
A microfluidic dual gradient generator for conducting cell-based drug combination assays†
Devrim
Kilinc
ab,
Jefrem
Schwab
a,
Stefano
Rampini
a,
Oshoke W.
Ikpekha‡
a,
Ashwin
Thampi
a,
Agata
Blasiak
a,
Peng
Li
a,
Robert
Schwamborn
c,
Walter
Kolch
bcd,
David
Matallanas
bcd and
Gil U.
Lee
*ab
aSchool of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: gil.lee@ucd.ie; Fax: +353-1-716-1178; Tel: +353-1-716-2399
bUCD Conway Institute, Belfield, Dublin 4, Ireland
cSystems Biology Ireland, UCD, Belfield, Dublin 4, Ireland
dSchool of Medicine and Medical Science, UCD, Belfield, Dublin 4, Ireland
First published on 10th November 2015
Abstract
We present a microfluidic chip that generates linear concentration gradients of multiple solutes that are orthogonally-aligned to each other. The kinetics of gradient formation was characterized using a fluorescent tracer matching the molecular weight of small inhibitory drugs. Live-cell signalling and motility experiments were conducted to demonstrate the potential uses and advantages of the device. A431 epidermoid carcinoma cells, where EGF induces apoptosis in a concentration-dependent manner, were simultaneously exposed to gradients of MEK inhibitor and EGF receptor (EGFR) inhibitor. By monitoring live caspase activation in the entire chip, we were able to quickly assess the combinatorial interaction between MEK and EGFR pathways, which otherwise would require costly and time consuming titration experiments. We also characterized the motility and morphology of MDA-MB-231 breast cancer cells exposed to orthogonal gradients of EGF and EGFR inhibitor. The microfluidic chip not only permitted the quantitative analysis of a population of cells exposed to drug combinations, but also enabled the morphological characterization of individual cells. In summary, our microfluidic device, capable of establishing concentration gradients of multiple compounds over a group of cells, facilitates and accelerates in vitro cell biology experiments, such as those required for cell-based drug combination assays.
Insight, innovation, integrationExposing A431 cells, in which EGF induces apoptosis, to orthogonal gradients of MEK inhibitor and EGF receptor inhibitor revealed the combinatorial interaction between MEK and EGF pathways in apoptotic signalling. Our novel microfluidic design enables adherent cells to be simultaneously exposed to linear gradients of multiple molecules that are formed in different directions. Thus, the combinatorial concentration response of a cell line to specific inhibitors of two distinct intracellular signalling pathways can be measured within the timeframe of a single experiment. Through integrating microfluidic technology with quantitative live-cell imaging, our device provides a novel in vitro platform for systems biology, where the systematic acquisition of quantitative cell signalling data is paramount. |
Introduction
Concentration gradients are common in cell physiology and developmental biology.1 Extracellular gradients, for example, are necessary in directing cell migration2 or guiding axonal growth cones.3 Intracellular gradients are necessary in regulating a range of physiological activities from measuring cell length prior to mitosis4 to maintaining the blood brain barrier.5 A number of methods have been developed for establishing concentration gradients in vitro, to mimic the physiological gradients experienced by cultured cells.6 These include micropipette discharge method,7 and Boyden,8 Zigmond,9 and Dunn10 chambers. However, these classical methods do not provide gradient stability or dynamics which may be required for biological experiments. Microfluidic cell culture devices claim to solve the problems associated with these devices by providing precise geometric constraints to form and maintain the concentration gradients.Flow-based microfluidic gradient generators rely on the diffusion of solutes through the boundaries of laminar flow streams that run parallel to each other. When a cascade of flow streams differing in starting concentrations are induced over a cell culture area, a concentration gradient forms orthogonal to the flow direction.11 By changing the flow rates of the inlet streams, one can manipulate the gradient steepness and the cell culture area that is exposed to gradients.12 The major disadvantage of flow-based gradient generators is that the cells are exposed to fluid shear stress, which may affect their physiology and behaviour.
Diffusion-based microfluidic gradient generators, on the other hand, rely on the passive diffusion of solutes from a source flow channel to a sink flow channel.13 Fick's law states that a linear concentration gradient forms between a source and a sink, provided that their respective concentrations remain constant. This principle has been applied to a variety of design configurations to suit experimental needs. These include point source devices,14,15 parallel compartments connected via microchannels13 or separated by hydrogel filling,16 a single compartment supplied by buried source channels,17 and devices designed for three-dimensional cell culture.18 Diffusion-based gradient devices do not expose cells to fluid shear stresses and hence provide a cell-benign system,19 suitable for cell biology research. Compared to flow-based gradient generators, diffusion-based gradient generators are much slower to form the gradients.
There is a need in biology for methods that monitor cell fate regulation through different concentrations of the same external stimulus, as well as, through combinations of multiple stimuli. Performing these types of studies using classical molecular biology methods involves titration experiments which are expensive and time consuming. Alternative methods include 96- and 384-well plate checkerboard technique combined with a spectrophotometer or a calorimeter, potentially employing digital dispensers to directly dispense drug combinations. However, while providing a wide dynamic range of concentrations (typically 3–4 logs), these methods require either high numbers of cells, large amounts of reagents, and many individual measurements, or specialized equipment. This issue is especially important for testing drug combinations in hard-to-obtain cells and for the new field of systems biology where there is a clear need of obtaining quantitative measurements in order to fit mathematical models and experimentally validate model predictions.20–24 With this motivation, we developed a diffusion-based gradient generator that forms multiple linear concentration gradients over a group of adherent cells. This device is suitable for quantitative live-cell microscopy and classical immunocytochemistry. We provide proof of concept experiments that assess apoptotic signalling and cell motility in cancer cell lines.
Experimental
Microfluidic device design and fabrication
The microfluidic circuit (Fig. 1A) consists of an extended square-shaped cell chamber (6 mm × 6 mm; 100 μm high) and four flow channels (200 μm wide; 100 μm high) running parallel to the edges of the square. Flow channels are connected to the cell chamber via parallel microchannels (200 μm long; 10 μm wide; 3 μm high; 40 μm spacing). The microfluidic master was fabricated through a two-step photolithography process on a 4 inch silicon wafer (University Wafer, Boston, MA): first, a 3 μm layer of SU-8 2002 (MicroChem, Newton, MA) was deposited using a spin-coater (Laurell Technologies, North Wales, PA) and exposed to UV light (365 nm; 14.8 mW) for 30 s through a photo-mask containing the microchannel geometries (Micro Lithography Services, Essex, UK). Following the development of the pattern in EC developer (Chestech, Bristol, UK), a 100 μm layer of SU-8 2010 (MicroChem) was deposited. Using a mask aligner (Suss MicroTec, Garching, Germany) the second layer of photoresist was exposed to UV light for 60 s through a second photo-mask (Micro Lithography) containing the rest of the geometry. Following development of the second layer in EC developer, the microfluidic master was treated with chlorotrimethylsilane (Sigma Aldrich, St. Louis, MO). The first generation of polydimethysiloxane (PDMS, Sylgard 184; Dow Corning, Midland, MI) pads were then fabricated via replica moulding from the master. Polyglass (Artificina, Puteaux, France) replicates were fabricated using the first generation pads. Polyglass replicates were routinely used to fabricate subsequent generations of PDMS pads.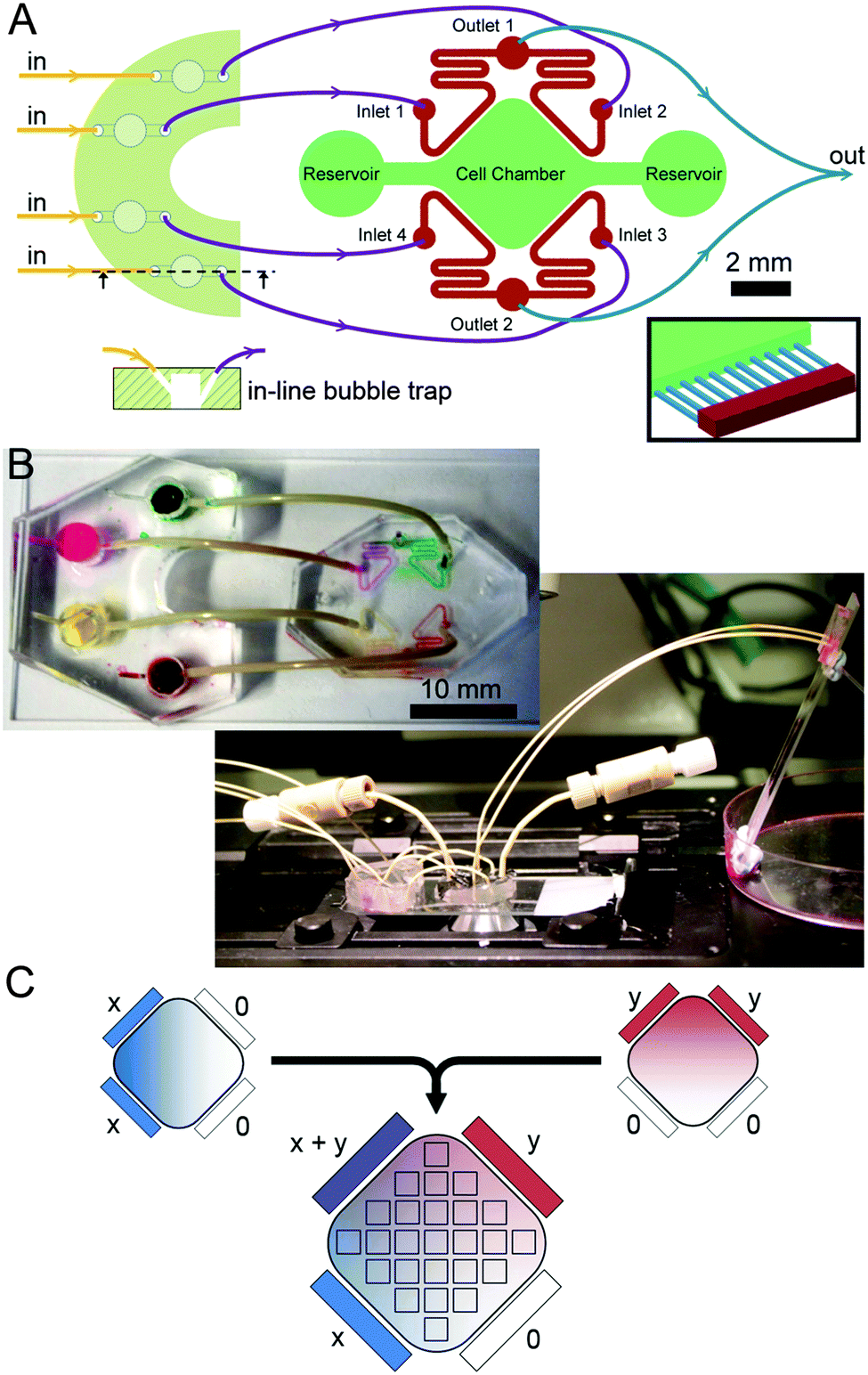 | ||
| Fig. 1 Design and operation principle of the microfluidic device. (A) Schematics of the microfluidic device and the bubble trap. The microfluidic device consists of a cell chamber and parallel channels interconnected via microchannels (inset). Bubble trap is not drawn to scale. (B) Photographs showing the assembled device during an experiment and the inlet tubing with coloured dyes. (C) The principle of generating orthogonal gradients. Linear gradients of x and y molecules are generated by flowing concentrated solutions of these molecules through two adjacent flow channels. By flowing concentrated solutions of x, y, and x + y in the given configuration leads to orthogonally-aligned gradients of two molecules. The array of squares indicates the 25 regions that are used to image the cell chamber. | ||
1.5 mm access holes were punched to the opposite ends of the cell chamber using biopsy punches (Ted Pella, Redding, CA). Likewise, 0.5 mm access holes were punched to the inlet and outlets of flow channels. The pad was then permanently bonded to a 50 × 24 mm glass coverslip via oxygen plasma (Plasma Etch, Carson City, NV). The cell chamber and the flow channels were wetted and the device was sterilized by exposing it to UV light for 20 min.
Bubble traps and fluidic assembly
Bubble trap devices, each containing four reservoirs, were placed in the inlet flow line to eliminate bubbles entering the microfluidic channels during experiments.25 Bubble traps were fabricated by first preparing rectangular PDMS pads of 4 mm thickness and punching out four through holes with a 4 mm diameter biopsy punch. Next, the pads were placed in contact with a semi-cured (15 min at 70 °C) PDMS layer of 1 mm thickness and then the composite was fully cured (30 min at 70 °C). Inlet and outlet holes were punched using 0.5 mm diameter biopsy punches with an angle of ca. 45°, where the outlet hole joined the reservoir at a lower position than the inlet hole (Fig. 1A). Bubble traps were bonded to the same glass coverslips next to the microfluidic devices (Fig. 1B).Inlet flows were induced by a syringe pump (KR Analytical, Cheshire, UK) with four identical syringe units. Syringes (Hamilton, Bonaduz, Switzerland) were connected to the inlet holes of the bubble traps with Luer connectors and PEEK tubing (Upchurch Scientific, Oak Harbor, WA) whose outer diameter is slightly larger than the inlet diameter of the hole. Outlets of the bubble traps were connected to the inlets of the microfluidic device. Outlet tubing from the microfluidic device were diverted to a third PDMS pad where the two flows merged, such that the outlet pressures were equal, and ceased at a waste collector. This third pad was bonded on a microscope slide coated with Pluronic F-127 (Sigma) to avoid droplet formation.
Medium reservoirs at either end of the cell chamber were blocked during experiments by directly inserting 1/16′′ O.D. tubing to the reservoir and plugging the other hand with a screw plug (Upchurch). A linear concentration gradient of a molecular species across the cell chamber was generated by delivering a bulk concentration of this species to two adjacent flow channels and not to the remaining flow channels. In order to induce orthogonal gradients of two different species, the same arrangement is juxtaposed with 90° angle (Fig. 1C).
Finite element simulations
Molecular diffusion in the microfluidic device was simulated using Comsol engineering platform. A three-dimensional finite-elements model was created that includes the cell culture chamber, diffusion barriers, and flow channels (Fig. S1, ESI†). To simplify the model, only those portions of the flow channels that are parallel to the cell chamber were included. Flow of solution carrying a diffusing species was simulated by calculating the average flow velocity (1 mm s−1) at the channel inlet for a given volumetric flow rate (1 μl min−1). The initial value of the concentration of this species was set to 1 only in the two inlet flow solutions that contained this species. A range of diffusion coefficients were tested to simulate the diffusion of Rhodamine, based on the diffusion coefficient reported for similarly-sized FITC in PBS (2.6 × 10−10 m2 s−1).26Characterization of concentration distribution
The distribution of the concentration within the microfluidic device was measured as a function of time using a fluorescent tracer with a molecular weight (MWs) matching small inhibitory drugs: Rhodamine-B (Rhod; Sigma). The microfluidic device was placed on the stage of an inverted microscope (Axioscope Z1; Zeiss, Cambridge, UK) equipped with an EMCCD camera (Hamamatsu, Herts, UK). The microscope was controlled by AxioVision imaging software (Zeiss) and maintained in the housing of a temperature controller (Life Imaging Services, Basel, Switzerland). First, the relationship between the concentration and signal intensity was established by filling the cell chamber with constant concentrations of tracers. The top surface of the microfluidic device was covered with light-absorbent vinyl film (VWR, Dublin, Ireland) to eliminate light reflecting from this surface. To induce gradients, two adjacent inlets were supplied with 125 μM Rhod in PBS, and the remaining two with PBS alone. 25 regions mapping the cell chamber were registered in the imaging software for automated acquisition (Fig. 1C). Images were acquired every 4 min for the first 4 h, every 6 min for the next 4 h, and every 10 min for the next 4 h.Induction of apoptosis in A431 cells
A431 epidermoid carcinoma cells were cultured in growth medium: DMEM containing 3.97 mM L-glutamine and 25 mM glucose (Gibco, Carlsbad, CA), 10% foetal bovine serum (FBS, Gibco), and an antibiotic mixture of 100 U per ml penicillin and 100 μg per ml streptomycin (Pen/Strep). Cells were passaged every 3–4 days. A set of exploratory experiments were conducted to assess combinatory interactions between signalling pathways using flow cytometry. A431 cells were cultured in multiwell plates, treated with 0.1 or 10 nM human epidermal growth factor (EGF; Roche, Clarecastle, Ireland) and co-treated with 10 μM of MEK inhibitor U0126 (Promega, Madison, WI), 5 μM EGF receptor inhibitor BIBX 1382 dihydrochloride (BIBX; Tocris Bioscience, Bristol, UK), 5 μM PI3K inhibitor LY294002 (Calbiochem, Hertfordshire, UK), 5 μM Src inhibitor PP2 (Sigma), 10 μM p38 inhibitor SB203580 (Cell Signaling Technology, Danvers, MA), or 50 μM JNK inhibitor SP600125 (Sigma). Apoptosis levels were measured as described previously.27 Briefly, the media were collected; the cells were trypsinised, pulled with the media and collected by centrifugation. The cell pellet was resuspended in PBS, divided in two and either (i) DNA fragmentation was measured by staining the cells with propidium iodide and the SubG1 fraction was determined via FACS analysis; or (ii) Caspase 3/7 activation was determined using zVAD-fmk-FITC (Promega) following the manufacturer's instructions. Experiments were repeated 3 times.Detection of live caspase activation
Caspase activation was monitored in live cells via the CellEvent caspase-3/7 green detection reagent (Invitrogen). This dye is a fluoreogenic substrate for activated caspase 3/7, whose activation is considered a crucial event in the induction of apoptosis. Off-chip titration experiments were conducted by culturing cells in glass-bottom multiwell plates and subjecting them to combinations of EGF (0.1 and 10 nM), BIBX (1 and 5 μM), and U0126 (2 and 10 μM). Cells were imaged every hour for 6 h using brightfield and green fluorescence. The percentage of caspase positive cells, as identified by the bright green fluorescence in their nuclei, was used to compare experimental groups.Live cell apoptotic induction in the microfluidic device
Two days before the experiment, A431 cells were harvested by trypsinisation and suspended in growth medium at a density of 7.5 × 105 cells per ml. 16 μl of cell suspension was then introduced into the cell chamber via one reservoir and sucked from the other side, such that the chamber was filled uniformly. For efficient cell attachment, the devices were incubated for 30 min before the reservoirs were topped with medium. Cells were incubated at 37 °C by placing the microfluidic device in a plastic Petri dish containing 1 ml 0.1% ethylendiaminetetraacetic acid (EDTA) in dH2O to minimize evaporation. One day after seeding, cells were serum-starved overnight by replacing the medium with DMEM containing Pen/Strep only. 2–6 h prior to the experiment, the medium was replaced again with Leibovitz's L15 medium (Gibco) containing Pen/Strep to compensate for the lack of CO2 during microscopy. 30 min prior to the experiment, A431 cells were labelled with CellEvent reagent by replacing the medium in the microfluidic device with 10 μM of dye solution in L15 medium. The cell chamber was imaged at 25 positions every 20 min for 12 h, using brightfield and green epifluorescence for caspase detection. Caspase-positive cells were identified by the sudden increase in their fluorescence intensity between two consecutive time points. Percentages of caspase-positive cells were calculated for all time points at all positions. The average of at least three independent experiments was reported for each condition. In combination experiments, A431 cells were treated with uniform concentrations of EGF which was added to the cell chamber, as well as included in all four flow media. Cells were subjected to orthogonal concentration gradients of BIBX and U0126. Bulk concentrations of U0126, BIBX, and EGF were 50 μM, 25 μM, and 0.1–10 nM, respectively.Characterization of cell motility in the microfluidic device
Random cell migration assays were performed on MDA-MB-231 breast carcinoma cell line stably transduced with a Lenti-virus containing H2B-eGFP (Addgene 21210).28 MDA-MB-231 cells were cultured in growth medium. Two days before the experiment, MDA-MB-231 cells were harvested by trypsinisation and suspended in growth medium at a density of 5 × 105 cells per ml. Cells were seeded in the microfluidic devices as described for A431 cells. Cells cultured in the microfluidic chips did not exhibit any morphological abnormalities (Fig. S2, ESI†). The cell chamber was imaged at 25 positions every 20 min for 16 h (12 h in control experiments), using brightfield and green epifluorescence. Movements of individual cells were analyzed by using the MTrackJ plugin29 of the ImageJ software (NIH, Bethesda, MD) by tracking their nuclei using a filter set suitable for GFP. Cell motility was expressed in terms of speed, velocity vector, and directionality ratio (speed divided by the magnitude of the velocity vector). Average of three independent experiments was reported. For EGF stimulation/inhibition experiments, MDA-MB-231 cells were subjected to orthogonal concentration gradients of EGF and BIBX. Bulk concentrations of EGF and BIBX in these experiments were 50 nM and 25 μM, respectively.Results and discussion
Linear concentration gradients form in the cell chamber
Fig. 2A shows the concentration distribution of Rhod at selected time points, demonstrating that a concentration gradient develops across the chamber in the x-direction. The experimentally-determined tracer concentration, C, within the cell chamber is expressed in terms of percentage of the bulk concentration supplied to the flow channels. For columns B, C, D, E, and F, one-way analysis of variance indicates that the differences between adjacent columns were statistically significant except between columns B and C at 30 min and 1 h time points (Table S1, ESI†). Analysis of the variation in concentration in the regions that make up each column confirmed that there was no statistically significant difference within columns for all time points analysed (Table S2, ESI†). These findings permitted the subsequent biological experiments to be analysed in terms of column (and row) averages.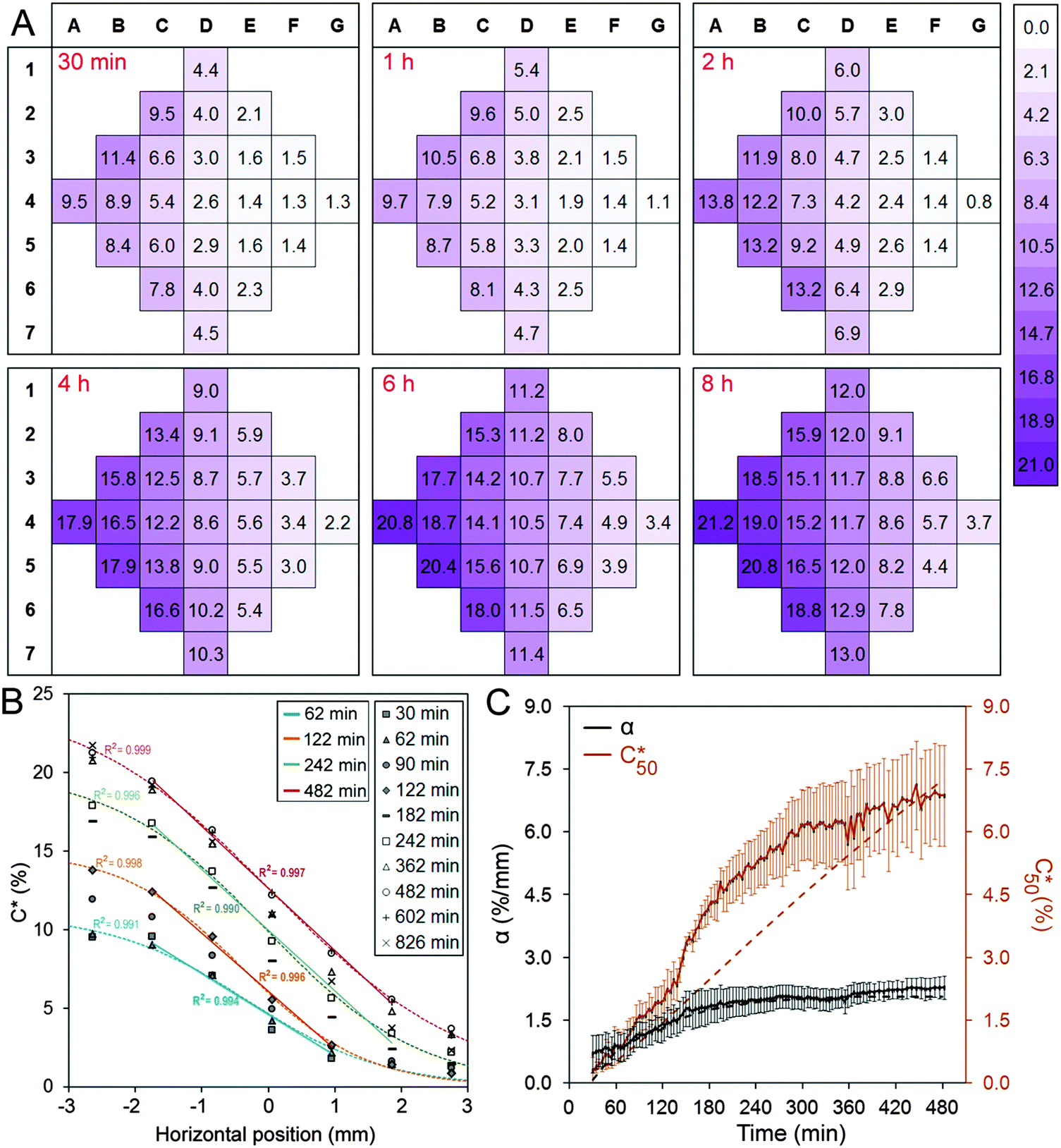 | ||
| Fig. 2 Establishment of the linear concentration gradient in the cell chamber of the microfluidic device. (A) 25-region maps of measured Rhod concentration (C*, normalized with bulk flow concentration) and finite-element simulation at different time points. (B) Column averages were plotted against x-distance from the centre of the cell chamber (column D) for indicated time points. For 1 h, 2 h, 4 h and 8 h time points, logistic curves (dashed) were fit to the concentration profiles and straight lines were fit to their central parts (straight lines). R2 values on the left hand side refer to the logistic curves. (C) Concentration profiles (columns 1–7 vs. x) were fit with straight lines using simple linear regression (C* = α·(x − L/2) + C50*). R2 > 0.95 for all time points after 1 h time point. Time profiles of α and C50* (C* value at x = L/2) are shown after background correction. Dashed lines show the time profiles of α and C50* obtained from finite-element simulation conducted for a molecule with a similar diffusion coefficient. Due to high measurement error in the fluorescent tracer data, time profiles of α and C50 are shown after background correction. Error bars = SEM. N = 4 independent experiments. | ||
Fig. 2B presents the concentration profile in the cell chamber as a function of time. A linear concentration gradient is established in the centre of the cell chamber in the early hours after the induction of the flow and remained throughout the duration of the experiment. The concentration profiles were fit with logistic curves, which are defined by the following sigmoidal function: 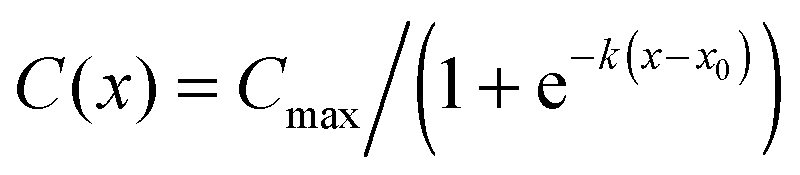 , where Cmax is the maximum concentration, k is the steepness, and x0 is the x-value of the midpoint. The media reservoirs at the two ends of the cell chamber act as sinks, which causes the tampering of the concentration profile thus resulting in the logistic curves. The central portions of the concentration profiles were also fit with straight lines using simple linear regression, which represents the data in terms of two variables: the slope α, expressed in % mm−1, and the mid-chamber concentration C50, expressed in %. To obtain the time profiles of α and C50, the concentration profile at each time point was fit with a straight line, which naturally resulted in a worse fit than the logistic curve; yet, R2 for these straight lines were greater than 0.90 between 30 min and 1 h time points and greater than 0.95 beyond 1 h.
, where Cmax is the maximum concentration, k is the steepness, and x0 is the x-value of the midpoint. The media reservoirs at the two ends of the cell chamber act as sinks, which causes the tampering of the concentration profile thus resulting in the logistic curves. The central portions of the concentration profiles were also fit with straight lines using simple linear regression, which represents the data in terms of two variables: the slope α, expressed in % mm−1, and the mid-chamber concentration C50, expressed in %. To obtain the time profiles of α and C50, the concentration profile at each time point was fit with a straight line, which naturally resulted in a worse fit than the logistic curve; yet, R2 for these straight lines were greater than 0.90 between 30 min and 1 h time points and greater than 0.95 beyond 1 h.
The concentration in the chamber along the x-coordinate was carefully measured during post-processing at different time points to analyze the dynamics of gradient formation (Fig. 2C). A volumetric flow rate of 1 μl min−1 was used to maintain a stable flow rate that is produced by the syringe pump, corresponding to an average flow velocity of 1 mm s−1 in the side channels. This resulted in a low Reynolds number (Re = 0.08) high Péclet number (Pe = 330) flow, suggesting that the flow rate is high enough to guarantee that the tracer is not diluted in the side channels. Accordingly, the concentration profile was similar for flow rates in the 0.1–10 μl min−1 range. The concentration distribution within the cell chamber under these conditions was simulated as a function of time using finite element analysis (Fig. S1, ESI†). The calculated concentration gradients and concentrations at the midpoint of the cell chamber are presented as dashed lines in Fig. 2C. It was found that a full simulation was required to capture the influence of the complicated geometry of the channels and the convective flow of Rhod at the inlets. The measured and simulated concentration gradients were found to be in reasonable agreement. However, the measured concentration appears to increase more rapidly than the simulated concentration. We attribute this discrepancy to the limited accuracy of the fluorescence measurements to determine the absolute concentration.
A number of insights result from the analysis of the simulations. First, the simulations confirm that a linear concentration gradient should form quickly in the cell chamber but it takes much longer to stabilize the mid-point concentration. The form of the concentration profiles are strongly influenced by the dimensions of the cell chamber and microchannels. Second, the time constants for α and C50 depended highly on the diffusion coefficient D of the tracer molecule used in the simulation (Fig. S1, ESI†). Importantly, the time profiles obtained by simulating the diffusion of a molecule with D = 2.5 × 10−10 m2 s−1 closely matched the experimentally-determined time profiles of Rhod, which is assumed to have a D that is similar to FITC (2.6 × 10−10 m2 s−1).26 This suggests that for a given device design, the concentration profiles and time constants for the molecules that are intended to be used can be estimated. Accordingly, the device design may be optimized to achieve the desired concentration profile and the time scale for the biological experiment at hand. For example, the dimensions of the cell chamber will determine the magnitude of α, and the relative sizes of the microchannels and the sizes and locations of side reservoirs will affect C50 and its temporal progression.
The dimensions of the cell chamber and microchannels in the prototype presented here are suitable for studying combinations of small molecules. Considering that most drugs used in the clinic and many known growth factors and cell stimulants are small molecules, gradient formation kinetics was measured for a small molecule tracer only. Gradients of larger molecules, such as the neuronal guidance molecules Slit-2 (100 kDa) and Netrin-1 (70 kDa), will take longer to establish in the presented device. To study the combinatory effect of such large molecules the dimensions of the device geometry is required to be optimized. The time required to form gradients of high MW molecules may be reduced by decreasing the length and the spacing of microchannels, and by decreasing the overall dimensions of the cell chamber.
The microfluidic device can be used to determine differential effects of drug combinations
Once we had observed the establishment of gradients in the cell chamber we tested if the microfluidic device could be used to conduct biological experiments using different cell line models. Our vision is that this device can facilitate and accelerate time consuming and expensive in vitro cell biology experiments, such as those required for drug combination studies. Compared to conventional multiwell plates, the microfludic device requires smaller amounts of compounds and a much smaller number of cells for conducting combination experiments. This is particularly important if the research involves hard-to-obtain cells such as stem cells or tumour-derived cells.30 In addition, the microfludic device allows performing studies at the single cell level which can be of great use in the field of systems biology. To demonstrate the advantages of the microfluidic device on both fronts, we decided to perform two different studies. For the first study, we used A431 cells, an epidermoid carcinoma cell line that over-expresses EGFR. EGF is a potent mitogen which induces cell proliferation, survival and motility upon binding and activating EGFR.31 It is well established that A431 cells respond differentially to different concentrations of EGF: low concentrations (1 nM) induce proliferation, whereas higher concentrations (10 nM) results in the activation of apoptosis.32 Thus, this is an ideal cell line for studying different cell fates caused by different concentrations of the same molecule. A431 cells are also highly relevant for the pathophysiology of certain malignant cancers that over-express EGFR.33 In fact, several EGFR inhibitors are currently used in the clinic as cancer therapeutics. Moreover, EGF dose has been shown to have drastic effects on the behaviour of cell signalling networks.34 We first reproduced the effect of EGF concentration in our cell line. Through flow cytometry, we observed that 16 hours of 10 nM EGF treatment resulted in cell death (Fig. S3, ESI†), which was confirmed to be through apoptosis, as indicated by caspase activation (Fig. S4, ESI†).The EGFR activates a complex signalling network that includes some of the best known signalling pathways involved in proliferation, cell survival, and apoptosis such as the Ras/MAPK, AKT, Src, and p38MAPK pathways. To understand the contribution of these different pathways to opposite cell responses caused by different EGF concentrations, we used specific inhibitors that target these pathways (Fig. S4, ESI†). Inhibition of EGFR by BIBX prevented the activation of apoptosis triggered by the high concentration of EGF. Importantly, this experiment showed that the Src inhibitor PP2 has the same effect as EGFR inhibitor BIBX suggesting that the signal mediated by EGF requires the activation of Src kinase. This experiment also indicated that the AKT and P38MAPK signalling pathways are not required for the proliferative signal triggered by low concentrations of EGF or for the pro-apoptotic signal caused by high concentrations of EGF. However, when we inhibited the MAPK pathway using anti-MEK inhibitors we saw an increase of apoptosis in non-stimulated cells and in cell stimulated with a low concentration of EGF, indicating that this pathway is mediating the pro-survival signal induced by the low concentration EGF in A431 cells. Moreover, MEK inhibition and high concentrations of EGF stimulation increased apoptosis in A431 cells confirming that the MAPK pathway exerts an anti-apoptotic effect in these cells. This experiment was conducted using entire populations of cells, a factor that prevents the observation of the effects of the treatments at the single cell level. Furthermore, this experiment assessed the effect of EGF at a single time point. Combining different inhibitory co-treatments and performing time course and titration experiments would require a very complex and time consuming experimental design.22 To exemplify this, we conducted a crude titration experiment for 0.1 nM and 10 nM EGF using two distinct concentrations of EGFR inhibitor BIBX and two distinct concentrations of MEK inhibitor U0126. With proper controls, this experiment comprises of 27 experimental groups, which deemed to be very costly in terms of cells and reagents. This crude multi-factorial checkerboard drug combination experiment confirmed the combinatory effect between EGF stimulation and MEK inhibition (Fig. S5, ESI†). MEK inhibition alone and EGF stimulation alone increased the rate of apoptotic induction. When EGF stimulation and MEK inhibition were combined, a further (but not significantly different) increase was observed. We propose that cell-based assays at such level of complexity can be greatly accelerated by the use of the microfluidic device described here. The first caveat to this proposition is that the microfluidic device produces scarce data (typically tens of cells) for a given concentration range and therefore multiple technical replicates are required. A second caveat is that our microfluidic device can provide linear gradients that span approximately one order of magnitude and therefore multiple experiments are required to obtain a logarithmic concentration range. As a demonstration of the proposed idea, we performed two sets of experiments in microfluidic devices by treating A431 cells with different concentrations of EGF and simultaneously subjecting them to combined EGFR inhibitor and MEK inhibitors. The concentration distribution in the cell chamber showed that the high end of the linear gradient reaches ca. 20% of the bulk (flow) concentration (Fig. 2B). The bulk concentration was therefore set as 5× the target concentration.
MEK inhibition-triggered apoptosis in A431 cells is enhanced by the activation of EGF receptor
A431 cells were exposed to uniform concentrations of EGF (0.1 nM or 10 nM) by adding EGF in the cell chamber as well as providing it through the four flow channels. As on-chip controls, we first exposed cells to 0.1 nM EGF without adding BIBX or U0126 to the side flows (Fig. S6, ESI†). The level of variance in this control dataset was not statistically significant (except for column 6 at 2 h time point; see Table S3, ESI†), confirming that the microfluidic device was suitable for quantifying apoptotic induction in A431 cells. Simultaneously, orthogonal linear gradients of BIBX and U0126, specific inhibitors of EGFR and MEK, respectively, were formed by supplying the flow channels with these molecules (Fig. 3A and 4A). Caspase 3/7 activation, an early marker of apoptotic induction, was monitored for 8 h at different locations in the cell chamber (Movie S1, ESI†). Apoptosis rate was expressed in terms of percentage of cells with activated caspase 3/7. In agreement with the flow cytometry data, inhibition of EGFR did not trigger apoptosis in cells treated with low concentrations of EGF. However, as shown in caspase activation heat maps for selected time points (Fig. 3B), the cells appear to undergo apoptosis in the left-hand side of the device, i.e., where the U0126 concentration is high. When the data is analysed in terms of column averages and row averages, a negative gradient in the x-direction can be seen (Fig. 3C). Apoptosis rate gradient in the x-direction, expressed as the increase in the percentage of apoptotic cells per mm, was constant during the course of experiment, suggesting the successful formation of the U0126 gradient. In the y-direction where the BIBX gradient was formed, no gradient was detected. This experiment shows that at low EGF concentrations, the EGFR activates the MAPK pathway which mediates a pro-survival signal necessary to prevent cell death. As expected, the rate of apoptosis increased considerably when A431 cells were exposed to EGF at high concentrations (Fig. 4; Movie S2, ESI†). Caspase activation heat maps (Fig. 4B) show a clear delay in apoptosis in cells receiving high concentrations of BIBX (top of the chamber), confirming our observations from off-chip experiments (Fig. S3–S5, ESI†). Accordingly, the y-gradient of the apoptosis rate has a negative value throughout the course of the experiment (Fig. 4C). This shows that EGF-induced apoptosis can be blocked by inhibiting EGF receptors in a dose-dependent manner. In the x-direction, no obvious difference can be detected between the cells receiving high U0126 and low U0126. However, when the analysis is constrained to the bottom half of the cell chamber (indicated with brown dashed line in Fig. 4A), a slight x-gradient becomes evident at later time points (apoptosis rate was 10% higher in cells receiving highest vs. lowest concentration of U0126 at 8 h time point), suggesting a small additive interaction between EGF stimulation and MEK inhibition. The microfluidic device allows the detection of such fine detail in the behaviour of different cell sub-populations, i.e., cells exposed to different combinations of inhibitor concentrations, which would not be detected using traditional techniques analysing entire populations.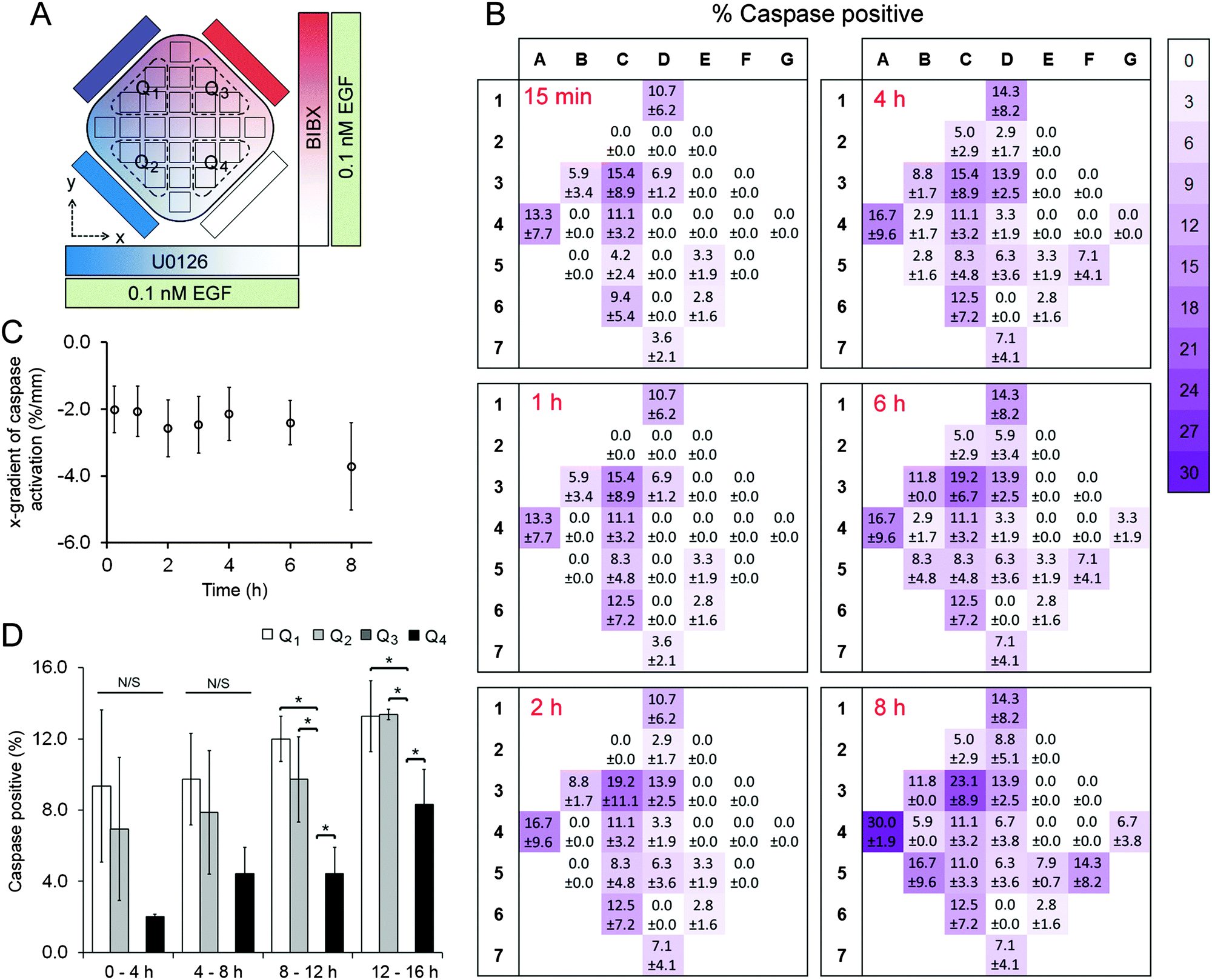 | ||
| Fig. 3 Live cell caspase activation experiments in A431 cells treated with 0.1 nM EGF. (A) The experimental paradigm showing orthogonal gradients of U0126 and BIBX in the x- and y-directions, respectively. Cells were treated with uniform concentrations of EGF. Dashed lines mark the four quadrants used in comparison. (B) 25-region maps showing the percentage of cells with activated caspase 3/7 at indicated time points during simultaneous U0126 (left-to-right, decreasing) and BIBX (top-to-bottom, decreasing) treatment. (C) Column averages of the apoptosis rate (percentage of cells with activated caspase) were fit with straight lines (R2 ranging from 0.59 to 0.72) to calculate the x-gradient. (D) Comparison of the apoptosis rate in four quadrants of the cell chamber. One-way ANOVA followed by t-test. *p < 0.05, N/S: not significant. Error bars = SEM. | ||
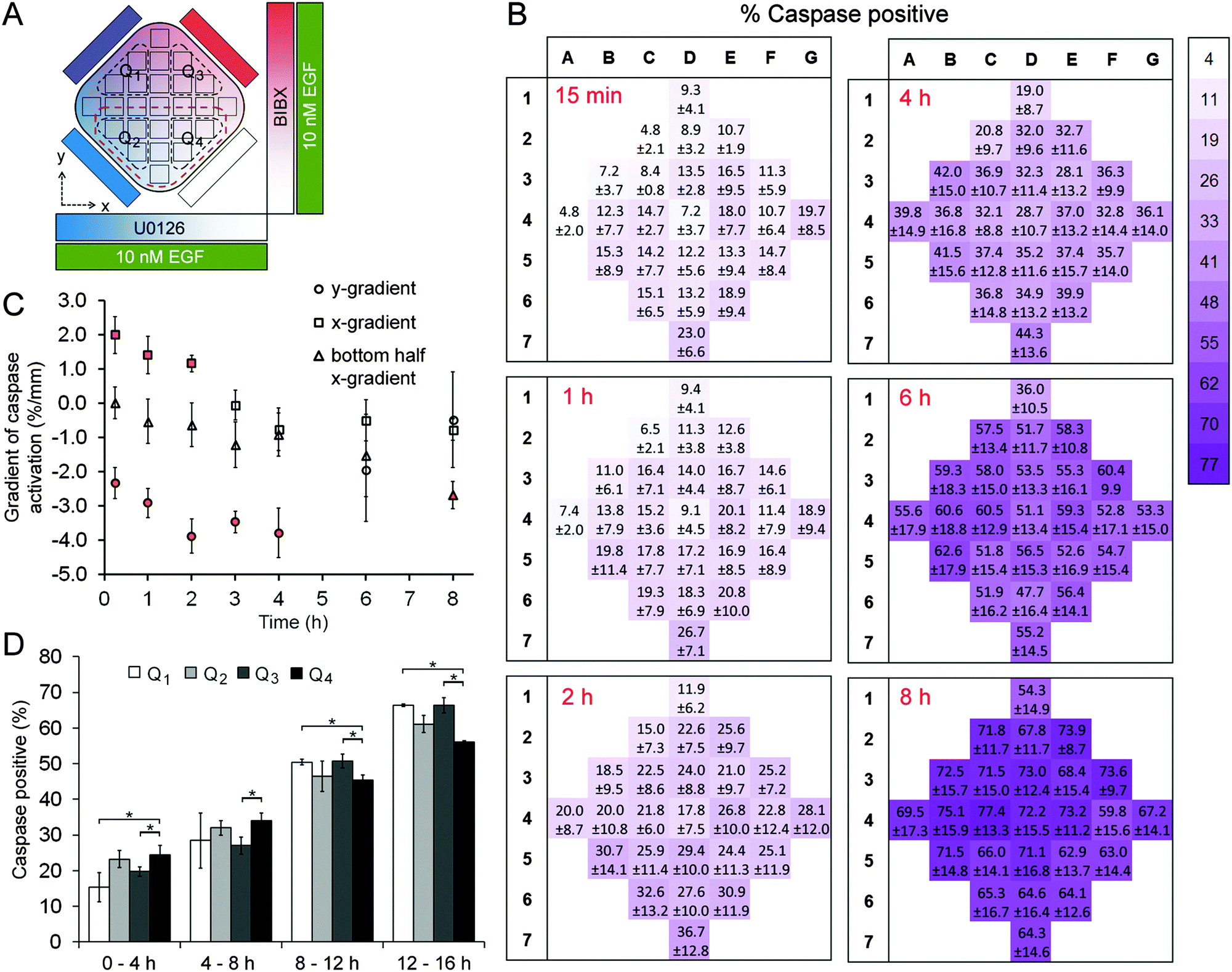 | ||
| Fig. 4 Live cell caspase activation experiments in A431 cells treated with 10 nM EGF. (A) The experimental paradigm showing orthogonal gradients of U0126 and BIBX in the x- and y-directions, respectively. Cells were treated with uniform concentrations of EGF. Black dashed lines mark the four quadrants used in comparison. Brown dashed line indicates the regions considered as the bottom half of the chamber. (B) 25-region maps showing the percentage of cells with activated caspase 3/7 at indicated time points during simultaneous U0126 (left-to-right, decreasing) and BIBX (top-to-bottom, decreasing) treatment. (C) Column and row averages of the apoptosis rate (percentage of cells with activated caspase) were fit with straight lines to calculate the x- and y-gradients, respectively. For the x-gradient R2 range was 0.59–0.82 in the first 2 h; for the y-gradient R2 range was 0.84–0.92 in the first 4 h (indicated by orange markers). (D) Comparison of the apoptosis rate in four quadrants of the cell chamber. One-way ANOVA followed by t-test. *p < 0.05, N/S: not significant. Error bars = SEM. | ||
Interestingly, a concentration dependent effect between EGFR inhibition and MEK inhibition was observed in early time points, as evidenced by the low rates of caspase activation in the top left quadrant, extending into the centre of the cell chamber (Fig. 4B). This demonstrates that at high EGF concentrations, the activation of the EGFR receptor signal does not only mediate an activation of the pro-survival MAPK pathway but also mediates the activation of a pro-apoptotic signal which is ultimately responsible for the apoptosis induced by high EGF concentrations. The activation of apoptosis in A431 exposed to high EGF concentrations is determined by the balance between this non-characterised pathway and the MAPK pathway. Altogether, the results described in this section indicate that in A431 cells exposed to low EGF concentrations, EGFR only activates the MAPK pathway; while in A431 cells exposed to high EGF concentrations, EGFR exhibits a stronger signal which triggers the activation of other pathways in the EGFR network. Among the pathways activated by high EGF concentrations there must be at least one pro-apoptotic pathway that is stronger than the pro-survival signal mediated by MAPK, and leads to the well-known increase in apoptosis shown in this cell line at high levels of EGF. In summary, using the microfluidic device we were able to determine the concentration and combination response of A431 cells to specific pathway inhibitors, within the timeframe of a single experiment, confirming the proposed advantages of the device for drug combination studies.
MDA-MB-231 cells respond to EGF stimulation and EGF receptor inhibition in dose-dependent fashion
The second set of experiments tested the idoneity of the microfluidic device to perform complex biological experiments using the MDA-MB-231 breast carcinoma cell line. Using these cells we aimed to demonstrate the capacity of the orthogonal gradient device for monitoring cell motility over extended periods. MDA-MB-231 cells with fluorescently-tagged nuclei were used as they can be easily tracked. Cell motility was expressed in terms of three parameters: cell speed (S), the average distance covered by the cell in each 20 min interval; velocity vector (V), the net distance that the cell moved during a period of time divided by that period of time, and directionality ratio (|V|/S). A set of 12 h-long control experiments were conducted that mimicked the orthogonal gradient experiments except the flow solutions did not have any stimulants or drugs (Fig. S7, ESI†). The average speed of the cells were 18.6 μm h−1, which means that it would take close to 40 h for a cell to traverse the 720 μm-wide region of interest even if it were to take a straight path. This finding justifies the analysis of cell motility in distinct regions of interest. Analysis of variance in the control data suggested that the variance in speed and in velocity were not statistically significant within the chip, within individual rows and columns, and among different rows and columns (Table S4, ESI†). In addition, these metrics were not different between the central and the peripheral regions of the cell chamber (t-test, p > 0.20 for speed, p > 0.18 for velocity). These findings suggested that the microfluidic device did not impose any regional variation to cell motility due to differences in their access to fresh medium or oxygen.In separate experiments, MDA-MB-231 cells were exposed to orthogonal gradients of EGF and BIBX (Fig. 5; Movie S3, ESI†). Heat maps of the cell motility parameters, as well as the magnitude of the velocity vector (|V|) in the central column and in the central row are shown for four consecutive 4 h-long intervals (Fig. S8, ESI†). The change in |V| with increasing y-coordinate suggested that cells receiving high BIBX exhibited reduced motility at later time points. In addition, using simple linear regression, straight lines were fit to column and row averages to obtain x and y gradients of speed and velocity magnitude, respectively (Fig. S9, ESI†). Due to inherent variations in the motility of non-synchronized cells, the data was also analyzed by dividing the cell chamber into four quadrants, indicated by broken lines in Fig. 5A. Cells in quadrant 1 (Q1) were exposed to high levels of EGF and high levels of BIBX, cells in quadrant 2 (Q2) were exposed to high levels of EGF and low levels of BIBX, cells in quadrant 3 (Q3) were exposed to low levels of EGF and high levels of BIBX, and cells in quadrant 4 (Q4) were exposed to low levels of EGF and low levels of BIBX. The motility parameters S and |V| were averaged within these quarters for each time interval (Fig. 5D). The relatively low motility in Q1 is evident within the first 4 h time period. The motility in Q1 further decreases between later periods and becomes statistically significant from 8 h on. In contrast, cells in Q3 exhibit a delayed decrease in their motility. Cell motility in Q3 was similar to Q4 levels during the first 12 h, but was similar to Q1 levels at the end of the 16 h experiment. This suggests that the inhibition of EGF receptor by BIBX blocks the motility of MDA-MB-231 cells. The dynamics of this process depends on the availability of EGF to the cells, where cells exposed to high EGF slowdown faster; however, eventually, cells receiving high and low EGF exhibit similar motilities. The effect of BIBX was confirmed by the positive |V| gradient in the y-direction, i.e., the direction of the BIBX gradient, at late time periods (Fig. S9, ESI†). In contrast, d|V|/dx was close to zero.
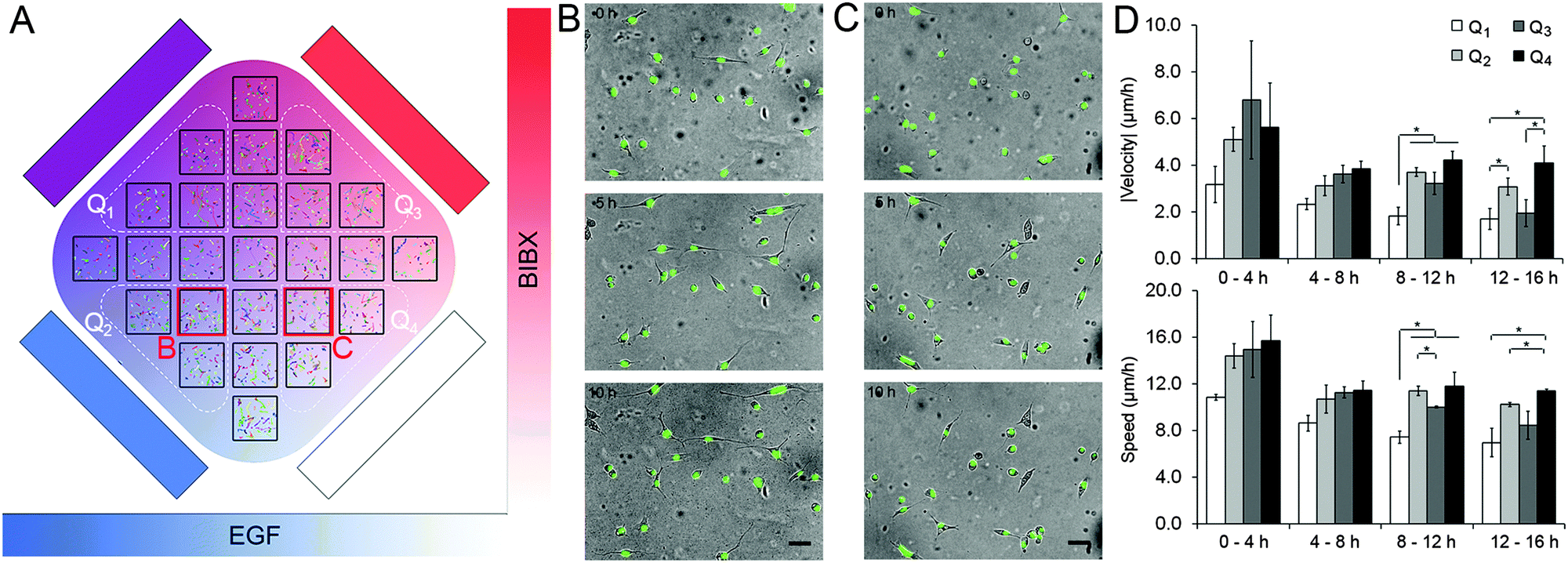 | ||
| Fig. 5 MDA-MB-231 live cell motility experiments. (A) The experimental paradigm showing orthogonal gradients of EGF and BIBX in the x- and y-directions, respectively. Colour overlays of 16-hour cell nuclei tracks displayed in 25 regions of the cell chamber. Regions that make up the four quadrants are marked with broken lines. (B and C) Exemplary cell images taken with 5 h intervals from regions C5 (B) and E5 (C). Phase contrast overlay with green fluorescence. Scale bars = 100 μm. (D) Average velocity magnitude and cell speed in each quadrant. Data presented is the average of three independent experiments. Error bars = SEM. | ||
The effect of EGF alone can be seen when Q2 and Q4 are compared since these quadrants receive low levels of BIBX but varying levels of EGF. Cell motility parameters were comparable in these quadrants suggesting that was independent of EGF. The lack of cell chemotaxis towards high EGF regions may be explained in the light of detailed studies showing that the EGF gradient detection in MDA-MB-231 cells require non-linear gradient profiles.35 This observation confirms that a linear EGF gradient has established in the cell chamber, thus not minimizing the cells' exposure to non-linear gradients. Nevertheless, these results suggest that the orthogonal gradient device provides a suitable platform for conducting experiments based on cell morphology and motility data. Importantly, careful observation of the cell morphology during the experiments allowed us to make an unanticipated observation which can also be related to the differences in the motility behaviour. As shown in Fig. 5B and C, respectively, cells in Q2 and Q4 exhibited striking morphological differences at late time points. Cells exposed to high EGF exhibited more “mesenchymal” morphologies, whereas cells exposed to low EGF were rounder. This finding is consistent with a recent study showing that EGF induces morphological changes associated with epithelial-to-mesenchymal transition in MDA-MB-231 cells.36 This effect would have not been easily observed using the most common cell migration assays currently used by cell biologist, such as Matrigel plugs, where entire cell populations are analysed. This result further highlights the advantage and the great potential of the microfluidic device in performing highly informative experiments.
Conclusions
We describe a microfluidic device that exposes adherent cells to orthogonally-aligned concentration gradients of multiple molecules. Through relevant examples from cancer biology, we demonstrate the implementation of the device for live-cell microscopy and provide proof-of-concept experiments where cell signalling, motility, and morphology was analysed. The device enabled us to determine, within the timeframe of a single experiment, the combinatorial concentration response of a cell line to specific inhibitors of two distinct yet related intracellular signalling pathways. Importantly, the device allowed for cell-based assays at population and at single-cell levels, and thereby provided a time- and cost-effective means to study drug combinations. We therefore argue that this microfluidic device is an excellent tool for systems biology where the systematic acquisition of quantitative cell signalling data is paramount.20–24Authors contributions
D. K., O. W. I., A. B. and P. L. designed, fabricated, and implemented the microfluidic system; D. K., J. S., S. R. and A. T. characterized the gradient formation; D. K., J. S. and R. S. conducted live cell assays; D. K. and D. M. designed the experiments and wrote the paper; W. K. and G. U. L. supervised the research.Acknowledgements
The authors thank Alex von Kriegsheim for MDA-MB-231 cells. PGK-H2BeGFP was a gift from Mark Mercola (Addgene 21210). This study was supported by the Science Foundation Ireland (08/RP1/B1376 and 08/IN1/B2072, G. U. L.; 06/CE/B1129, W. K. and D. M.), by the Irish Higher Education Authority (PRTLI Nanoremedies; G. U. L.), by a Marie Curie Intra-European Fellowship (D. K.), by an AXA Research Fund Doctoral Fellowship (A. B.), and by an Erasmus scholarship (J. S.).Notes and references
- A. Kicheva, T. Bollenbach and O. Wartlick, et al. , Curr. Opin. Genet. Dev., 2012, 22, 527–532 CrossRef CAS PubMed.
- J. Halkias, H. J. Melichar and K. T. Taylor, et al. , J. Clin. Invest., 2013, 123, 2131–2142 CAS.
- G. Gallo and P. C. Letourneau, J. Neurobiol., 2004, 58, 92–102 CrossRef CAS PubMed.
- S. G. Martin and M. Berthelot-Grosjean, Nature, 2009, 459, 852–856 CrossRef CAS PubMed.
- B. V. Zlokovic, Neuron, 2008, 57, 178–201 CrossRef CAS PubMed.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
- C. B. Guan, H. T. Xu and M. Jin, et al. , Cell, 2007, 129, 385–395 CrossRef CAS PubMed.
- S. Boyden, J. Exp. Med., 1962, 115, 453–466 CrossRef CAS PubMed.
- S. H. Zigmond, J. Cell Biol., 1977, 75, 606–616 CrossRef CAS PubMed.
- D. Zicha, G. Dunn and G. Jones, Methods Mol. Biol., 1997, 75, 449–457 CAS.
- D. L. Englert, M. D. Manson and A. Jayaraman, Nat. Protoc., 2010, 5, 864–872 CrossRef CAS PubMed.
- N. L. Jeon, H. Baskaran and S. K. Dertinger, et al. , Nat. Biotechnol., 2002, 20, 826–830 CrossRef CAS PubMed.
- A. Shamloo, N. Ma and M. M. Poo, et al. , Lab Chip, 2008, 8, 1292–1299 RSC.
- D. Kim, M. A. Lokuta and A. Huttenlocher, et al. , Lab Chip, 2009, 9, 1797–1800 RSC.
- W. K. Raja, B. Gligorijevic and J. Wyckoff, et al. , Integr. Biol., 2010, 2, 696–706 RSC.
- S. Y. Cheng, S. Heilman and M. Wasserman, et al. , Lab Chip, 2007, 7, 763–769 RSC.
- J. Atencia, G. A. Cooksey and L. E. Locascio, Lab Chip, 2012, 12, 309–316 RSC.
- R. L. Smith, C. J. Demers and S. D. Collins, Microfluid. Nanofluid., 2010, 9, 613–622 CrossRef.
- N. Bhattacharjee, N. Li and T. M. Keenan, et al. , Integr. Biol., 2010, 2, 669–679 RSC.
- D. K. Arrell and A. Terzic, Clin. Pharmacol. Ther., 2010, 88, 120–125 CrossRef CAS PubMed.
- X. Feng, W. Du and Q. Luo, et al. , Anal. Chim. Acta, 2009, 650, 83–97 CrossRef CAS PubMed.
- H. P. Fischer, Biotechnol. Annu. Rev., 2005, 11, 1–68 CAS.
- D. Wlodkowic and J. M. Cooper, Curr. Opin. Chem. Biol., 2010, 14, 556–567 CrossRef CAS PubMed.
- J. Zou, M. W. Zheng and G. Li, et al. , BioMed Res. Int., 2013, 2013, 742835 Search PubMed.
- D. T. Eddington, In-line microfluidic bubble trap, Lab Chip - Chips & Tips, http://blogs.rsc.org/chipsandtips/2006/11/22/in-line-microfluidic-bubble-trap/, Accessed Nov-22, 2006 Search PubMed.
- N. Periasamy and A. S. Verkman, Biophys. J., 1998, 75, 557–567 CrossRef CAS PubMed.
- D. Matallanas, D. Romano and K. Yee, et al. , Mol. Cell, 2007, 27, 962–975 CrossRef CAS PubMed.
- H. Kita-Matsuo, M. Barcova and N. Prigozhina, et al. , PLoS One, 2009, 4, e5046 Search PubMed.
- I. F. Sbalzarini and P. Koumoutsakos, J. Struct. Biol., 2005, 151, 182–195 CrossRef CAS PubMed.
- Y. Welte, C. Davies and R. Schafer, et al. , J. Visualized Exp., 2013, e50200 Search PubMed.
- X. Zhang, J. Meng and Z.-Y. Wang, PLoS One, 2012, 7, e41613 CAS.
- G. N. Gill and C. S. Lazar, Nature, 1981, 293, 305–307 CrossRef CAS PubMed.
- L. F. Gulli, K. C. Palmer and Y. Q. Chen, et al. , Cell Growth Differ., 1996, 7, 173–178 CAS.
- N. Borisov, E. Aksamitiene and A. Kiyatkin, et al. , Mol. Syst. Biol., 2009, 5, 256 CrossRef PubMed.
- S. J. Wang, W. Saadi and F. Lin, et al. , Exp. Cell Res., 2004, 300, 180–189 CrossRef CAS PubMed.
- Z. Zhang, M. Yang and R. Chen, et al. , Oncogene, 2014, 33, 3374–3382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: A supplementary information document including four tables and nine figures; three supplementary movies. See DOI: 10.1039/c5ib00209e |
| ‡ Current address: School of Electronic Engineering, Dublin City University, Dublin 9, Ireland. |
| This journal is © The Royal Society of Chemistry 2016 |