
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical phenotyping of primary human skeletal stem cells in heterogeneous populations by real-time deformability cytometry†
Miguel
Xavier
ab,
Philipp
Rosendahl
c,
Maik
Herbig
c,
Martin
Kräter
d,
Daniel
Spencer
a,
Martin
Bornhäuser
d,
Richard O. C.
Oreffo
b,
Hywel
Morgan
a,
Jochen
Guck
c and
Oliver
Otto
*c
aFaculty of Physical Sciences and Engineering, Institute for Life Sciences, University of Southampton SO17 1BJ, UK. E-mail: hm@ecs.soton.ac.uk
bCentre for Human Development, Stem Cells and Regeneration, Institute of Developmental Sciences, Southampton General Hospital, Tremona Road SO16 6YD Southampton, UK. E-mail: roco@soton.ac.uk
cBiotechnology Center, Technische Universität Dresden, Dresden, Germany. E-mail: oliver.otto@biotec.tu-dresden.de
dUniversitätsklinikum Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
First published on 8th March 2016
Abstract
Skeletal stem cells (SSCs) are a sub-population of mesenchymal stromal cells (MSCs) present in bone marrow with multipotent differentiation potential. A current unmet challenge hampering their clinical translation remains the isolation of homogeneous populations of SSCs, in vitro, with consistent regeneration and differentiation capacities. Cell stiffness has been shown to play an important role in cell separation using microfluidic techniques such as inertial focusing or deterministic lateral displacement. Here we report that the mechanical properties of SSCs, and of a surrogate human osteosarcoma cell line (MG-63), differ significantly from other cell populations found in the bone marrow. Using real-time deformability cytometry, a recently introduced method for cell mechanical characterization, we demonstrate that both MG-63 and SSCs are stiffer than the three primary leukocyte lineages (lymphocytes, monocytes and granulocytes) and also stiffer than HL-60, a human leukemic progenitor cell line. In addition, we show that SSCs form a mechanically distinct sub-population of MSCs. These results represent an important step towards finding the bio-physical fingerprint of human SSCs that will allow their label-free separation from bone marrow with significant physiological and therapeutic implications.
Insight, innovation, integrationUsing real-time deformability cytometry, an innovative microfluidic technique for high-throughput cell mechanical phenotyping, we characterised skeletal stem cells (SSCs) from human bone marrow within complex samples. SSCs were different in both cell size and deformability from granulocytes, monocytes and lymphocytes, and also from HL-60, a myeloid precursor commonly used as a model system for haematopoietic precursor cells. In addition we show that SSCs form a mechanically distinct sub-population of mesenchymal stromal cells. A factor still hampering SSC clinical translation is the need to obtain, in vitro, a large population of cells with homogeneous regeneration and differentiation capacity. This study should facilitate the development of a deformability-based, label-free isolation method of SSCs with improved purity and significant physiological and therapeutic implications. |
1. Introduction
Advances in medical care have led to a generalised increase in human life expectancy around the globe.1 However, an aging population results in an exacerbated burden for healthcare and increased bone trauma and bone disease are major contributors. There is thus an unmet need to search for innovative strategies for bone augmentation allowing bone repair and regeneration.2–4Skeletal stem cells (SSCs) are a rare multipotent stem cell population present in the bone marrow stroma. The term skeletal stem cell is used to refer specifically to the mesenchymal stromal cell (MSC) sub-population with self-renewing capability, which is responsible for the regenerative capacity inherent to bone.5–7 SSCs have osteogenic, chondrogenic and adipogenic potential, which has long been demonstrated,8,9 and have been extensively studied for tissue engineering and regenerative and stem cell-based therapies.5–7,10–12 For example, SSCs have been shown to improve bone graft integration contributing to the generation of new bone in the femoral heads of human patients.2–4 SSCs in combination with an appropriate scaffold and growth factors (bone morphogenetic protein – BMP) have also been shown to contribute to regenerating large calvarial defects in dogs and a large mandibular defect in a human patient.13 However, one of the issues restricting their broad clinical application is the need to isolate homogeneous populations of SSCs in vitro, that display consistent regeneration and differentiation capacities.14
The enrichment of SSCs from human bone marrow samples is typically achieved by a density centrifugation to remove most erythrocytes and granulocytes, followed by immuno-magnetic separation of bone marrow mononuclear cells (BMMNCs) labelled with microbeads against a cell surface marker, for example, the trypsin-resistant cell surface Stro-1 antigen.4,14,15 Stro-1 was first identified by Simmons and Torok-Storb and binds to approximately 10% of all BMMNCs, from which only a very small fraction of the Stro-1+ cells (<1%) represent progenitor cells.16,17 Nevertheless, positive selection of Stro-1+ cells followed by plastic culture adherence has led to marked enrichment of skeletal progenitor cells with enrichment factors reaching 950-fold and up to 2000-fold following combinatory approaches.18,19 However, given the Stro-1 marker is not a specific marker for the SSC, coupled with the scarcity of SSCs in bone marrow (estimated at <1 per 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 BMMNCs), current protocols are significantly limited in the purity of SSCs available following enrichment and selection by plastic culture adherence.20,21
000 BMMNCs), current protocols are significantly limited in the purity of SSCs available following enrichment and selection by plastic culture adherence.20,21
Consequently, in the absence of a specific SSC antigen, alternative isolation strategies exploiting distinct phenotypic features of SSCs offer the potential to improve both yield and purity. It has long been demonstrated that cell mechanical properties are intrinsically related to their cytoskeletal structure which translates directly to different cell states, function and subsequent differentiation paths.22–27 For example, MSCs stiffen with both passage number and population doubling; more deformable cells typically display enhanced differentiation potential.28 A recent study has also correlated different states of MSC differentiation potency with elastic modulus.29 However, there has been no study of the deformability properties of SSCs in the context of a label-free bio-marker for cell separation.
Microfluidic techniques, operating in laminar flow regime, are capable of precise and deterministic control over fluid samples taking advantage of unique physical phenomena for label-free cell isolation and characterisation.30 Thus, the application of microfluidic techniques could provide improved methods for enrichment as part of a suite of label-free approaches using e.g. inertial microfluidics or deterministic lateral displacement (DLD).
Both inertial microfluidics and DLD separate cells primarily based on size, but also in combination with other bio-physical features such as cell stiffness or deformability. Inertial microfluidic techniques rely on establishing different equilibrium positions for particles flowing within a microfluidic channel. The equilibria are created by the balance of inertial lift and shear-gradient forces. DLD uses arrays of row-shifted micropillars to divide a flow stream into parallel lamellae. Cells of different sizes are displaced within the devices leading to fractionation.30–35 Separation based on stiffness is based on the fact that two cells of similar size but with significantly different mechanical properties will have different effective radii. Accordingly, the more deformable or compliant cell will be squeezed by high shear forces and therefore appear smaller than its actual size. Holmes et al.32 have shown that by deforming cells in a DLD device operated at increasing pressure (flow rates), it is possible to discriminate between sub-populations of leukocytes (T-lymphocytes and neutrophils) through a combination of size and deformation differences.
Real-time deformability cytometry (RT-DC) is a contactless microfluidic technique for high-throughput screening of cell mechanical properties (Fig. 1a). Cells, suspended in a viscosity-adjusted medium, are deformed by shear and normal stresses as they pass through a narrow constriction (Fig. 1b and c).36,37 The technique determines the size and shape of each cell, with image analysis performed in real-time, at rates of 1000 s−1. The ability to study large populations of cells, in a relatively short time window, is an important advantage over other serial mechanical characterisation techniques such as atomic force microscopy,28 micropipette aspiration38 or optical stretching.26,28
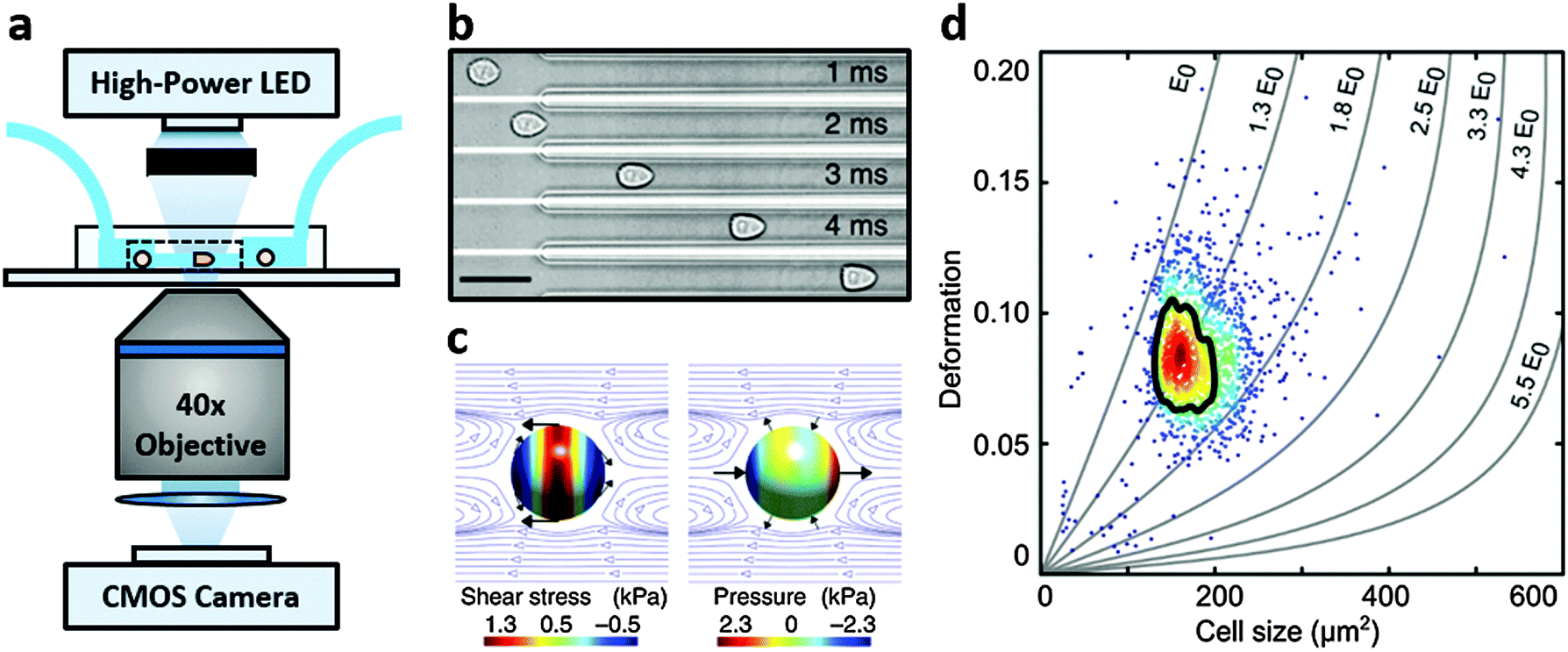 | ||
| Fig. 1 Real-time deformability cytometry. (a) Schematic of the RT-DC set-up. (b) Shows real images of a single cell deforming into the characteristic bullet-like shape in a 20 μm × 20 μm constriction within the microfluidic chip depicted in (a); scale bar: 50 μm. (c) Shows the shear and normal stresses acting on a cell flowing in the constriction channel; black arrows indicate stress directions; surface colour indicates magnitude and blue lines show the flow profile in a co-moving reference frame. (d) Shows deformation vs. projected size (in μm2) of HL-60 cells, measured at 0.32 μL s−1, including isoelasticity lines, which divide a typical deformation scatter plot from a 30 μm × 30 μm constriction channel into areas of identical stiffness for multiples of a given elastic modulus (E0). Solid black line highlights 50%-density contour. Adapted from Otto et al., Nat. Method, 2015, 12, 199–202. | ||
The current study has used RT-DC to measure the mechanical properties of a Stro-1+ human osteosarcoma cell line (MG-63) and enriched populations of primary human SSCs. Data was compared with other cell populations present in the bone marrow, including leukocytes from human blood, and a human myeloid progenitor cell line – HL-60.39 We hypothesise that deformability would provide a biophysical feature of SSCs that can be applied in a combinatorial approach, together with other discriminatory features such as size or surface marker expression, to improve the yield and specificity of sorted homogenous SSC populations.
2. Materials and methods
2.1 Real-time deformability cytometry
RT-DC measurements were performed as previously described.36,37 Briefly, a microfluidic chip was made out of PDMS using soft-lithography and sealed with a glass cover-slip after plasma surface activation. The microfluidic chip consisted of two reservoirs connected by a 300 μm long constriction channel with a 30 μm by 30 μm cross-section. A row of filter posts at the inlet prevents the channel from clogging by cell clumps and debris.For measurement, cells were harvested, mixed appropriately, centrifuged and re-suspended in a 0.5% methylcellulose solution in PBS at a concentration of 1–2 × 106 cells per mL. For measurements of SSCs, cells were first filtered through a 70 μm cell strainer. The cell suspension was drawn into 1 mL syringes and connected to the chip by polymer tubing. Cells were pumped with a syringe pump at a constant flow rate for 2 minutes before collecting data, to stabilise flow. Data was acquired in real-time with a high-speed CMOS camera (MC1362; Mikrotron, Unterschleissheim, Germany), operating at 2000 fps, and illuminated by a high-power LED (CBT-120, 462 nm; Luminus devices, Woburn, MA, USA) through a 40× objective from an inverted microscope (Axiovert 200M, Carl Zeiss, Oberkochen, Germany) at the end of the 300 μm long constriction channel where the cell shape has reached steady state (Fig. 1a and b). An image processing algorithm implemented on C/LabVIEW was used to determine the cell cross-sectional area and circularity, defined as 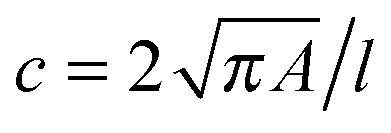 , where A is the projected area of the cells and l its perimeter. Cells deform into a characteristic bullet-like shape at the end of the constriction (Fig. 1b)36,37 and are characterised by their deformation (D = 1 − c), which defines the deviation of the cell shape from a perfect circle (c = 1). Typically, a minimum of 2000 or 5000 (for measurements with leukocytes) events was acquired at flow rates of 0.16 μL s−1 (0.04 μL s−1 sample + 0.12 μL s−1 sheath) and 0.32 μL s−1 (0.08 μL s−1 sample + 0.24 μL s−1 sheath). Measurements of all samples were repeated at least three times. As a reference, cell circularity was obtained in a section of the microfluidic chip with large cross section where deformation does not occur (ESI†).
, where A is the projected area of the cells and l its perimeter. Cells deform into a characteristic bullet-like shape at the end of the constriction (Fig. 1b)36,37 and are characterised by their deformation (D = 1 − c), which defines the deviation of the cell shape from a perfect circle (c = 1). Typically, a minimum of 2000 or 5000 (for measurements with leukocytes) events was acquired at flow rates of 0.16 μL s−1 (0.04 μL s−1 sample + 0.12 μL s−1 sheath) and 0.32 μL s−1 (0.08 μL s−1 sample + 0.24 μL s−1 sheath). Measurements of all samples were repeated at least three times. As a reference, cell circularity was obtained in a section of the microfluidic chip with large cross section where deformation does not occur (ESI†).
Both cell deformation and size are displayed using scatter plots and an example is shown in Fig. 1d for a flow rate of 0.32 μL s−1. Cell deformation is dependent on cell size for RT-DC measurements, i.e. having two cells that only differ in size, the larger will deform more as it is exposed to higher shear stresses. An analytical model was developed to disentangle both contributions by calculating the hydrodynamic flow profile around a moving cell in confinement, and coupling the resulting surface stress to a linear elastic model.40 The model predicts the resulting deformation and introduces isoelasticity lines to the scatter plots (Fig. 1d). These lines divide a typical deformation scatter plot into areas of identical stiffness for multiples of a given elastic modulus E0 (Fig. 1d).36,37,40 This enables the cell deformation to be determined independently of hydrodynamic stresses, thus permitting comparison between cells with significant size differences.36,40
2.2 Statistical analysis
A two-dimensional mixture model using a normal distribution for cell size and a lognormal distribution for deformation was applied using R and rebmix.41–43 These assumptions are justified since the lower boundary of D = 0 skews the probability density function towards larger deformation values. The algorithm allowed the unbiased assignment of measurement events into clusters that represent different cell populations. Subsequent 1-dimensional linear mixed model analysis based on the R-package lme4 (Fig. 2b) enabled a statistical comparison with respect to cell size and cell deformation.44,45 To represent the experimental situation, one fixed and one random effect were taken into account to model the difference between sub-populations of cells and to consider the variation of biological replicates, respectively. The random effect was allowed to lead to random intercepts and slopes. P-values were calculated by a likelihood ratio test comparing the full model with a model lacking the fixed effect term. The results were obtained from at least 3 independent measurements and all data analysis was treated equally.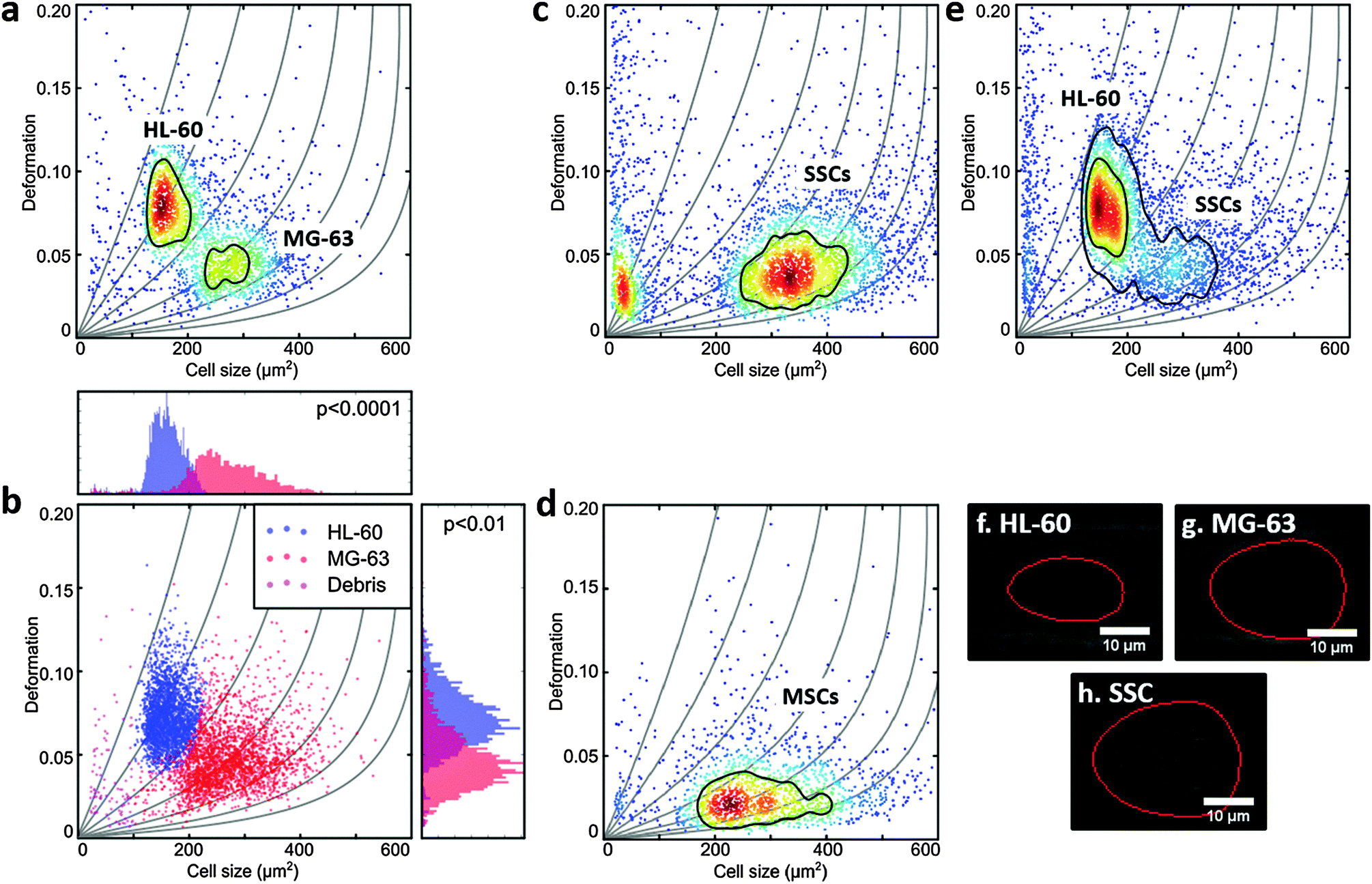 | ||
| Fig. 2 Real-time deformability cytometry of (a) MG-63 mixed 1:1 with HL-60; (b) shows deformation vs. projected size (in μm2) of MG-63 and HL-60 combining events from 3 measurements unbiasedly allocated using mixture models, with respective histograms demonstrating the cells size and deformation distributions. The p-values were calculated using linear mixed models. (c) Stro-1+-enriched skeletal stem cells; (d) human bone marrow mesenchymal stromal cells and (e) Stro-1+-enriched skeletal stem cells mixed 1:1 with HL-60; All scatter plots show deformation vs. cell size (cross-sectional area) and experiments were carried out at 0.32 μL s−1 through the 30 μm × 30 μm cross-sectional channel; colour indicates density scale and each dot is representative of a single event from a total of 2000 events. Images (f)–(h) show representative captures of the cells from (a, c and e) with the red line representing the contour determined by image analysis in real-time. | ||
2.3 Cell culture
Following extraction of cells from the bone marrow samples and extensive washes in plain α-MEM, the cell suspension was filtered through a 40 μm cell strainer and layered upon Lymphoprep™ for removal of erythrocytes and the majority of granulocytes by density centrifugation. The bone marrow mononuclear cell fraction was collected from the ‘buffy coat’ at the interphase between the Lymphoprep™ and the culture medium and incubated with a mouse hybridoma supernatant monoclonal (IgM) anti-human Stro-1 antibody produced in loco. The SSC-enriched Stro-1+ cell population was isolated by magnetic separation of cells labelled with anti-mouse IgM microbeads, as previously published.15
The enriched SSCs were cultured to confluence in monolayer cultures in α-MEM supplemented with 10% FCS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin and maintained in a humidified chamber at 37 °C and 5% CO2. For the RT-DC measurements, cells were harvested using a 0.025% (w/v) Trypsin–EDTA solution with 0.05% glucose for 5 minutes at 37 °C and pre-treated with collagenase IV (200 μg mL−1) when needed. Cells were not passaged during cell expansion.
:1 ratio. The reaction occurred under constant mixing for 6 seconds and was halted by immediate addition of 5.3 μL of an isosmotic quencher (0.6% w/v sodium carbonate, 3% sodium chloride solution in distilled water) per microliter of whole blood at the start of the procedure.46
2.4 Materials
BioWhittaker® Dulbecco's modified Eagle medium with glucose and L-glutamine (DMEM), Alpha minimum essential medium with deoxyribonucleotides, ribonucleotides and ultra-glutamine (α-MEM), Dulbecco's phosphate buffered saline (DPBS), foetal calf serum (FCS) and trypsin/EDTA were obtained from Lonza (Basel, Switzerland). RPMI-1640 culture medium was obtained from Life Technologies (Carlsbad, CA, USA). Penicillin-streptomycin 100×, AB human serum, Bovine serum albumin, collagenase IV, ethylenediamine tetra-acetic acid (EDTA) and methylcellulose were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lymphoprep™ was bought from Stem Cell Technologies (Vancouver, Canada). Anti-mouse IgM microbeads, LS MACS™ columns and the QuadroMACS™ separator were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). S-Monovette® 9NC tri-sodium citrate tubes were obtained from Sarstedt (Nümbrecht, Germany) and Sylgard 184 polydimethylsiloxane (PDMS) was purchased from VWR (Darmstadt, Germany). All reagents were used as received and according to the manufacturer's recommendations.3. Results and discussion
3.1 HL-60, MG-63, MSCs and skeletal stem cells
MG-63, MSCs and SSCs are large cells with considerable heterogeneity.47 The channel in the RT-DC device needed to be wide enough to allow the larger cells to flow through, but also not so large that smaller cells would not deform.36 HL-60 are generally measured using 20 μm × 20 μm cross-section channels, but for this work 30 μm × 30 μm channels were used to accommodate the larger MG-63 and SSCs. Consequently, the first experiments were to verify that the deformation of HL-60 cells (a model for human haematopoietic progenitor cells) could be measured in these larger channels at different flow rates (Fig. 1d and ESI,† Fig. S1).To compare the deformability of HL-60 and MG-63, both cells were mixed in approximately equal numbers and measured simultaneously. The deformation scatter plot for the two cell populations demonstrated a clear discrimination in size and deformation (Fig. 2a and ESI,† Fig. S2). Using a two-dimensional mixture model and linear mixed models, as described in the Materials and methods section of this manuscript, MG-63 cells were observed to be larger (p < 0.0001) and deform less (p < 0.01) than HL-60 cells (Fig. 2b). Analysis of captured cell images (Fig. 2f and g) confirmed that the HL-60 cells were smaller and observed to be more elongated (more deformed) than the larger MG-63. As previously described, larger cells are subject to higher shear gradient stresses as the cells flow closer to the channel walls. The fact that MG-63 are both larger and less deformed than HL-60 cells indicates that MG-63 have a higher Young's modulus.
SSC size and deformation were subsequently measured in isolation (Fig. 2c and ESI,† Fig. S3). The population of very small particles was debris from the high ECM production of confluent cells. SSCs displayed a much broader distribution reflecting the population heterogeneity. This was expected given these are primary cells from human samples enriched via Stro-1 antibody selection, but still represent a heterogeneous population. Despite the greater spread, the SSC population almost matches the MG-63 osteoblast-like cells in terms of size and deformation. This can also be seen for the representative images in Fig. 2g and h. Although it is hard to find agreement in the published values of the Young's modulus of MSCs, probably owing to their ambiguity and different tissue sources,48 studies report that MSCs are typically stiffer than their differentiation progenies or similar to osteoblasts.24,38,49,50
In fact, measurement of MSCs in isolation (Fig. 2d and ESI,† Fig. S4) shows a population that is heterogeneous in size and encompasses the SSC population shown in Fig. 2c. This supports the fact that SSCs are a sub-population found within the group of progenitors that represent the MSCs. Interestingly, Stro-1-enriched SSCs differ significantly in deformation from MSCs (p < 0.05), highlighting the potential of our method for label-free identification and sorting of skeletal stem cells.
Measurement of a mixture of SSCs and HL-60 cells together (Fig. 2d and ESI,† Fig. S5), displayed distinct populations though with a degree of overlap. SSCs were observed to be larger (p < 0.001), however, their deformation was generally lower than that of HL-60 (p < 0.01), indicating higher stiffness. In a previous study it was shown that HSCs derived from human bone marrow are softer than HL-60.36 As a consequence the current studies imply that SSCs are stiffer than human bone marrow-derived HSCs.
3.2 Mixed leukocyte and skeletal stem cell populations
For label-free isolation of SSCs from human bone marrow to reach successful clinical application, SSCs should be significantly different from leukocytes. Leukocytes arise from HSCs, which share a similar stem cell niche as SSCs in the bone marrow, and can be divided into three main populations: lymphocytes, monocytes and granulocytes, with neutrophils representing the vast majority of the latter.51–53 Leukocytes were isolated from human blood samples by selective lysis and measured by RT-DC. As shown in Fig. 3a (ESI,† Fig. S6), it was possible to discriminate between lymphocytes (Fig. 3b) and granulocytes/monocytes (Fig. 3c), with lymphocytes seen to be the smallest cells and deforming less than granulocytes and monocytes (p < 0.001).53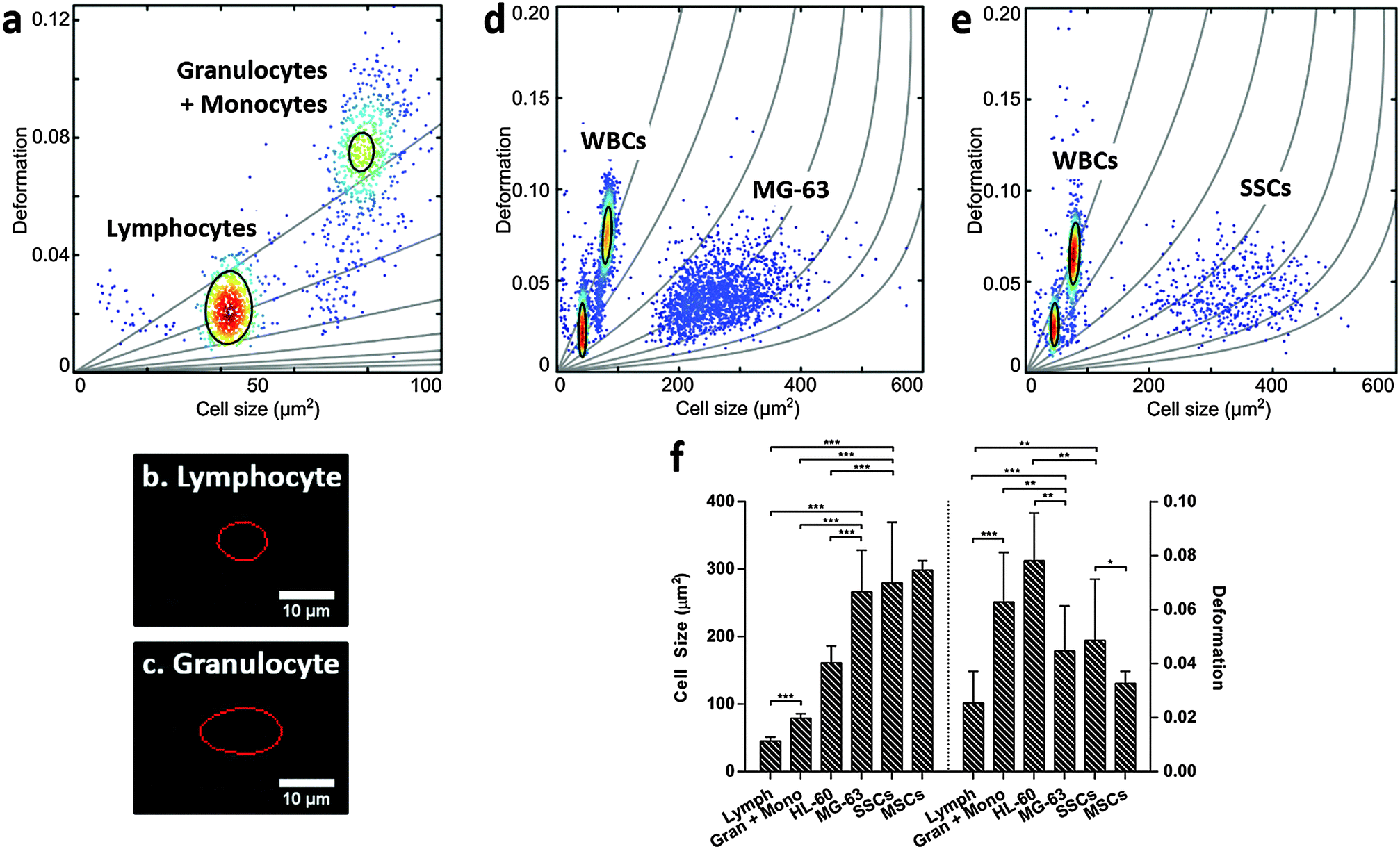 | ||
| Fig. 3 Real-time deformability cytometry of (a) leukocytes obtained from erythrocyte-lysed human whole blood; images (b) and (c) show representative captures of leukocytes when flowing at 0.32 μL s−1 through the 30 μm × 30 μm cross-sectional channel with the red line representing the contour determined by image analysis in real-time; (d) MG-63 mixed 1:2 with human leukocytes; and (e) Stro-1+-enriched skeletal stem cells mixed 1:2 with human leukocytes; showing scatter plots of deformation vs. cell size (cross-sectional area); colour indicates density scale and each dot is representative of a single event from a total of 5000 events. (f) Bar chart summarising size and deformation of leukocytes, HL-60, MG-63, Stro-1+-enriched skeletal stem cells and MSCs measured by real-time deformability cytometry. Values represent mean ± SD (N ≥ 3; *p < 0.05, **p < 0.01, ***p < 0.001). | ||
It is broadly accepted that white blood cells are highly deformable. Indeed, leukocytes' inherent ability to deform is important in their immune function and methods for determining impaired leukocyte deformability have been previously proposed for the diagnostics of certain conditions such as trauma or sepsis.25,54,55
The scatter plots in Fig. 3d (ESI,† Fig. S7) and Fig. 3e (ESI,† Fig. S8), show RT-DC measurements for human leukocytes mixed with MG-63 or SSCs, respectively. The data has been analysed using a two-dimensional mixture model and linear mixed models (Fig. 3f) and it reveals that all cell types can be discriminated based on size and deformation with statistical significance. Although the MG-63 and SSCs are the largest cells measured, they deform less (p < 0.01 for MG-63; not significant for SSCs) than granulocytes and monocytes. The two leukocyte sub-populations are difficult to resolve in Fig. 3d and e due to scaling, but comparison of these figures with Fig. 3a show that the two leukocyte sub-populations are located within isoelasticity lines (Fig. 1c) of lower Young's modulus indicating that both, MG-63 and SSCs, have a Young's modulus larger than any sub-population of leukocytes.
This provides a possibility for exploiting novel mechanical-based sorting strategies for SSCs. The fact that SSCs form a mechanically different sub-population within MSCs of higher deformability clearly points in this direction and is in agreement with current literature indicating that cells of higher potency tend to deform more.28 Taking advantage of the high-throughput of RT-DC, SSCs could be detected and sorted in real-time using an integrated cell sorting mechanism. Alternatively, passive label-free sorting techniques such as DLD or inertial microfluidics could be used as these have been shown to be sensitive to differences in both cell size and deformation.32–35 One successful example of label-free sorting of rare cells was the use of DLD to isolate circulating tumour cells from cancer patients to aid in diagnosis and guided therapies.56 The system completely depletes whole blood of red blood cells and platelets followed by negative selection of CD45+ and CD66b+ cells by magnetophoresis. It may be possible to use a similar approach to isolate SSCs out of heterogeneous samples such as the bone marrow.
4. Conclusions
The current study has demonstrated significant differences in size and deformability between skeletal stem cells, mesenchymal stromal cells and leukocytes. Critically, primary human SSCs are stiffer when compared to HL-60, a myeloid progenitor cell line, and to all main leukocyte populations (lymphocytes, monocytes and granulocytes) after RBC-lysis. SSCs were measured after expansion, following adherence on tissue culture plastic and thus it will be interesting to determine whether such large difference is also observed for freshly harvested cells. Since the bone marrow is a highly complex tissue with multiple cell types displaying overlapping features, SSC isolation will not depend solely on their separation from cells of the haematopoietic lineage but also from other adherent cell populations such as fibroblasts or endothelial cells.18,19,57 These cells may be similar in size and/or deformability.The findings reported in this paper demonstrate the potential for exploiting differences in cell stiffness within a new deformability-based strategy to improve SSC isolation with significant physiological and therapeutic implications and the potential for their clinical translation.
Acknowledgements
This work was supported by the European Commission through the Label-free particle sorting (LAPASO) ITN project from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013 under REA grant agreement no 607350, by the Alexander von Humboldt foundation (Alexander von Humboldt Professorship to JG) and the Sächsische Ministerium für Wissenschaft und Kunst (TG70 grant to OO and JG). The authors would also like to express their gratitude to the surgeons from Southampton General Hospital and the Spire Southampton Hospital for provision of patient bone marrow samples, and to Don and Ada Olis for providing the HL-60 cell line.Notes and references
- K. Christensen, G. Doblhammer, R. Rau and J. W. Vaupel, Lancet, 2009, 374, 1196–1208 CrossRef.
- A. Aarvold, J. O. Smith, E. R. Tayton, A. M. H. Jones, J. I. Dawson, S. Lanham, A. Briscoe, D. G. Dunlop and R. O. C. Oreffo, J. Tissue Eng. Regener. Med., 2014, 8, 779–786 CrossRef CAS PubMed.
- R. S. Tare, J. Kanczler, A. Aarvold, A. M. H. Jones, D. G. Dunlop and R. O. C. Oreffo, Proc. Inst. Mech. Eng., Part H, 2010, 224, 1455–1470 CrossRef CAS.
- J. I. Dawson, J. Kanczler, R. Tare, M. Kassem and R. O. C. Oreffo, Stem Cells, 2014, 32, 35–44 CrossRef CAS PubMed.
- P. Bianco, Bone, 2015, 70C, 2–9 CrossRef PubMed.
- P. Bianco, S. A. Kuznetsov, M. Riminucci and P. Gehron Robey, in Methods Enzymol., ed. I. K. and R. Lanza, Academic Press, 2006, vol. 419, pp. 117–148 Search PubMed.
- P. Bianco, P. G. Robey, I. Saggio and M. Riminucci, Hum. Gene Ther., 2010, 21, 1057–1066 CrossRef CAS PubMed.
- A. J. Friedenstein, K. V. Petrakova, A. I. Kurolesova and G. P. Frolova, Transplantation, 1968, 6, 230–247 CrossRef CAS PubMed.
- A. J. Friedenstein and M. Owen, Cell and Molecular Biology of Vertebrate Hard Tissues, CIBA Foundation Symposium, 1988, p. 321 Search PubMed.
- B. M. Abdallah and M. Kassem, J. Cell. Physiol., 2009, 218, 9–12 CrossRef CAS PubMed.
- C. Colnot, J. Bone Miner. Res., 2009, 24, 274–282 CrossRef PubMed.
- R. Tasso, F. Fais, D. Reverberi, F. Tortelli and R. Cancedda, Biomaterials, 2010, 31, 2121–2129 CrossRef CAS PubMed.
- P. G. Robey and P. Bianco, J. Am. Dent. Assoc., JADA, 2006, 137, 961–972 CrossRef CAS PubMed.
- D. Gothard, R. S. Tare, P. D. Mitchell, J. I. Dawson and R. O. C. Oreffo, Lab Chip, 2011, 11, 1206–1220 RSC.
- R. S. Tare, R. O. C. Oreffo, P. D. Mitchell and J. Kanczler, Bone Research Protocols, Springer Science & Business Media, 2012, vol. 816, pp. 83–99 Search PubMed.
- P. J. Simmons and B. Torok-Storb, Blood, 1991, 78, 55–62 CAS.
- R. S. Tare, J. C. Babister, J. Kanczler and R. O. C. Oreffo, Mol. Cell. Endocrinol., 2008, 288, 11–21 CrossRef CAS PubMed.
- S. Gronthos, A. C. W. Zannettino, S. J. Hay, S. Shi, S. E. Graves, A. Kortesidis and P. J. Simmons, J. Cell Sci., 2003, 116, 1827–1835 CrossRef CAS PubMed.
- S. Shi and S. Gronthos, J. Bone Miner. Res., 2003, 18, 696–704 CrossRef PubMed.
- D. Gothard, J. I. Dawson and R. O. C. Oreffo, Cell Tissue Res., 2013, 352, 237–247 CrossRef CAS PubMed.
- D. Gothard, J. Greenhough, E. Ralph and R. O. Oreffo, J. Tissue Eng., 2014, 5, 1–17, DOI:10.1177/2041731414551763.
- E. L. Elson, Annu. Rev. Biophys. Biophys. Chem., 1988, 17, 397–430 CrossRef CAS PubMed.
- R. D. González-Cruz, V. C. Fonseca and E. M. Darling, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1523–E1529 CrossRef PubMed.
- T. Bongiorno, J. Kazlow, R. Mezencev, S. Griffiths, R. Olivares-Navarrete, J. F. McDonald, Z. Schwartz, B. D. Boyan, T. C. McDevitt and T. Sulchek, J. Biomech., 2014, 47, 2197–2204 CrossRef PubMed.
- D. D. Carlo, J. Lab. Autom., 2012, 17, 32–42 CrossRef PubMed.
- F. Lautenschläger, S. Paschke, S. Schinkinger, A. Bruel, M. Beil and J. Guck, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15696–15701 CrossRef PubMed.
- H. T. K. Tse, D. R. Gossett, Y. S. Moon, M. Masaeli, M. Sohsman, Y. Ying, K. Mislick, R. P. Adams, J. Rao and D. D. Carlo, Sci. Transl. Med., 2013, 5, 212ra163 CrossRef PubMed.
- J. M. Maloney, D. Nikova, F. Lautenschläger, E. Clarke, R. Langer, J. Guck and K. J. Van Vliet, Biophys. J., 2010, 99, 2479–2487 CrossRef CAS PubMed.
- W. C. Lee, H. Shi, Z. Poon, L. M. Nyan, T. Kaushik, G. V. Shivashankar, J. K. Y. Chan, C. T. Lim, J. Han and K. J. V. Vliet, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4409–E4418 CrossRef CAS PubMed.
- H. Amini, W. Lee and D. D. Carlo, Lab Chip, 2014, 14, 2739–2761 RSC.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS PubMed.
- D. Holmes, G. Whyte, J. Bailey, N. Vergara-Irigaray, A. Ekpenyong, J. Guck and T. Duke, Interface Focus, 2014, 4, 20140011 CrossRef PubMed.
- J. P. Beech, S. H. Holm, K. Adolfsson and J. O. Tegenfeldt, Lab Chip, 2012, 12, 1048 RSC.
- G. Wang, W. Mao, R. Byler, K. Patel, C. Henegar, A. Alexeev and T. Sulchek, PLoS One, 2013, 8, e75901 CAS.
- S. C. Hur, N. K. Henderson-MacLennan, E. R. B. McCabe and D. D. Carlo, Lab Chip, 2011, 11, 912–920 RSC.
- O. Otto, P. Rosendahl, A. Mietke, S. Golfier, C. Herold, D. Klaue, S. Girardo, S. Pagliara, A. Ekpenyong, A. Jacobi, M. Wobus, N. Töpfner, U. F. Keyser, J. Mansfeld, E. Fischer-Friedrich and J. Guck, Nat. Methods, 2015, 12, 199–202 CrossRef CAS PubMed.
- C. J. Chan, A. E. Ekpenyong, S. Golfier, W. Li, K. J. Chalut, O. Otto, J. Elgeti, J. Guck and F. Lautenschläger, Biophys. J., 2015, 108, 1856–1869 CrossRef CAS PubMed.
- S. C. Tan, W. X. Pan, G. Ma, N. Cai, K. W. Leong and K. Liao, BMC Cell Biol., 2008, 9, 40 CrossRef PubMed.
- G. D. Birnie, Br. J. Cancer, Suppl., 1988, 9, 41–45 CAS.
- A. Mietke, O. Otto, S. Girardo, P. Rosendahl, A. Taubenberger, S. Golfier, E. Ulbricht, S. Aland, J. Guck and E. Fischer-Friedrich, Biophys. J., 2015, 109, 2023–2036, DOI:10.1016/j.bpj.2015.09.006.
- M. Nagode, J. Algorithms Optim., 2015, 3, 14–28, DOI:10.5963/JAO0302001.
- M. Nagode and M. Fajdiga, Commun. Stat. - Theory Methods, 2011, 40, 876–892 CrossRef.
- M. Nagode and M. Fajdiga, Commun. Stat. - Theory Methods, 2011, 40, 2022–2034 CrossRef.
- D. Bates, M. Mächler, B. Bolker and S. Walker, ArXiv14065823 Stat, 2014.
- D. Bates, M. Maechler, B. Bolker, S. Walker, R. H. B. Christensen, H. Singmann, B. Dai and G. Grothendieck, lme4: Linear Mixed-Effects Models using “Eigen” and S4, 2015 Search PubMed.
- D. Holmes, D. Pettigrew, C. H. Reccius, J. D. Gwyer, C. van Berkel, J. Holloway, D. E. Davies and H. Morgan, Lab Chip, 2009, 9, 2881–2889 RSC.
- A. Ismail, M. Hughes, H. Mulhall, R. Oreffo and F. Labeed, J. Tissue Eng. Regener. Med., 2015, 9, 162–168 CrossRef CAS PubMed.
- M. J. Whitfield, W. C. J. Lee and K. J. Van Vliet, Stem Cell Res., 2013, 11, 1365–1377 CrossRef CAS PubMed.
- E. M. Darling, M. Topel, S. Zauscher, T. P. Vail and F. Guilak, J. Biomech., 2008, 41, 454–464 CrossRef PubMed.
- I. Titushkin and M. Cho, Biophys. J., 2007, 93, 3693–3702 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- S. Méndez-Ferrer, T. V. Michurina, F. Ferraro, A. R. Mazloom, B. D. MacArthur, S. A. Lira, D. T. Scadden, A. Ma’ayan, G. N. Enikolopov and P. S. Frenette, Nature, 2010, 466, 829–834 CrossRef PubMed.
- S. M. Lewis, in Dacie and Lewis Practical Haematology (Tenth Edition), ed. S. M. L. J. B. Bates, Churchill Livingstone, Philadelphia, 2006, pp. 11–24 Search PubMed.
- D. B. Khismatullin, in Current Topics in Membranes, ed. K. Ley, Academic Press, 2009, vol. 64, pp. 47–111 Search PubMed.
- F. Moazzam, F. A. DeLano, B. W. Zweifach and G. W. Schmid-Schönbein, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5338–5343 CrossRef CAS.
- N. M. Karabacak, P. S. Spuhler, F. Fachin, E. J. Lim, V. Pai, E. Ozkumur, J. M. Martel, N. Kojic, K. Smith, P. Chen, J. Yang, H. Hwang, B. Morgan, J. Trautwein, T. A. Barber, S. L. Stott, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed.
- D. Fawcett and W. Bloom, A Textbook of Histology, Chapman & Hall, 12th edn, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00304k |
| This journal is © The Royal Society of Chemistry 2016 |