
Stiff substrates enhance monocytic cell capture through E-selectin but not P-selectin†
Joanna L.
MacKay
a and
Daniel A.
Hammer
*ab
aDepartment of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, 311A Towne Building, 220 S. 33rd Street, Philadelphia, PA 19104, USA. E-mail: hammer@seas.upenn.edu; Fax: +1-215-573-2091; Tel: +1-215-573-6761
First published on 13th November 2015
Abstract
The stiffening of blood vessel walls is associated with inflammatory diseases, including atherosclerosis, diabetes, and obesity. These diseases are driven by the excessive recruitment of inflammatory leukocytes out of the bloodstream and into tissues, but whether vascular stiffening plays a direct role in this process is not clear. In this study, we investigated the possibility that leukocyte capture from blood flow is enhanced on stiffer substrates. We modeled blood flow in vitro by perfusing monocytic cells over hydrogels that matched the stiffness of healthy and diseased arteries. The hydrogels were coated with either E-selectin or P-selectin, which are the endothelial adhesion proteins known to mediate immune cell capture from flow. Interestingly, we discovered that cell attachment to P-selectin coated gels was not dependent on substrate stiffness, while attachment through E-selectin was enhanced on stiffer gels. Specifically we found that on E-selectin coated gels, cells attached in greater numbers, remained attached for longer time periods, and rolled more slowly on stiff gels than soft gels. These results suggest that vascular stiffening could promote leukocyte adhesion to vessel walls where E-selectin is expressed, but may have less of an effect when P-selectin is also present.
Insight, innovation, integrationChronic inflammation is driven by the recruitment of immune cells out of the bloodstream and is also associated with blood vessel stiffening. However, whether vessel stiffening directly contributes to immune cell recruitment is not known. In this study, we developed an in vitro assay to study the effects of substrate stiffness on immune cell capture from flow. We perfused monocytic cells over compliant hydrogels functionalized with adhesion proteins E-selectin and P-selectin. This approach enabled us to discover that cell attachment to E-selectin is enhanced on stiffer substrates, while cell attachment to P-selectin is not. Both selectins are expressed on endothelium but with different spatiotemporal patterns, suggesting that vessel stiffening may promote immune cell recruitment only when E-selectin is present. |
Introduction
Many inflammatory diseases are associated with increased stiffening of blood vessels, which results from extracellular matrix remodeling that includes collagen deposition and degradation of elastin fibers. Arterial stiffness can be measured non-invasively in patients from blood pulse-wave velocity measurements, and numerous studies have found correlations between arterial stiffening and the development of atherosclerosis, diabetes, obesity, rheumatoid arthritis, and chronic obstructive pulmonary disease.1–8 Each of these diseases is characterized by chronic inflammation, which involves the recruitment of leukocytes (primarily monocytes and neutrophils) out of the blood and into tissues, where they release pro-inflammatory cytokines and reactive oxygen species that can damage tissues. The molecular mechanisms by which vascular stiffening might promote inflammation and vice versa are not clear.In order to carry out their immune functions in tissues, circulating leukocytes are recruited out of the bloodstream through a multistep adhesion cascade.9 First, endothelial cells lining the vasculature become activated in response to inflammatory stimuli. Within minutes of exposure to secretagogues (e.g., thrombin or histamine), the adhesion protein P-selectin is transported to the endothelial cell surface, as storage granules called Weibel–Palade bodies fuse with the plasma membrane. Exposure to inflammatory cytokines, including TNF-alpha and IL1, leads to increased transcription of P-selectin, as well as the adhesion proteins E-selectin, ICAM-1, and VCAM-1, which appear on the surface of the endothelium within a few hours. Next, P-selectin and E-selectin mediate the initial attachment of circulating leukocytes as they flow across the endothelial surface. Cell capture from blood flow is possible due to the fast binding kinetics between selectins and their ligands on the leukocyte surface. The primary ligand for P-selectin is P-selectin glycoprotein ligand 1 (PSGL-1), while E-selectin can bind several different ligands as long as they are properly glycosylated, including PSGL-1, CD44, and L-selectin.10,11 After initial cell attachment, shear flow causes the leukocytes to roll across the endothelium, which is mediated by selectin bonds rapidly forming at the cell front, flow-induced rotation of the cell, and rapid dissociation of selectin bonds at the cell rear.12 Leukocyte rolling is then slowed down by interactions between leukocyte integrins and ICAM-1 and VCAM-1 on the endothelial surface. Finally, full integrin activation leads to leukocyte arrest, spreading, and transmigration through the endothelium and into the surrounding tissue.
Selectins and their ligands have been shown to exhibit “catch-slip” bond behavior, which has a unique dependence on applied force. Most molecular bonds are considered to be slip bonds, in which applying force to the bond decreases the bond lifetime and promotes dissociation, as described by the Bell model.13 On the other hand, the idea of catch bonds was first hypothesized by Dembo et al. as bonds that resist dissociation when force is applied.14 This could be due to force-induced structural changes in proteins that alter the conformation or the orientation of the ligand binding pocket.15,16 Using atomic force microscopy (AFM) to interrogate the behavior of the P-selectin/PSGL-1 bond, Marshall and coworkers demonstrated that in low force regimes, P-selectin/PSGL-1 behaved as catch bonds where increasing the applied force resulted in longer bond lifetimes. At higher applied forces, they found that P-selectin/PSGL-1 bonds behaved as slip bonds.17 This catch-slip behavior of P-selectin bonds has been confirmed in other studies using AFM,18,19 biomembrane force probes,20 and optical traps,21 and E-selectin bonds have been shown to exhibit catch bond behavior as well.22 Interestingly, Zhang et al. have shown that the stiffness of the probe used for such force measurements also impacts selectin bond behavior. By varying the spring constant of the optical trap used to pull on P-selectin/PSGL-1 bonds, they found that increasing the probe stiffness led to longer bond lifetimes.21 Presumably this could be explained by stiffer probes imparting greater force on the bonds and thereby enhancing the “catch” behavior. We expect that substrate stiffness would have a similar effect.
The goal of this study was to determine whether leukocyte capture from flow could be sensitive to the stiffness of the underlying substrate. We studied monocytic cell attachment to P-selectin and E-selectin using an in vitro model for blood flow, since modulating vascular stiffness in an animal model would be difficult to accomplish without inadvertently influencing other experimental factors. By comparing cell capture on polyacrylamide gels with elastic moduli spanning the stiffness of healthy and diseased arteries, we found that the influence of substrate stiffness was markedly different for P-selectin versus E-selectin mediated adhesion.
Materials and methods
Cell culture
The human monocytic cell lines THP-1 (ATCC# TIB-202) and U937 (ATCC# CRL-1593.2) were acquired from the University of Pennsylvania Cell Center. U937 cells were cultured in RPMI 1640 (Life Technologies 22400, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich F2442, St. Louis, MO) and 100 units per mL penicillin and 100 μg mL−1 streptomycin (Life Technologies 15140). THP-1 cells were cultured in the same medium with the addition of 0.05 mM 2-mercaptoethanol (Bio-Rad 161-0710, Hercules, CA). Both cell lines were maintained at 2–10 × 105 cells per mL in a 37 °C humidified incubator with 5% CO2.Polyacrylamide gels
Aqueous solutions of acrylamide and bis-acrylamide (Bio-Rad) were prepared according to Yeung et al.23 to formulate gels with elastic moduli around 1 kPa (3% acrylamide, 0.2% bis-acrylamide), 5 kPa (5.5% acrylamide, 0.15% bis-acrylamide), 10 kPa (7.5% acrylamide, 0.15% bis-acrylamide), 24 kPa (7.5% acrylamide, 0.3% bis-acrylamide), and 84 kPa (12% acrylamide, 0.28% bis-acrylamide). The acrylamide solutions were sterilized by passing through a 0.2 μm filter and degassed by placing under vacuum for 30 minutes. Polymerization was initiated by adding ammonium persulfate (Bio-Rad 161-0700) at a final concentration of 0.1% w/v and TEMED (Bio-Rad 161-0800) at a final concentration of 0.1%. After gentle mixing, 50 μL of solution was pipetted onto 25 mm glass coverslips that had been activated with 3-aminopropyltrimethoxysilane (Sigma-Aldrich 281778) and glutaraldehyde (Sigma-Aldrich G7776) as previously described.24 RainX-treated 25 mm glass coverslips were then carefully placed on top, and the gels were allowed to polymerize for 5 minutes. To aid removal of the top coverslip, polymerized gels were submerged in water for at least 5 minutes. The top coverslip was then carefully pried off with tweezers, and the gels were washed three time with water.In our experiments, we found that gels must be smooth and free of defects (e.g., wrinkles, tears, debris) so that fluid flow is not disturbed near the gel surface. We believe the following details may have been important for creating smooth gels. All solutions were sterile filtered to remove debris (including water and PBS used for washing) and were handled within a tissue culture hood. Before gel polymerization, the RainX-treated coverslips were wiped with clean gloves until they were spotless and free of debris, and a stream of nitrogen was used to remove debris from the silane-activated coverslips. To easily detach the top coverslip without tearing the polymerized gel, we found it best to use the same size top and bottom coverslips. We then held the coverslip-gel sandwich in one hand and used sharp tweezers to pry between the two coverslips and carefully lift the top coverslip off of the gel. We found that soft gels with less than 0.1% w/v bis-acrylamide often had small tears on the gel surface after removing the top coverslip, which was why we chose formulations containing 0.15–0.3% w/v bis-acrylamide.
Protein conjugation
Recombinant human P-selectin and E-selectin (R&D Systems ADP3 and ADP1, Minneapolis, MN) were conjugated to the surface of polyacrylamide gels using the crosslinker sulfo-SANPAH (Life Technologies 22589), which was stored at a stock concentration of 100 mg mL−1 in DMSO at −20 °C with desiccant in the dark. A set of four gels (two 1 kPa and two 24 kPa) was prepared together in a 6-well plate (not tissue culture treated). First, excess water was aspirated from the surface of the gels, and then 200 μL of sulfo-SANPAH solution (0.25 mg mL−1 in ultrapure water) was added to each gel in the dark. The plate was placed uncovered under a UV lamp (UVP B-100AP, Upland, CA) for 8 minutes, and then the gels were washed three times with ultrapure water to remove excess sulfo-SANPAH. After aspirating the water, 50 μL of 4 μM P-selectin or 1 μM E-selectin in PBS was added to each gel and incubated on a rocking table overnight at 4 °C. The gels were then washed with PBS and incubated with 1% ethanolamine (Sigma-Aldrich E0135) in PBS for 30 minutes to react with unreacted sulfo-SANPAH. The gels were again washed with PBS and kept at 4 °C until used for experiments later that day.For experiments utilizing protein A/G and selectin–Fc chimeras, the same conjugation protocol was followed except for minor changes. A set of five gels (one of each stiffness) was prepared together. The sulfo-SANPAH solution was at 0.5 mg mL−1 in 50 mM HEPES (pH 8.5), and the gels were placed under a UV lamp (Spectroline EN-160L, Westbury, NY) for 15 minutes. The gels were washed three times with 50 mM HEPES and then 200 μL of 10 μg mL−1 protein A/G (BioVision 6502, Milpitas, CA) in 50 mM HEPES was added to each gel. After incubating overnight at 4 °C, the gels were washed with water, incubated with 1% ethanolamine in 50 mM HEPES for 30 minutes, and then washed with PBS. Recombinant human P-selectin/Fc or E-selectin/Fc (R&D Systems 137-PS and 724-ES), which form disulfide-linked homodimers, were added to each gel at 0.5–10 nM in 100 μL of PBS and incubated for 1 hour at 37 °C. The gels were then washed with PBS and kept at 4 °C.
Comparison of protein density across gel formulations
To compare the levels of protein conjugation across gels with different elastic moduli, we used confocal microscopy to image fluorescently-labeled protein at the surface of each gel. To detect E-selectin or P-selectin, we stained with the mouse monoclonal antibody BBIG-E6 (R&D Systems BBA1) and Alexa Fluor 488 goat anti-mouse secondary antibody (Life Technologies A-11001). However, for gels coated with protein A/G and selectin–Fc chimeras, antibody staining led to high background fluorescence, since the antibodies could directly bind protein A/G through their Fc domain. We therefore added human IgG1 (Abcam 90283, Cambridge, UK) labelled with Alexa Fluor 555 to gels coated with protein A/G, as a surrogate for P-selectin/Fc and E-selectin/Fc. Four sets of each gel stiffness were prepared on different days, and the gels were imaged using a Leica TCS SP5 confocal microscope at four different positions on each gel, while maintaining the same laser, PMT, and scan settings. To account for the gels not being level relative to the objective, z-stacks were acquired near the gel surface and processed using the built-in Z-project function in ImageJ to select the maximum intensity value of each pixel within the stack. This produced an image representing the surface of the gel, where fluorescence was highest. The average fluorescence intensity of each gel was calculated relative to the 1 kPa gel prepared in parallel.Quantification of selectin site densities
To quantify the molecular site density of P-selectin/Fc and E-selectin/Fc on polyacrylamide gels, we used a horse radish peroxidase (HRP)-tagged mouse IgG1 monoclonal antibody that recognizes the hinge domain of human IgG1 (Southern Biotech 9052-05, Birmingham, AL). The hinge domain is preserved in the selectin/Fc fusion proteins and is not blocked by protein A/G binding. 24 kPa gels were prepared as described above and coated with 0.5 or 10 nM P-selectin/Fc, 1 or 10 nM E-selectin/Fc, or no protein for 1 hour at 37 °C. The gels were washed and then incubated with 25 μg mL−1 mouse IgG1 isotype control for 2 hours at 37 °C to occupy available binding sites on protein A/G, and thereby block subsequent binding of the HRP-tagged antibody through its Fc domain. Non-specific adhesion was blocked by incubating with 1% bovine serum albumin (BSA) for 1 hour at room temperature, and then the HRP-tagged anti-Fc antibody was added at a saturating concentration of 2.5 μg mL−1 for 1 hour at room temperature. After extensive washing, 1-Step Turbo TMB ELISA substrate (Thermo Fisher Scientific 34022, Waltham, MA) was added to each gel for 15 minutes. The solution was then transferred to a 96 well plate and an equal volume of 2 N sulfuric acid was added to stop the reaction. A Tecan Infinite M200 plate reader was used to measure absorbance at 450 nm. The absorbance of control gels coated with protein A/G but no Fc protein was quite high (0.15–0.2), perhaps due to non-specific binding or the inability to completely wash out HRP-tagged antibody. We therefore normalized the absorbance values of the selectin/Fc coated gels by subtracting the absorbance of the control gels. We did try this ELISA protocol with 1 kPa gels, but the background absorbance of control gels was too high (greater than 0.6).To convert absorbance into a molecular site density, we created a standard curve using human IgG1 labelled with Alexa Fluor 555 (abbreviated as IgG1-AF555) as follows. A stock of IgG1-AF555 at 1 mg mL−1 (confirmed by absorbance at 280 nm) was used to create serial dilutions in water from 0.16–10 nM. Known volumes of each dilution were incubated in a 96-well tissue culture-treated plate for 30 minutes. The solutions were then removed from the wells, and the remaining fluorescence was compared to the fluorescence of fresh dilutions of IgG1-AF555, by using a Tecan Infinite M200 plate reader (excitation: 555 nm, emission: 580 nm). The loss of fluorescence (before and after incubation in the 96-well plate) was used to calculate the number of IgG1-AF555 molecules that must have adsorbed to the plate. These wells were then blocked with 1% BSA, incubated with 2.5 μg mL−1 HRP-tagged antibody, washed extensively, and then incubated with 1-Step Turbo TMB ELISA substrate, as described above for the polyacrylamide gels. The experiment was repeated on four different days, and a plot of the absorbance values at 450 nm versus the number of IgG1-AF555 molecules adsorbed per well was fit using linear regression. For all experiments, we ensured that fluorescence and absorbance values were within the linear detection range of the assay, and we also confirmed by fluorescence that adsorbed IgG1-AF555 was not removed during the incubation and washing steps. We assumed that one HRP-tagged anti-Fc antibody binds one IgG1-AF555 molecule, since the molecules are adsorbed to the plastic surface and likely have one binding site available. We assumed that two HRP-tagged anti-Fc antibodies bind each homodimer of P- or E-selectin/Fc (i.e., one HRP-tagged antibody for each monomer), since the molecules are oriented upwards on the gels. The selectin densities are listed as monomer molecules per μm2, in order to compare to values cited from previous studies.
Flow adhesion assay
To perfuse cells across the polyacrylamide gels, we utilized a circular parallel-plate flow chamber (GlycoTech 31-001, Gaithersburg, MD). The flow chamber was designed to fit inside a 35 mm culture dish, with a silicon rubber gasket placed in between to create a rectangular flow path with a height of 127 μm and width of 0.25 cm. Typically the apparatus is held together tightly under vacuum, but we found that soft gels were often squeezed into the flow path. We therefore carefully assembled the flow chamber on top of the polyacrylamide gel so that vacuum was not necessary. First we placed the flow chamber on the gasket, flipped it over, and ensured that the gasket was centered with no air bubbles or debris. We then held the polyacrylamide gel with tweezers, removed excess PBS from the surface of the gel by touching the edge to a Kimwipe, and carefully placed the gel face-down on the gasket. A small droplet of PBS was left on the other side of the glass coverslip, which was necessary for acquiring clear images. We then gently placed the flow chamber inside a dry 35 mm culture dish and wrapped paraffin film around the edges to prevent the chamber from sliding within the dish. We inspected the bottom of the apparatus to ensure that the gel remained centered and that the small droplet of liquid between the coverslip and the dish did not spread beyond the area of the coverslip. We found that if a large droplet was left on the glass coverslip when the apparatus was assembled, the liquid would spread beyond the area of the coverslip and cause leaking during the experiment. The apparatus was placed on a Zeiss Axiovert 200 microscope enclosed by a chamber heated to 37 °C. Before each experiment, we perfused PBS through the flow chamber and removed any air bubbles caught in the chamber or the inlet or outlet tubing, and then perfused through cell culture medium. THP-1 or U937 cells (2 × 105 cells per mL in culture medium) were perfused over polyacrylamide gels for 10 minutes at 40.3 μL min−1 using a syringe pump, which corresponds to a shear rate of 100 s−1 and a shear stress of 0.79 dyn cm−2, assuming the viscosity of the culture medium is 0.79 cP.25 A single field of view of 974 × 731 μm2 was observed at the center of the flow path using a 10× phase objective and recorded continuously with a CCD camera (COHU 4195-4000, Poway, CA) and DVD recorder. For experiments using blocking antibodies, 5 x 104 cells in 25 μL of culture medium were pre-incubated with 50 μg mL−1 of antibody for 30 minutes at 37 °C. Then 475 μL of medium was added to the cells, and the flow experiments were performed as described above. The following antibodies were used: anti-PSGL-1 mouse IgG1 (clone KPL-1, BioLegend 328802, San Diego, CA), anti-FcγRI/CD64 mouse IgG1 (clone 10.1, BioLegend 305015), mouse IgG1 isotype control (clone MOPC-21, BioLegend 400123), and pooled human IgG1 (Abcam 90283).Analysis of cell attachment
Videos were converted into tiffs using HandBrake (https://handbrake.fr) and QuickTime at a frame rate of 30 fps and image size of 320 × 236 pixels. ImageJ was used to process the images, which included background subtraction and conversion to binary images using a uniform intensity threshold. The ImageJ plugin MTrack2 (http://valelab.ucsf.edu/∼nstuurman/ijplugins/MTrack2.html) was used to track the XY positions of cells traveling near the gel surface that had instantaneous velocities below 8 pixels per frame, which corresponds to 730 μm s−1. We considered cells to be attached to the gel if their instantaneous velocity dropped below 180 μm s−1 for 3 consecutive frames or 0.1 s. Since the total number of cells that entered the field of view varied between experiments, we reported the number of attached cells as the percentage of total cells that were tracked (i.e., with instantaneous velocities<730 μm s−1). We considered attached cells to be detached if their instantaneous velocity increased above 180 μm s−1 for 6 consecutive frames or 0.2 s. The rolling speed of attached cells was calculated as the end-to-end distance divided by the time that cells remained attached to the gels. We considered attached cells to be arrested if their instantaneous velocity dropped below 0.4 μm s−1 for 5 seconds. Cell trajectories were plotted as the distance traveled versus time, beginning when cells first attached to the surface.Statistical analysis
We used the two-tailed Welch's t-test to determine whether conditions were significantly different, because sample sizes were small and had unequal variances. Statistically significant differences at p < 0.05 are marked with an asterisk on plots (*), and actual p values are noted in the figure legends.Results
To explore whether substrate stiffness could influence selectin-mediated capture of leukocytes from fluid flow, we employed a simple in vitro model for blood flow. We first fabricated thin polyacrylamide gels on coverslips, varying the elastic moduli of the gels from that of healthy blood vessels (1–5 kPa) to that of diseased vessels (>10 kPa).26,27 We then conjugated purified E-selectin or P-selectin protein to the surface of each gel and carefully assembled a parallel plate flow chamber on top. This strategy allowed us to precisely control the stiffness of the substrate without altering other parameters such as ligand density and surface topography. In this study, we used the human monocytic cell lines THP-1 and U937, because monocytes are the key leukocytes driving chronic inflammation and these cell lines are often utilized to study leukocyte adhesion.28,29 We perfused the cells across each gel surface at a relatively low shear stress of 0.79 dyn cm−2, because very few cells attached to the gels at higher flowrates. While the average shear stress in arteries in vivo is over 10 dyn cm−2, some regions of the arterial wall experience much lower shear stress (below 1 dyn cm−2), particularly near bends and bifurcations where fluid flow is disturbed.30,31 These low stress regions promote leukocyte attachment and are particularly prone to atherosclerotic plaque formation.32,33We first investigated whether THP-1 and U937 cells could attach to gels coated with E-selectin, and whether the level of cell attachment depended on substrate stiffness. We perfused cells over 1 kPa or 24 kPa gels, and continuously imaged a single field of view focused at the gel surface for 10 minutes. Since a large fraction of cells traveled with the free stream velocity too far above the gel to interact with the surface, we only tracked the positions of cells that were traveling near the gel surface, and we calculated the number of attached cells as the percentage of cells that were tracked. Interestingly, we found that far more THP-1 and U937 cells attached to stiff gels coated with E-selectin than soft gels (Fig. 1A). Furthermore, attached cells rolled for longer periods of time on stiff gels before detaching, while on soft gels, cells only briefly attached (Fig. 1B). To ensure that this result was not simply due to differences in E-selectin conjugation, we confirmed that the surfaces of soft and stiff gels had similar levels of E-selectin by immunofluorescence staining (Fig. S1, ESI†).
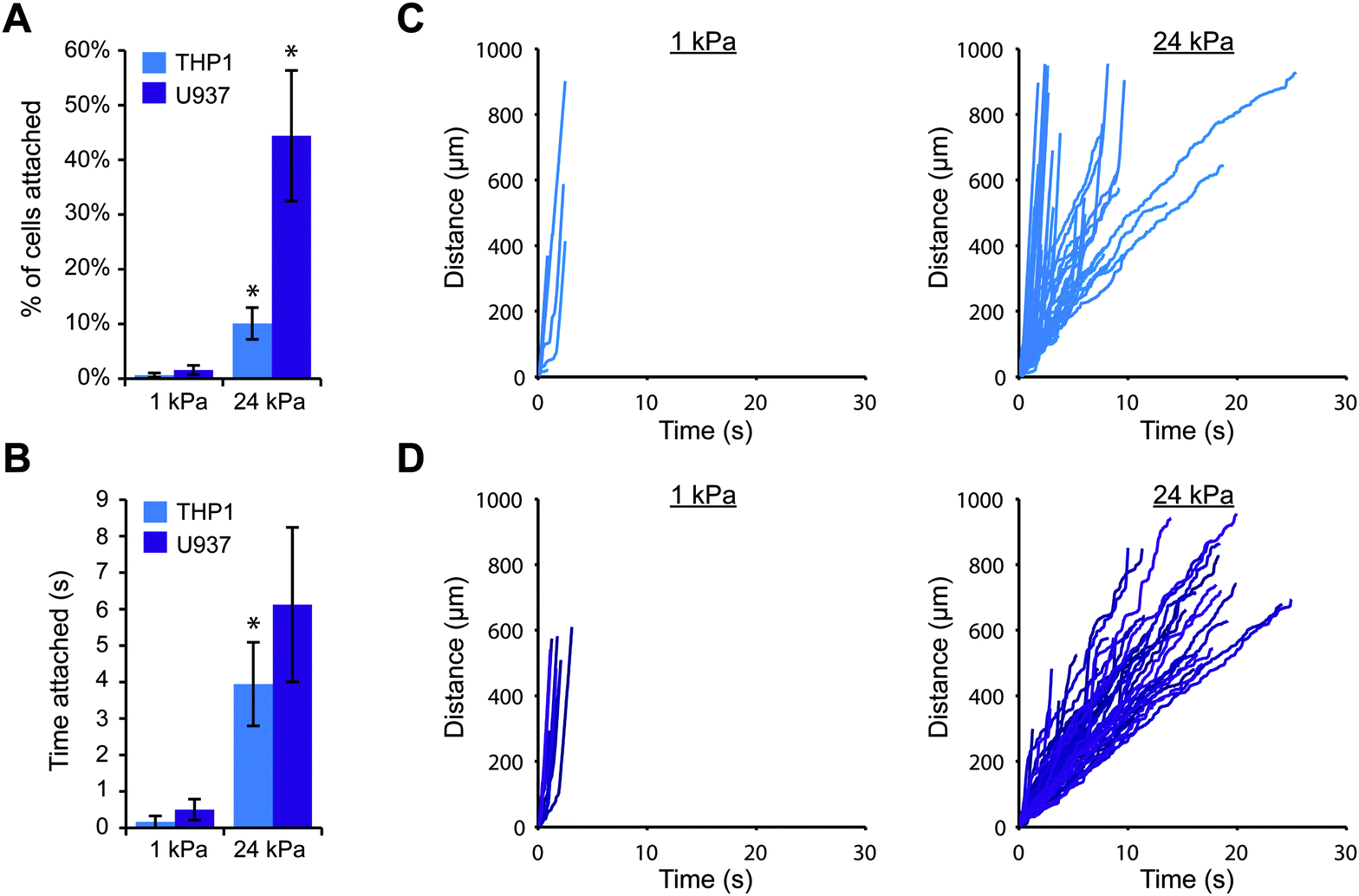 | ||
| Fig. 1 Cell attachment through E-selectin is enhanced on stiff substrates. THP-1 or U937 cells were perfused at 100 s−1 across 1 kPa or 24 kPa gels coated with 1 μM E-selectin. (A) More cells attached to stiff gels than soft gels (n = 3 experiments, mean ± s.e., t-test: p = 0.059 for THP1 and p = 0.017 for U937). (B) Attached cells remained for longer periods of time on stiff gels (n = 3 experiments, mean ± s.e., t-test: p = 0.08 for THP1 and p = 0.12 for U937). (C) Representative tracks of THP-1 cells plotted as distance traveled versus time. (D) Representative tracks of U937 cells. | ||
We then attempted to perform the same experiments on gels coated with P-selectin, but we were unable to achieve the same level of protein conjugation. Even with a four-fold higher coating concentration of P-selectin (4 μM for P-selectin versus 1 μM for E-selectin), the resulting density of P-selectin on the gels was less than half the density attained with E-selectin coating (Fig. S1, ESI†). As a result, very few THP-1 or U937 cells attached to P-selectin coated gels, regardless of stiffness (Fig. S2, ESI†). While this may suggest that P-selectin binding to PSGL-1 is not influenced by substrate stiffness, it is likely that any difference in cell attachment would be difficult to detect since only 0 to 2 cells had attached to each gel.
We therefore switched to a different strategy to attach P-selectin to polyacrylamide gels, in which we first used sulfo-SANPAH to conjugate protein A/G to the gel surface. Protein A/G is a recombinant protein that binds the Fc domain of antibodies or fusion proteins containing the Fc domain. This strategy then allowed us to coat the gels with P-selectin/Fc, a chimeric protein consisting of the Fc domain of human IgG1 fused to the C-terminus of P-selectin. There are several benefits to this approach. First, the P-selectin/Fc protein is presented in the proper orientation for ligand binding, since its C-terminus is bound to protein A/G and the N-terminus is pointed upwards, as it would be on endothelial cells. Second, P-selectin/Fc protein forms disulfide-linked homodimers, and several studies have shown that dimeric P-selectin binds PSGL-1 with a higher affinity than monomeric P-selectin.34,35 Third, the reaction between sulfo-SANPAH and primary amines on protein is relatively inefficient due to competing hydrolysis of the N-hydroxysuccinimide ester. Since protein A/G is easier to acquire than purified P-selectin, we can first use an abundance of protein A/G to react with sulfo-SANPAH and then a low concentration of P-selectin/Fc (which readily binds protein A/G). Importantly, there is one major drawback to using Fc fusion proteins for these experiments. Most leukocytes, including monocytic cells, have Fc receptors and could potentially bind the Fc region of P-selectin/Fc. We controlled for this possibility in the following experiments by blocking Fc receptors on the cells with either soluble human IgG1 or antibody against the high affinity Fc receptor, FcγRI (aka CD64). We also confirmed that THP-1 and U937 cells do not attach to gels coated with protein A/G alone or to gels coated with protein A/G and human IgG1. To ensure that P-selectin/Fc would be present on soft and stiff gels at similar densities, we used fluorescently labelled IgG1 to show that comparable levels of Fc bind to gels coated with protein A/G regardless of stiffness (Fig. S3, ESI†).
Upon switching to a coating strategy employing protein A/G, we discovered that we could achieve much higher cell attachment on gels with P-selectin/Fc (while using 1000-fold less P-selectin protein), which allowed us to more thoroughly investigate the effects of substrate stiffness. We created polyacrylamide gels with elastic moduli of 1, 5, 10, 24 and 84 kPa, and we coated them first with protein A/G and then with three different concentrations of P-selectin/Fc: 0.5, 1, and 10 nM, which corresponded to site densities of 16 ± 3, 32 ± 6, and 178 ± 3 selectin molecules per μm2, respectively. As before, we perfused THP-1 cells for 10 minutes and tracked cells that attached within the field of view. Surprisingly, we found that for each P-selectin/Fc density, substrate stiffness had no obvious effect on cell attachment (Fig. 2). For gels coated with the lowest density of P-selectin/Fc, a small percentage of cells attached briefly to each gel, regardless of substrate stiffness (Fig. 2A and Fig. S4, ESI†). More cells attached to gels coated with the intermediate density of P-selectin/Fc (Fig. 2B), and they remained rolling for an average of 4–5 seconds on each gel before detaching again (Fig. S5, ESI†). On gels coated with the highest density of P-selectin/Fc, most attached cells remained rolling on the gels until exiting the field of view. The average rolling speed did not vary with substrate stiffness (Fig. 2C), except for on 84 kPa gels where cells rolled about 25% slower (Fig. S6, ESI†). To confirm that cells attached to the gels only through P-selectin/PSGL-1 binding, we pre-treated THP-1 cells with either blocking antibody against PSGL-1, blocking antibody against FcγRI, or soluble human IgG1 (to functionally block Fc receptors), and then performed flow experiments on gels coated with P-selectin/Fc at 178 molecules per μm2. As expected, blocking PSGL-1 inhibited cell attachment, while blocking Fc receptors had no effect (Fig. S7, ESI†). We also conducted flow experiments with U937 cells on gels coated with P-selectin/Fc at 16 or 32 molecules per μm2 and similarly found that cell attachment did not vary with substrate stiffness (Fig. S8, ESI†).
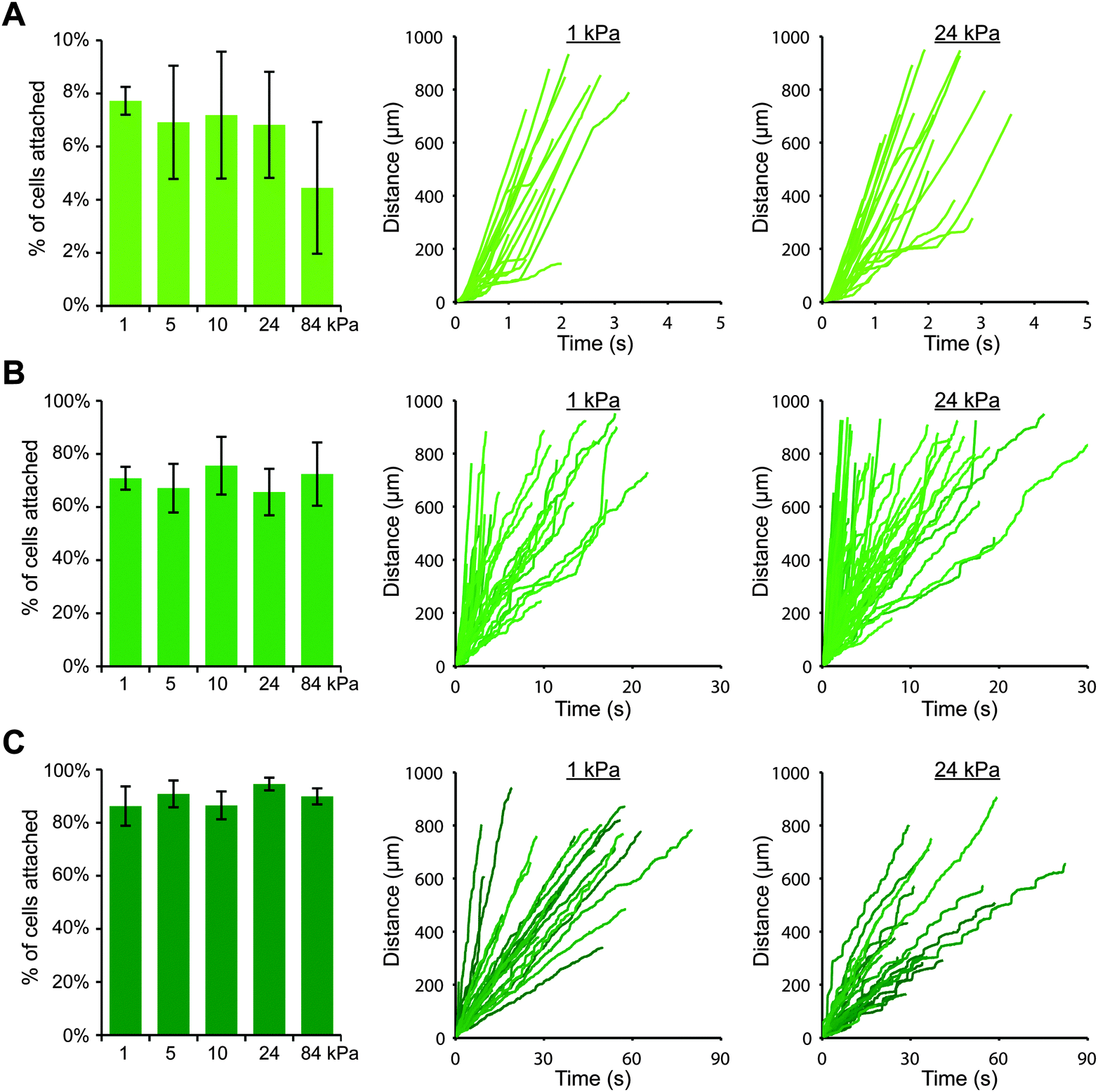 | ||
| Fig. 2 Cell attachment through P-selectin/Fc is not sensitive to substrate stiffness. THP-1 cells were perfused at 100 s−1 across gels coated with different densities of P-selectin/Fc: (A) 16 molecules per μm2, (B) 32 molecules per μm2, and (C) 178 molecules per μm2. For each P-selectin/Fc density, the percentage of attached cells did not vary with substrate stiffness (n = 3 or 4 experiments, mean ± s.e., t-test for 1 vs. 24 kPa: p = 0.7 (A), p = 0.61 (B), p = 0.4 (C)). Representative cell tracks are shown for each condition, plotted as distance traveled versus time. | ||
Since the attachment of THP-1 and U937 cells was greater on stiff gels coated with E-selectin than soft gels (Fig. 1), we wanted to confirm that cell attachment through E-selectin/Fc would also be dependent on substrate stiffness. We again created polyacrylamide gels with elastic moduli of 1, 5, 10, 24 and 84 kPa, coated them first with protein A/G, and then added E-selectin/Fc at two coating concentrations: 1 and 10 nM, which corresponded to site densities of 20 ± 6 and 152 ± 16 selectin molecules per μm2, respectively. We then perfused cells for 10 minutes and tracked cells near the gel surface. We found that at the lower density of E-selectin/Fc, both cell types behaved in a similar stiffness-dependent manner as they had on gels coated directly with E-selectin (Fig. 3 and Fig. S9, ESI†). On the softest gels, very few cells attached and remained only briefly before detaching again. With increasing gel stiffness, more cells attached and remained rolling for longer periods of time. To confirm that this result was due to cells binding the E-selectin portion of the fusion protein and not the Fc domain, we pretreated THP-1 cells with anti-FcγRI antibody or human IgG1 before flow experiments on soft and stiff gels and found no differences in cell attachment compared to controls (Fig. S10, ESI†).
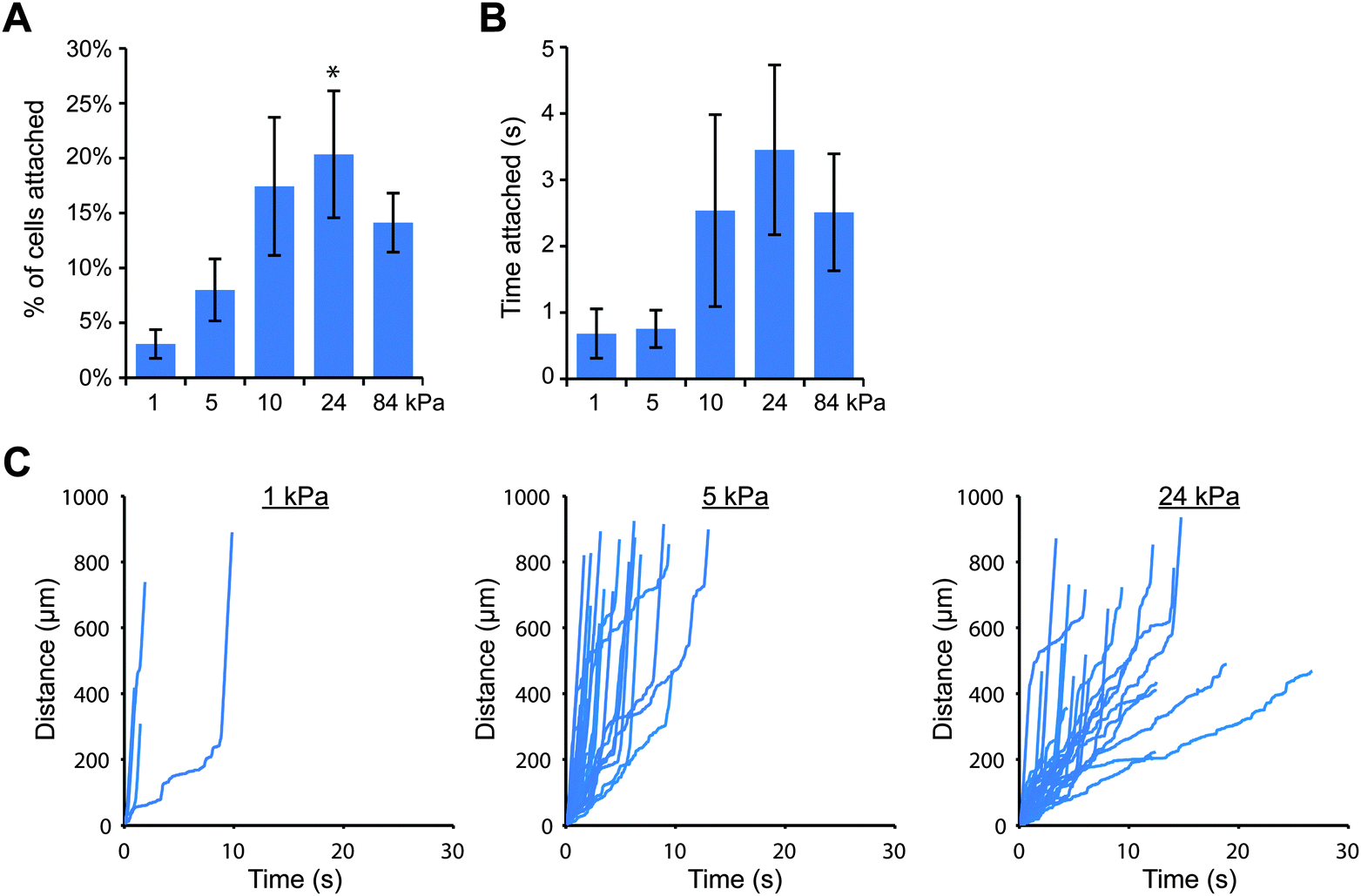 | ||
| Fig. 3 Cell attachment through E-selectin/Fc is enhanced on stiff substrates. THP-1 cells were perfused at 100 s−1 across gels coated with E-selectin/Fc at 20 molecules per μm2. (A) The percentage of attached cells increased with increasing substrate stiffness (n = 5 experiments, mean ± s.e., t-test for 1 vs. 24 kPa: p = 0.015). (B) Attached cells remained for longer periods of time on stiffer gels (n = 5 experiments, mean ± s.e., t-test for 1 vs. 24 kPa: p = 0.11). (C) Representative cell tracks plotted as distance traveled versus time. | ||
We were then curious as to whether this stiffness-dependence would occur at a higher E-selectin density. Interestingly, we found that a high percentage of THP-1 cells attached to gels coated with E-selectin/Fc at 152 molecules per μm2 regardless of substrate stiffness (Fig. S11, ESI†); however cells began to arrest on stiff gels, which we discovered was mediated by Fc binding. Pre-treating cells with human IgG1 for 30 minutes before flow experiments completely prevented arrest on stiff gels, while pre-treating with anti-FcγRI antibody partially blocked arrest (Fig. S12, ESI†). We therefore chose to block Fc binding by pre-treating cells with human IgG1 and repeated the flow experiments on 1, 5, 10, 24, and 84 kPa gels coated with E-selectin/Fc at 152 molecules per μm2. We again found that similar percentages of cells attached to each of the gels, but attached cells rolled more slowly on stiff gels than soft gels (Fig. 4).
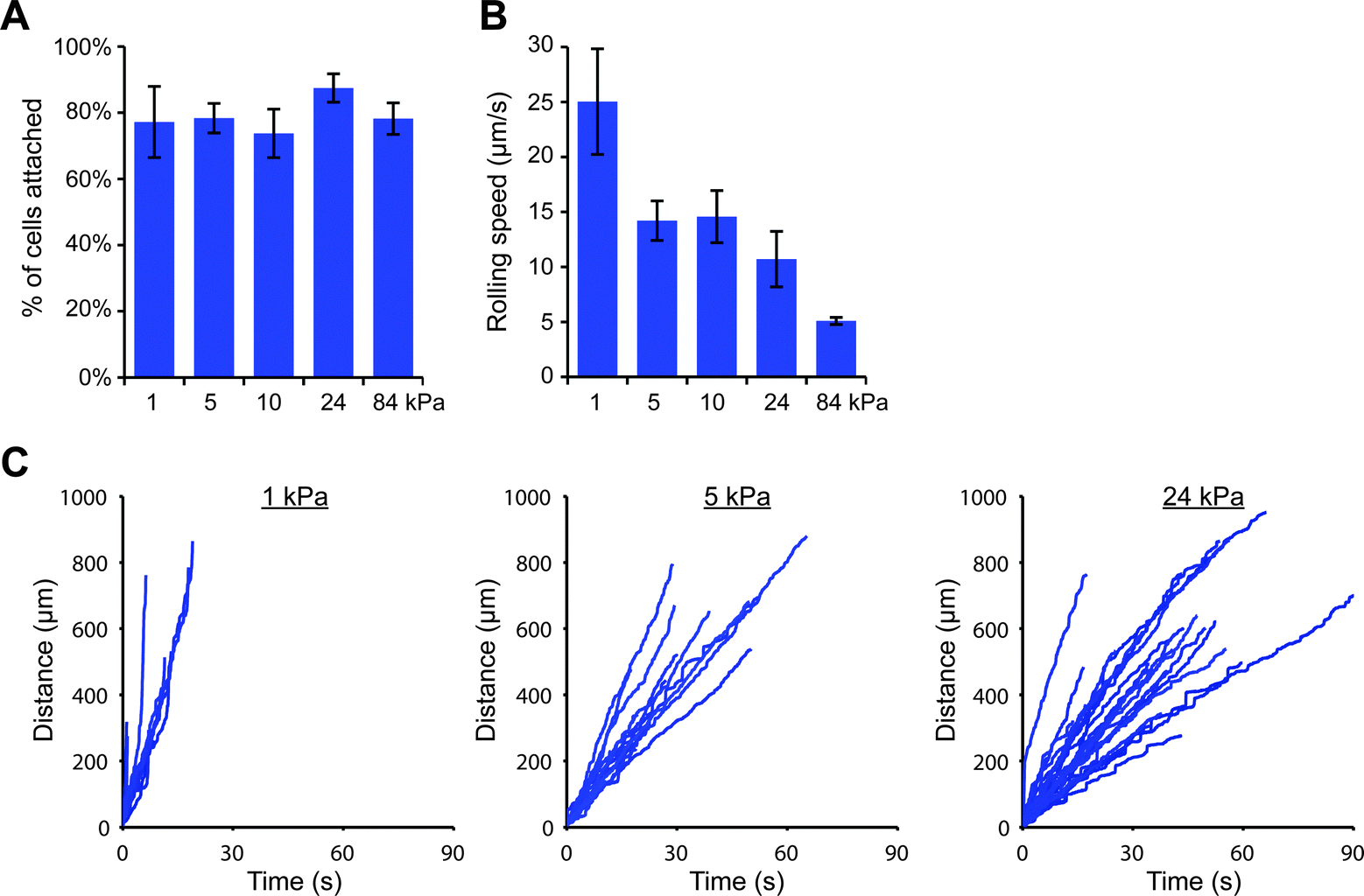 | ||
| Fig. 4 On a high density of E-selectin/Fc, cells roll slower on stiff gels. THP-1 cells were perfused at 100 s−1 across gels coated with E-selectin/Fc at 152 molecules per μm2. (A) The percentage of attached cells did not vary with substrate stiffness (n = 3 experiments, mean ± s.e., t-test for 1 vs. 24 kPa: p = 0.44). (B) The rolling speed of attached cells decreased with increasing substrate stiffness (n = 3 experiments, mean ± s.e., t-test for 1 vs. 24 kPa: p = 0.078). (C) Representative cell tracks plotted as distance traveled versus time. | ||
Discussion
Given that vascular stiffening is strongly correlated with inflammation,2 we investigated whether leukocyte capture via selectins is sensitive to the stiffness of the underlying substrate. We fabricated polyacrylamide gels with elastic moduli ranging from 1 to 84 kPa and conjugated E-selectin or P-selectin to the gel surface, either directly or through the use of Fc chimeras and protein A/G to improve cell attachment. Using a flow chamber, we then perfused human monocytic cells across each gel and studied the dynamics of cell attachment. Surprisingly, we found that whether cell attachment was sensitive to substrate stiffness depended on the selectin that was present. On gels coated with P-selectin, cell attachment was not stiffness-dependent. Approximately equal numbers of cells attached to both soft and stiff gels, and the cells rolled at similar speeds. In contrast, cell attachment through E-selectin was enhanced on stiffer gels. Higher percentages of cells attached to E-selectin coated stiff gels than soft gels, and the cells rolled more slowly and for longer periods of time on stiff gels. Since slow leukocyte rolling on endothelium enables detection of chemokines and integrin-binding proteins, stiff blood vessels expressing E-selectin may promote leukocyte arrest, in addition to enhancing initial cell attachment.One question that arose from our findings was why cell attachment through E-selectin depended on substrate stiffness, while attachment through P-selectin did not. The answer could lie in their different requirements for ligand binding. The primary ligand for P-selectin is PSGL-1, which is heavily glycosylated and constitutively expressed on circulating leukocytes. P-selectin binding requires that PSGL-1 contain a sialyl Lewis X (sLex) on a specific core 2 O-glycan at the N-terminus,36 and binding is enhanced when specific tyrosine residues on PSGL-1 are sulfated.37–39 E-selectin also binds PSGL-1 but with a 50-fold lower affinity,40 and binding can occur at several different sLex sites and is not enhanced by tyrosine sulfation. In addition, E-selectin can bind other glycosylated proteins, as long as they are decorated with sLex or the stereoisomer sialyl Lewis a. For human leukocytes, E-selectin ligands include PSGL-1, CD44, L-selectin, CD43, glycolipids, and possibly additional ligands still to be identified.10,11 We suspect that these differences in ligand affinity between E-selectin and P-selectin could potentially explain the different responses to substrate stiffness. For example, the affinity between E-selectin and its ligands could have been too weak to support cell attachment on soft gels, while stiffer gels perhaps enhanced E-selectin binding through a catch-bond mechanism by providing greater resistive force. Since P-selectin has a much higher affinity for PSGL-1, maybe P-selectin/PSGL-1 binding was already optimal on soft gels and could not be further strengthened by increasing substrate stiffness. Another possibility is that PSGL-1 bound both P-selectin and E-selectin in a stiffness-independent manner, but a specific E-selectin ligand (that does not bind P-selectin) was responsible for the stiffness-dependent response. Perhaps this specific ligand behaves differently from PSGL-1 due to differences in affinity for E-selectin, how far the ligand protrudes past the leukocyte glycocalyx, or how the ligand is anchored to the cytoskeleton. Cell-free rolling experiments using beads could be useful for testing whether specific E-selectin ligands have different dependencies on substrate stiffness.41
The expression patterns of P-selectin and E-selectin on endothelium are known to vary over time in response to inflammatory stimuli, which is why we studied cell attachment on polyacrylamide gels at several different selectin densities (P-selectin: 16, 32, or 178 molecules per μm2; E-selectin: 20 or 152 molecules per μm2). In mice, baseline expression of E-selectin is undetectable, while P-selectin is constitutively expressed at low levels in some tissues, including the lung and mesentery.42 After histamine treatment, P-selectin expression increases several-fold within minutes before returning to baseline levels, reflecting the transient release of P-selectin from Weibel–Palade bodies.42 In cultured human umbilical vein endothelial cells (HUVECs), the peak P-selectin site density after histamine treatment was found to be 16–40 molecules per μm2.43 On the other hand, LPS or cytokine treatment leads to increased transcription of both P-selectin and E-selectin for several hours.42 Estimates of the peak E-selectin site density on cultured endothelial cells after interleukin-1α or interleukin-1β treatment range between 100 and 750 molecules per μm2,35,44,45 which suggests that at some intermediate time point, the E-selectin density would be near the values used in this study (20 and 152 molecules per μm2). Since endothelial cells express both P-selectin and E-selectin simultaneously at varying ratios in vivo, it would be interesting to repeat our experiments on polyacrylamide gels coated with P-selectin and E-selectin together at different densities. We would imagine that the effects of P-selectin and E-selectin mediated adhesion would be additive, and thus lead to increased cell attachment on stiff gels relative to soft gels. However, another possibility is that P-selectin binding could lead to such robust adhesion on both soft and stiff gels that any stiffness effect from E-selectin mediated adhesion would be obscured. Including VCAM-1 would also be interesting, since VCAM-1 has been shown to mediate initial cell capture in the absence of selectins.46,47
In this study, we modeled the endothelium by conjugating purified P-selectin or E-selectin to the surface of polyacrylamide gels. We could have instead cultured endothelial cell monolayers on polyacrylamide gels so that the monolayer stiffness increased with gel stiffness,48 which would have been more physiologically relevant. However, our approach provided two major advantages. First, coating gels with a single protein at a defined density allowed us to discover that cell attachment to E-selectin was stiffness-dependent, while cell attachment to P-selectin was not. In contrast, controlling the expression levels of P-selectin, E-selectin, ICAM-1, and VCAM-1 on cultured endothelial cells would be very difficult, and this type of in vitro experiment would be unlikely to recapitulate the complex spatiotemporal expression patterns seen in vivo. Second, another advantage to not using endothelial cells was that we could be sure that varying substrate stiffness did not alter other surface properties, such as topography and ligand density. Others have shown that endothelial monolayers cultured on stiff gels are more contractile and have weaker cell–cell junctions than if cultured on soft gels,48,49 which could result in topographical changes that alter fluid flow patterns near the endothelial surface and potentially alter the velocity of approaching leukocytes. Endothelial cells cultured on soft and stiff gels could also express different levels of adhesion proteins or those proteins could be clustered differently within the plasma membrane. Of course, it would still be valuable in future studies to confirm that our results on selectin-coated gels are also true on endothelial monolayers. We imagine that the best way to study leukocyte attachment to a specific adhesion protein on endothelium would be to either block all other adhesion proteins on activated endothelial cells (e.g., with blocking antibodies against ICAM-1, VCAM-1, and the other selectin) or to genetically overexpress only one adhesion protein in un-activated endothelial cells.
It is worth mentioning that a previous study by Huynh et al. showed that neutrophils attached in equal numbers to TNF-alpha treated endothelial monolayers cultured on 2.5, 5, and 10 kPa polyacrylamide gels, but the cells transmigrated more frequently through monolayers on stiff gels.49 The authors suggested that the increased transmigration on stiff gels was due to destabilized endothelial cell–cell junctions (which agrees with other studies48,50), although they did not compare rolling velocities or the ability of cells to arrest. We speculate that cell attachment did not appear stiffness-dependent in their case, because P-selectin, E-selectin, ICAM-1, and VCAM-1 were all highly expressed due to TNF-alpha activation. Another possibility is that neutrophil attachment is less sensitive to substrate stiffness than monocytic cells, which would be interesting to directly compare in future studies.
Conclusions
The recruitment of inflammatory leukocytes into tissues is a complex process that is known to depend on multiple factors, including endothelial expression of adhesion proteins and chemokines, leukocyte expression of adhesion receptors and chemokines receptors, and the level of fluid shear stress at the vessel wall. In this study, we explored the effects of an additional factor: substrate stiffness. Utilizing a simplified in vitro model, we have shown that the capture of monocytic cells is enhanced on stiffer substrates when cells attach through E-selectin but is stiffness-independent when cells attach through P-selectin. Whether the same behavior occurs in vivo remains to be seen, but our results suggest the possibility that stiffer blood vessels could promote leukocyte capture through E-selectin and potentially contribute to the pathogenesis of chronic inflammation.Acknowledgements
We thank Richard K. Assoian and Ellen Puré for insightful discussions. The Hammer laboratory acknowledges support from the NIH AI082292 and HL18208. JLM acknowledges support from the NIH 1F32GM110961.References
- M. J. Roman, R. B. Devereux, J. E. Schwartz, M. D. Lockshin, S. A. Paget, A. Davis, M. K. Crow, L. Sammaritano, D. M. Levine, B. A. Shankar, E. Moeller and J. E. Salmon, Hypertension, 2005, 46, 194–199 CrossRef CAS PubMed.
- S. Jain, R. Khera, V. F. Corrales-Medina, R. R. Townsend and J. A. Chirinos, Atherosclerosis, 2014, 237, 381–390 CrossRef CAS PubMed.
- F. U. S. Mattace-Raso, T. J. M. van der Cammen, A. Hofman, N. M. van Popele, M. L. Bos, M. A. D. H. Schalekamp, R. Asmar, R. S. Reneman, A. P. G. Hoeks, M. M. B. Breteler and J. C. M. Witteman, Circulation, 2006, 113, 657–663 CrossRef PubMed.
- C. Vlachopoulos, K. Aznaouridis and C. Stefanadis, J. Am. Coll. Cardiol., 2010, 55, 1318–1327 CrossRef PubMed.
- S. B. Prenner and J. A. Chirinos, Atherosclerosis, 2015, 238, 370–379 CrossRef CAS PubMed.
- R. P. Wildman, R. H. Mackey, A. Bostom, T. Thompson and K. Sutton-Tyrrell, Hypertension, 2003, 42, 468–473 CrossRef CAS PubMed.
- K. M. Mäki-Petäjä, F. C. Hall, A. D. Booth, S. M. L. Wallace, Yasmin, P. W. P. Bearcroft, S. Harish, A. Furlong, C. M. McEniery, J. Brown and I. B. Wilkinson, Circulation, 2006, 114, 1185–1192 CrossRef PubMed.
- R. Sabit, C. E. Bolton, P. H. Edwards, R. J. Pettit, W. D. Evans, C. M. McEniery, I. B. Wilkinson, J. R. Cockcroft and D. J. Shale, Am. J. Respir. Crit. Care Med., 2007, 175, 1259–1265 CrossRef PubMed.
- P. Sundd, M. K. Pospieszalska, L. S.-L. Cheung, K. Konstantopoulos and K. Ley, Biorheology, 2011, 48, 1–35 Search PubMed.
- S. D. Chase, J. L. Magnani and S. I. Simon, Ann. Biomed. Eng., 2012, 40, 849–859 CrossRef CAS PubMed.
- A. Zarbock, K. Ley, R. P. McEver and A. Hidalgo, Blood, 2011, 118, 6743–6751 CrossRef CAS PubMed.
- R. Alon, D. A. Hammer and T. A. Springer, Nature, 1995, 374, 539–542 CrossRef CAS PubMed.
- G. Bell, Science, 1978, 200, 618–627 CAS.
- M. Dembo, D. C. Torney, K. Saxman and D. Hammer, Proc. R. Soc. London, Ser. B, 1988, 234, 55–83 CrossRef CAS.
- W. E. Thomas, V. Vogel and E. Sokurenko, Annu. Rev. Biophys., 2008, 37, 399–416 CrossRef CAS PubMed.
- S. Rakshit and S. Sivasankar, Phys. Chem. Chem. Phys., 2014, 16, 2211–2223 RSC.
- B. T. Marshall, M. Long, J. W. Piper, T. Yago, R. P. McEver and C. Zhu, Nature, 2003, 423, 190–193 CrossRef CAS PubMed.
- B. T. Marshall, K. K. Sarangapani, J. Lou, R. P. McEver and C. Zhu, Biophys. J., 2005, 88, 1458–1466 CrossRef CAS PubMed.
- K. K. Sarangapani, J. Qian, W. Chen, V. I. Zarnitsyna, P. Mehta, T. Yago, R. P. McEver and C. Zhu, J. Biol. Chem., 2011, 286, 32749–32761 CrossRef CAS PubMed.
- E. Evans, A. Leung, V. Heinrich and C. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11281–11286 CrossRef CAS PubMed.
- Y. Zhang, S. Lü and M. Long, Sci. China: Phys., Mech. Astron., 2011, 54, 923–929 CrossRef CAS.
- A. M. Wayman, W. Chen, R. P. McEver and C. Zhu, Biophys. J., 2010, 99, 1166–1174 CrossRef CAS PubMed.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Cell Motil. Cytoskeleton, 2005, 60, 24–34 CrossRef PubMed.
- V. Damljanovic, B. C. Lagerholm and K. Jacobson, BioTechniques, 2005, 39, 847–851 CrossRef CAS PubMed.
- K. D. Rinker, V. Prabhakar and G. A. Truskey, Biophys. J., 2001, 80, 1722–1732 CrossRef CAS PubMed.
- D. Kothapalli, S. L. Liu, Y. H. Bae, J. Monslow, T. Xu, E. A. Hawthorne, F. J. Byfield, P. Castagnino, S. Rao, D. J. Rader, E. Pure, M. C. Phillips, S. Lund-Katz, P. A. Janmey and R. K. Assoian, Cell Rep., 2012, 2, 1259–1271 CrossRef CAS PubMed.
- J.-Y. Chen, P.-J. Tsai, H.-C. Tai, R.-L. Tsai, Y.-T. Chang, M.-C. Wang, Y.-W. Chiou, M.-L. Yeh, M.-J. Tang, C.-F. Lam, S.-C. Shiesh, Y.-H. Li, W.-C. Tsai, C.-H. Chou, L.-J. Lin, H.-L. Wu and Y.-S. Tsai, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 839–846 CrossRef CAS PubMed.
- Z. Qin, Atherosclerosis, 2012, 221, 2–11 CrossRef CAS PubMed.
- P. Harris and P. Ralph, J. Leukocyte Biol., 1985, 37, 407–422 CAS.
- I. Pantos, G. Patatoukas, E. P. Efstathopoulos and D. Katritsis, Expert Rev. Cardiovasc. Ther., 2007, 5, 927–938 CrossRef PubMed.
- G. Dai, M. R. Kaazempur-Mofrad, S. Natarajan, Y. Zhang, S. Vaughn, B. R. Blackman, R. D. Kamm, G. Garcia-Cardena and M. A. Gimbrone, Jr., Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14871–14876 CrossRef CAS PubMed.
- K. S. Cunningham and A. I. Gotlieb, Lab. Invest., 2004, 85, 9–23 CrossRef PubMed.
- P. A. VanderLaan, C. A. Reardon and G. S. Getz, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 12–22 CrossRef CAS PubMed.
- V. Ramachandran, T. Yago, T. K. Epperson, M. M. A. Kobzdej, M. U. Nollert, R. D. Cummings, C. Zhu and R. P. McEver, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10166–10171 CrossRef CAS PubMed.
- Y. Zhang, N. Jiang, V. I. Zarnitsyna, A. G. Klopocki, R. P. McEver and C. Zhu, PLoS One, 2013, 8, e57202 CAS.
- F. Li, H. P. Erickson, J. A. James, K. L. Moore, R. D. Cummings and R. P. McEver, J. Biol. Chem., 1996, 271, 6342–6348 CrossRef CAS PubMed.
- D. J. Goetz, D. M. Greif, H. Ding, R. T. Camphausen, S. Howes, K. M. Comess, K. R. Snapp, G. S. Kansas and F. W. Luscinskas, J. Cell Biol., 1997, 137, 509–519 CrossRef CAS PubMed.
- V. Ramachandran, M. U. Nollert, H. Qiu, W. J. Liu, R. D. Cummings, C. Zhu and R. P. McEver, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13771–13776 CrossRef CAS.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS PubMed.
- K. L. Moore, S. F. Eaton, D. E. Lyons, H. S. Lichenstein, R. D. Cummings and R. P. McEver, J. Biol. Chem., 1994, 269, 23318–23327 CAS.
- D. K. Brunk and D. A. Hammer, Biophys. J., 1997, 72, 2820–2833 CrossRef CAS PubMed.
- M. J. Eppihimer, B. Wolitzky, D. C. Anderson, M. A. Labow and D. N. Granger, Circ. Res., 1996, 79, 560–569 CrossRef CAS PubMed.
- R. Hattori, K. K. Hamilton, R. D. Fugate, R. P. McEver and P. J. Sims, J. Biol. Chem., 1989, 264, 7768–7771 CAS.
- J. D. Levin, H. P. Ting-Beall and R. M. Hochmuth, Biophys. J., 2001, 80, 656–667 CrossRef CAS PubMed.
- N. Mondal, A. Buffone, Jr., G. Stolfa, A. Antonopoulos, J. T. Lau, S. M. Haslam, A. Dell and S. Neelamegham, Blood, 2015, 125, 687–696 CrossRef CAS PubMed.
- R. Alon, P. D. Kassner, M. W. Carr, E. B. Finger, M. E. Hemler and T. A. Springer, J. Cell Biol., 1995, 128, 1243–1253 CrossRef CAS PubMed.
- C. Berlin, R. F. Bargatze, J. J. Campbell, U. H. von Andrian, M. C. Szabo, S. R. Hasslen, R. D. Nelson, E. L. Berg, S. L. Erlandsen and E. C. Butcher, Cell, 1995, 80, 413–422 CrossRef CAS PubMed.
- K. M. Stroka and H. Aranda-Espinoza, Blood, 2011, 118, 1632–1640 CrossRef CAS PubMed.
- J. Huynh, N. Nishimura, K. Rana, J. M. Peloquin, J. P. Califano, C. R. Montague, M. R. King, C. B. Schaffer and C. A. Reinhart-King, Sci. Transl. Med., 2011, 3, 112ra122 Search PubMed.
- K. M. Stroka, I. Levitan and H. Aranda-Espinoza, J. Biomech., 2012, 45, 1828–1834 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00199d |
| This journal is © The Royal Society of Chemistry 2016 |