
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Genetically modified bacteriophages
Antonia P.
Sagona
a,
Aurelija M.
Grigonyte
ab,
Paul R.
MacDonald
ac and
Alfonso
Jaramillo
 *ad
*ad
aWarwick Integrative Synthetic Biology Centre and School of Life Sciences, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Alfonso.Jaramillo@synth-bio.org
bSynthetic Biology Centre for Doctoral Training, University of Warwick, Coventry, CV4 7AL, UK
cMOAC DTC, Senate House, University of Warwick, Coventry, CV4 7AL, UK
diSSB, Genopole, CNRS, UEVE, Université Paris-Saclay, Évry, France
First published on 15th February 2016
Abstract
Phages or bacteriophages, viruses that infect and replicate inside bacteria, are the most abundant microorganisms on earth. The realization that antibiotic resistance poses a substantial risk to the world's health and global economy is revitalizing phage therapy as a potential solution. The increasing ease by which phage genomes can be modified, owing to the influx of new technologies, has led to an expansion of their natural capabilities, and a reduced dependence on phage isolation from environmental sources. This review will discuss the way synthetic biology has accelerated the construction of genetically modified phages and will describe the wide range of their applications. It will further provide insight into the societal and economic benefits that derive from the use of recombinant phages in various sectors, from health to biodetection, biocontrol and the food industry.
 Antonia P. Sagona | Dr Antonia P. Sagona received her PhD (2012) in cancer cell biology in the laboratory of Professor Harald Stenmark, in the Centre for Cancer Biomedicine, Faculty of Medicine, University of Oslo (Norway). She then received an EMBO Postdoctoral long-term fellowship (2013), which was hosted in the Division of Biomedical Cell Biology, Warwick Medical School (UK). She is currently a Research Fellow in the School of Life Sciences, University of Warwick (UK) and her work focuses in establishing an in vitro novel system for phage therapy, consisting of recombinant phages designed to target a specific pathogen in a mammalian cell environment. |
 Aurelija M. Grigonyte | Aurelija Marija Grigonyte is a PhD candidate in synthetic biology at SynBio Centre for Doctoral Training, University of Warwick, UK. Originally trained in biochemistry she is now working on engineering synthetic bacteriophages. Her main research interests include: understanding the key variants responsible for bacteriophage to its target binding; broadening bacteriophage host range; interactions between modified bacteriophage and mammalian cells; using engineered bacteriophages for treatment against variety of pathogens. |
 Paul R. MacDonald | Paul MacDonald is a PhD candidate in MOAC (Molecular Organisation and Assembly in Cells) Doctoral Training Centre, University of Warwick, UK interested in developing proof of concepts for phage therapeutics. Trained in mathematics at the University of Birmingham, UK, his research interests include phage genome engineering, receptor binding proteins and computational modeling. |
 Alfonso Jaramillo | Professor Alfonso Jaramillo holds a PhD in Particle Physics (1999) and a Habilitation in Biology (2007). After postdoctoral appointments with Prof. Wodak (ULB, Brussels) and Prof. Karplus (ULP, France and Harvard, USA), he started his academic career in 2003 as Assistant Professor at the Ecole Polytechnique (France), becoming tenured in 2005. There, he further developed computational synthetic biology. In 2009, after moving to Genopole (France) as CNRS-senior researcher, he started his experimental synthetic biology lab, utilizing directed evolution, microscopy, microfluidics and 3D printing. In 2013, he opened a second lab at the University of Warwick (UK), where he holds the Chair of Synthetic Biology. |
Insight, innovation, integrationIn this review, we address the technological advances of synthetic biology that enable the genetic engineering of bacteriophages. Further, we discuss the wide spectrum of applications of genetically engineered bacteriophages. |
1. Introduction
1.1 Why use recombinant phages?
Current advances in synthetic biology have facilitated the rational design, modification and construction of recombinant phages – that contain genetically engineered DNA and/or have been through genetic recombination – enabling the extension of their innate phenotypes. The host specificity of phages is evolutionarily refined, with most phages targeting one species. This host recognition specificity is conferred by receptor binding domains (RBDs) that are found in either the tail-spike or tail fiber protein assemblies of the virions. Many researchers have altered the specificity of phages towards non-native hosts.1–6 In one such example, the host range of fd filamentous phage is altered by fusing a RBD from another filamentous phage (IKe), onto the infection-mediating protein of fd. Moving on to lytic phages, a genetically engineered T4 phage repository, was curated by randomizing the T4 RBD using polymerases with less fidelity to PCR amplify non-conserved regions. The repository was found to propagate in Yersinia ruckeri and Pseudomonas aeruginosa, indicating that the host range of T4 had been re-directed from its native Escherichia coli host.2 Similarly, T3 (which naturally infects E. coli) and T7 phage (which infects E. coli and some species of Yersinia) was assembled in Saccharomyces cerevisiae with exogenous phage protein domains in order to alter its host range. As a proof of concept, it was demonstrated that modified T3 and T7 phage scaffolds could target pathogenic Yersinia and Klebsiella bacteria respectively. In addition, Klebsiella phage scaffolds were retargeted against Escherichia coli by swapping their phage tail components.3Recombinant phages also function as vehicles for antimicrobials that are either incorporated into the phage7 or attached to its surface.8 For instance, they have been used to deliver light-activated antimicrobial agents (photosensitizers), which are seen as promising alternatives to antibiotics for treatment of common skin infections.9 Phages can also be engineered to suppress host SOS DNA repair system, enhancing the effectiveness of broad-spectrum antibiotics in vitro.10 Understandably, a natural concern about using engineered organisms is that the balance between natural phages in the environment may be affected. However, a recent study illustrated that phages endowed with gain of function mutations were out-competed by natural phages specific to the same host, suggesting that engineered phage genomes might not persist in the wild.11 Additionally, one strategy to commercialize phages is to re-engineer them to be non-replicative or non-lytic.12,13
1.2 Synthetic biology technologies for phage engineering
a. In vitro methods. Restriction endonuclease-based methods have been used to construct recombinant genomes in vitro. For instance, the genome of T7 was redesigned to remove overlapping genetic elements among other modifications. The new version of the genome (T7.1) is divided into 73 ‘parts’ belonging to six sections. The first two sections of T7.1 were synthesized and shown to be viable.14
An additional, in vitro method is reported for the genetic modification of lytic phages, called genome recombineering with electroporated DNA (BRED), where they used the P1vir phage as a proof of concept. BRED is based on the use of recombinases (obtained from bacterial heterologous overexpression) to assemble phage genomes from purified phage genome and given synthetic DNA fragments.15 BRED has been recently used to genetically modify a P1vir phage. Specifically, a copy of the mobile element IS1 was removed from the genome of P1vir phage, with the use of BRED.16
b. In vivo methods. In vivo methods have been used for bacteriophage engineering involving marker-based or marker-less selections of genetically modified bacteriophages. The most commonly used in vivo method is homologous recombination (HR), where the sequence to be inserted is cloned into a vector with flanking regions matching upstream and downstream of phage genome sequence. Phage that has undergone HR can then be selected from the phage population, with the methods described below.
Marker-based selection methods exploit genes promoting phage propagation. They could be genes encoded in the phage genome or host factors required for phage propagation. For the former, the gene has to be previously deleted from the genome, where the phage may require a strain expressing such gene to propagate. A first step would consist of the homologous recombination of the insert and the gene marker. Later, a marker-deficient strain could be used for selection. Alternatively, a host factor can be used. For instance, in order to identify a marker for T7 selection, E. coli BW25113 is screened for genes that promote T7 phage growth, and these can be used as selection markers when editing T7 phage genome. Two potential genes are identified: cmk and trxA. Deletion of the trxA gene appears to confer phage infection inhibition, whereas deletion of the cmk gene in the bacterial host shows a lower efficiency of plating of T7 bacteriophage in comparison to the control bacteria. The reduced efficiency of plating, but not a complete absence of plaques, indicates that despite the gene deletion, some degree of T7 infection is obtained. These false positives can be removed by serial dilutions. In this study the E. coli gene cmk, which encodes for CMP/dCMP kinase, was inserted instead of T7 gp5 by HR between wild-type T7 and a plasmid containing the cmk gene. The plated recombined phage was shown to be negative for growth on cells that were deficient in cmk, and which did not contain a plasmid expressing gp5. This expected phenotype was confirmed to be true at the genotypic level by sequencing.17
False positives may occur in the marker-less selection method, CRISPR/Cas.18 In a recent report, CRISPR/Cas system, and specifically type I-E CRISPR/Cas system, was used to select for engineered T7 bacteriophage (see Fig. 1). The T7 phage genome was edited by homologous recombination and the recombinant phages were selected by targeting wild-type phages with the CRISPR/Cas system.19
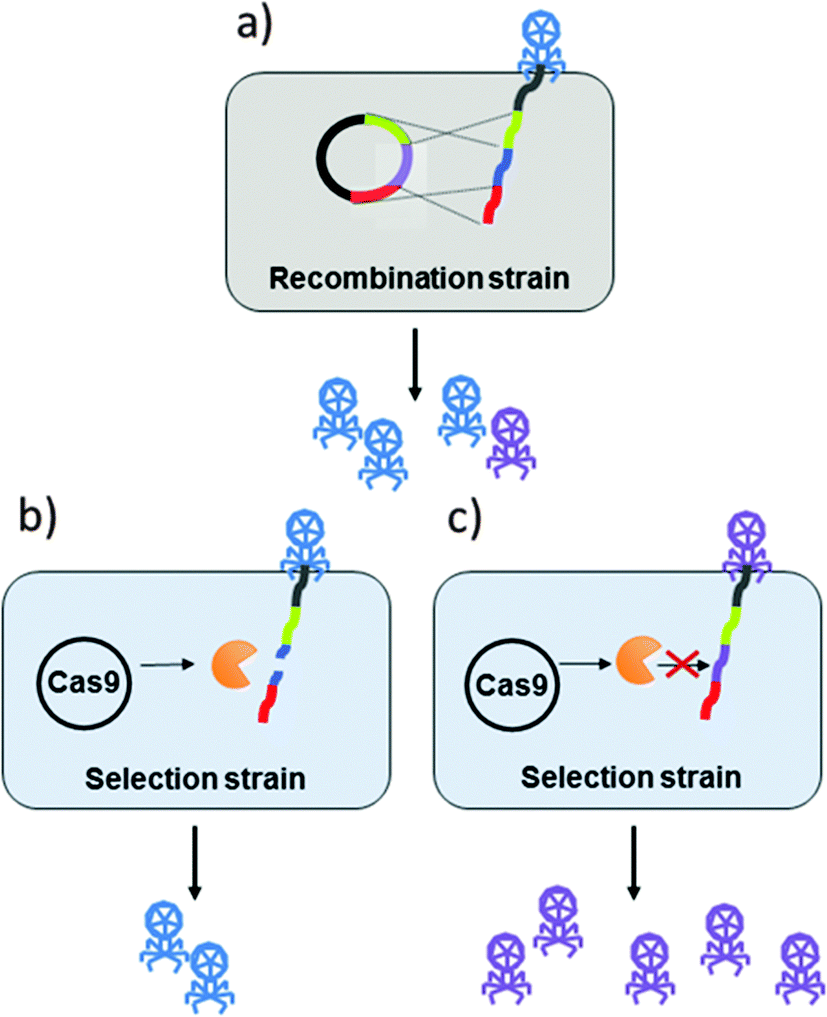 | ||
| Fig. 1 Summary of CRISPR/Cas system used for recombinant phage selection via non-edited genome targeting. (a) The DNA of wild type (WT) bacteriophage (in light blue) undergoes homologous recombination following infection of the recombinant strain. The recombination strain contains a plasmid with a sequence (in violet) with flanking homology regions that replaces the WT phage genome sequence (in blue). This infection results in mixed population of phage progeny, producing recombinant phage (in violet) in addition to the WT phage (in blue). (b) The selection strain contains a CRISPR-Cas system, which targets and cleaves the WT sequence. Phages can still propagate in this strain, either because of inefficiencies associated with the CRISPR machinery, or because they contain one or more SNPs in the region targeted by the Cas-gRNA complex (deemed escape mutants). (c) The CRISPR-Cas system does not complex with the recombinant phage, resulting in relatively more progeny. | ||
In another report, in addition to using CRISPR/Cas for the selection of engineered phage, CRISPR/Cas II-A system was used to in vivo modify phage 2972. Phage genome editing included gene exchange, point mutation and small or large deletions. For the gene exchange, orf33 in the phage 2972 was replaced with methyltransferase gene of the type II restriction/modification (R/M) system LlaDCHI from L. lactis. Since the results showed a successful gene swap and a fully functioning methyltransferase it was concluded that the CRISPR/Cas engineering system can be used for gene insertions into the phage's genome.20 CRISPR/Cas technology could be adapted to other phage genomes.
In another example, bacteriophage engineering is accomplished using an in vivo yeast platform as an alternative host for bacteriophage assembly. This platform is used to engineer phage with novel host ranges by swapping viral tail fibre scaffolds. Phage genomes are placed in Saccharomyces cerevisiae, allow gene modifications and result in generation of engineered phage. Each fragment of the phage genome is first amplified by PCR while retaining a homologous overhang. First and last fragments of the phage genome have a homology region with the yeast artificial chromosome (YAC). All amplified phage genome fragments as well as YAC are then transformed into yeast where gap repair facilitated joining of all the fragments and the YAC according to homology regions. After the purification of the vector the phage can then be initiated to form functional phages when transformed into bacteria. This yeast phage-engineering platform has a great potential because any genomic loci can be modified even by adding genes toxic to E. coli.3 One possible inconvenience is that the phage may have repeats at their ends (as it is the case for T7) and recombination may produce excision of the phage from the vector. This may be overcome by including a selective marker for yeast inside the phage genome.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold greater capacity of the phage to evade immune system and remain in the circulatory system. This was a result of a single mutation which led to amino acid substitution in the major lambda phage capsid protein E.22
000 fold greater capacity of the phage to evade immune system and remain in the circulatory system. This was a result of a single mutation which led to amino acid substitution in the major lambda phage capsid protein E.22
PACE (phage-assisted continuous evolution) combines continuous culture with increased mutagenesis to accelerate the evolution of M13 phagemids (non-replicative phages that require a strain carrying a helper system). PACE is used to evolve regulatory molecules rather than structural proteins in the virion. This is done by removing an essential gene (gpIII) from the phage and placing it, in an inactivated form, in the host strain. The gene responsible for the initiation of the evolution is introduced into the phage. The product of this gene must activate gpIII to produce infectious progeny. It is possible to use a similar methodology to evolve proteins from the phage such as those conferring host specificity or involved in replication.23 This method has been further expanded to evolve biomolecules with altered or highly specific new activities, using negative selection and modulation of selection stringency.24
2. Applications of engineered phages
Herein, we discuss how genetically modified phages (listed in Table 1) are used in different fields (presented in Fig. 2).| Engineered bacteriophage | Application |
|---|---|
| M13 | Phage display,25–33 lethal delivery agent,83 engineered protein purification,84 nanomaterials,87 vaccinology109,110 |
| M13KE | Pathogen detection100 |
| T7 | Phage display,39–43 gene therapy,69 biofilm control114 |
| Lambda (λ) | Phage display44–47 vaccinology,46,70 biocontrol116 |
| T4 | Phage display,37,38 vaccinology72,112 |
| ϕA1122 | Pathogen detection106 |
| A511 | Pathogen detection104 |
| HK620 | Pathogen detection98 |
| PBSPCA1 | Agriculture108 |
| fd | Phage display,34–36 nanodevice fabrication and bottom-up manufacturing85,86 |
 | ||
| Fig. 2 A schematic representation of the synthetic bacteriophage applications discussed in this review. | ||
2.1 Therapeutic applications
Natural phages have multiple barriers that could prevent them from being developed into viable phage therapeutic products. Issues can arise both from their entry to mammalian cells and from circumventing the immune response of the host. Modified phages can be developed to avoid inactivation by the host defense system and persist in the body, thus enhancing their therapeutic potential.In this section, examples of recombinant phages as therapeutic agents for a variety of diseases are given. This includes phage therapy in mammalian hosts as well as phages as lethal delivery vehicles for prokaryotic hosts.
The T7 bacteriophage was genetically modified for a potential therapy of hepatitis B. Globally HCC (hepatocellular carcinoma) is the fifth most common cancer in men, and the eighth most common in women, and it was estimated that during the year 2000, more than 500000 new cases arose.68 The first step of hepatitis B virus (HBV) infection is the interaction between a cell surface receptor and the HBV envelope protein (specifically the PreS1 region). T7 phage was modified to display polypeptides of varying length (up to fifty amino acids) of the PreS1 region. This was achieved by fusing the peptides of interest (PreSi region variants) to the C-terminus of the capsid protein gp10B from T7. It was suggested that this system could be enhanced by displaying amino acids of the first half of the PreS1 region, which would increase modified phage uptake and hence the efficiency of desired gene transfer into the cell. This study introduces T7 display as a model system for gene delivery into mammalian host cells and elucidates recombinant phages expand upon natural phage therapy.69
Another example of therapeutic bacteriophage applications in mammalian systems is the display of a membrane glycoprotein, of H5 influenza virus, on bacteriophage lambda major capsid protein gpD.70 Here the lambda bacteriophage is assembled without its major capsid protein first in vivo, and then a fusion of gpD and H5 influenza membrane glycoprotein is added in vitro. This approach can be used to ‘decorate’ gpD deficient phage with recombinant gpD fused proteins (which are heterologously expressed in insect cells). The recombinant phages bind to the receptors on red blood cells and create a lattice structure of interconnected virus and red blood cells. It is observed that two H5 influenza-specific monoclonal antibodies inhibit the binding of red blood cells to the recombinant phages. These modified phages mimic H5 influenza virus behavior by attaching to the red blood cells; this could potentially be exploited for the detection of influenza virus-specific antibodies in vaccine trials.71
T4 phage is another example of engineered phage use in vaccinology. The outer capsid protein of T4 phage was fused to a protective antigen from Bacillus anthracis, to develop a vaccine for anthrax. The fusion protein was expressed in E. coli, purified, and in vitro added to the assembled phage. The PA-T4 particles presented immunogenicity in mice in the absence of an adjuvant. This study provides a promising system for construction of customized vaccines against anthrax.72
Phages target bacteria more specifically than most antibiotics, and consequently have less effect on the human gut microbiome. However, lytic phages, whose concomitant cell lysis may result in the release of toxic substances (endotoxins) have encouraged the development of lysis-deficient phages. These lysis-deficient phages can be engineered by harnessing the phage machinery responsible for cell lysis. This consists of a membrane protein (normally deemed, holin) and endolysin or murein hydrolase. Holins form holes in the cell membrane, thus letting endolysin cross the membrane and degrade the peptidoglycan layer of the cell wall.73 An example of engineering a recombinant phage that is lysis-deficient involves Staphylococcus aureus phage P954 where its endolysin gene was inactivated by a loss of function insertion.74 Endolysin-deficient phages encoding lethal but non-lytic proteins are able to kill bacteria while reducing the endotoxin release.75 Engineered non-lytic phages have been shown to be efficient in treating mice infected with P. aeruginosa, E. coli or S. aureus which present higher survival rates, due to the lower levels of endotoxin release.13,74,76,77 Additionally, filamentous phages (which do not lyse the host) have been used for the specific delivery of lethal substances or genes to the site of infection.78 Among possible genes, those encoding for modified holin,79 lethal transcription regulator80 and addiction toxins (which induce programmed cell death) have been reported.81,82
Modified phage particles can also be used as lethal delivery agents for efficient pathogen killing. A recent example is the engineering of the filamentous phage M13 to carry an integrin binding peptide and a fragment of the polymorphic membrane protein D from the sexually transmitted pathogen Chlamydia trachomatis (Ct), as a possible way to eliminate Ct infection. Based on this report, the engineered phage was able to significantly reduce Ct infection in both primary endocervical and HeLa cells, addressing the current lack of treatments against Chlamydia trachomatis.83
2.2 Industrial applications
High-gradient magnetic fishing (HGMF) partially purifies target products from heterogeneous bioprocess liquors. In HGMF, the target product is captured using magnetic adsorbent particles in combination with high-gradient magnetic separation equipment. HGMF binding capacity of microbeads was increased by placing an engineered M13 bacteriophage monolayer on a superparamagnetic (SPM) core of microbeads. This was achieved by genetic and chemical modification of the M13 coat proteins, pIII (minor coat) and pVIII (major coat) respectively. pIII protein was modified at its N-terminus to enable its binding onto nitrilotriacetic acid or silica coated SPMs, respectively. pVIII protein subunits of wild-type M13 were chemically cross-linked to a carboxyl-functionalized bead permitting side-on linkage of the phage to the SPMs. The phage-SPM particles, when used to fish the desired antibodies, led to >90% purified product from high protein solutions in one purification step.84fd filamentous phage has been engineered to increase the affinity of gold (Au) to its protein coat. This was achieved by substituting five amino acids on the N-terminal region of p8, the fd phage's major coat protein.85,86 These studies show how recombinant phages can be coated with a metal of interest, demonstrating the potential of recombinant phages as self-assembling templates for applications in bottom up manufacturing.
In addition, M13 filamentous bacteriophage has been used as a scaffold for the self-assembly of cobalt manganese oxide nanowires to make LiO2 battery electrodes. Here, the phage coat protein gpVIII is modified to display peptides of negatively charged amino acids, and is able to interact with cationic metal precursors (such as cobalt and manganese) resulting in high production yield oxides. These oxides formed LiO2 battery electrodes which were more porous and had a higher specific heat capacity in comparison to carbon electrodes.87
2.3 Biodetection applications
One area in which synthetic phages are proving useful is biodetection, where one of their main advantages is that they can be quickly amplified in the targeted live bacteria, compared to PCR or antibody-wash detection systems (which subsequently incur more false positives from the detection of dead bacteria).88 Early adoptions of biodetection with synthetic phages have focused on the insertion of reporter genes into naturally isolated phages.89–93 Most of these methods comprise of either inserting luciferase genes into the phage genome, or of fusing fluorescent proteins to the phage capsid, mimicking the engineering strategy employed in phage display.94 It should be emphasized that these systems are far from just academic enquires. Sample6 recently launched its DETECT/L kit on the back of luciferase-based recombinant phage technologies.93 The kit correctly identifies 50 Listeria species, as well as correctly excluding 30 non-listeria species that were subjected to testing. However, converting luciferase-based assays to more optimal multiplex assays (i.e. assays that detect more than one bacterial species in a given sample) may not be straightforward,95 which is an issue that could be circumvented by fluorescent-based reporter systems with compatible emission and excitation wavelengths.96The precision of phage host recognition has been explored for pathogen detection from environmental samples. Water quality control is one of the major concerns for public health as well as marine environment, and rapid methods need to be developed to allow accurate pathogen identification. One such method used a phage-based fluorescent biosensor ‘phagosensor’ prototype for enteric bacteria detection.97 In this system, the synthesis of the fluorescent protein only occurs after it is delivered to E. coli TD2158 by temperate bacteriophage HK620 carrying the fluorescent gene.98 The recombinant phages were incubated with the sample for one hour, followed by flow cytometry, which allowed sensitive detection of environmental E. coli TD2158 strain in diluted samples, and in mixed co-cultures. The established template was also successfully adapted to phage P22 to detect Salmonella enterica Typhimurium.99
Colorimetry is another method for detecting pathogenic bacteria in water. In this instance, the target bacterial strain, ER2378, is trapped on a syringe filter followed by infection of a specific phage, M13KE.100 ER2378 is a lacZα-complementing strain of E. coli that expresses the ω-domain of the β-gal (ωGal) and the αGal peptide is cloned in an intergenetic region of the M13KE genome.101,102 Upon αGal peptide delivery to the bacterial strain, αGal is converted to the β-gal active form of αGal, which is detectable by colorimetric assay.100
Another example of bacteriophage-based pathogen detection is the high-intensity fluorophage that can detect Mycobacterium tuberculosis.103 Phage A511 was modified to report cells of Listeria genus upon synthesis of a bacterial luciferase gene. The modified gene was placed downstream of the major capsid protein (cps) and was expressed upon phage infection of Listeria cells.104
Yersinia pestis is the etiological agent of the plague, which has seen a breakout of cases in America recently.105 To detect Yersinia pestis in blood samples, a recombinant reporter phage containing bacterial luxAB reporter genes was inserted in an early-transcribed noncoding region of the plague-diagnostic lytic phage ϕA1122 by homologous recombination. Upon infection of Yersinia pestis with the recombinant ϕA1122, the bioluminescent phenotype was observed after 10 to 15 min.106 In a following study, the reporter phage was assessed as a diagnostic tool for Yersinia pestis in samples taken directly from the blood. Even though it displayed 100% inclusivity for Yersinia pestis, some non-pestis Yersinia strains and Enterobacteriaceae also showed signal transduction. The reporter phage demonstrated rapid detection of antimicrobial susceptibility profiling upon antibiotic incubation of the blood samples. As a consequence these results suggest that the application of lytic reporter phages to detect bacterial pathogens in blood samples could reduce the time to diagnosis for patients afflicted with.107
Phage-reporter systems were also developed for agricultural settings. Pseudomonas cannabina pv. alisalensis and Pseudomonas syringae pv. maculicola are both causative agents for diseases of Brassicaceae family. In a recent study, phage-based diagnostic was developed to identify the cause of bacterial blight. P. cannabina pv. alisalensis or PBSPCA1 phage was modified by integration of bacterial luxAB genes in the place of nonessential phoH gene using homologous recombination upon wild-type PBSPCA1 phage infection of P. cannabina pv. alisalensis BS91 containing luxAB expression cassette. A successful detection of Pseudomonas cannabina pv. alisalensis versus Pseudomonas syringae pv. maculicola resulted in more than 100-fold increase in bioluminescence within 4 hours of tissue harvesting.108
2.4 Veterinary applications
Genetically engineered phages have a wide range of applications in veterinary science and medicine. In the vast majority of cases, recombinant phages deliver antigens to be used for vaccination against animal diseases.Recombinant M13 bacteriophage has been used to vaccinate pigs against the tapeworm Taenia solum, which causes cysticercosis, a disease to which humans and pigs are susceptible. KETc1, KETc12 and GK1 peptides were fused to coat protein gpVIII and a recombinant antigen KETc7 was displayed on coat protein gpIII. The pooled phages were successful in reducing the number of cysts in the murine model (990 in mice receiving M13 vs. 338 immunized with the pool of recombinant phage). Preliminary work demonstrated sporadic effectiveness for a small sample of pigs, but further studies were needed to corroborate this evidence.109 Such a study was performed on a larger sample size, and significantly reduced the occurrence of cysticercosis in vaccinated pigs by 54.2%.110
Phages lambda and T4, as mentioned earlier in this review, have been used to vaccinate against human diseases. In this section, we briefly mention their application as possible vaccines against pathogens that target animals. Recombinant lambda phage can be used as a potential vaccine against porcine Circovirus 2,46 a virus that causes post-weaning multisystemic wasting syndrome (PMWS), and has proved costly in the swine industry.111
In another study, the T4 bacteriophage was used to develop a vaccine against infectious bursal disease virus (vvIBDV), a virus that causes infectious bursal disease (IBD) in chickens. Immunization of chickens with the recombinant T4-VP2 phage resulted in no clinical death; however, some temporary bursal damages were observed.112
2.5 Biocontrol applications
Biocontrol is the regulation of pest/pathogen levels by biological means; a strategy that is being progressively favored by industry. One of the major problems in industrial processing is biofilm formation, and this is especially apparent in food industry.113 To this end, T7 bacteriophage was modified to express dispersin B (DspB),114 an enzyme produced by Actinobacillus actinomycetemcomitans.115 DspB acts via β-1,6-N-acetyl-D-glucosamine hydrolysis which disrupts biofilm formation and integrity. DspB was placed downstream of capsid gene 10B under the control of the strong T7 10 promoter. This allowed DspB to be expressed intracellularly so that its release would occur during cell lysis. The results showed that DspB expressing phage was significantly more effective at killing E. coli in comparison to wild-type T7 and wild-type T3 bacteriophages, and that this phage reduced the amount of biofilm by a factor of 2.6 in comparison to non-engineered T7 control phage.114In another study lambda phage was engineered to deliver CRISPR/Cas system to sensitize bacteria with antibiotic resistance genes. Once delivered to a pathogen, the CRISPR/Cas system was transferable between bacterial hosts so that bacteria with antibiotic genes would be outcompeted. In addition, pathogens with acquired CRISPR/Cas were no longer susceptible to engineered lytic phage infection. This system showed effective reduction in infections of antibiotic-resistant pathogens and thus offering a potential biocontrol system for hospital surface treatment.116
3. Discussion
Natural phages may be a solution to a myriad of issues in agriculture, biocontrol, and medicine: in agriculture, they are applied against plant infections; in biocontrol, in the protection and control of crops and food products; and in medicine, a number of trials have explored their safety and efficacy.Phage therapy has reached a critical juncture in its development. Natural phages are, according to current legislature, unpatentable, as they are no longer considered novel, having first been introduced over a century ago.117 This prevents them from being commercially viable in industry, especially for big pharma corporations that have to undergo expensive clinical trials, and so is an impediment to the development of infrastructure that can deliver treatments to patients. The intellectual property of recombinant phages, on the other hand, can be secured, thus overcoming these issues. Yet recombinant phages do more than to provide opportunities for profit; they extend upon natural phages by creating additional functionalities (such as delivery of variety of cargos, e.g. depolymerase114) as well as overcoming some of their limitations.
One such limitation is that natural phages can induce a mammalian immune response upon their entry, a response that could be avoided by modifying the phage's coat protein. Nonetheless, in some instances, an overexcited immune response is desired, for example, when developing vaccines against viral particles. In the case of HBV, phage-displayed viral peptides can evoke immune response, and help identify the antibodies specific to these viral peptides.
Furthermore, by refactoring phage genomes,14 phages could have their sequences rejigged to avoid host restriction systems, by removing the palindromic sequences targeted by type II restriction enzymes, or by appending peptides that inhibit the CRISPR/Cas proteins. One can also envisage the swapping or addition of exogenous endo/exonucleases to cleave host genomes faster. Further work demonstrated that rational engineering of tail fibres can change host tropism of bacteriophages1–6 and pyocins118,119 (phage-like particles called bacteriocins, produced by some strains of Pseudomonas aeruginosa for use in intra-species warfare). Employing this in combination with directed evolution, phage cocktails could be generated to target a given pathogen through separate, or a combination of, receptors, and adapted to circumvent the host-immune system. Importantly, new techniques are constantly being developed in the fields of genome engineering and gene synthesis, decreasing also the cost of gene synthesis. It is expected that also because of that, the engineering of recombinant bacteriophages will be further facilitated and will enable the corresponding broadening of their applications.
In summary, although there are still ethical, socio-economical, and experimental issues to resolve, the groundwork of phages appears promising, and will surely come to establish itself at the forefront of personalized therapeutics and diagnostics.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work is supported by the grants FP7-ICT-FET 610730 (EVOPROG), EPSRC-BBSRC BB/M017982/1 (WISB centre) and the startup allocation grant from the School of Life Sciences to A. J. A. P. S. is funded by FP7-ICT-FET 610730 (EVOPROG). A. G. is funded by a doctoral fellowship from the EPSRC & BBSRC Centre for Doctoral Training in Synthetic Biology (grant EP/L016494/1). P. M. is funded by a doctoral fellowship from the EPSRC Doctoral Training Centre Molecular Organisation and Assembly in Cells (Grant No. EP/F500378/1). We also wish to acknowledge Matthew Tridgett for contributing to Fig. 1.References
- R. Marzari, D. Sblattero, M. Righi and A. Bradbury, Gene, 1997, 185, 27–33 CrossRef CAS PubMed.
- F. Pouillot, H. Blois and F. Iris, Biosecur Bioterror, 2010, 8, 155–169 CrossRef PubMed.
- H. Ando, S. Lemire, D. P. Pires and T. K. Lu, Cell Syst., 2015, 1, 187–196 CrossRef.
- J. R. Meyer, D. T. Dobias, J. S. Weitz, J. E. Barrick, R. T. Quick and R. E. Lenski, Science, 2012, 335, 428–432 CrossRef CAS PubMed.
- F. Mahichi, A. J. Synnott, K. Yamamichi, T. Osada and Y. Tanji, FEMS Microbiol. Lett., 2009, 295, 211–217 CrossRef CAS PubMed.
- M. Yoichi, M. Abe, K. Miyanaga, H. Unno and Y. Tanji, J. Biotechnol., 2005, 115, 101–107 CrossRef CAS PubMed.
- E. W. Kovacs, J. M. Hooker, D. W. Romanini, P. G. Holder, K. E. Berry and M. B. Francis, Bioconjugate Chem., 2007, 18, 1140–1147 CrossRef CAS PubMed.
- I. Yacoby, H. Bar and I. Benhar, Antimicrob. Agents Chemother., 2007, 51, 2156–2163 CrossRef CAS PubMed.
- M. L. Embleton, S. P. Nair, W. Heywood, D. C. Menon, B. D. Cookson and M. Wilson, Antimicrob. Agents Chemother., 2005, 49, 3690–3696 CrossRef CAS PubMed.
- T. K. Lu and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4629–4634 CrossRef CAS PubMed.
- E. G. Gladstone, I. J. Molineux and J. J. Bull, J. Biol. Eng., 2012, 6, 13 CrossRef CAS PubMed.
- S. P. Denyer, N. A. Hodges and S. P. Gorman, Hugo and Russell's pharmaceutical microbiology, John Wiley & Sons, 8th edn, 2008 Search PubMed.
- V. D. Paul, S. Sundarrajan, S. S. Rajagopalan, S. Hariharan, N. Kempashanaiah, S. Padmanabhan, B. Sriram and J. Ramachandran, BMC Microbiol., 2011, 11, 195 CrossRef CAS PubMed.
- L. Y. Chan, S. Kosuri and D. Endy, Mol. Syst. Biol., 2005, 1, 0018 CrossRef PubMed.
- L. J. Marinelli, M. Piuri, Z. Swigonova, A. Balachandran, L. M. Oldfield, J. C. van Kessel and G. F. Hatfull, PLoS One, 2008, 3, e3957 Search PubMed.
- T. Feher, I. Karcagi, F. R. Blattner and G. Posfai, J. Microb. Biotechnol., 2012, 5, 466–476 CrossRef PubMed.
- U. Qimron, B. Marintcheva, S. Tabor and C. C. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19039–19044 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- R. Kiro, D. Shitrit and U. Qimron, RNA Biol, 2014, 11, 42–44 CrossRef CAS PubMed.
- B. Martel and S. Moineau, Nucleic Acids Res., 2014, 42, 9504–9513 CrossRef PubMed.
- Y. Husimi, Adv. Biophys., 1989, 25, 1–43 CrossRef CAS PubMed.
- C. R. Merril, B. Biswas, R. Carlton, N. C. Jensen, G. J. Creed, S. Zullo and S. Adhya, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3188–3192 CrossRef CAS.
- K. M. Esvelt, J. C. Carlson and D. R. Liu, Nature, 2011, 472, 499–503 CrossRef CAS PubMed.
- J. C. Carlson, A. H. Badran, D. A. Guggiana-Nilo and D. R. Liu, Nat. Chem. Biol., 2014, 10, 216–222 CrossRef CAS PubMed.
- W. Markland, B. L. Roberts, M. J. Saxena, S. K. Guterman and R. C. Ladner, Gene, 1991, 109, 13–19 CrossRef CAS PubMed.
- D. Bhardwaj, S. S. Singh, S. Abrol and V. K. Chaudhary, J. Immunol. Methods, 1995, 179, 165–175 CrossRef CAS PubMed.
- B. L. Roberts, W. Markland and R. C. Ladner, Methods Enzymol., 1996, 267, 68–82 CAS.
- J. A. Chappel, M. He and A. S. Kang, J. Immunol. Methods, 1998, 221, 25–34 CrossRef CAS PubMed.
- T. Belien, K. Hertveldt, K. Van den Brande, J. Robben, S. Van Campenhout and G. Volckaert, J. Biotechnol., 2005, 115, 249–260 CrossRef CAS PubMed.
- S. S. Sidhu, B. K. Feld and G. A. Weiss, Methods Mol. Biol., 2007, 352, 205–219 CAS.
- K. Hertveldt, T. Belien and G. Volckaert, Methods Mol. Biol., 2009, 502, 321–339 CAS.
- D. Ghosh, Y. Lee, S. Thomas, A. G. Kohli, D. S. Yun, A. M. Belcher and K. A. Kelly, Nat. Nanotechnol., 2012, 7, 677–682 CrossRef CAS PubMed.
- Y. Ivarsson, R. Arnold, M. McLaughlin, S. Nim, R. Joshi, D. Ray, B. Liu, J. Teyra, T. Pawson, J. Moffat, S. S. Li, S. S. Sidhu and P. M. Kim, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2542–2547 CrossRef CAS PubMed.
- M. Tornetta, S. Baker, B. Whitaker, J. Lu, Q. Chen, E. Pisors, L. Shi, J. Luo, R. Sweet and P. Tsui, J. Immunol. Methods, 2010, 360, 39–46 CrossRef CAS PubMed.
- J. W. Gillespie, A. L. Gross, A. T. Puzyrev, D. Bedi and V. A. Petrenko, Front Microbiol, 2015, 6, 628 Search PubMed.
- A. Prisco and P. De Berardinis, Int. J. Mol. Sci., 2012, 13, 5179–5194 CrossRef CAS PubMed.
- V. P. Efimov, I. V. Nepluev and V. V. Mesyanzhinov, Virus Genes, 1995, 10, 173–177 CrossRef CAS PubMed.
- M. Gamkrelidze and K. Dabrowska, Arch. Microbiol., 2014, 196, 473–479 CrossRef CAS PubMed.
- H. M. Li, K. Guo, Z. Yu, R. Feng and P. Xu, Thorac. Cancer, 2015, 6, 469–474 CrossRef CAS PubMed.
- C. L. Wong, C. C. Sieo and W. S. Tan, J. Virol. Methods, 2013, 193, 611–619 CrossRef CAS PubMed.
- M. Tsuboyama and I. Maeda, J. Biosci. Bioeng., 2013, 116, 28–33 CrossRef CAS PubMed.
- S. Liu, Y. Sun, H. Chen, S. Song and Y. Xu, Appl. Biochem. Biotechnol., 2010, 162, 1206–1213 CrossRef CAS PubMed.
- M. Dai, J. Temirov, E. Pesavento, C. Kiss, N. Velappan, P. Pavlik, J. H. Werner and A. R. Bradbury, Protein Eng., Des. Sel., 2008, 21, 413–424 CrossRef CAS PubMed.
- N. Sternberg and R. H. Hoess, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1609–1613 CrossRef CAS.
- E. Pavoni, P. Vaccaro, V. D'Alessio, R. De Santis and O. Minenkova, BMC Biotechnol., 2013, 13, 79 CrossRef CAS PubMed.
- L. N. Gamage, J. Ellis and S. Hayes, Vaccine, 2009, 27, 6595–6604 CrossRef CAS PubMed.
- R. Levy, I. J. Molineux, B. L. Iverson and G. Georgiou, J. Immunol. Methods, 2007, 321, 164–173 CrossRef CAS PubMed.
- G. N. Sundell and Y. Ivarsson, BioMed Res. Int., 2014, 2014, 176172 Search PubMed.
- N. Lonberg, Curr. Opin. Immunol., 2008, 20, 450–459 CrossRef CAS PubMed.
- D. J. Christensen, E. B. Gottlin, R. E. Benson and P. T. Hamilton, Drug Discovery Today, 2001, 6, 721–727 CrossRef CAS PubMed.
- L. F. Wang and M. Yu, Curr. Drug Targets, 2004, 5, 1–15 CrossRef CAS PubMed.
- J. M. Gershoni, A. Roitburd-Berman, D. D. Siman-Tov, N. Tarnovitski Freund and Y. Weiss, BioDrugs, 2007, 21, 145–156 CrossRef CAS PubMed.
- P. Molek, B. Strukelj and T. Bratkovic, Molecules, 2011, 16, 857–887 CrossRef CAS PubMed.
- M. E. Kokoszka and B. K. Kay, Methods Mol. Biol., 2015, 1248, 173–188 Search PubMed.
- B. Addepalli, S. Rao and A. G. Hunt, Methods Mol. Biol., 2015, 1255, 147–158 Search PubMed.
- C. Blikstad and Y. Ivarsson, Cell Commun. Signaling, 2015, 13, 38 CrossRef PubMed.
- H. J. Ren, R. D. Liu, Z. Q. Wang and J. Cui, Parasitol. Res., 2013, 112, 1857–1863 CrossRef PubMed.
- E. A. Coelho, M. A. Chavez-Fumagalli, L. E. Costa, C. A. Tavares, M. Soto and L. R. Goulart, Rev. Soc. Bras. Med. Trop., 2015, 48, 370–379 CrossRef PubMed.
- K. A. Henry, M. Arbabi-Ghahroudi and J. K. Scott, Front Microbiol, 2015, 6, 755 Search PubMed.
- M. Hyvonen and P. Laakkonen, Methods Mol. Biol., 2015, 1324, 205–222 Search PubMed.
- H. Shim, BMB Rep., 2015, 48, 489–494 CrossRef CAS PubMed.
- K. Omidfar and M. Daneshpour, Expert Opin. Drug Discovery, 2015, 10, 651–669 CrossRef CAS PubMed.
- D. Sanchez-Martin, M. D. Sorensen, S. Lykkemark, L. Sanz, P. Kristensen, E. Ruoslahti and L. Alvarez-Vallina, Trends Biotechnol., 2015, 33, 292–301 CrossRef CAS PubMed.
- G. A. Loset, G. Berntzen, T. Frigstad, S. Pollmann, K. S. Gunnarsen and I. Sandlie, Front Oncol, 2014, 4, 378 Search PubMed.
- E. C. Roncolato, L. B. Campos, G. Pessenda, L. Costa e Silva, G. P. Furtado and J. E. Barbosa, Toxicon, 2015, 93, 79–84 CrossRef CAS PubMed.
- A. Zhao, M. R. Tohidkia, D. L. Siegel, G. Coukos and Y. Omidi, Crit. Rev. Biotechnol., 2015, 1–14, DOI:10.3109/07388551.2014.958978.
- C. E. Chan, A. P. Lim, P. A. MacAry and B. J. Hanson, Int. Immunol., 2014, 26, 649–657 CrossRef CAS PubMed.
- F. X. Bosch, J. Ribes, M. Diaz and R. Cleries, Gastroenterology, 2004, 127, S5–S16 CrossRef.
- K. H. Tang, K. Yusoff and W. S. Tan, J. Virol. Methods, 2009, 159, 194–199 CrossRef CAS PubMed.
- J. Mattiacio, S. Walter, M. Brewer, W. Domm, A. E. Friedman and S. Dewhurst, Vaccine, 2011, 29, 2637–2647 CrossRef CAS PubMed.
- W. Domm, M. Brewer, S. F. Baker, C. Feng, L. Martinez-Sobrido, J. Treanor and S. Dewhurst, J. Virol. Methods, 2014, 197, 47–50 CrossRef CAS PubMed.
- S. B. Shivachandra, M. Rao, L. Janosi, T. Sathaliyawala, G. R. Matyas, C. R. Alving, S. H. Leppla and V. B. Rao, Virology, 2006, 345, 190–198 CrossRef CAS PubMed.
- I. Young, I. Wang and W. D. Roof, Trends Microbiol., 2000, 8, 120–128 CrossRef CAS PubMed.
- T. Matsuda, T. A. Freeman, D. W. Hilbert, M. Duff, M. Fuortes, P. P. Stapleton and J. M. Daly, Surgery, 2005, 137, 639–646 CrossRef PubMed.
- R. Young and U. Blasi, FEMS Microbiol. Rev., 1995, 17, 191–205 CrossRef CAS PubMed.
- S. Hagens, A. Habel, U. von Ahsen, A. von Gabain and U. Blasi, Antimicrob. Agents Chemother., 2004, 48, 3817–3822 CrossRef CAS PubMed.
- M. J. Catalao, F. Gil, J. Moniz-Pereira, C. Sao-Jose and M. Pimentel, FEMS Microbiol. Rev., 2013, 37, 554–571 CrossRef CAS PubMed.
- M. Russel, N. A. Linderoth and A. Sali, Gene, 1997, 192, 23–32 CrossRef CAS PubMed.
- S. Hagens and U. Blasi, Lett. Appl. Microbiol., 2003, 37, 318–323 CrossRef CAS PubMed.
- Z. Moradpour, Z. Sepehrizadeh, F. Rahbarizadeh, A. Ghasemian, M. T. Yazdi and A. R. Shahverdi, FEMS Microbiol. Lett., 2009, 296, 67–71 CrossRef CAS PubMed.
- C. Westwater, L. M. Kasman, D. A. Schofield, P. A. Werner, J. W. Dolan, M. G. Schmidt and J. S. Norris, Antimicrob. Agents Chemother., 2003, 47, 1301–1307 CrossRef CAS PubMed.
- T. M. Viertel, K. Ritter and H. P. Horz, J. Antimicrob. Chemother., 2014, 69, 2326–2336 CrossRef CAS PubMed.
- S. R. Bhattarai, S. Y. Yoo, S. W. Lee and D. Dean, Biomaterials, 2012, 33, 5166–5174 CrossRef CAS PubMed.
- J. Muzard, M. Platt and G. U. Lee, Small, 2012, 8, 2403–2411 CrossRef CAS PubMed.
- N. Korkmaz Zirpel, T. Arslan and H. Lee, J. Colloid Interface Sci., 2015, 454, 80–88 CrossRef CAS PubMed.
- N. Korkmaz, Colloids Surf., B, 2013, 112, 219–228 CrossRef CAS PubMed.
- D. Oh, J. Qi, B. Han, G. Zhang, T. J. Carney, J. Ohmura, Y. Zhang, Y. Shao-Horn and A. M. Belcher, Nano Lett., 2014, 14, 4837–4845 CrossRef CAS PubMed.
- N. K. Petty, T. J. Evans, P. C. Fineran and G. P. Salmond, Trends Biotechnol., 2007, 25, 7–15 CrossRef CAS PubMed.
- S. Kim, M. Kim and S. Ryu, Anal. Chem., 2014, 86, 5858–5864 CrossRef CAS PubMed.
- M. Oda, M. Morita, H. Unno and Y. Tanji, Appl. Environ. Microbiol., 2004, 70, 527–534 CrossRef CAS PubMed.
- C. P. Kodikara, H. H. Crew and G. S. Stewart, FEMS Microbiol. Lett., 1991, 67, 261–265 CrossRef CAS PubMed.
- T. Funatsu, T. Taniyama, T. Tajima, H. Tadakuma and H. Namiki, Microbiol. Immunol., 2002, 46, 365–369 CrossRef CAS PubMed.
- M. Cappillino, R. P. Shivers, D. R. Brownell, B. Jacobson, J. King, P. Kocjan, M. Koeris, E. Tekeian, A. Tempesta, J. Bowers, E. Crowley, P. Bird, J. Benzinger and K. Fisher, J. AOAC Int., 2015, 98, 436–444 CrossRef CAS PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 CAS.
- S. D. Alcaine, L. Tilton, M. A. Serrano, M. Wang, R. W. Vachet and S. R. Nugen, Appl. Microbiol. Biotechnol., 2015, 99, 8177–8185 CrossRef CAS PubMed.
- A. E. Smartt, T. Xu, P. Jegier, J. J. Carswell, S. A. Blount, G. S. Sayler and S. Ripp, Anal. Bioanal. Chem., 2012, 402, 3127–3146 CrossRef CAS PubMed.
- M. Vinay, N. Franche, G. Gregori, J. R. Fantino, F. Pouillot and M. Ansaldi, PLoS One, 2015, 10, e0131466 Search PubMed.
- B. P. Cormack, R. H. Valdivia and S. Falkow, Gene, 1996, 173, 33–38 CrossRef CAS PubMed.
- K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef CAS PubMed.
- R. Derda, M. R. Lockett, S. K. Tang, R. C. Fuller, E. J. Maxwell, B. Breiten, C. A. Cuddemi, A. Ozdogan and G. M. Whitesides, Anal. Chem., 2013, 85, 7213–7220 CrossRef CAS PubMed.
- K. A. Noren and C. J. Noren, Methods, 2001, 23, 169–178 CrossRef CAS PubMed.
- J. Norrander, T. Kempe and J. Messing, Gene, 1983, 26, 101–106 CrossRef CAS PubMed.
- P. Jain, T. E. Hartman, N. Eisenberg, M. R. O'Donnell, J. Kriakov, K. Govender, M. Makume, D. S. Thaler, G. F. Hatfull, A. W. Sturm, M. H. Larsen, P. Moodley and W. R. Jacobs, Jr., J. Clin. Microbiol., 2012, 50, 1362–1369 CrossRef CAS PubMed.
- M. J. Loessner, M. Rudolf and S. Scherer, Appl. Environ. Microbiol., 1997, 63, 2961–2965 CAS.
- H. A. Hill, L. D. Elam-Evans, D. Yankey, J. A. Singleton and M. Kolasa, MMWR Morb Mortal Wkly Rep, 2015, 64, 889–896 CrossRef PubMed.
- D. A. Schofield, I. J. Molineux and C. Westwater, J. Clin. Microbiol., 2009, 47, 3887–3894 CrossRef CAS PubMed.
- J. P. Vandamm, C. Rajanna, N. J. Sharp, I. J. Molineux and D. A. Schofield, J. Clin. Microbiol., 2014, 52, 2998–3003 CrossRef CAS PubMed.
- D. A. Schofield, C. T. Bull, I. Rubio, W. P. Wechter, C. Westwater and I. J. Molineux, Appl. Environ. Microbiol., 2012, 78, 3592–3598 CrossRef CAS PubMed.
- K. Manoutcharian, A. Diaz-Orea, G. Gevorkian, G. Fragoso, G. Acero, E. Gonzalez, A. De Aluja, N. Villalobos, E. Gomez-Conde and E. Sciutto, Vet. Immunol. Immunopathol., 2004, 99, 11–24 CrossRef CAS PubMed.
- J. Morales, J. J. Martinez, K. Manoutcharian, M. Hernandez, A. Fleury, G. Gevorkian, G. Acero, A. Blancas, A. Toledo, J. Cervantes, V. Maza, F. Quet, H. Bonnabau, A. S. de Aluja, G. Fragoso, C. Larralde and E. Sciutto, Vaccine, 2008, 26, 2899–2905 CrossRef CAS PubMed.
- J. C. Harding, C. D. Baker, A. Tumber, K. A. McIntosh, S. E. Parker, D. M. Middleton, J. E. Hill, J. A. Ellis and S. Krakowka, J. Vet. Diagn. Invest., 2008, 20, 274–282 CrossRef PubMed.
- Y. C. Cao, Q. C. Shi, J. Y. Ma, Q. M. Xie and Y. Z. Bi, Acta Biochim. Biophys. Sin., 2005, 37, 657–664 CrossRef CAS PubMed.
- R. Van Houdt and C. W. Michiels, J. Appl. Microbiol., 2010, 109, 1117–1131 CrossRef CAS PubMed.
- T. K. Lu and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11197–11202 CrossRef CAS PubMed.
- Y. Itoh, X. Wang, B. J. Hinnebusch, J. F. Preston and T. Romeo, 3rd, J. Bacteriol., 2005, 187, 382–387 CrossRef CAS PubMed.
- I. Yosef, M. Manor, R. Kiro and U. Qimron, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7267–7272 CrossRef CAS PubMed.
- K. Thiel, Nat. Biotechnol., 2004, 22, 31–36 CrossRef CAS PubMed.
- S. R. Williams, D. Gebhart, D. W. Martin and D. Scholl, Appl. Environ. Microbiol., 2008, 74, 3868–3876 CrossRef CAS PubMed.
- D. Scholl, M. Cooley, S. R. Williams, D. Gebhart, D. Martin, A. Bates and R. Mandrell, Antimicrob. Agents Chemother., 2009, 53, 3074–3080 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |