
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The appeasement of Doug: a synthetic approach to enhancer biology†
Ben J.
Vincent
,
Javier
Estrada
and
Angela H.
DePace
*
Department of Systems Biology, Harvard Medical School, 200 Longwood Avenue, Boston, MA 02115, USA. E-mail: angela_depace@hms.harvard.edu
First published on 25th February 2016
Abstract
Genetic approaches have been instrumental in dissecting developmental enhancers by characterizing their transcription factor binding sites. Though some enhancers have been well-studied in this regard, we cannot currently build developmental enhancers from scratch. Reconstitution experiments can provide important complementary tests of our understanding of enhancer function, but these experiments are exceedingly rare in the literature, possibly due to the difficulty of publishing negative results. In this perspective, we argue that the time is right for a synthetic approach to enhancer biology. Focusing primarily on Drosophila enhancers as examples, we review classic and modern methods for dissecting enhancer function as well as computational tools for enhancer design. We include our own negative results from attempts to reconstitute the stripe 2 enhancer from the even-skipped locus and discuss possible ways forward. We believe that with a communal effort in open data sharing, we can make substantial progress toward a complete understanding of enhancer function.
Insight, innovation, integrationSynthetic biology tests our knowledge of biology through building. By trying to build biological elements with defined functions, we can rigorously test our knowledge of the underlying design principles. In this approach, successes and failures are informative. In this review, we discuss synthetic approaches to decipher the function of developmental enhancers that control gene expression patterns in space and time. We describe both successes and failures and advocate for an iterative cycle of experiment and modeling enabled by sharing negative results and development of computational frameworks to contextualize results. |
Introduction
The beloved parable of Doug, Bill, and their quest to understand the inner workings of the automobile is a scientific fable in two parts, first from the perspective of a geneticist, and then from the perspective of a biochemist.1 In “The Salvation of Doug,” geneticist Bill investigates the function of individual automotive components by randomly selecting factory workers to incapacitate and observing the effects on the cars rolling off the lot. Biochemist Doug is forced to concede that Bill's methods are more successful than his own attempts to reconstitute a car from its component fractions of glass, plastic and steel. However, in “The Demise of Bill,” Doug finds his own success through painstaking efforts to uncover how individual ‘pathways’ function in his car, such as the fuel injection and electrical systems. Doug's hard won expertise helps him fix Bill's car when it breaks because he understands how the parts work together. While written in a spirit of friendly competition and good fun, these parables teach serious lessons about the limitations of genetic and biochemical approaches. Genetics relies on a simple principle: break individual parts and catalog the resulting phenotypes. This approach is analogous to building a parts list and defining their interrelationships in abstract epistatic terms that may or may not imply direct physical interactions. In contrast, biochemistry is about building systems from their component parts. This approach makes these abstract relationships mechanistically concrete. Biochemist Doug sums up this contrast quite nicely when he challenges geneticist Bill: “Now that you have learned so much, tell me how the car works.”Like automobiles, developmental enhancers are complex machines built of many parts. Enhancers are thought to be the main determinants of cell-type specific gene expression in space and time and play critical roles in evolution and human disease.2–5 Geneticists like Bill have dominated the study of developmental enhancers for years. Genetic approaches are incredibly effective at identifying enhancer components – binding sites for the transcription factors (TFs) that regulate them. However, we cannot yet interpret regulatory sequence variation within enhancers because it remains complicated to determine the contribution of each TF binding site. Unlike variation in coding regions, where a given change will generally have the same effect on protein sequence no matter the cell type or organism, disrupting or adding a TF binding site within an enhancer will only affect cells where the TF is expressed. Even in cells expressing a given TF, the phenotypic effects of binding site changes may be buffered or augmented by interactions within or between enhancers.6–10 In the context of interpreting variation within regulatory DNA, Doug's challenge rings true in the ears of many experimentalists – we know a great deal about developmental enhancers, but we still can't tell you how they work.
How can we satisfy Doug? We argue that we need to take a “biochemical” approach and build enhancers from their component parts. The field has taken promising steps in this direction. Synthetic approaches have been successful in cell lines, where arrays of TF binding sites have been used to decipher their interactions and mechanisms of action.11–14 Arrays of binding sites can also drive patterned expression in Drosophila embryos.15,16 This approach forms the basis of the Gal4/UAS system, which has become an indispensable part of the genetic toolkit in animal systems.17–19 Systematic characterization of binding site arrays is also an effective way to interrogate the grammatical rules of TF binding site arrangement.16,20
To appease Doug, however, we must take a specific enhancer and reconstitute it from scratch. This has not yet been achieved. How can we design this experiment? At one extreme, we could use an unbiased empirical approach, where we synthesize large libraries of random sequence and screen them for the activity we want. While this approach would eventually yield a new sequence that generates a target pattern, the combinatorial space is vast, the experiment expensive, and we would need many successes to derive any general rules that govern enhancer function. Instead, Doug would use rational design to reconstitute a developmental enhancer from its component binding sites. This approach requires that we determine an enhancer's complete regulatory logic – the identity and function of all TFs that bind it. Once this criterion is met, we can identify and parameterize interactions between binding sites by altering their number, affinity and spacing. Armed with this information, we should be able to generate any number of functional sequences that produce the expression pattern of interest. This level of understanding may also allow us to design sequences that generate any expression pattern we want, which may be useful as experimental or therapeutic reagents.21 Because our goal is reconstituting a particular enhancer, the gold standard would be to test whether a reconstituted enhancer can rescue deletions of its endogenous counterpart.22 This type of rescue experiment is now tractable in animal systems due to the recent explosion of genome editing technology, and would provide the best argument for convincing Doug that we understand how developmental enhancers work.23
In this perspective, we discuss how close we are to appeasing Doug using illustrative examples primarily from Drosophila. We outline how regulatory logic is experimentally determined and the limitations inherent in these approaches. We also examine rules governing the interactions between TF binding sites. Finally, we discuss what we have learned from recent attempts to build developmental enhancers. We argue that both the experimental and computational groundwork has been laid for a synthetic, “bottom up” approach to enhancer biology.
Classic bill: identifying the regulators of eve stripe 2
Identifying the TFs that regulate an enhancer takes years of assiduous work in experimental genetics. While genetic methods have proven successful, they are fundamentally limited in their ability to characterize an enhancer's complete regulatory logic. To reveal these successes and limitations, we consider a single case study – the enhancer that controls the second stripe of the even-skipped (eve) expression pattern in Drosophila blastoderm embryos (eve2) (Fig. 1). Even after 25 years of research on this 484 base pair sequence, we are still discovering putative eve2 regulators using a combination of developmental genetics, functional genomics and quantitative biology.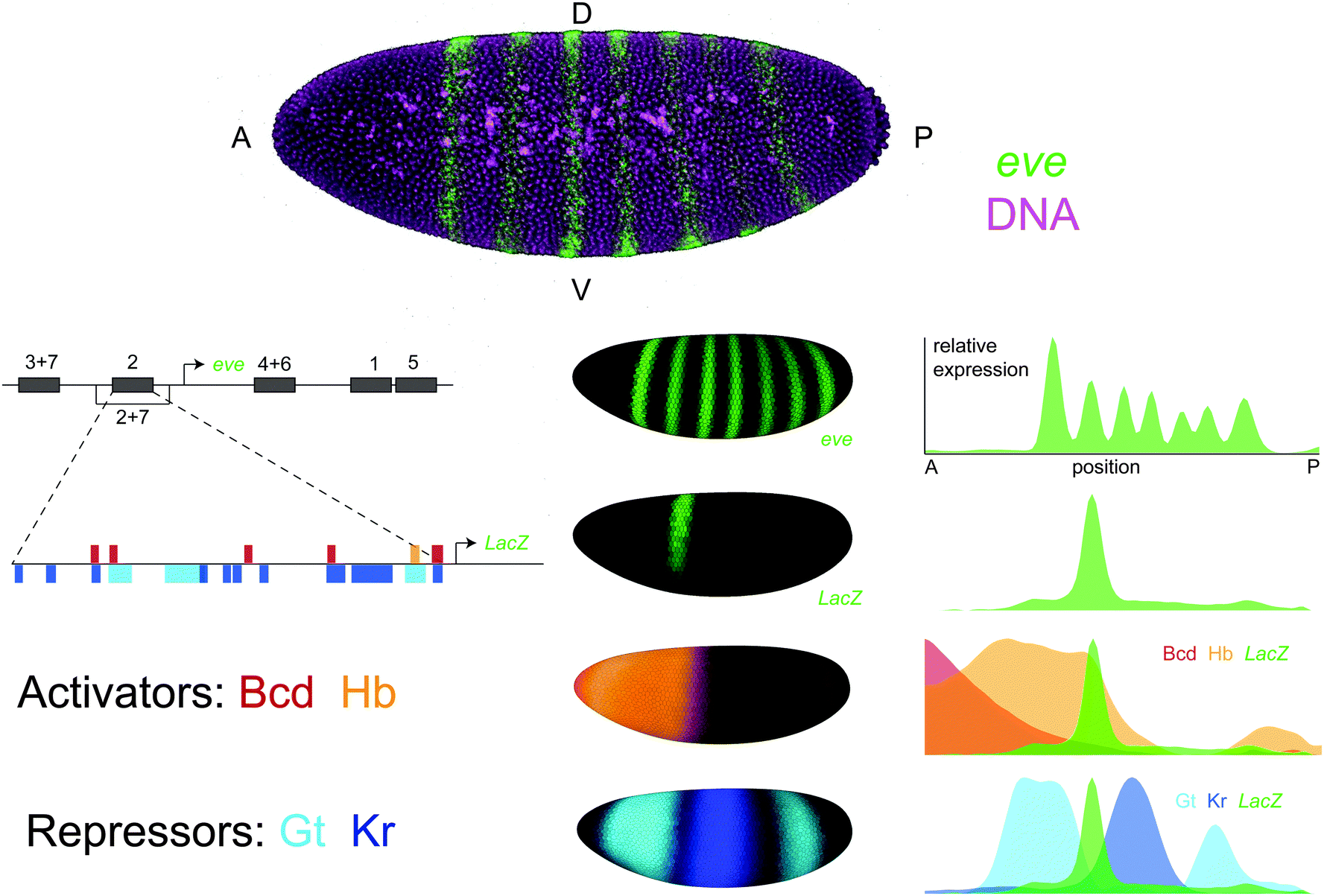 | ||
| Fig. 1 The even-skipped stripe 2 enhancer in Drosophila melanogaster. even-skipped (eve) is expressed in seven transverse stripes along the anterior-posterior axis of the blastoderm embryo. The eve locus contains five enhancers that control expression during this stage, each of which generates one or two individual stripes. The eve stripe 2 enhancer (eve2) is a 484 bp sequence that drives expression of the second eve stripe in transgenic animals when cloned into a LacZ reporter construct. eve2 contains binding sites for the activators bicoid (Bcd) and hunchback (Hb) as well as the repressors giant (Gt) and Krüppel (Kr). Top: Maximum intensity projection of a Drosophila melanogaster blastoderm embryo imaged with 2-photon confocal microscopy where eve mRNA has been stained using fluorescent in situ hybridization and outer nuclei and yolk cells have been labelled with SYTOX Green.116 Left: A schematic of the eve locus and footprinted binding sites within eve2. Sites are represented as horizontal bars where color indicates TF identity.107 Middle: The 3D expression patterns of eve, eve2, and its regulators. Shown are visual renderings of quantitative expression data from gene expression atlases.109,123 These images were generated using PointCloudXplore.124 Right: Expression patterns of eve, eve2, and its regulators are displayed as line traces where relative expression along the lateral side of the embryo is plotted as a function of anterior-posterior position. Plots were generated in MATLAB using the PointCloudToolbox (http://bdtnp.lbl.gov/Fly-Net/) and normalized by the maximum of each expression pattern. | ||
The story of eve2 begins with the seminal screens that identified genes controlling embryonic patterning in Drosophila melanogaster embryos. Larval cuticles derived from eve mutants lacked every other abdominal segment, which placed eve in the ‘pair rule’ class of mutants affecting segmentation.24 Genetic mapping, in situ hybridization and immunostaining revealed the characteristic seven-stripe eve expression pattern, which beautifully mirrored the mutant phenotype.25,26 Painstaking molecular dissection of the eve regulatory region with transgenic reporter constructs identified the eve2 enhancer region as the sequence necessary27 and sufficient28 to drive expression of the second eve stripe in embryos. Candidate regulators were identified by examining mutants that altered the eve2 expression pattern,29 and binding site mutations in eve2 confirmed that these interactions were direct.28,30,31 These early experiments produced a working model of enhancer function that persists in cell and developmental biology classrooms to this day.32,33 In this model, eve2 is regulated by direct binding of the anterior activators bicoid and hunchback as well as the gap gene repressors giant and Krüppel (Fig. 1).
Aficionados know that the story of eve2 did not end there. First, this model failed to explain the absence of expression in the region anterior to giant, where both activators are present in high concentrations without a repressor. Genetic experiments identified additional direct and indirect regulators that account for this discrepancy.34 The ubiquitously expressed activator zelda also directly binds eve2,35 and computational work has proposed additional roles for the activator caudal and the repressors knirps and tailless.36,37 Finally, while the minimal eve2 element is sufficient to drive expression of stripe 2, surrounding sequences are critical for robustness to genetic and environmental perturbation38 as well as the response of eve stripe 7 to TF misexpression.39
The eve2 story demonstrates the power of genetic approaches for identifying and validating enhancer regulators. However, these methods are candidate-driven: they can only determine whether a given regulator has a role or not. They cannot tell us whether we know all relevant regulators; this question can only be resolved by reconstitution. In other words, while we have come far in characterizing the regulatory logic of eve2, genetic approaches cannot tell us how much further we have to go.
New age Bill: leveraging big data to define regulatory logic
The methods we use to define an enhancer's regulatory logic have transformed in the last decade. Genetics and molecular biology are now complemented by high-throughput measurements of chromatin state and TF binding. These functional genomic methods can complement classic genetic approaches in two ways. First, we can systematically identify candidate regulators by measuring binding genome-wide for all known TFs.40–44 As with the classic approach, we must then validate these candidates by mutating sites using experimentally derived TF binding preferences.45–47 However, we can also circumvent a candidate approach altogether by screening large libraries of mutated enhancer reporter constructs for expression and analyzing them retrospectively.48–50These new methods carry their own limitations. First, it is difficult to capture spatial information in high-throughput. This limitation applies to measurements of TFs (inputs) as well as enhancer-driven expression patterns (outputs). For inputs, we can measure gene expression and chromatin accessibility in single cells,51,52 but in vivo measurements of TF binding must be performed in homogenized tissue or cell populations. New methods may soon address this shortcoming.53 For outputs, measurements of enhancer reporter libraries are typically performed in cell populations or in disaggregated tissue,54 although high-throughput imaging of whole animals after transfection is an emerging technology.55 Reporter libraries can only incorporate DNA fragments of a certain size, which is currently less than 200 bp.56 Therefore, an unbiased approach cannot be applied to larger enhancers. Many endogenous developmental enhancers exceed this size limit,43,57 including some that have been experimentally ‘minimized’, such as our example, eve2.31,58,59 In addition, this experimental design requires reporters to drive transcription, and thus does not detect fragments that repress or modulate the activity of other sequences.38,60 This is part of a larger issue of assaying regulatory DNA in reporter constructs outside of its native context. Finally, making transgenic animals is slow compared to transfection of isolated cells. This constraint limits throughput to tens or hundreds of constructs by an individual, and thousands of constructs by consortia or larger projects.61,62 In the long-term, genomic technologies may advance to the point where we can determine the regulatory logic of developmental enhancers in an unbiased, data-centric way. For the moment, however, enhancer reconstitution through rational design provides a critical reciprocal test of an enhancer's complete regulatory logic.
Some general rules governing binding site interactions
A complete understanding of enhancer logic requires not only identifying all of its TF binding sites, but also characterizing their interactions. Here, we review some rules governing binding site interactions within enhancers, with an emphasis on case studies in Drosophila. Many other flagship systems have revealed mechanistically similar interactions between TFs,63–66 and these have been reviewed elsewhere.67,68Cooperativity, competition and context
Interactions between TFs are common in animal enhancers. TFs can exhibit cooperativity by influencing one another's binding directly through protein–protein interactions, or indirectly through effects on nucleosome occupancy or other co-factors.69 In the simplest case, cooperativity occurs between binding sites for the same TF, as has been proposed for the activator Bicoid binding to the hunchback P2 enhancer.70 Cooperativity can also occur between different TFs, which has been suggested for Dorsal and twist in Drosophila as well as their homologs in mice.71,72 In the extreme, cooperative physical interactions between many TFs underlie the formation of a complex structure such as the enhanceosome that regulates human interferon-beta expression during viral infection.73 TFs can also physically interact with each other to interfere with binding. Repressors can directly compete with activators for overlapping sites within enhancers, as was originally proposed for regulation of eve2.30 However, this does not appear to be the dominant mode of repressor function.69 Finally, bifunctional TFs can act as activators or repressors depending on context.39,74,75 Context-dependent function may be controlled by dimerization on DNA,76 interactions with neighboring TFs,37 or inputs from signalling cascades.77Long-range interactions allow design flexibility
Cooperative binding and TF competition occur at short DNA length scales (tens of base pairs). However, TFs bound to enhancers also interact over longer length scales through other mechanisms. TFs can collaborate to expel nucleosomes, leading to indirect “collaborative cooperativity” on the length-scale of ∼150 bp.78 Activators have been proposed to functionally interact due to effects on nucleosomes or the core transcriptional machinery.13,79 Repressors can be classed by their range of influence on neighboring activators, with short-range repressors operating at a scale of ∼100 bp and long-range repressors operating at a scale of more than 1 kb;80 these different classes of repressors induce distinct chromatin changes when bound.81 Finally, cofactors and architectural proteins can mediate interactions between pieces of regulatory DNA at various length-scales ranging from intergenic to interchromosomal contacts.82Given that there are so many different types of TF interactions, it may seem hopeless to design an enhancer from scratch. However, the wide variety of mechanisms allows for flexible enhancer design with many ways to be successful.83 Indeed, orthologous enhancers drive similar expression patterns and are functionally conserved despite extensive turnover in TF binding sites.15,22,84 These general categories of interactions have been known for quite some time, and have been explored computationally, as we discuss below. However, we have not yet effectively leveraged computational tools to reconstitute enhancers that function in vivo.
Tools for rational design
Mathematical models can be useful tools for reasoning about complex systems. They formalize our assumptions and can generate testable predictions.85 In tackling the problem of building developmental enhancers, mathematical models can be useful in multiple ways. Regression models on imaging or sequencing data can suggest candidate regulators for a given expression pattern.86–89 More complex models based on fractional occupancy of TFs on enhancers have been used to analyze how expression patterns are generated.90 This type of model has been widely used for bacterial sequences91–95 and has been extended to developmental enhancers.36,37,96–102Multiple groups have studied how TF binding site arrangement affects gene expression by measuring the output of synthetic enhancers and using this data to fit mathematical models. The Arnosti lab used this approach to examine the function of short-range repressors.20 They constructed a set of synthetic enhancers where binding sites for short-range repressors were placed in configurations that altered their number, affinity and spacing relative to a common set of activator binding sites. By fitting parameters for each binding site within a fractional occupancy model, they uncovered a non-monotonic relationship between a repressor's efficiency and its distance from activator sites. They used their results to dissect the relative contributions of individual binding sites within an endogenous enhancer. More recently, the Furlong lab used an analogous approach to study activator function in the Drosophila mesoderm.16 In this case, the authors measured expression patterns driven by homotypic and heterotypic binding site arrays using computationally designed spacer sequences that prevented the creation of additional sites. They found that some arrays of single activators were sufficient to drive expression in embryos; these arrays recapitulated the expression pattern of the activator. Some heterotypic arrays also drove expression; these arrays varied in their sensitivity to changes in the spacing and orientation of the sites. Finally, the authors used fractional site occupancy modeling to explore the potential impact of ‘higher-order’ cooperativity between binding site clusters. Both case studies demonstrate how TF interactions can be interrogated by combining careful measurements, highly controlled synthetic reporter constructs and computational modeling.
Even these systematic efforts will not satisfy Doug, who remains perhaps unreasonably demanding. Both studies employ the common practice of post hoc analysis, where models are used to analyze existing data to explain their experimental results. We advocate for a complementary approach, where models are used at the outset to predict new, functional arrangements of binding sites, and these predictions are explicitly tested by embedding binding sites in ‘neutral’ DNA sequence. This model-first approach would test the predictive power of models for synthetic applications, as well as the completeness of our understanding of the rules governing enhancer function.103
Instructive failures in enhancer reconstitution
Surprisingly few studies have attempted to reconstitute enhancer function from their component binding sites. One notable exception from the Barolo lab merits a detailed discussion.104 For two candidate enhancers – the proneural enhancer of E(spl)m4 and the sparkling enhancer of dPax2 – arrays of high affinity binding sites for key activators were found to be insufficient to drive expression in vivo. These results contrast with the studies discussed above, where binding site arrays drive patterned expression in the Drosophila blastoderm, mesoderm and neurogenic ectoderm.15,16 Furthermore, reconstituted enhancers from dPax2 and dpp that maintained the endogenous arrangement, affinity and spacing of known binding sites also failed to drive expression in vivo. Finally, an in-depth study of the dppVM enhancer revealed that regulatory sequence between known binding sites is essential for function. By including mapped binding sites as well as evolutionarily conserved sequences, the authors generated an enhancer that drove expression in the correct location, but not to the correct level. This study represents one of the most comprehensive attempts to build a specific enhancer from its component parts, but even after the creation and measurement of over 25 constructs in transgenic animals, we still do not know the complete regulatory logic of the dppVM enhancer.Legend has it that Steve Small attempted to build eve2 from scratch following its initial characterization. This legend is true. Small fragments of eve2 recapitulate TF-specific activation and repression in cell culture reporter constructs.28 However, when these fragments were introduced into embryos, they failed to drive expression on their own. Additional constructs tested whether function could be restored by multimerization of the elements or the introduction of a sequence that restored endogenous spacing between them (Fig. 2A). These additional constructs also failed to recapitulate eve2 expression in embryos, and the results were never published (Steve Small, personal communication).
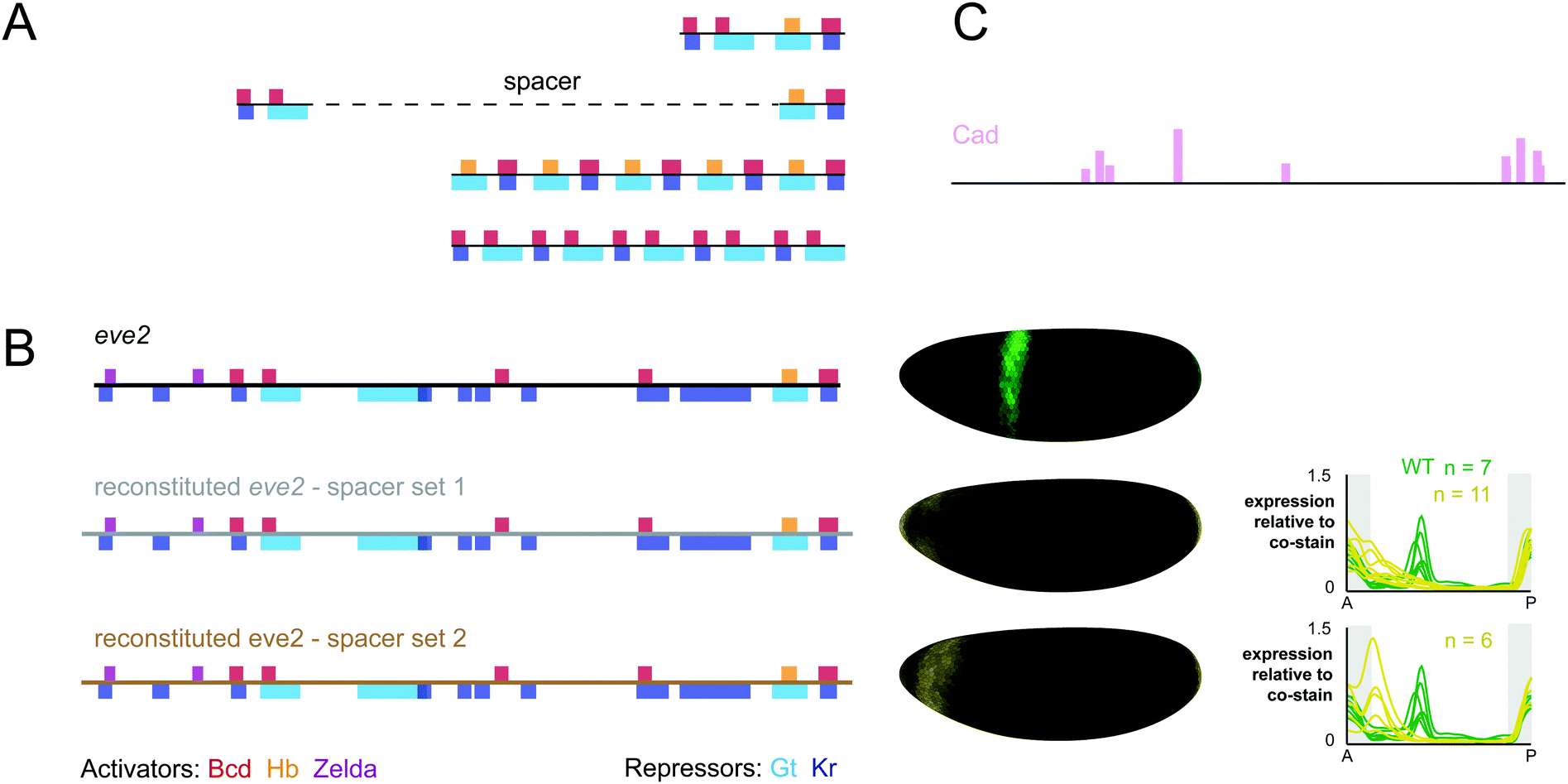 | ||
| Fig. 2 Reconstituted eve2 enhancers do not drive correct expression patterns in embryos. (A) Constructs designed by Steve Small to reconstitute eve2 function from enhancer fragments. As in Fig. 1, footprinted binding sites are represented as bars where color indicates TF identity. Red: bicoid; orange: hunchback; violet: zelda; light blue: giant; dark blue: Krüppel. A fusion of two ∼50 bp fragments which drives regulated expression in cell lines fails to drive expression of stripe 2 in embryos. Followup constructs that tested the impact of maintaining endogenous spacing or multimerization also failed to drive expression of stripe 2 in embryos (Steve Small, personal communication). (B) Left: We used SiteOut to design two sets of spacer sequences that did not create binding sites for these TFs or other known eve regulators.108 We note that reconstituted enhancers disrupted a footprinted binding site for the repressor sloppy paired 1, which was not contained in Redfly, as well as a single base pair within a footprinted bicoid site (see annotated sequences in ESI†). However, this single base pair change does not disrupt any bicoid sites predicted using binding preferences derived from bacterial one-hybrid experiments.45 Middle: The 3D expression patterns generated by wild-type eve2 (green) and two reconstituted enhancers (yellow). Shown here are visual representations of gene expression atlases from embryos stained for lacZ mRNA and co-stained for hkb mRNA to normalize for lacZ levels between lines.125 Right: Expression patterns for all three constructs are displayed as line traces normalized to the hkb co-stain. hkb is expressed in the poles of the embryo, as indicated by the shaded region in grey. (C) Binding sites for caudal were predicted using PATSER software and displayed as vertical bars, where height is proportional to the predicted affinity of the site (see Methods, ESI†). | ||
We attempted to reconstitute eve2 again using some newly available resources. eve2 contains many binding sites for known regulators including zelda, a recently discovered activator that is critical for function of bicoid-dependent enhancers in Drosophila embryos.35,105–107 We were able to design our constructs while rigorously controlling for the inadvertent creation of other binding sites using a computational tool, SiteOut.108 We commercially synthesized two synthetic eve enhancers that included all footprinted binding sites from the RedFly database separated by two different sets of spacers at their endogenous locations relative to one another (Fig. 2). We integrated both constructs into the same genomic location (attP2) using the phiC31 system and measured their expression patterns at cellular resolution relative to a control construct containing wild-type eve2 (see Supplemental Methods in the ESI†). We have deposited quantitative data in individual embryos as well as an averaged gene expression atlas for these three constructs on figshare.org (see Supplemental Data in the ESI†).109
As Fig. 2B shows, we were unsuccessful in reconstituting eve2 function from its footprinted sites in RedFly. However, our experiment ‘failed well’ in that it suggested some specific ways forward. Both synthetic constructs drive expression in the anterior of the embryo, outside the area of eve stripe 2. We hypothesize this ectopic expression is due to disruption of a footprinted site for the repressor sloppy-paired, which was not included in the RedFly annotation.34 We also hypothesize that our synthetic construct is missing sites for one or more additional activators which are expressed in the region of eve stripe 2. caudal is an attractive candidate: caudal sites have been included in computational models of eve2 flanking sequences36 and we can find predicted caudal sites within the boundaries of eve2 using position weight matrices derived from bacterial 1-hybrid experiments45 (Fig. 2C). Our results also suggest that the two different sets of spacer sequences drive slightly different levels of expression, despite our attempts to control binding site content with SiteOut. This discrepancy may help identify other regulating TFs or relevant sequence features in future experiments. Other explanations for our results are also possible. For example, the predicted binding sites that we used may not be accurate enough for this purpose. We used footprinted binding sites;107,110 but other data sources are available,45,111 and not all binding preferences are the same across methods. Finally, we many have disrupted local sequence features outside transcription factor binding sites that affect TF binding, such as DNA shape or nucleosome positioning.68,112–115 All of these possibilities can be tested with additional constructs, and we hope that the quantitative data we provide will be useful in designing follow up experiments and testing computational models.
One thing is clear from all of these reconstitution experiments: we still have much to learn, even for the most well-characterized animal enhancers.
Doug needs us to share our negative results
In this perspective, we have argued that our understanding of developmental enhancers is incomplete, and experiments that attempt to reconstitute enhancer function from individual binding sites are an important test of how much we know thus far. In enhancer reconstitution, both successes and failures are informative. Successes confirm what we know, but do not always identify the sources of our success. Failures illuminate what we do not know, and converting a failure into a success pinpoints a new critical feature of enhancer design. Due to the explosion of functional genomic data generated from large consortia, the low cost of DNA synthesis and the availability of computational tools for rational design, we believe the time is right to perform reconstitution experiments for endogenous enhancers in animal systems. We advocate for an iterative cycle of enhancer reconstitution, where computational models are used to predict active enhancer sequences, which are then synthesized and experimentally tested using quantitative techniques.116–120 Results are then used to train models to improve design for the next round. For a given enhancer, the first rounds of this cycle are likely to fail, as we have shown with our own constructs. However, in order for this approach to succeed, failures must be shared publically so that the greater community can learn from the mistakes. Despite the known difficulties of publishing negative data,121,122 new online resources such as http://bioRXiv.org and http://figshare.com make this type of open data sharing possible. We believe that with a communal investment in synthetic approaches, we can finally meet Doug's challenge and learn the basic principles governing how enhancers work.Acknowledgements
The authors gratefully acknowledge the public financial support that makes our work possible, the environment of the Department of Systems Biology at Harvard Medical School, the Biological and Biomedical Science graduate program and current and formers members of the DePace Lab for helping to develop ideas. In particular, we acknowledge Clarissa Scholes and Max Staller for their detailed input on the manuscript. Finally, we wish to thank our anonymous reviewers, who provided critical comments that substantially improved the clarity and breadth of this work.References
- W. T. Sullivan, The salvation of Doug, Generations, 1993, 1(3), 1439–1443 Search PubMed.
- M. Levine, Transcriptional enhancers in animal development and evolution, Curr. Biol., 2010, 20(17), R754–R763 CrossRef CAS PubMed.
- S. B. Carroll, Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution, Cell, 2008, 134(1), 25–36 CrossRef CAS PubMed.
- J. Zeitlinger and A. Stark, Developmental gene regulation in the era of genomics, Dev. Biol., 2010, 339(2), 230–239 CrossRef CAS PubMed.
- M. T. Maurano, R. Humbert, E. Rynes, R. E. Thurman, E. Haugen and H. Wang, et al. Systematic localization of common disease-associated variation in regulatory DNA, Science, 2012, 337(6099), 1190–1195 CrossRef CAS PubMed.
- M. Z. Ludwig, C. Bergman, N. H. Patel and M. Kreitman, Evidence for stabilizing selection in a eukaryotic enhancer element, Nature, 2000, 403(6769), 564–567 CrossRef CAS PubMed.
- A. M. Moses, D. A. Pollard, D. A. Nix, V. N. Iyer, X.-Y. Li and M. D. Biggin, et al. Large-scale turnover of functional transcription factor binding sites in Drosophila, PLoS Comput. Biol., 2006, 2(10), e130 Search PubMed.
- S. W. Doniger and J. C. Fay, Frequent gain and loss of functional transcription factor binding sites, PLoS Comput. Biol., 2007, 3(5), e99 Search PubMed.
- S. Barolo, Shadow enhancers: frequently asked questions about distributed cis-regulatory information and enhancer redundancy, BioEssays, 2012, 34(2), 135–141 CrossRef CAS PubMed.
- C. D. Arnold, D. Gerlach, D. Spies, J. A. Matts, Y. A. Sytnikova and M. Pagani, et al. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution, Nat. Genet., 2014, 46(7), 685–692 CrossRef CAS PubMed.
- J. Gertz, E. D. Siggia and B. A. Cohen, Analysis of combinatorial cis-regulation in synthetic and genomic promoters, Nature, 2009, 457(7226), 215–218 CrossRef CAS PubMed.
- J. Gertz and B. A. Cohen, Environment-specific combinatorial cis-regulation in synthetic promoters, Mol. Syst. Biol., 2009, 5, 244 CrossRef PubMed.
- A. J. Keung, C. J. Bashor, S. Kiriakov, J. J. Collins and A. S. Khalil, Using targeted chromatin regulators to engineer combinatorial and spatial transcriptional regulation, Cell, 2014, 158(1), 110–120 CrossRef CAS PubMed.
- G. Stampfel, T. Kazmar, O. Frank, S. Wienerroither, F. Reiter and A. Stark, Transcriptional regulators form diverse groups with context-dependent regulatory functions, Nature, 2015, 528(7580), 147–151 CAS.
- L. M. Liberman and A. Stathopoulos, Design flexibility in cis-regulatory control of gene expression: synthetic and comparative evidence, Dev. Biol., 2009, 327(2), 578–589 CrossRef CAS PubMed.
- J. Erceg, T. E. Saunders, C. Girardot, D. P. Devos, L. Hufnagel and E. E. M. Furlong, Subtle changes in motif positioning cause tissue-specific effects on robustness of an enhancer's activity, PLoS Genet., 2014, 10(1), e1004060 Search PubMed.
- A. H. Brand and N. Perrimon, Targeted gene expression as a means of altering cell fates and generating dominant phenotypes, Development, 1993, 118(2), 401–415 CAS.
- D. M. Ornitz, R. W. Moreadith and P. Leder, Binary system for regulating transgene expression in mice: targeting int-2 gene expression with yeast GAL4/UAS control elements, Proc. Natl. Acad. Sci. U. S. A., 1991, 88(3), 698–702 CrossRef CAS.
- M. E. Halpern, J. Rhee, M. G. Goll, C. M. Akitake, M. Parsons and S. D. Leach, Gal4/UAS transgenic tools and their application to zebrafish, Zebrafish, 2008, 5(2), 97–110 CrossRef CAS PubMed.
- W. D. Fakhouri, A. Ay, R. Sayal, J. Dresch, E. Dayringer and D. N. Arnosti, Deciphering a transcriptional regulatory code: modeling short-range repression in the Drosophila embryo, Mol. Syst. Biol., 2010, 6, 341 CrossRef PubMed.
- S. Goverdhana, M. Puntel, W. Xiong, J. M. Zirger, C. Barcia and J. F. Curtin, et al. Regulatable gene expression systems for gene therapy applications: progress and future challenges, Mol. Ther., 2005, 12(2), 189–211 CrossRef CAS PubMed.
- M. Z. Ludwig, A. Palsson, E. Alekseeva, C. M. Bergman, J. Nathan and M. Kreitman, Functional evolution of a cis-regulatory module, PLoS Biol., 2005, 3(4), e93 Search PubMed.
- A. V. Wright, J. K. Nuñez and J. A. Doudna, Biology and Applications of CRISPR Systems: Harnessing Nature's Toolbox for Genome Engineering, Cell, 2016, 164(1–2), 29–44 CrossRef CAS PubMed.
- C. Nüsslein-Volhard and E. Wieschaus, Mutations affecting segment number and polarity in Drosophila, Nature, 1980, 287(5785), 795–801 CrossRef.
- K. Harding, C. Rushlow, H. J. Doyle, T. Hoey and M. Levine, Cross-regulatory interactions among pair-rule genes in Drosophila, Science, 1986, 233(4767), 953–959 CAS.
- M. Frasch, T. Hoey, C. Rushlow, H. Doyle and M. Levine, Characterization and localization of the even-skipped protein of Drosophila, EMBO J., 1987, 6(3), 749–759 CAS.
- T. Goto, P. Macdonald and T. Maniatis, Early and late periodic patterns of even skipped expression are controlled by distinct regulatory elements that respond to different spatial cues, Cell, 1989, 57(3), 413–422 CrossRef CAS PubMed.
- S. Small, R. Kraut, T. Hoey, R. Warrior and M. Levine, Transcriptional regulation of a pair-rule stripe in Drosophila, Genes Dev., 1991, 5(5), 827–839 CrossRef CAS PubMed.
- M. Frasch and M. Levine, Complementary patterns of even-skipped and fushi tarazu expression involve their differential regulation by a common set of segmentation genes in Drosophila, Genes Dev., 1987, 1(9), 981–995 CrossRef CAS PubMed.
- D. Stanojevic, S. Small and M. Levine, Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo, Science, 1991, 254(5036), 1385–1387 CAS.
- S. Small, A. Blair and M. Levine, Regulation of even-skipped stripe 2 in the Drosophila embryo, EMBO J., 1992, 11(11), 4047–4057 CAS.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff and K. Roberts, et al. Molecular Biology of the Cell, Sixth Edition, Garland Science, 2014, 1464 Search PubMed.
- S. F. Gilbert, Developmental Biology, Sinauer Associates, Inc., 10th ed, 2013 Search PubMed.
- L. P. M. Andrioli, V. Vasisht, E. Theodosopoulou, A. Oberstein and S. Small, Anterior repression of a Drosophila stripe enhancer requires three position-specific mechanisms, Development, 2002, 129(21), 4931–4940 CAS.
- P. Struffi, M. Corado, L. Kaplan, D. Yu, C. Rushlow and S. Small, Combinatorial activation and concentration-dependent repression of the Drosophila even skipped stripe 3 + 7 enhancer, Development, 2011, 138(19), 4291–4299 CrossRef CAS PubMed.
- H. Janssens, S. Hou, J. Jaeger, A.-R. Kim, E. Myasnikova and D. Sharp, et al. Quantitative and predictive model of transcriptional control of the Drosophila melanogaster even skipped gene, Nat. Genet., 2006, 38(10), 1159–1165 CrossRef CAS PubMed.
- A.-R. Kim, C. Martinez, J. Ionides, A. F. Ramos, M. Z. Ludwig and N. Ogawa, et al. Rearrangements of 2.5 kilobases of noncoding DNA from the Drosophila even-skipped locus define predictive rules of genomic cis-regulatory logic, PLoS Genet., 2013, 9(2), e1003243 CAS.
- M. Z. Ludwig, Manu, R. Kittler, K. P. White and M. Kreitman, Consequences of eukaryotic enhancer architecture for gene expression dynamics, development, and fitness, PLoS Genet., 2011, 7(11), e1002364 CAS.
- M. V. Staller, B. J. Vincent, M. D. J. Bragdon, T. Lydiard-Martin, Z. Wunderlich and J. Estrada, et al. Shadow enhancers enable Hunchback bifunctionality in the Drosophila embryo, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(3), 785–790 CrossRef CAS PubMed.
- M. Slattery, L. Ma, R. F. Spokony, R. K. Arthur, P. Kheradpour and A. Kundaje, et al. Diverse patterns of genomic targeting by transcriptional regulators in Drosophila melanogaster, Genome Res., 2014, 24(7), 1224–1235 CrossRef CAS PubMed.
- C. L. Araya, T. Kawli, A. Kundaje, L. Jiang, B. Wu and D. Vafeados, et al. Regulatory analysis of the C. elegans genome with spatiotemporal resolution, Nature, 2014, 512(7515), 400–405 CrossRef CAS PubMed.
- ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome, Nature, 2012, 489(7414), 57–74 CrossRef PubMed.
- F. Yue, Y. Cheng, A. Breschi, J. Vierstra, W. Wu and T. Ryba, et al. A comparative encyclopedia of DNA elements in the mouse genome, Nature, 2014, 515(7527), 355–364 CrossRef CAS PubMed.
- A. P. Boyle, C. L. Araya, C. Brdlik, P. Cayting, C. Cheng and Y. Cheng, et al. Comparative analysis of regulatory information and circuits across distant species, Nature, 2014, 512(7515), 453–456 CrossRef CAS PubMed.
- M. B. Noyes, X. Meng, A. Wakabayashi, S. Sinha, M. H. Brodsky and S. A. Wolfe, A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system, Nucleic Acids Res., 2008, 36(8), 2547–2560 CrossRef CAS PubMed.
- D. E. Newburger and M. L. Bulyk, UniPROBE: an online database of protein binding microarray data on protein-DNA interactions, Nucleic Acids Res., 2009, 37(database issue), D77–D82 CrossRef CAS PubMed.
- E. Portales-Casamar, S. Thongjuea and A. T. Kwon, JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles, Nucleic Acids, 2009, Available from: http://nar.oxfordjournals.org/content/early/2009/11/11/nar.gkp950.short Search PubMed.
- J. C. Kwasnieski, I. Mogno, C. A. Myers, J. C. Corbo and B. A. Cohen, Complex effects of nucleotide variants in a mammalian cis-regulatory element, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(47), 19498–19503 CrossRef CAS PubMed.
- P. Kheradpour, J. Ernst, A. Melnikov, P. Rogov, L. Wang and X. Zhang, et al. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay, Genome Res., 2013, 23(5), 800–811 CrossRef CAS PubMed.
- M. C. Canver, E. C. Smith, F. Sher, L. Pinello, N. E. Sanjana and O. Shalem, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis, Nature, 2015, 527(7577), 192–197 CrossRef CAS PubMed.
- J. D. Buenrostro, P. G. Giresi, L. C. Zaba, H. Y. Chang and W. J. Greenleaf, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position, Nat. Methods, 2013, 10(12), 1213–1218 CrossRef CAS PubMed.
- Q. Deng, D. Ramsköld, B. Reinius and R. Sandberg, Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells, Science, 2014, 343(6167), 193–196 CrossRef CAS PubMed.
- A. Rotem, O. Ram, N. Shoresh, R. A. Sperling, A. Goren and D. A. Weitz, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state, Nat. Biotechnol., 2015, 33(11), 1165–1172 CrossRef CAS PubMed.
- S. S. Gisselbrecht, L. A. Barrera, M. Porsch, A. Aboukhalil, P. W. Estep and A. Vedenko, et al. Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos, Nat. Methods, 2013, 10(8), 774–780 CrossRef CAS PubMed.
- E. K. Farley, K. M. Olson, W. Zhang, A. J. Brandt, D. S. Rokhsar and M. S. Levine, Suboptimization of developmental enhancers, Science, 2015, 350(6258), 325–328 CrossRef CAS PubMed.
- M. Levo and E. Segal, In pursuit of design principles of regulatory sequences, Nat. Rev. Genet., 2014, 15(7), 453–468 CrossRef CAS PubMed.
- J. W. K. Ho, Y. L. Jung, T. Liu, B. H. Alver, S. Lee and K. Ikegami, et al. Comparative analysis of metazoan chromatin organization, Nature, 2014, 512(7515), 449–452 CrossRef CAS PubMed.
- S. Small, A. Blair and M. Levine, Regulation of two pair-rule stripes by a single enhancer in the Drosophila embryo, Dev. Biol., 1996, 175(2), 314–324 CrossRef CAS PubMed.
- M. Fujioka, Y. Emi-Sarker, G. L. Yusibova, T. Goto and J. B. Jaynes, Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multi-stripe positioning by gap gene repressor gradients, Development, 1999, 126(11), 2527–2538 CAS.
- C. I. Swanson, N. C. Evans and S. Barolo, Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer, Dev. Cell., 2010, 18(3), 359–370 CrossRef CAS PubMed.
- A. Jenett, G. M. Rubin, T.-T. B Ngo, D. Shepherd, C. Murphy and H. Dionne, et al. A GAL4-driver line resource for Drosophila neurobiology, Cell Rep., 2012, 2(4), 991–1001 CrossRef CAS PubMed.
- E. Z. Kvon, T. Kazmar, G. Stampfel, J. O. Yáñez-Cuna, M. Pagani and K. Schernhuber, et al. Genome-scale functional characterization of Drosophila developmental enhancers in vivo, Nature, 2014, 512(7512), 91–95 CAS.
- W. Lee, A. Haslinger, M. Karin and R. Tjian, Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40, Nature, 1987, 325(6102), 368–372 CrossRef CAS PubMed.
- J. W. Dolan and S. Fields, Cell-type-specific transcription in yeast, Biochim. Biophys. Acta., 1991, 1088(2), 155–169 CrossRef CAS.
- E. Shaulian and M. Karin, AP-1 as a regulator of cell life and death, Nat. Cell Biol., 2002, 4(5), E131–E136 CrossRef CAS PubMed.
- Y. Yamamoto and R. B. Gaynor, IκB kinases: key regulators of the NF-κB pathway, Trends Biochem. Sci., 2004, 29(2), 72–79 CrossRef CAS PubMed.
- D. S. Latchman, Eukaryotic Transcription Factors, Academic Press, 5 edn, 2010 Search PubMed.
- S. Weingarten-Gabbay and E. Segal, The grammar of transcriptional regulation, Hum. Genet., 2014, 133(6), 701–711 CrossRef CAS PubMed.
- F. Spitz and E. E. M. Furlong, Transcription factors: from enhancer binding to developmental control, Nat. Rev. Genet., 2012, 13(9), 613–626 CrossRef CAS PubMed.
- W. Driever, G. Thoma and C. Nüsslein-Volhard, Determination of spatial domains of zygotic gene expression in the Drosophila embryo by the affinity of binding sites for the bicoid morphogen, Nature, 1989, 340(6232), 363–367 CrossRef CAS PubMed.
- J. M. Shirokawa and A. J. Courey, A direct contact between the dorsal rel homology domain and Twist may mediate transcriptional synergy, Mol. Cell Biol., 1997, 17(6), 3345–3355 CrossRef CAS PubMed.
- D. Šošić, J. A. Richardson, K. Yu, D. M. Ornitz and E. N. Olson, Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity, Cell, 2003, 112(2), 169–180 CrossRef.
- M. Carey, The enhanceosome and transcriptional synergy, Cell, 1998, 92(1), 5–8 CrossRef CAS PubMed.
- D. Shore and K. Nasmyth, Purification and cloning of a DNA binding protein from yeast that binds to both silencer and activator elements, Cell, 1987, 51(5), 721–732 CrossRef CAS PubMed.
- Z. Deng, P. Cao, M. M. Wan, G. Sui and Y. Yang, a multifaceted protein beyond a transcription factor, Transcription, 2010, 1(2), 81–84 CrossRef PubMed.
- D. Papatsenko and M. S. Levine, Dual regulation by the Hunchback gradient in the Drosophila embryo, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(8), 2901–2906 CrossRef CAS PubMed.
- S. Barolo and J. W. Posakony, Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling, Genes Dev., 2002, 16(10), 1167–1181 CrossRef CAS PubMed.
- L. A. Mirny, Nucleosome-mediated cooperativity between transcription factors, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(52), 22534–22539 CrossRef CAS PubMed.
- J. Blau, H. Xiao, S. McCracken, P. O'Hare, J. Greenblatt and D. Bentley, Three functional classes of transcriptional activation domains, Mol. Cell Biol., 1996, 16(5), 2044–2055 CrossRef CAS PubMed.
- A. J. Courey and S. Jia, Transcriptional repression: the long and the short of it, Genes Dev., 2001, 15(21), 2786–2796 CAS.
- L. M. Li and D. N. Arnosti, Long- and short-range transcriptional repressors induce distinct chromatin states on repressed genes, Curr. Biol., 2011, 21(5), 406–412 CrossRef CAS PubMed.
- H. Q. Nguyen and G. Bosco, Gene Positioning Effects on Expression in Eukaryotes, Annu. Rev. Genet., 2015, 49, 627–646 CrossRef CAS PubMed.
- D. N. Arnosti and M. M. Kulkarni, Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards?, J. Cell. Biochem., 2005, 94(5), 890–898 CrossRef CAS PubMed.
- E. E. Hare, B. K. Peterson, V. N. Iyer, R. Meier and M. B. Eisen, Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation, PLoS Genet., 2008, 4(6), e1000106 Search PubMed.
- J. Gunawardena, Models in biology: “accurate descriptions of our pathetic thinking”, BMC Biol., 2014, 12(1), 29 CrossRef PubMed.
- G. R. Ilsley, J. Fisher, R. Apweiler, A. H. De Pace and N. M. Luscombe, Cellular resolution models for even skipped regulation in the entire Drosophila embryo, eLife, 2013, 2, e00522 Search PubMed.
- E. Segal, M. Shapira, A. Regev, D. Pe'er, D. Botstein and D. Koller, et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data, Nat. Genet., 2003, 34(2), 166–176 CrossRef CAS PubMed.
- G. Karlebach and R. Shamir, Modelling and analysis of gene regulatory networks, Nat. Rev. Mol. Cell Biol., 2008, 9(10), 770–780 CrossRef CAS PubMed.
- I. Amit, A. Regev and N. Hacohen, Strategies to discover regulatory circuits of the mammalian immune system, Nat. Rev. Immunol., 2011, 11(12), 873–880 CAS.
- H. D. Kim, T. Shay, E. K. O'Shea and A. Regev, Transcriptional regulatory circuits: predicting numbers from alphabets, Science, 2009, 325(5939), 429–432 CAS.
- P. H. von Hippel, A. Revzin, C. A. Gross and A. C. Wang, Non-specific DNA binding of genome regulating proteins as a biological control mechanism: I. The lac operon: equilibrium aspects, Proc. Natl. Acad. Sci. U. S. A., 1974, 71(12), 4808–4812 CrossRef CAS.
- G. K. Ackers, A. D. Johnson and M. A. Shea, Quantitative model for gene regulation by lambda phage repressor, Proc. Natl. Acad. Sci. U. S. A., 1982, 79(4), 1129–1133 CrossRef CAS.
- M. A. Shea and G. K. Ackers, The OR control system of bacteriophage lambda. A physical-chemical model for gene regulation, J. Mol. Biol., 1985, 181(2), 211–230 CrossRef CAS PubMed.
- H. G. Garcia, A. Sanchez, J. Q. Boedicker, M. Osborne, J. Gelles and J. Kondev, et al. Operator sequence alters gene expression independently of transcription factor occupancy in bacteria, Cell Rep., 2012, 2(1), 150–161 CrossRef CAS PubMed.
- R. C. Brewster, F. M. Weinert, H. G. Garcia, D. Song, M. Rydenfelt and R. Phillips, The transcription factor titration effect dictates level of gene expression, Cell, 2014, 156(6), 1312–1323 CrossRef CAS PubMed.
- R. P. Zinzen, K. Senger, M. Levine and D. Papatsenko, Computational models for neurogenic gene expression in the Drosophila embryo, Curr. Biol., 2006, 16(13), 1358–1365 CrossRef CAS PubMed.
- E. Segal, T. Raveh-Sadka, M. Schroeder, U. Unnerstall and U. Gaul, Predicting expression patterns from regulatory sequence in Drosophila segmentation, Nature, 2008, 451(7178), 535–540 CrossRef CAS PubMed.
- X. He, M. A. H. Samee, C. Blatti and S. Sinha, Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression, PLoS Comput. Biol., 2010, 6(9), e1000935 Search PubMed.
- M. A. White, D. S. Parker, S. Barolo and B. A. Cohen, A model of spatially restricted transcription in opposing gradients of activators and repressors, Mol. Syst. Biol., 2012, 8, 614 CrossRef PubMed.
- C. A. Martinez, K. A. Barr, A.-R. Kim and J. Reinitz, A synthetic biology approach to the development of transcriptional regulatory models and custom enhancer design, Methods, 2013, 62(1), 91–98 CrossRef CAS PubMed.
- A. H. Samee and S. Sinha, Evaluating thermodynamic models of enhancer activity on cellular resolution gene expression data, Methods, 2013, 62(1), 79–90 CrossRef PubMed.
- M. A. H. Samee and S. Sinha, Quantitative modeling of a gene's expression from its intergenic sequence, PLoS Comput. Biol., 2014, 10(3), e1003467 Search PubMed.
- R. Phillips, Theory in Biology: Fig. 1 or Fig. 7?, Trends Cell Biol., 2015, Available from: http://www.sciencedirect.com/science/article/pii/S0962892415001944 Search PubMed.
- L. A. Johnson, Y. Zhao, K. Golden and S. Barolo, Reverse-Engineering a Transcriptional Enhancer: A Case Study in Drosophila, Tissue Eng., Part A, 2008, 14(9), 1549–1559 CrossRef PubMed.
- H.-L. Liang, C.-Y. Nien, H.-Y. Liu, M. M. Metzstein, N. Kirov and C. Rushlow, The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila, Nature, 2008, 456(7220), 400–403 CrossRef CAS PubMed.
- Z. Xu, H. Chen, J. Ling, D. Yu, P. Struffi and S. Small, Impacts of the ubiquitous factor Zelda on Bicoid-dependent DNA binding and transcription in Drosophila, Genes Dev., 2014, 28(6), 608–621 CrossRef CAS PubMed.
- S. M. Gallo, D. T. Gerrard, D. Miner, M. Simich, B. Des Soye and C. M. Bergman, et al. REDfly v3. 0: toward a comprehensive database of transcriptional regulatory elements in Drosophila, Nucleic Acids Res., 2011, 39(suppl 1), D118–D123 CrossRef CAS PubMed.
- J. Estrada, T. Ruiz-Herrero, C. Scholes, Z. Wunderlich, A. DePace, SiteOut: an online tool to design binding site-free DNA sequences [Internet]. 2015 Oct. Available from: http://biorxiv.org/lookup/doi/10.1101/029645.
- C. C. Fowlkes, C. L. L. Hendriks, S. V. E. Keränen, G. H. Weber, O. Rübel and M.-Y. Huang, et al. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm, Cell, 2008, 133(2), 364–374 CrossRef CAS PubMed.
- D. A. Pollard, C. M. Bergman, J. Stoye, S. E. Celniker and M. B. Eisen, Benchmarking tools for the alignment of functional noncoding DNA, BMC Bioinf., 2004, 5, 6 CrossRef PubMed.
- S. MacArthur, X.-Y. Li, J. Li, J. B. Brown, H. C. Chu and L. Zeng, et al. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions, Genome Biol., 2009, 10(7), R80 CrossRef PubMed.
- R. Rohs, S. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, The role of DNA shape in protein-DNA recognition, Nature, 2009, 461(7268), 1248–1253 CrossRef CAS PubMed.
- M. A. White, C. A. Myers, J. C. Corbo and B. A. Cohen, Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(29), 11952–11957 CrossRef CAS PubMed.
- I. Barozzi, M. Simonatto, S. Bonifacio, L. Yang, R. Rohs and S. Ghisletti, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers, Mol. Cell, 2014, 54(5), 844–857 CrossRef CAS PubMed.
- M. Levo, E. Zalckvar, E. Sharon, A. C. Dantas Machado, Y. Kalma and M. Lotam-Pompan, et al. Unraveling determinants of transcription factor binding outside the core binding site, Genome Res., 2015, 25(7), 1018–1029 CrossRef CAS PubMed.
- C. L. Luengo Hendriks, S. V. E. Keränen, C. C. Fowlkes, L. Simirenko, G. H. Weber and A. H. DePace, et al. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution I: data acquisition pipeline, Genome Biol., 2006, 7(12), R123 CrossRef PubMed.
- A. Raj and A. van Oudenaarden, Single-molecule approaches to stochastic gene expression, Annu. Rev. Biophys., 2009, 38, 255–270 CrossRef CAS PubMed.
- S. C. Little, M. Tikhonov and T. Gregor, Precise developmental gene expression arises from globally stochastic transcriptional activity, Cell, 2013, 154(4), 789–800 CrossRef CAS PubMed.
- H. G. Garcia, M. Tikhonov, A. Lin and T. Gregor, Quantitative imaging of transcription in living Drosophila embryos links polymerase activity to patterning, Curr. Biol., 2013, 23(21), 2140–2145 CrossRef CAS PubMed.
- N. Crosetto, M. Bienko and A. van Oudenaarden, Spatially resolved transcriptomics and beyond, Nat. Rev. Genet., 2015, 16(1), 57–66 CrossRef CAS PubMed.
- D. Fanelli, Negative results are disappearing from most disciplines and countries, Scientometrics, 2012, 90(3), 891–904 CrossRef.
- N. Matosin, E. Frank, M. Engel, J. S. Lum and K. A. Newell, Negativity towards negative results: a discussion of the disconnect between scientific worth and scientific culture, Dis. Models & Mech., 2014, 7(2), 171–173 Search PubMed.
- M. V. Staller, C. C. Fowlkes, M. D. J. Bragdon, Z. Wunderlich, J. Estrada and A. H. DePace, A gene expression atlas of a bicoid-depleted Drosophila embryo reveals early canalization of cell fate, Development, 2015, 142(3), 587–596 CrossRef CAS PubMed.
- O. Rübel, G. H. Weber, S. V. E. Keränen, C. C. Fowlkes, C. L. L. Hendriks, L. Simirenko, et al., PointCloudXplore: Visual Analysis of 3D Gene Expression Data Using Physical Views and Parallel Coordinates. In: EuroVis. 2006. p. 203–210.
- Z. Wunderlich, M. D. Bragdon and A. H. DePace, Comparing mRNA levels using in situ hybridization of a target gene and co-stain, Methods, 2014, 68(1), 233–241 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00321k |
| This journal is © The Royal Society of Chemistry 2016 |