
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifluoro- and trifluoro diazoalkanes – complementary approaches in batch and flow and their application in cycloaddition reactions†
Katharina J.
Hock‡
,
Lucas
Mertens‡
,
Friederike K.
Metze
,
Clemens
Schmittmann
and
Rene M.
Koenigs
 *
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 15th December 2016
Abstract
Herein we report on applications of fluorinated diazoalkanes in cycloaddition reactions, with the emphasis on studying subtle differences between diverse fluorinated diazo compounds. These differences led to two major synthetic protocols in batch and flow that allow the safe and scalable synthesis of fluoroalkyl-, sulfone-substituted pyrazolines.
Fluoroalkyl-substituted diazocompounds are a versatile class of reagents for the introduction of fluorinated groups and their synthetic potential has attracted significant interest in the past decade.1 However, safe and scalable applications of fluoroalkyl-substituted diazo compounds are still highly desired, as this class of diazo compounds requires stringent safety precautions.2
Trifluoro diazoethane is probably the best-studied reagent among these diazo compounds and several groups reported its application.3,4 Interestingly, difluoro diazoethane only recently found its way into the organic synthesis repertoire, although it differs only in a single fluorine atom from well-studied trifluoro diazoethane.5 Additionally, fluoroalkyl-substituted donor–acceptor diazo compounds are studied only to a very limited extent.5b,6
As part of our continuous interest in fluorinated diazocompounds, we decided to investigate the differences of these diazoalkanes, which give important information on potential synthetic routes, required synthesis technology and subsequent scale-up of transformations using these versatile reagents.
Against this background, we started our investigations by studying the dipolar cycloaddition reaction of these diazoalkanes with vinyl sulfones (Scheme 1c). This transformation is of particular importance as the reaction product is a highly valuable sulfone- and fluoroalkyl-disubstituted pyrazoline (Scheme 1a).7 This fascinating class of heterocycles finds regular application,8 although current synthetic methodology does not allow an atom-economic, streamlined access to this class of compounds.8,9
 | ||
| Scheme 1 Applications and synthetic approaches towards sulfonated pyrazoles. | ||
Sulfone-substituted heterocycles are typically prepared by late-stage introduction of the sulfone moiety and more importantly, the fluorinated analogues can only be accessed using hazardous fluorinating reagents on decagram-scale, which is far from being practical and scalable (Scheme 1b).8c
We initiated our investigations towards fluorinated, sulfone-substituted pyrazolines using a phase-transfer protocol for the generation of trifluoro diazoethane.10 Different retrosynthetically feasible precursors were investigated, though exclusively vinyl sulfones (7a and 7b) yielded the desired sulfonated heterocycles (15a/b). We also tested alkynyl sulfones (10) and vinyl sulfoxides (13) under the phase transfer conditions, yet both failed to give the desired product (Scheme 2).
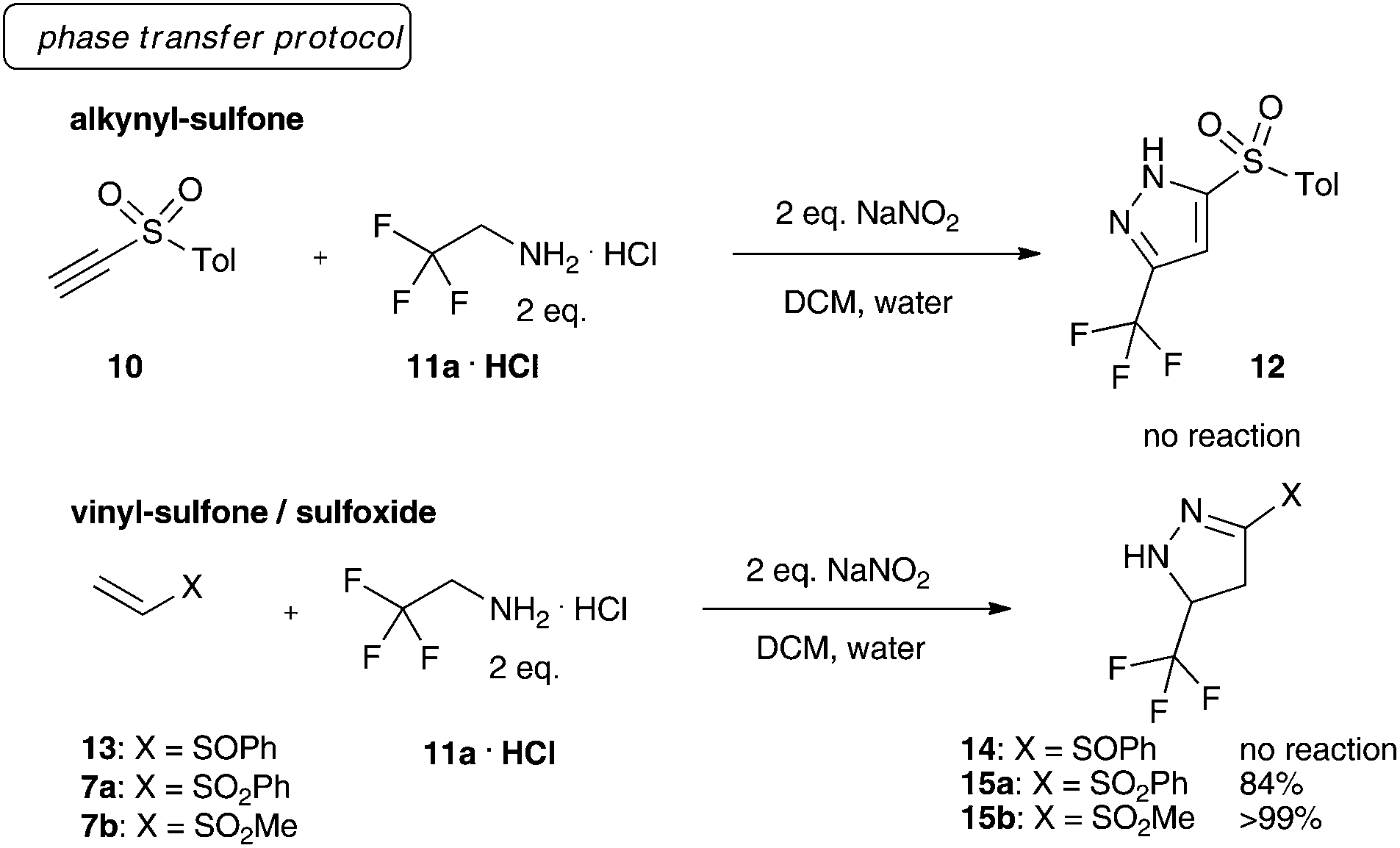 | ||
| Scheme 2 Preliminary investigations on sulfur-substituted pyrazolines. | ||
Next, we examined a range of different organic solvents under phase transfer conditions and were delighted to observe that the desired trifluoromethyl substituted pyrazolines 15a and 15b could be obtained in different polar, and non-polar solvents in good to excellent yield. Dichloromethane and three equivalents of sodium nitrite proved to be the optimum reaction conditions to yield both aryl- and alkyl-sulfone substituted pyrazolines in excellent yield.11
We next studied the substrate scope of the reaction of trifluoro diazoethane with vinyl sulfones under phase transfer conditions. To our delight a range of different aromatic and aliphatic vinyl sulfones (7a–j) could be converted to the corresponding trifluoromethyl- and sulfone-substituted pyrazolines (Table 1) in excellent yield. The sulfone substituent had only little influence on this transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Yield | Entry | R | Yield |
| Reaction conditions: vinyl sulfone (0.5 mmol, 1 eq.), trifluoroethyl amine hydrochloride (2 eq.) and NaNO2 (3 eq.) in DCM (8 mL)/water (2 mL) was vigourosly stirred for 14 h at room temperature; isolated yields. | |||||
| 1 | Phenyl | 84% | 6 | 4-Fluorophenyl | 93% |
| 2 | Methyl | >99% | 7 | 4-Chlorophenyl | 91% |
| 3 | Ethyl | >99% | 8 | 4-MeO-phenyl | 93% |
| 4 | Benzyl | 93% | 9 | 3-Methylphenyl | >99% |
| 5 | 4-Methylphenyl | 98% | 10 | 2-Methylphenyl | 94% |
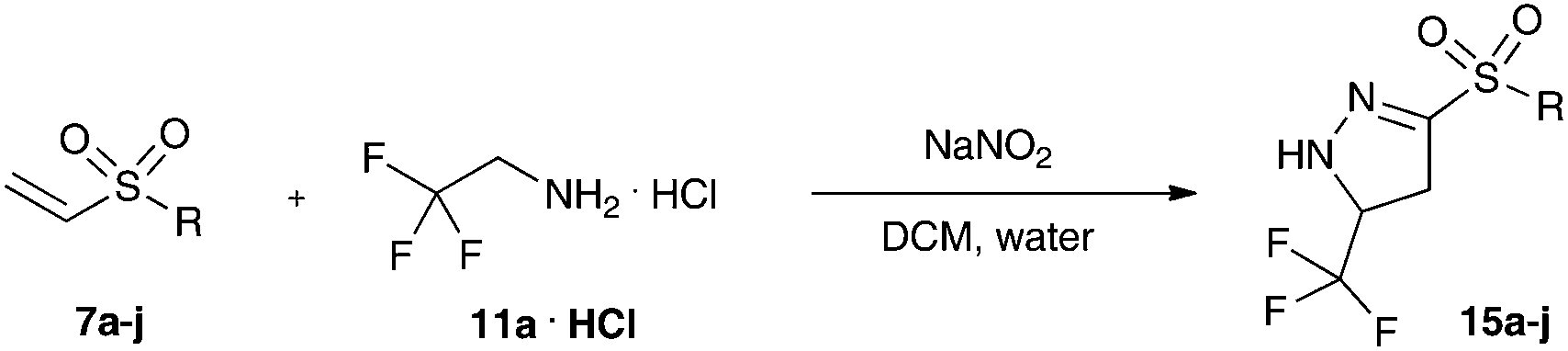
It should be noted, that we observed the initial formation of both Δ1- and Δ2-pyrazoline, after purification on silica gel we exclusively observed the Δ2-pyrazoline (Scheme 3).
 | ||
| Scheme 3 Preliminary investigations on sulfur-substituted pyrazolines. | ||
In further investigations, we probed the closely related difluoro diazoethane. The difluoromethyl group differs only in a single fluorine atom from the well-studied trifluoromethyl group; still, its introduction, physical-chemical and biological properties are very much different.12
Hence, we investigated difluoro ethylamine hydrochloride as a source of difluoro diazoethane. Yet, we could never observe the formation of the desired reaction product (Scheme 4).
 | ||
| Scheme 4 Attempts to the synthesis of difluoromethyl substituted pyrazolines. | ||
We hypothesized this surprising observation with a significantly reduced stability in aqueous solution of difluoro diazoethane and hydrolysis of the diazoalkane. Recently, Mykhailiuk and our group reported on tert-butyl nitrite as an efficient organic nitrite source for the formation of difluoro diazoethane.5 We thus examined this transformation using a purely organic medium (Scheme 4) and both trifluoro and difluoro ethylamine for comparison. We could observe formation of the corresponding trifluoromethylated pyrazoline 15a only in moderate yield (48%), the difluoromethyl-substituted pyrazoline 16a could be isolated in 8% yield.
Thus, we focused our efforts for the dipolar cycloaddition reaction of difluoro diazoethane using continuous flow technology.4j,5b,c,13 Using a 200 μL micromixer (Little Things Factory, MR-LAB MST) and an 800 μL microreactor (PTFE tubing) we were able to mix a stream of difluoroethyl amine with a second stream of tert-butyl nitrite and catalytic amounts of acetic acid providing a continuous access to difluoro diazoethane. After optimization of the reaction conditions, we were able to obtain the desired pyrazolines 16a and 16b in excellent yield. Under these reaction conditions we next probed trifluoro diazoethane but unexpectedly, the desired pyrazoline 15a could only be obtained in significantly decreased yield.11
Similarly, as in the phase-transfer protocol, we observed the formation of Δ1- and Δ2-pyrazoline and subsequent isomerization to the Δ2-pyrazoline after chromatography.
Next, we examined the flow protocol in detail and evaluated different vinyl sulfones. Under the optimum flow conditions for this transformation, we were able to show that difluoromethyl- and sulfone-substituted pyrazolines could be obtained in excellent yield. The effect of different linear aliphatic, or differently substituted aromatic substituents had only a negligible effect on the yield of the desired pyrazolines (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Yield | Entry | Sustrate | Yield |
| Reaction conditions: a solution of amine (0.2 M in CHCl3, 4 eq.) and of tBuONO plus AcOH (2.4 M/0.08 M in CHCl3) were placed in two separate syringes and added by syringe pump at a flow rate of 50 μL min−1 for each syringe into a microreactor (residence time 10 min, temperature = 55 °C). The outlet was connected via a back pressure regulator (20 psi) to a reaction flask containing the vinyl sulfone (0.5 mmol) in CHCl3 and stirred for 14 h at room temperature. Isolated yields. | |||||
| 1 | Phenyl | 93% | 6 | 4-Fluorophenyl | 97% |
| 2 | Methyl | 92% | 7 | 4-Chlorophenyl | 89% |
| 3 | Ethyl | 99% | 8 | 4-MeO-phenyl | 83% |
| 4 | Benzyl | 98% | 9 | 3-Methylphenyl | >99% |
| 5 | 4-Methylphenyl | 95% | 10 | 2-Methylphenyl | >99% |
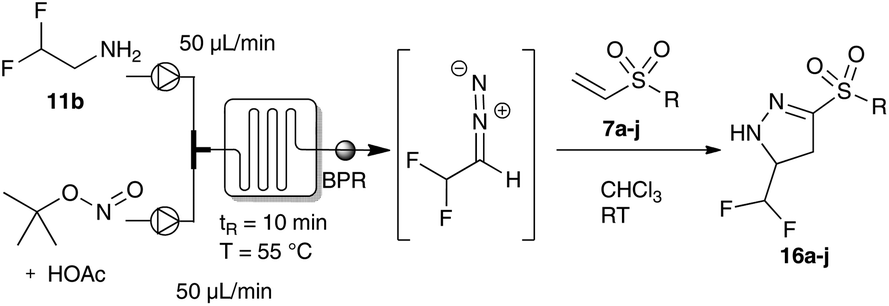
For a comprehensive evaluation of fluoroalkyl-substituted diazoalkanes in this cycloaddition reaction, we next investigated fluoroalkyl-aryl substituted diazoalkanes. These diazoalkanes are closely related to donor–acceptor diazoalkanes due to the strong electron-withdrawing nature of the fluoroalkyl group (Fig. 1).14
 | ||
| Fig. 1 Donor–acceptor substituted diazoalkanes. | ||
In initial experiments we examined both protocols described above for the synthesis of fluorinated donor–acceptor diazo compounds. Yet, the phase transfer protocol and the water-free domino reaction protocol both failed to give the desired product in acceptable yield.11 However, if the diazo compound is prepared in situ and methyl vinyl sulfone is added subsequently after three hours in a one-pot protocol, the desired pyrazoline can be isolated in excellent yield.
This observation prompted us to examine the continuous-flow protocol for these substrates. To our delight the desired pyrazolines could be obtained in excellent yield (Table 3, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 (diazo compound) | Yield batch | Yield flow |
| Reaction conditions: for the batch reactions: the diazo compound is prepared by heating the amine (4 eq.), tBuONO (4.8 eq.) and AcOH (1.6 eq.) in CHCl3 to 55 °C for 3 h, then after cooling to RT, vinyl sulfone (0.5 mmol) is added; isolated yields after chromatography. For the flow reactions: a solution of amine (0.2 M in CHCl3, 4 eq.) and of tBuONO plus AcOH (2.4 M/0.8 M in CHCl3) were placed in two separate syringes and added by syringe pump at a flow rate of 50 μL min−1 for each syringe into a microreactor (residence time 10 min). The outlet was connected via a back pressure regulator (20 psi) to a reaction flask containing the vinyl sulfone (0.5 mmol) in CHCl3 and stirred for 14 h; isolated yields. | ||||
| 1 | Methyl | Phenyl | 97% | 99% |
| 2 | Methyl | 4-Methylphenyl | 75% | 97% |
| 3 | Methyl | 4-Fluorophenyl | 80% | 98% |
| 4 | Methyl | –CH2–CH2–Ph | 67% | 97% |
| 5 | Methyl | Methyl | 96% | 99% |
| 6 | Phenyl | Methyl | 99% | 99% |
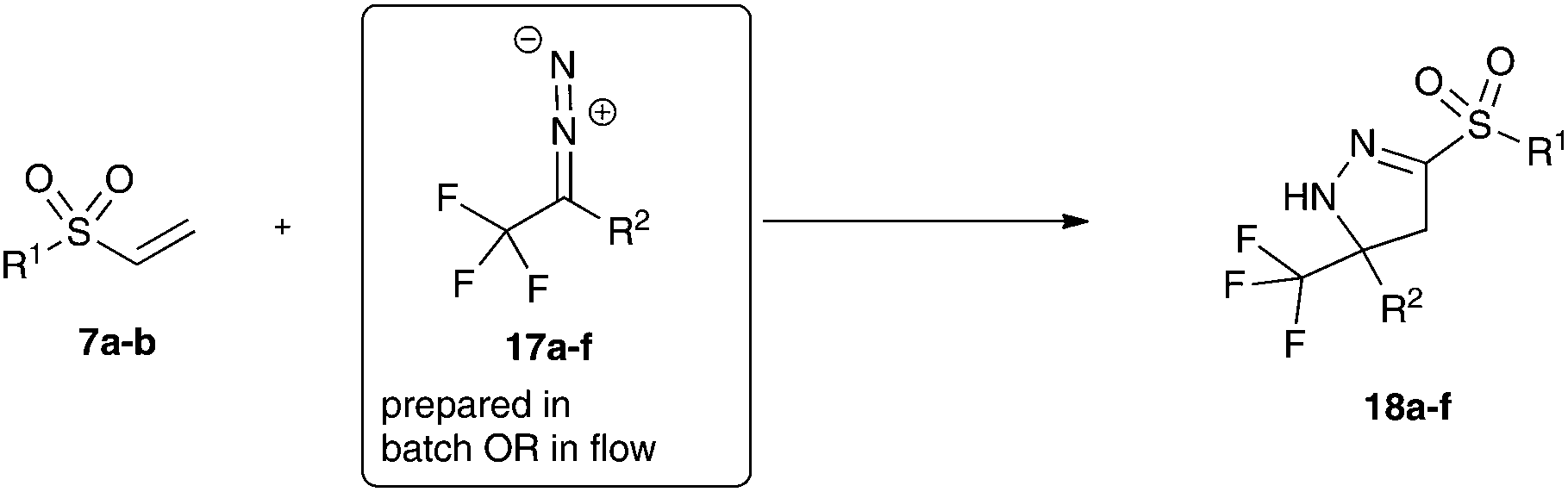
We next investigated the substrate scope for this transformation and could demonstrate, that both the batch and the continuous-flow protocol can be used to deliver the desired pyrazolines in excellent yield. In general, different donor–acceptor substituted fluorinated diazoalkanes can be prepared very efficiently using either batch or flow conditions. The advantages of the batch transformation being: no isolation of the diazo compound as well as fast and routine reaction setup for small-scale transformations. The advantages of the flow protocol being: process safety and a little better performance in terms of yield.
In further experiments, we investigated the robustness and scalability of these transformations. This is of particular importance, as organic synthesis with diazo compounds needs to be conducted with greatest care to prevent serious incidents.
Firstly, we examined the phase transfer protocol for the reaction of trifluoro diazoethane with vinyl sulfones on a gram-scale reaction. To our delight, the sulfone-substituted pyrazoline 15a could be isolated in excellent yield. Similarly, difluoro diazoethane can be prepared in a continuous flow fashion for more than 10 hours and the subsequent cycloaddition reaction was scaled-up to gram scale yielding 1.08 g of the difluoromethyl substituted pyrazoline 16a (Scheme 5).
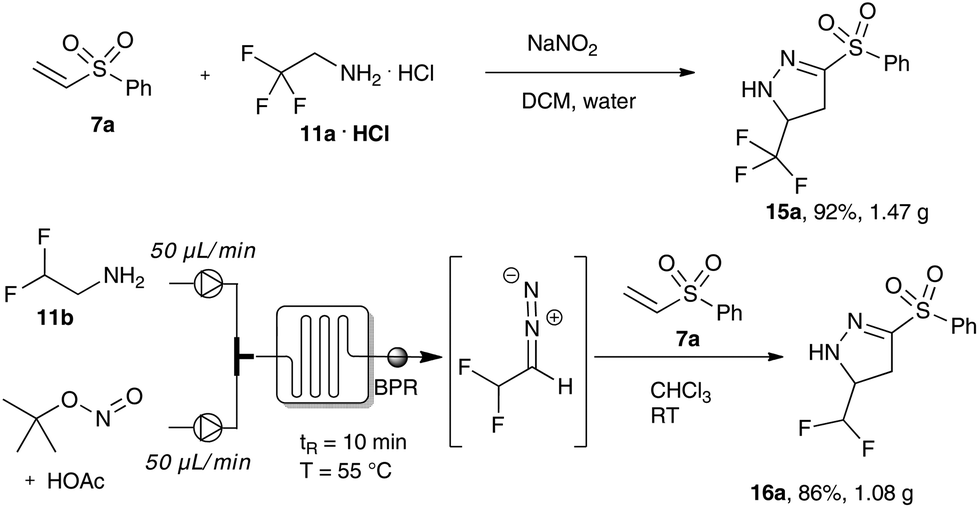 | ||
| Scheme 5 Gram-scale synthesis of sulfone-substituted fluorinated pyrazolines. | ||
In summary, we have reported on robust and atom-economic batch and flow protocols for the preparation of fluoroalkyl-substituted, sulfonated pyrazolines in one synthetic step without the need for hazardous fluorinating reagents. Small subtle differences in reactivity and stability between fluorinated diazo compounds have been observed resulting in two complementary synthetic protocols for the synthetic application of these diazo compounds.
Fluorinated donor–acceptor diazo compounds can be handled using either batch chemistry or flow technology. Trifluoro diazoethane can be handled on gram-scale most-efficiently in batch reactions. Contrarily, difluoro diazoethane necessitates the application of continuous-flow technology to access (a) new synthetic opportunities and (b) safe and scalable handling of this reagent.
The authors gratefully thank the Fonds der Chemischen Industrie for financial support (Sachkostenzuschuss).
Notes and references
- For a review article on fluoroalkyl-substituted diazoalkanes: L. Mertens and R. M. Koenigs, Org. Biomol. Chem., 2016, 14, 10547–10556 CAS.
- (a) R. Fields and J. P. Tomlinson, J. Fluorine Chem., 1979, 13, 147–158 CrossRef CAS; (b) R. Fields and R. N. Haszeldine, J. Chem. Soc., 1964, 1881–1889 RSC.
- H. Gilman and R. G. Jones, J. Am. Chem. Soc., 1943, 65, 1458–1460 CrossRef CAS.
- Selected articles on trifluoro diazoethane. (a) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS PubMed; (b) F. Li, J. Nie, L. Sun, Y. Zheng and J.-A. Ma, Angew. Chem., Int. Ed., 2013, 52, 6255–6258 CrossRef CAS PubMed; (c) A.-J. Cai, Y. Zheng and J.-A. Ma, Chem. Commun., 2015, 51, 8946–8949 RSC; (d) Z. Chen, S.-Q. Fan, Y. Zheng and J.-A. Ma, Chem. Commun., 2015, 51, 16545–16548 RSC; (e) J.-J. Shen, S.-F. Zhu, Y. Cai, H. Xu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13188–13191 CrossRef CAS PubMed; (f) O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed; (g) C. B. Liu, W. Meng, F. Li, S. Wang, J. Nie and J. A. Ma, Angew. Chem., Int. Ed., 2012, 51, 6227–6230 CrossRef CAS PubMed; (h) B. Morandi, B. Mariampillai and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 1101–1104 CrossRef CAS PubMed; (i) S. Hyde, J. Veliks, B. Liegault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55, 3785–3789 CrossRef CAS PubMed; (j) B. Pieber and O. C. Kappe, Org. Lett., 2016, 18, 1076–1079 CrossRef CAS PubMed; (k) A. V. Arkhipov, V. V. Arkhipov, J. Cossy, V. O. Kovtunenko and P. K. Mykhailiuk, Org. Lett., 2016, 18, 3406–3409 CrossRef CAS PubMed.
- Articles on difluoro diazoethane: (a) P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2015, 54, 6558–6561 CrossRef CAS PubMed; (b) L. Mertens, K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2016, 22, 9542–9545 CrossRef CAS PubMed; (c) K. J. Hock, L. Mertens and R. M. Koenigs, Chem. Commun., 2016, 52, 13783–13786 RSC.
- Selected references: (a) R. Barroso, A. Jimenez, M. C. Perez-Aguilar, M.-P. Cabal and C. Valdes, Chem. Commun., 2016, 52, 3677 RSC; (b) F. G. Adly, M. G. Gardiner and A. Ghanem, Chem. – Eur. J., 2016, 22, 3447–3461 CrossRef CAS PubMed; (c) E. Emer, J. Twilton, M. Tredwell, S. Calderwood, T. L. Collier, B. Liegault, M. Taillefer and V. Gouverneur, Org. Lett., 2014, 16, 6004–6007 CrossRef CAS PubMed; (d) J. F. Briones and H. M. L. Davies, Org. Lett., 2011, 13, 3984–3987 CrossRef CAS PubMed; (e) M. Uehara, H. Suematsu, Y. Yasotumi and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 170–171 CrossRef CAS PubMed; (f) J. R. Denton, D. Sukumaran and H. M. L. Davies, Org. Lett., 2007, 9, 2625–2628 CrossRef CAS PubMed; (g) G. Shi and Y. Xu, J. Fluorine Chem., 1990, 46, 173–178 CrossRef CAS.
- For selected reviews on pyrazoles: (a) S. Güniz Küçükgüzel and S. Senkardes, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed; (b) M. L. Quan and J. M. Smallheer, Curr. Opin. Drug Discovery Dev., 2004, 7, 460–469 CAS; (c) D. B. Solit and G. Chiosis, Drug Discovery Today, 2008, 13, 38–43 CrossRef CAS PubMed; (d) F. K. Keter and J. Darkwa, BioMetals, 2012, 25, 9–21 CrossRef CAS PubMed; (e) J. Shonberg, P. J. Scammells and B. Capuano, ChemMedChem, 2011, 6, 963–974 CrossRef CAS PubMed; (f) H. Kumar, D. Saini, S. Jain and N. Jain, Eur. J. Med. Chem., 2013, 70, 248–258 CrossRef CAS PubMed; (g) P. Chauhan, S. Mahajan and D. Enders, Chem. Commun., 2015, 51, 12890–12907 RSC.
- (a) C. J. Mathews and D. R. Baker, (Zeneca) WO9718196, 1997 Search PubMed; (b) C. Hassig, T. C. Gahman, M. R. Herbert and A. M. Thayer, (Kalypsys), WO2007084868, 2007 Search PubMed; (c) J. Oslob, D. Aubele, J. Kim, R. McDowell, Y. Song, A. Sran and M. Zhong, (MyoKardia) WO2016118774, 2016 Search PubMed.
- For selected references: (a) A. Padwa, M. Meske and A. Rodriguez, Heterocycles, 1995, 40, 191–204 CrossRef CAS; (b) A. Padwa and M. W. Wannamaker, Tetrahedron, 1990, 46, 1145–1162 CrossRef CAS; (c) A. Padwa, S. P. Craig, U. Chicchio and D. N. Kline, J. Org. Chem., 1988, 53, 2232–2238 CrossRef CAS; (d) V. A. Vasin, V. V. Razin, E. V. Bezrukova, D. Y. Korovin, P. S. Petrov and N. V. Somov, Russ. J. Org. Chem., 2015, 51, 1144–1154 CrossRef CAS.
- (a) E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 2487–2495 CrossRef CAS; (b) P. K. Mykhailiuk, Org. Biomol. Chem., 2015, 13, 3438–3445 RSC; (c) P. K. Mykhailiuk, Chem. – Eur. J., 2014, 20, 4942–4947 CrossRef CAS PubMed; (d) P. K. Mykhailiuk, Beilstein J. Org. Chem., 2015, 11, 16–24 CrossRef PubMed.
- For details see ESI.†.
- Selected references on CF2H groups: (a) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed; (b) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC; (c) J. Hu and C. Ni, Science of Synthesis: C-1 Building Blocks in Organic Synthesis, 2014, vol. 2, p. 409 Search PubMed; (d) C. Ni, J. Liu, L. Zhang and J. Hu, Angew. Chem., Int. Ed., 2007, 46, 786 CrossRef CAS PubMed; (e) C. Ni and J. Ju, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- Selected review articles: (a) S. T. R. Mueller and T. Wirth, ChemSusChem, 2015, 8, 245–250 CrossRef CAS PubMed; (b) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed; (c) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC; (d) S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed; (e) C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC; (f) T. H. Rehm, Chem. Eng. Technol., 2016, 39(1), 66–68 CrossRef; (g) B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298–2308 CrossRef CAS PubMed; (h) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- Selected review articles: (a) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC; (b) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061–3071 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc03187k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |