
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent- and halide-free synthesis of pyridine-2-yl substituted ureas through facile C–H functionalization of pyridine N-oxides†
Valentin A.
Rassadin
*a,
Dmitry P.
Zimin
 a,
Gulnara Z.
Raskil'dina
ab,
Alexander Yu.
Ivanov
c,
Vadim P.
Boyarskiy
a,
Semen S.
Zlotskii
b and
Vadim Yu.
Kukushkin
*a
a,
Gulnara Z.
Raskil'dina
ab,
Alexander Yu.
Ivanov
c,
Vadim P.
Boyarskiy
a,
Semen S.
Zlotskii
b and
Vadim Yu.
Kukushkin
*a
aInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, 199034 Saint Petersburg, Russia. E-mail: v.rassadin@spbu.ru; v.kukushkin@spbu.ru
bUfa State Petroleum Technological University, Kosmonavtov 1, Ufa, Bashkortostan, Russia
cResearch Park SPbSU, Center for Magnetic Resonance, Saint Petersburg State University, Universitetskaya Nab. 7/9, 199034 Saint Petersburg, Russia
First published on 7th October 2016
Abstract
A novel solvent- and halide-free atom-economical synthesis of practically useful pyridine-2-yl substituted ureas utilizes easily accessible or commercially available pyridine N-oxides (PyO) and dialkylcyanamides. The observed C–H functionalization of PyO is suitable for the good-to-high yielding synthesis of a wide range of pyridine-2-yl substituted ureas featuring electron donating and electron withdrawing, sensitive, or even fugitive functional groups at any position of the pyridine ring (63–92%; 19 examples). In the cases of 3-substituted PyO, the C–H functionalization occurs regioselectively providing a route for facile generation of ureas bearing a 5-substituted pyridine-2-yl moiety.
Introduction
Ureas, especially those functionalized with a heterocyclic moiety, are widely applied in drug design1,2 and demonstrate antimicrobial,3,4 antimalarial,5–7 antivirus,8 and anticancer9–14 activities. Moreover, ureas act as kinase (LIM, VEGFR2, FGFR, FLT3) inhibitors,15–20 they control gastric acid secretion,21 and are used as plant growth regulators.22,23 All known syntheses of ureas employ either various organic (in particular, chlorinated) solvents or heavy metals. In many instances, the reported methods start from toxic and/or halide-containing substrates or require a special laboratory set up to perform the reaction under high pressure. The most straightforward approach to ureas includes the reaction of amines with poisoning phosgene24,25 or hazardous isocyanates26–28 that leads – apart from the target products – to huge amounts of halide-containing wastes (Scheme 1).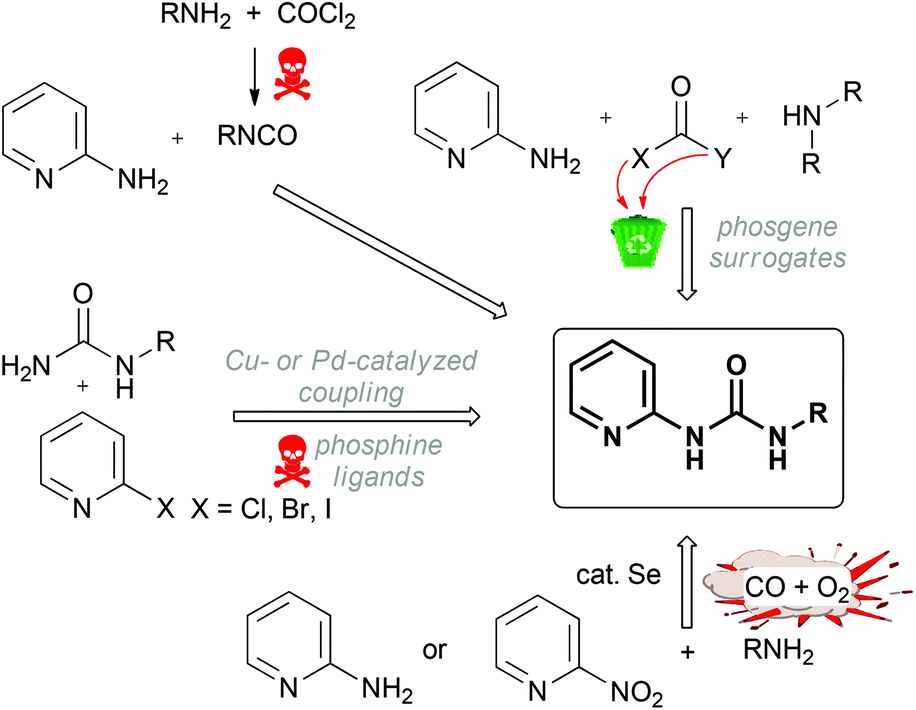 | ||
| Scheme 1 Environmentally unfriendly syntheses of pyridine-2-yl substituted ureas. | ||
It is clear that the employment of volatile and highly toxic phosgene is a serious drawback of this method, especially for large-scale industrial processes. Therefore several “phosgene surrogates”,29viz. trichloromethylchloroformate (diphosgene),30 bis(trichloromethy1)carbonate (triphosgene),31,32 diethyl carbonate,33S,S-dimethyl dithiocarbonate,34,35 bis(4-nitrophenyl)carbonate,36 carbonyldiimidazole,37,38 methanol,39 and 1,1′-carbonylbisbenzotriazole,40 have been applied for the preparation of ureas (Scheme 1). Although the listed surrogates are less dangerous than phosgene, these solvent-involving methods could not be considered as atom-economical as they lead to substantial amounts of waste and this contradicts with one of the cornerstones of green chemistry.41
Another halide-involving synthesis of bisaryl-substituted ureas is based on metal-catalyzed cross-coupling of aryl halides and unsubstituted urea. Despite good yields of the target ureas, the cross-coupling requires the employment of a toxic heavy metal (i.e. palladium) and occurs either in DME or in dioxane in the presence of toxic xanthene-based bidentate ligands (Scheme 1).42–44
Yet another protocol employs Se-catalyzed oxidative carbonylation of amines with a mixture of CO and O2 (Scheme 1). It is widely used for the preparation of symmetrically substituted ureas and for the synthesis of pyridine-2-yl unsymmetrically substituted ureas in toluene in the presence of selenium.45 In most cases, the carbonylation of amines requires elevated temperatures and moderate to high pressures of CO. Not only is CO toxic, but a risk of explosion of the mixture of CO and O2 should also be taken into account.46 In another method, unsymmetrically substituted pyridyl ureas can be selectively obtained by Se- or SeO2-catalyzed reductive carbonylation of nitropyridines with CO in the presence of various amines (Scheme 1).47–50
In view of the current ecological requirements, on the one hand, and the significance of ureas, on the other hand, the development of solvent- and halide-free sustainable reactions giving these species is a challenging task. It is not surprising that a few efforts have recently been carried out to find out green chemical processes for the synthesis of ureas and the obtained results have been published in this journal (Scheme 2).51–53 The suggested routes start from amine and CO2 and they were conducted under extremely high pressure of CO2 (25–55 atm) and, in some instances, performed in highly reprotoxic solvents such as N-methylpyrrolidinone;54 ureas were isolated in low yields and the scope of the reaction includes only rather simple alkylamines such as butyl- or benzylamine. Although it is obvious that certain progress in the elaboration of green routes to ureas has already been reached, the developed approaches still need further improvement.
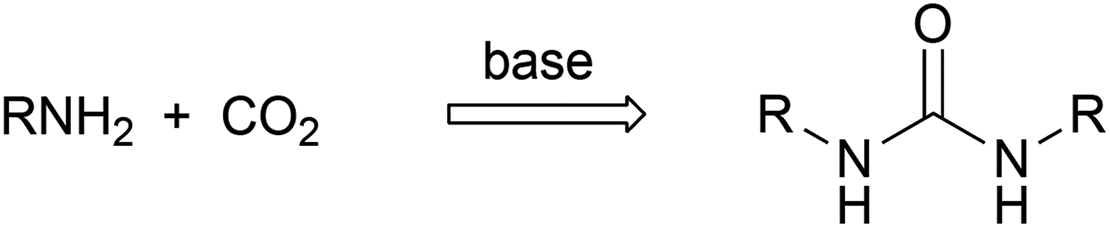 | ||
| Scheme 2 Attempted green synthesis of ureas. | ||
Upon our studies on gold-catalyzed generation of 2-amino-1,3-oxazoles from terminal alkynes and cyanamides in the presence of 2-picoline N-oxide,55 we observed that when excess of 2-picoline N-oxide is used in the reaction, heterocyclization is complicated with a side reaction furnishing 1,1-dimethyl-3-(6-methylpyridin-2-yl)urea. Being interested in understanding this unusual C–H functionalization of 2-picoline N-oxide, in this work, we found a way that turns the side reaction into a high yielding green approach to pyridine-2-yl substituted ureas. We now report on solvent- and halide-free atom-economical synthesis of N,N-dialkyl-N′-pyridine-2-yl ureas based on C–H functionalization between pyridine N-oxides and dialkylcyanamides.
Results and discussion
Toward environmentally benign conditions of the C–H functionalization
In our previous work,55 we reported that the formation of 1,1-dimethyl-3-(6-methylpyridin-2-yl)urea from 2-picoline N-oxide and Me2NCN proceeds under acid catalysis. This result indicates that the first step of the studied reaction is most likely the activation of the cyanamide by protonation. To the best of our knowledge, acid- or metal-catalyzed addition of pyridine N-oxide to nitriles or cyanamides is yet an unknown reaction and only alkylnitrilium salts react with pyridine N-oxide accomplishing different amide derivatives.56,57 The optimization of the reaction conditions was performed with unsubstituted pyridine N-oxide (1a) and Me2NCN (2a). Initially the reaction was performed in neat dimethylcyanamide (2a) (10 equiv.) in the presence of 1.0 equiv. of methane sulfonic acid, MeSO3H (3), at 60 °C for 3 h. Although we achieved full conversion of pyridine N-oxide (1a) (Table 1, entry 1) and the isolated yield of urea 4a was 93% (NMR based conversion is 100%), this approach does not meet the requirements of green chemistry as it far from being atom-economical.|
|
||||
|---|---|---|---|---|
| Entry | Molar ratio of reagents | Conditions (conversion,b %) | ||
| 2a | Acid | |||
| a For more information related to optimization of the reaction conditions see the ESI. b Conversion of the PyO was estimated by 1H NMR. | ||||
| 1 | 10 | MeSO3H | 1.0 | 60 °C, 3 h (100) |
| 2 | 1.0 | MeSO3H | 1.0 | 60 °C, 3 h (85) |
| 3 | 1.5 | MeSO3H | 1.0 | 60 °C, 3 h (98) |
| 4 | 2.0 | MeSO3H | 1.0 | 60 °C, 3 h (98) |
| 5 | 1.5 | MeSO3H | 0.1 | 60 °C, 3 h (46) |
| 6 | 1.5 | MeSO3H | 0.1 | 60 °C, 8 h (74) |
| 7 | 1.5 | H3PO4 | 0.1 | 60 °C, 3 h (7) |
| 8 | 1.5 | CF3SO3H | 0.1 | 60 °C, 3 h (92) |
| 9 | 1.5 | MeSO3H | 1.0 | 40 °C, 3 h (56) |
| 10 | 1.5 | MeSO3H | 1.0 | 60 °C, 1 h (78) |
| 11 | 1.5 | MeSO3H | 1.0 | 60 °C, 2 h (98) |

In the next step the amount of dimethylcyanamide (2a) was reduced to 1.0, 1.5, and 2.0 equiv. (Table 1, entries 2–4), and it appears that 1.5 equiv. of 2a is optimal for achieving almost full conversion of the starting pyridine N-oxide (1a) to substituted urea 4a and the reaction takes 3 h. Further, we attempted the reaction with catalytic amounts of MeSO3H. In the case of 0.1 equiv. of methane sulfonic acid, conversion of 1a was 46% and 74% after 3 h and 8 h, respectively (Table 1, entries 5 and 6). However, a small amount of the yet unidentified by-product (10 and 5% for entries 5 and 6, respectively) was detected in the reaction mixture.
We assumed that changing of the acid would have an effect on the reaction rate and probably would decrease the amount of the undesirable by-product. For rather weak H3PO4, the conversion of the pyridine N-oxide was only 7% after 3 h (Table 1, entry 7), although the employment of the stronger CF3SO3H gave a better result. Conversion of 1a was 92% after 3 h and the amount of the by-product was less than 6% (Table 1, entry 8). However, we continued our study with an equimolar amount of MeSO3H, because the employment of a catalytic amount of either methane sulfonic or trifluoromethane sulfonic acid led to the formation of a small amount of the by-product and required a longer reaction time. The formed pyridinium salt can be easily transformed into the corresponding free base by treatment with potassium carbonate and thus the formed CF3SO3K can be utilized in the preparation of antiperspirants.58
The effect of temperature and reaction time was then studied. The reaction was slow at 40 °C and the conversion of 1a was only 56% after 3 h (Table 1, entry 9). Higher temperatures were not tested insofar as our idea was to find out environmentally friendly conditions that do not anticipate elevated temperatures. With respect to the reaction time, we have found that keeping the mixture at 60 °C for 1 h resulted in poor conversion of 1a, whereas stirring for 2 h is sufficient to achieve almost quantitative conversion of 1a to urea 4a, like in the case when the reaction was performed for 3 h (Table 1, entries 3, 10, and 11).
To demonstrate the possibility of the scale up synthesis of target urea 4a, we carried out the reaction starting from 1.90 g of 1a and the isolated yield of 4a was 3.04 g (92%). We succeeded in recycling 550 mg (78% of excess Me2NCN) of 2a from the reaction mixture.
To summarize the optimization of the reaction conditions, we found that the employment of 1.5 equiv. of cyanamide and 1.0 equiv. of methane sulfonic acid leads to the best synthetic results. It is noteworthy that the excess of cyanamide was recycled by conventional vacuum distillation.
Reaction scope and limitation of the green synthesis of pyridine-2-yl substituted ureas
To verify the scope and limitations of the developed approach several pyridine N-oxides and dialkylcyanamides were tested (Scheme 3).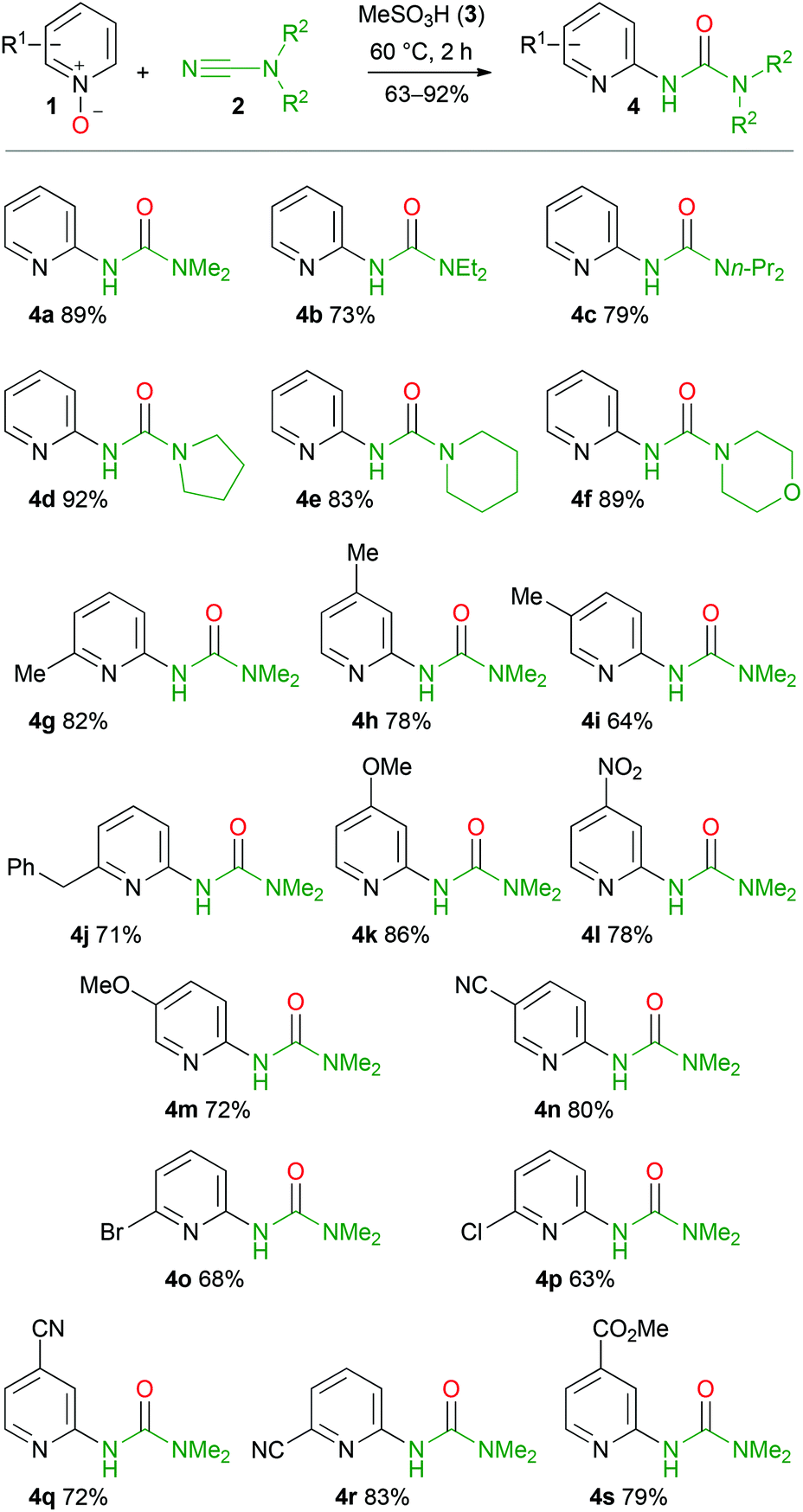 | ||
| Scheme 3 Reaction scope with various pyridine N-oxides and cyanamides. | ||
In most cases, pyridine N-oxides 1 were prepared by oxidation of the corresponding pyridine with a mixture of hydrogen peroxide and acetic acid according to a conventional protocol.59 Alternatively, green oxidation of N-heteroaromatic amines based on a lipase–glucose oxidase system can also be applied in these syntheses.60 Firstly, we tested several dialkylcyanamides, whose intriguing chemistry becomes increasingly popular in recent years.61–68 In all cases, target ureas 4a–f were obtained in 73–92% yields (Scheme 3). Even for 4-morpholinecarbonitrile, urea 4f was isolated in 89% yield. To check the effect of substitution on the pyridine rings and to demonstrate the stability of a wide range of functional groups under the reaction conditions, several N-oxides were tested. Firstly, we checked 2- and 4-substituted pyridine N-oxides and no significant difference between unsubstituted pyridine N-oxide (1a) and its derivatives bearing strong electron donating (4-MeO 1k), weak electron donating (4-Me 1h, 2-Me 1g, 2-PhCH21j), and strong electron withdrawing (4-NO21l) groups was observed. In the case of 3-substituted pyridine derivatives the situation was slightly different. Thus, 3-cyanopyridine N-oxide (1n) reacts similarly to the other substituted pyridine N-oxides and target urea 4n was isolated in 80% yield. At the same time 3-methoxypyridine N-oxide (1m) reacted comparatively slower and full conversion of 1m was achieved only after stirring the reaction mixture at 60 °C for 5 h. The isolated yield of urea 4m was 72%.
Surprisingly, in the case of 3-substituted pyridine N-oxides (3-Me 1i, 3-MeO 1m, and 3-CN 1n), only 2,5-disubstituted pyridine ureas were formed in good yields. We were unable to detect even the traces of the isomeric 2,3-disubstituted pyridine urea in the reaction mixture by 1H NMR. It means that the C–H functionalization proceeds regioselectively and could be used for the synthesis of 5-substituted ureas 4 starting from meta-substituted derivatives of pyridine N-oxides.
The halogen substituted pyridine N-oxides (2-Br 1o, 2-Cl 1p) gave appropriate ureas 4o and 4p in 68 and 63% yields, respectively. Pyridine N-oxides featuring cyano (3-NC 1n, 4-NC 1q, 2-NC 1r) and methoxycarbonyl (4-MeO2C 1s) groups efficiently undergo the reaction and in all cases the corresponding ureas were isolated in 63–83% yields. It is noteworthy that obtained ureas 4n–s are potentially suitable for further modifications. Thus, the halogen atom in the pyridine ring of 4o and 4p could be substituted with various nucleophiles through SNAr69–74 or metal-catalyzed reactions,75–82 whereas the methoxycarbonyl group in 4s could be converted to amides and esters, reduced to alcohols or aldehydes as well as undergo the Barton decarboxylation.83
Based on the above discussion a plausible mechanism for the formation of the urea is given in Scheme 4.
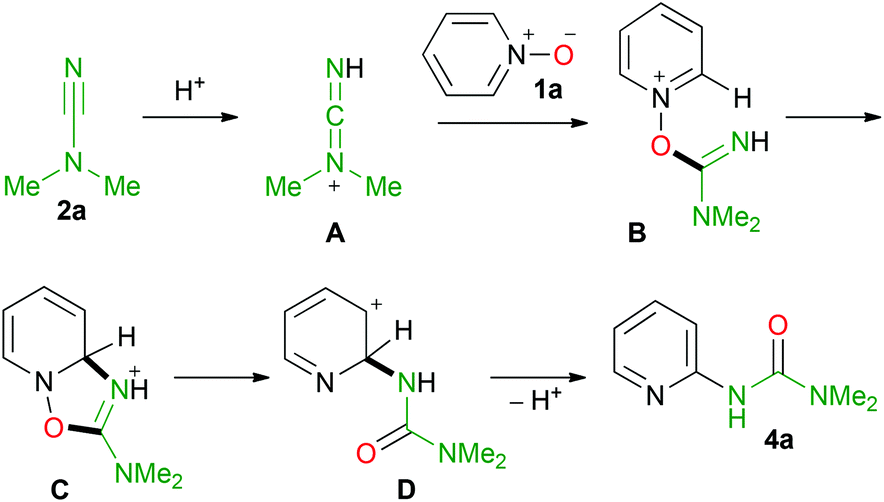 | ||
| Scheme 4 Plausible mechanism for formation of urea 4a. | ||
The first step of the reaction most likely includes the activation of the cyanamide by protonation (A in Scheme 4) followed by nucleophilic addition of pyridine N-oxide to cation A giving B, which then undergoes intramolecular cyclization furnishing C. In C, the heterolytic N–O bond cleavage results in the ring opening reaction giving D, which restores the aromaticity via proton elimination thus accomplishing target urea 4a.
Unexpected reaction pathway for 2-methoxypyridine N-oxide
An interesting and unexpected result has been obtained when the C–H functionalization was performed with 2-methoxypyridine N-oxide (1t). Based on the LC-HRMS data two isomeric compounds were formed and the brutto-formula corresponds to the desired urea 4t. The 1H and 13C as well as 1H–13C HMBC and 1H–13C HSQC spectra clearly indicate that both isomers feature the same fragments, but the position of the substituents on the pyridine ring needs to be specified; the 1H–15N HSQC and 1H–15N HMBC NMR experiments allowed the identification of both products (Scheme 5). A mixture of ureas 5t and 4t (molar ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was formed in 90% overall yield.
:1) was formed in 90% overall yield.
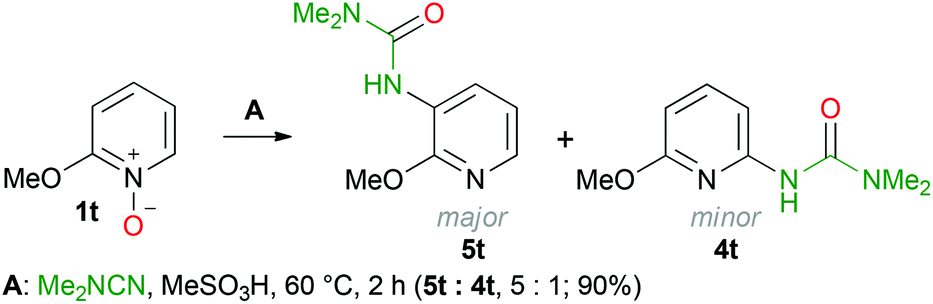 | ||
| Scheme 5 Formation of ureas 5t and 4t. | ||
The cross-peaks corresponding to the coupling between the urea nitrogen and the H-4 proton of the pyridine ring and also between the pyridine nitrogen and the H-5 and H-6 protons were observed in the 1H–15N HMBC spectra of the major isomer. In the case of the minor isomer, we observed the cross-peaks corresponding to the coupling of the pyridine nitrogen and the H-3 and H-5 protons of the pyridine ring (for more details see the ESI†). Moreover, for the major isomer, the signal of the H-5 proton (6.84 ppm) appears as a doublet of a doublet (J = 5.0, 7.8 Hz), whereas for the minor isomer the signal of the H-4 proton (7.39 ppm) appears as a triplet (J = 7.9 Hz). These data agree with the well-known fact that for pyridines the value of the H-2/H-3 coupling constant is smaller than that of H-3/H-4 (for pyridine: 3JH-2/H-3 = 4.88 Hz and 3JH-3/H-4 = 7.67 Hz).84 Unfortunately column chromatography on silica did not allow the separation of a mixture of ureas 5t and 4t, because of their similar retention times. We isolated pure urea 5t (328 mg, 42%) by the repeated recrystallization of a mixture of 5t and 4t from hexane/Et2O. The structure of 3-(2-methoxypyridin-3-yl)-1,1-dimethylurea (5t) in the solid state has been additionally confirmed by single-crystal X-ray diffraction (Fig. 1) The bond length values in the C(2B)–N(2B)–C(7B)–(O(2B))–N(3B) moiety of urea 5t are typical for pyridine-3-yl substituted ureas and are in agreement with the reported data.85–88
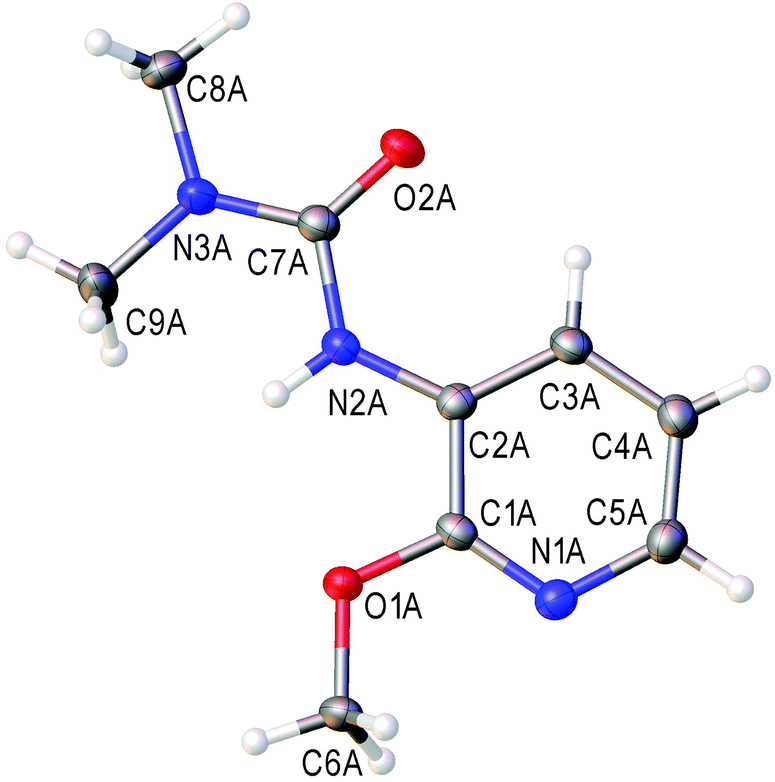 | ||
| Fig. 1 View of the molecular structure of 5t (CCDC 1473655). Thermal ellipsoids are drawn at the 50% probability level. Only one of the two crystallographically independent molecules is presented. Selected bond lengths (Å): N(2B)–C(2B) 1.403(2); N(2B)–C(7B) 1.385(2); O(2B)–C(7B) 1.226(2); N(3B)–C(7B) 1.361(2). | ||
Such unusual results obtained for 2-methoxypyridine N-oxide (1t) forced us to pay more attention to the structures of ureas obtained from pyridine N-oxide bearing either the OMe group or, especially, the halogen atom at the second position, which can react similarly to N-oxide 1t. In the case of 3-(4-methoxypyridin-2-yl)-1,1-dimethylurea (4k), the structure was confirmed by NOESY NMR. We observed two cross-peaks due to the NOE between the protons of the methoxy group and two protons (H-3 and H-5) of the pyridine ring. For bromo- and chlorosubstituted ureas (4o and 4p) the structures were additionally confirmed by single-crystal X-ray diffraction (Fig. 2).
 | ||
| Fig. 2 View of the molecular structure of 4o (CCDC 1473656) and 4p (CCDC 1473657). Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å): N(2)–C(1) 1.400(4); N(2)–C(6) 1.401(5); O(1)–C(6) 1.233(4); N(3)–C(6) 1.350(5) for 4o; N(2)–C(1) 1.396(2); N(2)–C(6) 1.396(2); O(1)–C(6) 1.235(2); N(3)–C(6) 1.349(2) for 4p. | ||
For both ureas 4o and 4p bond length values in the C(1)–N(2)–C(6)–O(1)–N(3) moiety are typical for pyridine-2-yl substituted ureas and correspond to the reported data.85,89–91 All these results indicate that 2-methoxypyridine N-oxide (1t) reacts differently with all other studied pyridine N-oxides and, in our opinion, the detailed investigation of a plausible mechanism goes beyond the scope of this work. We are going to provide a full account report (including both experimental and theoretical studies) on the mechanism of the observed transformation, and the corresponding research work is underway in our group.
Conclusions
We have developed solvent- and halide-free green synthesis of pyridine-2-yl substituted ureas that is based on the facile C–H functionalization of various pyridine N-oxides with a wide range of dialkylcyanamides. The observed C–H functionalization of the pyridine moiety is suitable for the good-to-high yielding synthesis of a broad spectrum of pyridine-2-yl substituted ureas featuring either electron donating or electron withdrawing groups at any position of the pyridine ring. Labile functional groups such as halogen atoms, cyano or methoxycarbonyl groups survive the reaction conditions and obtained ureas could be used for the synthesis of more complex structures.Experimental section
Experimental procedures and analytical data of all compounds (1H and 13C{H} NMR, IR, HRESIMS), copy of the 1H, 13C{H}, and 2D NMR spectra and also X-ray data are available in the ESI.†Acknowledgements
V. A. R. gratefully acknowledges support from the Scientific Council of the President of the Russian Federation (Grant MK-3228.2015.3) and he also thanks Saint Petersburg State University for postdoctoral grant (12.50.1190.2014). G. Z. R. expresses her gratitude towards the Russian Foundation for Basic Research for sabbatical grant (15-33-50913 mol_nr). Structural studies of the obtained compounds were supported by Saint Petersburg State University (research grant 12.37.214.2016). All physicochemical measurements were performed at the Center for Magnetic Resonance, the Center for Chemical Analysis and Material Research, the Center for X-ray Diffraction Methods, and the Educational Center of Chemistry (all belong to Saint Petersburg State University).Notes and references
- I. Gallou, Org. Prep. Proced. Int., 2007, 39, 355–383 CrossRef CAS.
- K. Matsuda, Med. Res. Rev., 1994, 14, 271–305 CrossRef CAS PubMed.
- G. D. Francisco, Z. Li, J. D. Albright, N. H. Eudy, A. H. Katz, P. J. Petersen, P. Labthavikul, G. Singh, Y. Yang, B. A. Rasmussen, Y.-I. Lin and T. S. Mansour, Bioorg. Med. Chem. Lett., 2004, 14, 235–238 CrossRef CAS PubMed.
- K. Pandurangan, J. A. Kitchen, S. Blasco, F. Paradisi and T. Gunnlaugsson, Chem. Commun., 2014, 50, 10819–10822 RSC.
- Y. Zhang, M. Anderson, J. L. Weisman, M. Lu, C. J. Choy, V. A. Boyd, J. Price, M. Sigal, J. Clark, M. Connelly, F. Zhu, W. A. Guiguemde, C. Jeffries, L. Yang, A. Lemoff, A. P. Liou, T. R. Webb, J. L. Derisi and R. K. Guy, ACS Med. Chem. Lett., 2010, 1, 460–465 CrossRef CAS PubMed.
- S. Jiang, S. T. Prigge, L. Wei, Y. Gao, T. H. Hudson, L. Gerena, J. B. Dame and D. E. Kyle, Antimicrob. Agents Chemother., 2001, 45, 2577–2584 CrossRef CAS PubMed.
- E. Comer, B. Munoz, J. A. Beaudoin, S. T. Le Quement, C. Scherer, J. Duvall, N. Kato, M. Maetani and B. E. Braibant, WO2015002755, 2015 Search PubMed; Chem. Abstr. 2015 34782 Search PubMed.
- T. Hu, X. Han, B. Kou, H. Shen, S. Yan and Z. Zhang, WO2016113273, 2016 Search PubMed; Chem. Abstr. 2016 1192981 Search PubMed.
- A. Bader, J. Zhao and A. Guerrero, WO2016081773, 2016 Search PubMed; Chem. Abstr. 2016 851904 Search PubMed.
- M. E. Pacold, K. R. Brimacombe, S. H. Chan, J. M. Rohde, C. A. Lewis, L. J. Y. M. Swier, R. Possemato, W. W. Chen, L. B. Sullivan, B. P. Fiske, S. Cho, E. Freinkman, K. Birsoy, M. Abu-Remaileh, Y. D. Shaul, C. M. Liu, M. Zhou, M. J. Koh, H. Chung, S. M. Davidson, A. Luengo, A. Q. Wang, X. Xu, A. Yasgar, L. Liu, G. Rai, K. D. Westover, M. G. Vander Heiden, M. Shen, N. S. Gray, M. B. Boxer and D. M. Sabatini, Nat. Chem. Biol., 2016, 12, 452–458 CrossRef CAS PubMed.
- Y. Nakazawa, S. Kawano, J. Matsui, Y. Funahashi, O. Tohyama, H. Muto, T. Nakagawa and T. Matsushima, Cancer Sci., 2015, 106, 201–207 CrossRef CAS PubMed.
- J. Lim, E. H. Kelley, J. L. Methot, H. Zhou, A. Petrocchi, H. Chen, S. E. Hill, M. C. Hinton, A. Hruza, J. O. Jung, J. K. F. Maclean, M. Mansueto, G. N. Naumov, U. Philippar, S. Raut, P. Spacciapoli, D. Sun and P. Siliphaivanh, J. Med. Chem., 2016, 59, 6501–6511 CrossRef CAS PubMed.
- L.-Y. Ma, B. Wang, L.-P. Pang, M. Zhang, S.-Q. Wang, Y.-C. Zheng, K.-P. Shao, D.-Q. Xue and H.-M. Liu, Bioorg. Med. Chem. Lett., 2015, 25, 1124–1128 CrossRef CAS PubMed.
- M. O. Anderson, H. Yu, C. Penaranda, B. A. Maddux, I. D. Goldfine, J. F. Youngren and R. K. Guy, J. Comb. Chem., 2006, 8, 784–790 CrossRef CAS PubMed.
- Y. Yin, K. Zheng, N. Eid, S. Howard, J.-H. Jeong, F. Yi, J. Guo, C. M. Park, M. Bibian, W. Wu, P. Hernandez, H. Park, Y. Wu, J.-L. Luo, P. V. LoGrasso and Y. Feng, J. Med. Chem., 2015, 58, 1846–1861 CrossRef CAS PubMed.
- S. Göring, D. Bensinger, E. C. Naumann and B. Schmidt, ChemMedChem, 2015, 10, 511–522 CrossRef PubMed.
- Y. Oguro, N. Miyamoto, K. Okada, T. Takagi, H. Iwata, Y. Awazu, H. Miki, A. Hori, K. Kamiyama and S. Imamura, Bioorg. Med. Chem., 2010, 18, 7260–7273 CrossRef CAS PubMed.
- R. Boulahjar, A. Ouach, S. Bourg, P. Bonnet, O. Lozach, L. Meijer, C. Guguen-Guillouzo, R. Le Guevel, S. Lazar, M. Akssira, Y. Troin, G. Guillaumet and S. Routier, Eur. J. Med. Chem., 2015, 101, 274–287 CrossRef CAS PubMed.
- Y. Dai, K. Hartandi, Z. Ji, A. A. Ahmed, D. H. Albert, J. L. Bauch, J. J. Bouska, P. F. Bousquet, G. A. Cunha, K. B. Glaser, C. M. Harris, D. Hickman, J. Guo, J. Li, P. A. Marcotte, K. C. Marsh, M. D. Moskey, R. L. Martin, A. M. Olson, D. J. Osterling, L. J. Pease, N. B. Soni, K. D. Stewart, V. S. Stoll, P. Tapang, D. R. Reuter, S. K. Davidsen and M. R. Michaelides, J. Med. Chem., 2007, 50, 1584–1597 CrossRef CAS PubMed.
- Y. Oguro, N. Miyamoto, T. Takagi, K. Okada, Y. Awazu, H. Miki, A. Hori, K. Kamiyama and S. Imamura, Bioorg. Med. Chem., 2010, 18, 7150–7163 CrossRef CAS PubMed.
- W. A. Bolhofer, A. A. Deana, C. N. Habecker, J. M. Hoffman, N. P. Gould, A. M. Pietruszkiewicz, J. D. Prugh, M. Lou Torchiana, E. J. Cragoe and R. Hirschmann, J. Med. Chem., 1983, 26, 538–544 CrossRef CAS PubMed.
- X. Le Yin, L. Jiang, N. H. Song and H. Yang, J. Agric. Food Chem., 2008, 56, 4825–4831 CrossRef PubMed.
- J. G. Kim, Y. Takami, T. Mizugami, K. Beppu, T. Fukuda and I. Kataoka, Sci. Hortic., 2006, 110, 219–222 CrossRef CAS.
- V. Papesch and E. F. Schroeder, J. Org. Chem., 1951, 16, 1879–1890 CrossRef CAS.
- U. Petersen, Methoden der Org. Chemie, Houben-Weyl, 1983, vol. E4, pp. 335–337 Search PubMed.
- E. A. German, J. E. Ross, P. C. Knipe, M. F. Don, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2015, 54, 2649–2652 CrossRef CAS PubMed.
- C. L. Cioffi, N. Dobri, E. E. Freeman, M. P. Conlon, P. Chen, D. G. Stafford, D. M. C. Schwarz, K. C. Golden, L. Zhu, D. B. Kitchen, K. D. Barnes, B. Racz, Q. Qin, E. Michelotti, C. L. Cywin, W. H. Martin, P. G. Pearson, G. Johnson and K. Petrukhin, J. Med. Chem., 2014, 57, 7731–7757 CrossRef CAS PubMed.
- G. S. Basarab, J. I. Manchester, S. Bist, P. A. Boriack-Sjodin, B. Dangel, R. Illingworth, B. A. Sherer, S. Sriram, M. Uria-Nickelsen and A. E. Eakin, J. Med. Chem., 2013, 56, 8712–8735 CrossRef CAS PubMed.
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140–148 RSC.
- K. Kurita, T. Matsumura and Y. Iwakura, J. Org. Chem., 1976, 41, 2070–2071 CrossRef CAS.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef.
- L. Cotarca, P. Delogu, A. Nardelli and V. Šunjić, Synthesis, 1996, 553–576 CrossRef CAS.
- R. Ballini, D. Fiorini, R. Maggi, P. Righi, G. Sartori and R. Sartorio, Green Chem., 2003, 5, 396–398 RSC.
- M. Leung, J.-L. Lai, K.-H. Lau, H. Yu and H.-J. Hsiao, J. Org. Chem., 1996, 61, 4175–4179 CrossRef CAS.
- E. Artuso, I. Degani, R. Fochi and C. Magistris, Synthesis, 2007, 3497–3506 CAS.
- J. lzdebski and D. Pawlak, Synthesis, 1989, 423–425 CrossRef.
- K. Otrubova, V. Srinivasan and D. L. Boger, Bioorg. Med. Chem. Lett., 2014, 24, 3807–3813 CrossRef CAS PubMed.
- R. A. Batey, V. Santhakumar, C. Yoshina-Ishii and S. D. Taylor, Tetrahedron Lett., 1998, 39, 6267–6270 CrossRef CAS.
- S. H. Kim and S. H. Hong, Org. Lett., 2016, 18, 212–215 CrossRef CAS PubMed.
- A. R. Katritzky, D. P. M. Pleynet and B. Yang, J. Org. Chem., 1997, 62, 4155–4158 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC.
- G. A. Artamkina, A. G. Sergeev and I. P. Beletskaya, Tetrahedron Lett., 2001, 42, 4381–4384 CrossRef CAS.
- A. Abad, C. Agulló, A. C. Cuñat and C. Vilanova, Synthesis, 2005, 915–924 CrossRef CAS.
- B. J. Kotecki, D. P. Fernando, A. R. Haight and K. A. Lukin, Org. Lett., 2009, 11, 947–950 CrossRef CAS PubMed.
- X. Zhang, D. Li, X. Ma, Y. Wang and G. Zhang, Synthesis, 2013, 1357–1363 CrossRef CAS.
- D. J. Díaz, A. K. Darko and L. McElwee-White, Eur. J. Org. Chem., 2007, 4453–4465 CrossRef.
- S. Lu, Tetrahedron Lett., 1999, 40, 4845–4846 CrossRef.
- J. Chen, G. Ling and S. Lu, Tetrahedron, 2003, 59, 8251–8256 CrossRef CAS.
- J. Mei, Y. Yang, Y. Xue and S. Lu, J. Mol. Catal. A: Chem., 2003, 191, 135–139 CrossRef CAS.
- J. Chen and S. Lu, Appl. Catal., A, 2004, 261, 199–203 CrossRef CAS.
- A. Ion, V. Parvulescu, P. Jacobs and D. De Vos, Green Chem., 2007, 9, 158–161 RSC.
- C. Wu, H. Cheng, R. Liu, Q. Wang, Y. Hao, Y. Yu and F. Zhao, Green Chem., 2010, 12, 1811–1816 RSC.
- T. Jiang, X. Ma, Y. Zhou, S. Liang, J. Zhang and B. Han, Green Chem., 2008, 10, 465–469 RSC.
- J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmer and A. J. Hunt, Green Chem., 2016, 18, 3990–3996 RSC.
- V. A. Rassadin, V. P. Boyarskiy and V. Y. Kukushkin, Org. Lett., 2015, 17, 3502–3505 CrossRef CAS PubMed.
- M. G. Hitzler, C. C. Freyhardt and J. C. Jochims, J. Prakt. Chem., 1996, 338, 243–250 CrossRef CAS.
- S. N. Karad and R.-S. Liu, Angew. Chem., Int. Ed., 2014, 53, 5444–5448 CrossRef CAS PubMed.
- B. Banowski and M. Claas, WO2015055200, 2015 Search PubMed; Chem. Abstr. 2015 691565 Search PubMed.
- A. R. Katritzky, J. A. T. Beard and N. A. Coats, J. Chem. Soc., 1959, 3680–3683 RSC.
- F. Yang, X. Zhang, F. Li, Z. Wang and L. Wang, Green Chem., 2016, 18, 3518–3521 RSC.
- M.-H. Larraufie, G. Maestri, M. Malacria, C. Ollivier, L. Fensterbank and E. Lacôte, Synthesis, 2012, 1279–1292 CAS.
- M.-H. Larraufie, C. Ollivier, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 2178–2181 CrossRef CAS PubMed.
- N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2013, 257, 2293–2316 CrossRef CAS.
- D. D. Nekrasov, Chem. Heterocycl. Compd., 2004, 40, 1107–1123 CrossRef CAS.
- R. Crutchley, Coord. Chem. Rev., 2001, 219–221, 125–155 CrossRef CAS.
- A. S. Smirnov, E. S. Yandanova, N. A. Bokach, G. L. Starova, V. V. Gurzhiy, M. S. Avdontceva, A. A. Zolotarev and V. Y. Kukushkin, New J. Chem., 2015, 39, 9330–9344 RSC.
- D. S. Bolotin, V. A. Rassadin, N. A. Bokach and V. Y. Kukushkin, Inorg. Chim. Acta, 2016 DOI:10.1016/j.ica.2016.02.025.
- M. Y. Demakova, D. S. Bolotin, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, Inorg. Chim. Acta, 2015, 425, 114–117 CrossRef CAS.
- T. Lister, R. H. Prager, M. Tsaconas and K. L. Wilkinson, Aust. J. Chem., 2003, 56, 913–916 CrossRef CAS.
- S. J. Connon and A. F. Hegarty, Eur. J. Org. Chem., 2004, 3477–3483 CrossRef CAS.
- Q. Wang, H. Sun, H. Cao, M. Cheng and R. Huang, J. Agric. Food Chem., 2003, 51, 5030–5035 CrossRef CAS PubMed.
- J. E. Argüello, L. C. Schmidt and A. B. Peñéñory, Org. Lett., 2003, 5, 4133–4136 CrossRef PubMed.
- F. Ghelfi, M. Pattarozzi, F. Roncaglia, A. Parsons, F. Felluga, U. Pagnoni, E. Valentin, A. Mucci and F. Bellesia, Synthesis, 2008, 3131–3141 CrossRef CAS.
- X. Jia, L. Yu, J. Liu, Q. Xu, M. Sickert, L. Chen and M. Lautens, Green Chem., 2014, 16, 3444–3449 RSC.
- L. Nicolas, P. Angibaud, I. Stansfield, L. Meerpoel, S. Reymond and J. Cossy, RSC Adv., 2013, 3, 18787–18790 RSC.
- S. M. Crawford, C. B. Lavery and M. Stradiotto, Chem. – Eur. J., 2013, 19, 16760–16771 CrossRef CAS PubMed.
- Q. Shen, S. Shekhar, J. P. Stambuli and J. F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371–1375 CrossRef CAS PubMed.
- C. Salomé, M. Schmitt and J.-J. Bourguignon, Tetrahedron Lett., 2009, 50, 3798–3800 CrossRef.
- L. Alcaraz, C. Bennion, J. Morris, P. Meghani and S. M. Thom, Org. Lett., 2004, 6, 2705–2708 CrossRef CAS PubMed.
- X. Wang, A. Guram, M. Ronk, J. E. Milne, J. S. Tedrow and M. M. Faul, Tetrahedron Lett., 2012, 53, 7–10 CrossRef CAS.
- A. Reichelt, J. R. Falsey, R. M. Rzasa, O. R. Thiel, M. M. Achmatowicz, R. D. Larsen and D. Zhang, Org. Lett., 2010, 12, 792–795 CrossRef CAS PubMed.
- J. B. Arterburn, C. Corona, K. V. Rao, K. E. Carlson and J. A. Katzenellenbogen, J. Org. Chem., 2003, 68, 7063–7070 CrossRef CAS PubMed.
- D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- V. M. S. Gil and A. J. L. Pinto, Mol. Phys., 2006, 19, 573–575 CrossRef.
- P. Byrne, D. R. Turner, G. O. Lloyd, N. Clarke and J. W. Steed, Cryst. Growth Des., 2008, 8, 3335–3344 CAS.
- K. Yamaguchi and K. Shudo, J. Agric. Food Chem., 1991, 39, 793–796 CrossRef CAS.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2006, 6, 161–173 CAS.
- R. W. Troff, R. Hovorka, T. Weilandt, A. Lützen, M. Cetina, M. Nieger, D. Lentz, K. Rissanen and C. A. Schalley, Dalton Trans., 2012, 41, 8410–8420 RSC.
- B. Ośmiałowski, K. Mroczyńska, E. Kolehmainen, M. Kowalska, A. Valkonen, M. Pietrzak and K. Rissanen, J. Org. Chem., 2013, 78, 7582–7593 CrossRef PubMed.
- V. Velikova, O. Angelova and K. Kossev, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1273–1275 Search PubMed.
- Y. Sun, Z. Zhang, X. Wang, X. Li, L. Weng and X. Zhou, Organometallics, 2009, 28, 6320–6330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1473655–1473657. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc02556k |
| This journal is © The Royal Society of Chemistry 2016 |