossRef CAS.
- B. Song, J. Ryu, H. S. Shin and K. Lee, J. Agric. Food Chem., 2014, 62, 9232–9238 CrossRef CAS PubMed.
- I. Rodushkin, E. Engström, D. Sörlin, C. Pontér and D. C. Baxter, Sci. Total Environ., 2007, 386, 159–168 CrossRef CAS PubMed.
- I. Rodushkin, E. Engström, D. Sörlin, C. Pontèr and D. C. Baxter, Sci. Total Environ., 2007, 386, 145–158 CrossRef CAS PubMed.
- N. Pallavicini, F. Ecke, E. Engström, D. C. Baxter and I. Rodushkin, J. Anal. At. Spectrom., 2013, 28, 1591–1599 RSC.
- I. Rodushkin, E. Engström, D. Sörlin, D. Baxter, B. Hörnfeldt, E. Nyholm and F. Ecke, Water, Air, Soil Pollut., 2011, 218, 603–610 CrossRef CAS.
- N. Pallavicini, E. Engström, D. C. Baxter, B. Öhlander, J. Ingri and I. Rodushkin, J. Anal. At. Spectrom., 2014, 29, 1570–1584 RSC.
- L. S. Sherman, J. D. Blum, J. T. Dvonch, L. E. Gratz and M. S. Landis, Sci. Total Environ., 2015, 502, 362–374 CrossRef CAS PubMed.
- L. S. Ruhl, G. S. Dwyer, H. Hsu-kim, J. C. Hower and A. Vengosh, Environ. Sci. Technol., 2014, 48, 14790–14798 CrossRef CAS PubMed.
- T. Fujii, F. Moynier, J. Blichert-Toft and F. Albarède, Geochim. Cosmochim. Acta, 2014, 140, 553–576 CrossRef CAS.
- K. Jaouen, M. L. Pons and V. Balter, Earth Planet. Sci. Lett., 2013, 374, 164–172 CrossRef CAS.
- C. Rodrigues, M. Brunner, S. Steiman, G. J. Bowen, J. M. F. Nogueira, L. Gautz, T. Prohaska and C. Máguas, J. Agric. Food Chem., 2011, 59, 10239–10246 CrossRef CAS PubMed.
- D. Houben, P. Sonnet, G. Tricot, N. Mattielli, E. Couder and S. Opfergelt, Environ. Sci. Technol., 2014, 48, 7866–7873 CrossRef CAS PubMed.
- Y. T. Tang, C. Cloquet, T. Sterckeman, G. Echevarria, J. Carignan, R. L. Qiu and J. L. Morel, Environ. Sci. Technol., 2012, 46, 9972–9979 CAS.
- D. Jouvin, D. J. Weiss, T. F. M. Mason, M. N. Bravin, P. Louvat, F. Zhao, F. Ferec, P. Hinsinger and M. F. Benedetti, Environ. Sci. Technol., 2012, 46, 2652–2660 CrossRef CAS PubMed.
- M. Rosner, W. Pritzkow, J. Vogl and S. Voerkelius, Anal. Chem., 2011, 83, 2562–2568 CrossRef CAS PubMed.
- T. Deng, C. Cloquet, Y. Tang and T. Sterckeman, Environ. Sci. Technol., 2014, 48, 11926–11933 CrossRef CAS PubMed.
- M. Guelke and F. Von Blanckenburg, Environ. Sci. Technol., 2007, 41, 1896–1901 CrossRef CAS PubMed.
- J. Viers, A. S. Prokushkin, O. S. Pokrovsky, A. V. Kirdyanov, C. Zouiten, J. Chmeleff, M. Meheut, F. Chabaux, P. Oliva and B. Dupré, Geochem. Trans., 2015, 16, 1–15 CrossRef CAS PubMed.
- B. M. Ryan, J. K. Kirby, F. Degryse, H. Harris, M. J. McLaughlin and K. Scheiderich, New Phytol., 2013, 199, 367–378 CrossRef CAS PubMed.
- C. R. Quétel, E. Ponzevera, I. Rodushkin, A. Gerdes, R. Williams and J. Woodhead, J. Anal. At. Spectrom., 2009, 24, 407–412 RSC.
- R. Wei, Q. Guo, H. Wen, J. Yang, M. Peters, C. Zhu, J. Ma, G. Zhu, H. Zhang, L. Tian, C. Wang and Y. Wan, Anal. Methods, 2015, 7, 2479–2487 RSC.
- V. Migeon, B. Bourdon, E. Pili and C. Fitoussi, J. Anal. At. Spectrom., 2015, 30, 1988–1996 RSC.
- J. Aggarwal, F. Böhm, G. Foster, S. Halas, B. Hönisch, S.-Y. Jiang, J. Kosler, A. Liba, I. Rodushkin, T. Sheehan, J. Jiun-San Shen, S. Tonarini, Q. Xie, C.-F. You, Z.-Q. Zhao and E. Zuleger, J. Anal. At. Spectrom., 2009, 24, 825–831 RSC.
- B. C. Reynolds, J. Aggarwal, L. Andre, D. Baxter, C. Beucher, M. A. Brzezinski, E. Engstrom, R. B. Georg, M. Land, M. J. Leng, S. Opfergelt, I. Rodushkin, H. J. Sloane, S. H. J. M. van den Boorn, P. Z. Vroon and D. Cardinal, J. Anal. At. Spectrom., 2007, 22, 561–568 RSC.
- J. F. Carter and B. Fry, Anal. Bioanal. Chem., 2013, 405, 2799–2814 CrossRef CAS PubMed.
- M. Lodenius, Environ. Res., 2013, 125, 113–123 CrossRef CAS PubMed.
- K. Tarricone, G. Wagner and R. Klein, Ecol. Indic., 2015, 57, 341–359 CrossRef.
- I. Rodushkin, P. Nordlund, E. Engström and D. C. Baxter, J. Anal. At. Spectrom., 2005, 20, 1250–1255 RSC.
- D. Malinovsky, I. Rodushkin, D. Baxter and B. Öhlander, Anal. Chim. Acta, 2002, 463, 111–124 CrossRef CAS.
- I. Rodushkin, F. Ödman and H. Holmström, Sci. Total Environ., 1999, 231, 53–65 CrossRef CAS.
- L. Ericson, M. Fabricius, E. Danielsson, B. Hultman, H. Juto, and C. Huhtasaari, De odlade jordarna i norrbottens och västerbottens län. The cultivated soils of Norrbotten and Västerbotten, 1985 Search PubMed.
- I. Rodushkin, E. Engström and D. C. Baxter, Anal. Bioanal. Chem., 2010, 396, 365–377 CrossRef CAS PubMed.
- E. Engström, A. Stenberg, S. Senioukh, R. Edelbro, D. C. Baxter and I. Rodushkin, Anal. Chim. Acta, 2004, 521, 123–135 CrossRef.
- I. Rodushkin, T. Ruth and Å. Huhtasaari, Anal. Chim. Acta, 1999, 378, 191–200 CrossRef CAS.
- R. Sun, M. Enrico, L.-E. E. Heimbürger, C. Scott and J. E. Sonke, Anal. Bioanal. Chem., 2013, 405, 6771–6781 CrossRef CAS PubMed.
- H. Lin, D. Yuan, B. Lu, S. Huang, L. Sun, F. Zhang and Y. Gao, J. Anal. At. Spectrom., 2015, 30, 353–359 RSC.
- Q. Huang, Y. Liu, J. Chen, X. Feng, W. Huang, S. Yuan, H. Cai and X. Fu, J. Anal. At. Spectrom., 2015, 30, 957–966 RSC.
- D. C. Baxter, I. Rodushkin, E. Engström and D. Malinovsky, J. Anal. At. Spectrom., 2006, 21, 427–430 RSC.
- L. van Heghe, E. Engström, I. Rodushkin, C. Cloquet and F. Vanhaecke, J. Anal. At. Spectrom., 2012, 27, 1327–1334 RSC.
- A. Olofsson and I. Rodushkin, Archaeometry, 2011, 53, 1142–1170 CrossRef CAS.
- E. P. Horwitz, R. Chiarizia and M. L. Dietz, Solvent Extr. Ion Exch., 1992, 10, 313–336 CrossRef CAS.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, A. M. Essling and D. Graczyk, Anal. Chim. Acta, 1992, 266, 25–37 CrossRef CAS.
- E. P. Horwitz, in International Workshop on the Application of Extraction Chromatography in Radionuclide Measurement, IRMM, Geel, 1998, vol. E, pp. 27–37 Search PubMed.
- B. Gao, Y. Liu, K. Sun, X. Liang, P. Peng, G. Sheng and J. Fu, Anal. Chim. Acta, 2008, 612, 114–120 CrossRef CAS PubMed.
- C. N. Maréchal, P. Télouk and F. Albarède, Chem. Geol., 1999, 156, 251–273 CrossRef.
- F. Larner, M. Rehkämper, B. J. Coles, K. Kreissig, D. J. Weiss, B. Sampson, C. Unsworth and S. Strekopytov, J. Anal. At. Spectrom., 2011, 26, 1627–1632 RSC.
- P. A. Sossi, G. P. Halverson, O. Nebel and S. M. Eggins, Geostand. Geoanal. Res., 2015, 39, 129–149 CrossRef CAS.
- I. Smet, D. de Muynck, F. Vanhaecke and M. Elburg, J. Anal. At. Spectrom., 2010, 25, 1025–1032 RSC.
- R. C. Weast, in CRC Handbook of Chemistry and Physics, CRC Press, Inc., Cleveland, 58th edn, 1977–1978, 1978, p. B–97 Search PubMed.
- P. Roux, D. Lemarchand, H. J. Hughes and T. Marie-Pierre, Geostand. Geoanal. Res., 2015 DOI:10.1111/j.1751-908X.2014.00328.x.
- T. Fujii, F. Moynier, M. Abe, K. Nemoto and F. Albarède, Geochim. Cosmochim. Acta, 2013, 110, 29–44 CrossRef CAS.
- C. N. Maréchal, E. Nicolas, C. Douchet and F. Albarède, Geochem., Geophys., Geosyst., 2000, 1, 1–15 CrossRef.
- V. Balter, A. Zazzo, A. P. Moloney, F. Moynier, O. Schmidt, F. J. Monahan and F. Albarede, Rapid Commun. Mass Spectrom., 2010, 24, 605–612 CrossRef CAS PubMed.
- B. Markert, Fresenius' Z. Anal. Chem., 1989, 335, 562–565 CrossRef CAS.
- M. Chaussidon and F. Albarède, Earth Planet. Sci. Lett., 1992, 108, 229–241 CrossRef CAS.
- M. Kiczka, J. G. Wiederhold, S. M. Kraemer, B. Bourdon and R. Kretzschmar, Environ. Sci. Technol., 2010, 44, 6144–6150 CrossRef CAS PubMed.
- N. Pérez Rodríguez, F. Langella, I. Rodushkin, E. Engström, E. Kothe, L. Alakangas and B. Öhlander, Environ. Sci. Pollut. Res., 2014, 21, 6836–6844 CrossRef PubMed.
- F. Moynier, T. Fujii, K. Wang and J. Foriel, C. R. Geosci., 2013, 345, 230–240 CrossRef CAS.
- C. Reimann, B. Flem, A. Arnoldussen, P. Englmaier, T. E. Finne, F. Koller and Ø. Nordgulen, Appl. Geochem., 2008, 23, 705–722 CrossRef CAS.
- G. Gaal and R. Gorbatschev, Precambrian Res., 1987, 35, 15–52 CrossRef CAS.
- G. Åberg, G. Jacks, T. Wickman and P. J. Hamilton, Catena, 1990, 17, 1–11 CrossRef.
- T. Nakano and T. Tanaka, in Tracers in Hydrology, 1993, pp. 73–78 Search PubMed.
- J. Irrgeher, T. Prohaska, R. E. Sturgeon, Z. Mester and L. Yang, Anal. Methods, 2013, 5, 1687–1694 RSC.
- M. Kersten, T. Xiao, K. Kreissig, A. Brett, B. J. Coles and M. Rehkämper, Environ. Sci. Technol., 2014, 48, 9030–9036 CrossRef CAS PubMed.
- S. Ripperger, M. Rehkämper, D. Porcelli and A. N. Halliday, Earth Planet. Sci. Lett., 2007, 261, 670–684 CrossRef CAS.
- A. E. Shiel, D. Weis and K. J. Orians, Sci. Total Environ., 2010, 408, 2357–2368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ja00274e |
| This journal is © The Royal Society of Chemistry 2016 |