
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of 6-amyl-α-pyrone as a potential biomass-derived platform molecule†
Md. Imteyaz
Alam‡
*a,
Shelaka
Gupta‡
a,
Ashish
Bohre
b,
Ejaz
Ahmad
a,
Tuhin S.
Khan
a,
Basudeb
Saha
*bc and
M. Ali
Haider
*a
aRenewable Energy and Chemicals Laboratory, Department of Chemical Engineering, Indian Institute of Technology Delhi, Hauz Khas, Delhi 110016, India. E-mail: imteyaz84@gmail.com; haider@iitd.ac.in; Fax: +91-11-2658-2037; Tel: +91-11-26591016
bLaboratory of Catalysis, Department of Chemistry, University of Delhi, Delhi 110007, India. E-mail: bsaha@udel.edu
cCatalysis Center for Energy Innovation, University of Delaware, Newark, DE 19713, USA
First published on 19th October 2016
Abstract
Catalytic transformation routes for the valorization of biomass-derived 6-amyl-α-pyrone (6PP) were explored for the first time. Ring-opening and decarboxylation of 6PP in water yielded non-2-en-4-one with 95% conversion and nearly 60% selectivity at 498 K, without the requirement of an acid catalyst. The decarboxylated product was further hydrogenated to yield 4-nonanone over a Pd/C catalyst. In order to produce longer chain hydrocarbons of diesel and jet fuel range, C–C coupling via aldol condensation of nonanone with furfural and 5-hydroxymethyl furfural (HMF) was experimented over a mixed oxide (CaO–MgO) catalyst measuring up to 40% and 60% yield of the branched aldol products at 443 K and 393 K respectively.
In an effort to produce fuels and chemicals from biomass, considerable attention has been given to the development of novel processes, producing long-chain and branched hydrocarbons. One prominent strategy proposed by Dumesic and co-workers1,2 involves the synthesis of ketones from biomass-derived platform molecules (i.e. biomass-based building block molecules utilised to produce a variety of chemicals and fuels).3–8 In this method, catalytic upgrading of biomass was carried out to produce C9 linear ketones having direct application as a fuel additive or upgradation to gasoline and diesel.2 The reported method essentially requires the integration of several steps, which include the conversion of lignocellulosic biomass into C6 sugars, dehydration of sugars to HMF, rehydration and ring-opening of HMF to levulinic acid (LA), hydrogenation and cyclization of LA to γ-valerolactone (GVL), ring-opening and hydrogenation of GVL to pentanoic acid and subsequent ketonization via a C–C coupling reaction to produce 5-nonanone.2,9 Understandably, for commercial success, the method needs to be simpler, the one that involves fewer steps and utilizes inexpensive catalysts. Thus, in this study, a novel strategy is proposed to produce unsaturated/saturated C9 linear ketones directly from biomass in two or three steps. The proposed method is based on the ring-opening and decarboxylation of 6PP yielding unsaturated C9 linear ketones. 6PP is obtained directly from solid-state fermentation of the lignocellulosic biomass.10 At present, 6PP is used in the food industry as a coconut aroma ingredient11 for enhancing the flavour of soft drinks, yogurts etc. Species of Trichoderma are known to produce 6PP on solid-state fermentation of waste biomass such as sugarcane bagasse or industrial agricultural waste.10 Since 6PP is toxic to the fungal growth, extractive fermentation techniques have been applied to increase the yield up to 7.1 g l−1.12 Interestingly, 6PP can also serve as a potential platform chemical to yield fuel-range hydrocarbons via further processing. We have performed ring-opening and decarboxylation of 6PP in the aqueous phase to achieve a high yield of non-2-en-4-one without the requirement of an acid catalyst. The reactivity of 6PP towards ring-opening and decarboxylation resembled the structurally analogous triacetic acid lactone (TAL), a biomass-derived platform molecule.13 6PP, having unsaturation in the lactone ring, underwent this reaction under relatively mild conditions compared to the saturated lactones (e.g. GVL), wherein high temperatures (>573 K) and an acid catalyst are required.14
Experimental results showing the representative reactions of 6PP undergoing ring-opening and decarboxylation in water–THF solvent, with and without an acid catalyst, are presented in Table 1. Complete conversion (up to 95%) of 6PP was obtained at 498 K in a water–THF mixture in 8 hours, without the requirement of an acid catalyst (Table 1, entry 1). The selectivity towards the product (non-2-en-4-one) was calculated to be 58%. Reaction in water was not tested due to the immiscible nature of 6PP. On performing this reaction in THF as the solvent and in the presence of a silica–alumina catalyst, 6PP conversion was reduced to 18% (Table 1, entry 2), which was due to the residual water present in THF. Therefore, water was considered to be absolute essential to carry out this reaction, which appears as the reactant in reaction stoichiometry as shown in Scheme 1.
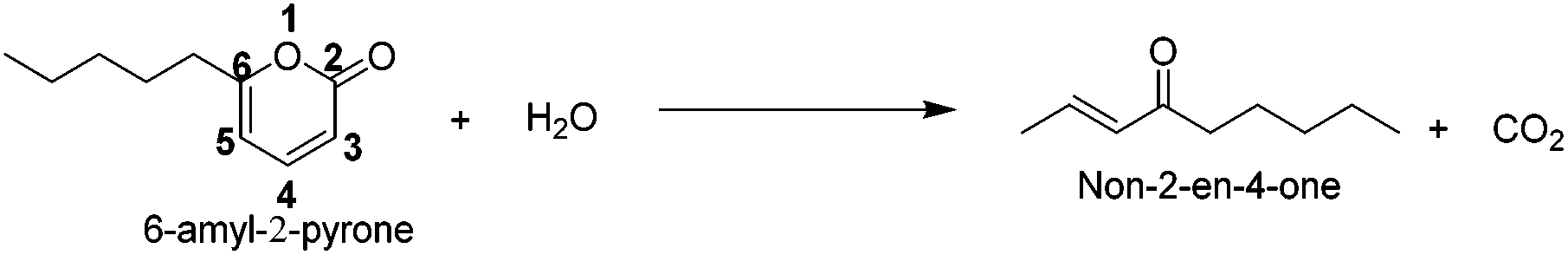 | ||
| Scheme 1 Ring-opening and decarboxylation of 6PP into non-2-en-4-one. | ||
| Entry | Substrate | T (K) | t (h) | Catalyst | % C | % S |
|---|---|---|---|---|---|---|
Substrate = 2 wt% of solvent; total volume of the solvent = 50 ml; solvent THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 3:1; catalyst = 50 wt% of substrate; T, temperature; t, time; C, conversion; S, selectivity.a THF alone as the solvent medium. :water = 3:1; catalyst = 50 wt% of substrate; T, temperature; t, time; C, conversion; S, selectivity.a THF alone as the solvent medium. |
||||||
| 1 | 6PP | 498 | 8 | — | 95 | 58 |
| 2 | 6PPa | 498 | 8 | Silica–alumina | 18 | 78 |
| 3 | 6PP | 463 | 8 | — | 55 | 41 |
| 4 | 6PP | 473 | 10 | Silica–alumina | >99 | 65 |
| 5 | 6PP | 463 | 8 | Silica–alumina | 77 | 65 |
| 6 | 6PP | 463 | 8 | γ-Alumina | 75 | 65 |
Compared to TAL, 6PP requires a relatively higher temperature for ring-opening and decarboxylation, the reasons for which could be attributed to the absence of a β-keto group which facilitates ring-opening and decarboxylation in TAL.15 Interestingly, earlier work on TAL had considered the hydroxyl (or ketone functionality) substitution at the β-position to be essential for the ring-opening and decarboxylation of the 2-pyrone-ring.15,16 Remarkably, 6PP without having any hydroxyl substitution showed relatively easier ring-opening and decarboxylation at a lower temperature compared to the saturated lactone. On reducing the reaction temperature to 463 K, the conversion and product selectivity were reduced to 55% and 41% respectively (Table 1, entry 3). 6PP conversion in water was fitted to the Arrhenius equation (Fig. S5†) and the apparent activation energy was calculated to be 166 kJ mol−1. As suggested for TAL, the rate-determining step is likely to be ring-opening.15Fig. 1a shows the elementary steps required for ring-opening of 6PP in water. The ring-opening reaction proceeds in two steps, nucleophilic addition of water on the carbonyl-carbon (C2) which leads to the ring-opening via a proton transfer to the ring-oxygen (O1). DFT calculations estimated the intrinsic activation barrier for the water addition step to be 180 kJ mol−1 (Fig. 1a), which is comparable to the experimentally measured value.
 | ||
| Fig. 1 Reaction diagram for the ring-opening of 6PP in (a) non-catalytic aqueous system and (b) acid catalysed system of γ-alumina. * indicates the atom by which species is making a bonding interaction with the surface. | ||
In the presence of an acid catalyst, the ring-opening and decarboxylation are likely to be accelerated. The Brønsted acidic protons facilitate proton transfer to the ring (O1) or carbonyl oxygen (O2), leading to the formation of stable carbenium or oxocarbenium ions in water for subsequent ring-opening.17 In contrast, Lewis acidic systems are expected to provide greater control on the product selectivity with reduced conversion as observed in the case of GVL ring-opening and decarboxylation.18 Indeed, on performing the reaction in a water–THF solvent mixture on a silica–alumina catalyst, an appreciable increase in conversion (>99%, Table 1, entry 4) was measured at 473 K compared to the non-catalytic aqueous system (Table 1, entry 1). The acid catalyzed conversion remained high (∼77%) even at a reduced temperature of 463 K compared to the non-catalytic system (Table 1, entry 3). Interestingly, the reaction on the Lewis acidic γ-alumina catalyst showed similar results. The conversion and product selectivity values (Table 1, entry 6) measured at 463 K for γ-alumina are comparable to the values measured for silica–alumina (Table 1, entry 5). The Arrhenius fits for both the catalysts estimated an apparent activation energy of 99 kJ mol−1 for γ-alumina and 125 kJ mol−1 for silica–alumina (Fig. S5†). The results, however; stand in-contrast to the reported reactivity trend for GVL ring-opening and decarboxylation, wherein varying total yields measuring 89% and 59% of the decarboxylated product were observed over the silica–alumina and γ-alumina catalysts, respectively.18
The ring-opening and decarboxylation of 6PP was accelerated with lower activation barriers in acid catalysts compared to the non-catalytic system. An insight into the mechanism of the acid catalyzed reaction may be obtained from the DFT calculations for ring-opening of 6PP performed on the γ-alumina surface. The {110} surface of γ-alumina was chosen for studying the ring-opening reaction.19 The 6PP molecule is preferentially adsorbed at the tri-coordinated aluminum center, Al(III) on the γ-alumina (110) surface by forming a bond between the carbonyl oxygen (O2) and surface Al(III) atoms. The ring-opening of 6PP proceeds with a nucleophilic water addition to the carbonyl carbon (C2) leading to acyl cleavage and hydrogen transfer (from the water) to the surface oxygen. In the ring-opened structure (Fig. 1b, 2b) the molecule was adsorbed on the surface with both carbonyl-oxygen (O2) and ring-oxygen (O1) forming a bond with two neighboring surface Al(III) atoms. The intrinsic activation energy for the ring-opening step was calculated to be 36 kJ mol−1 (Fig. 1b, 2a to TS2a). In the next step, the ring-oxygen was observed to receive back the proton from the surface oxygen to yield the same ring-opened intermediate (Fig. 1b, 2c) as in the non-catalytic aqueous system (Fig. 1a, 1c). The proton-transfer step is endothermic and the activation energy for this step is likely to be higher than 98 kJ mol−1 (Fig. 1, 2b to 2c). The apparent activation energy measured for the γ-alumina catalyzed reaction was 99 kJ mol−1, which is comparable to the DFT calculated value. The unsaturated keto product obtained was hydrogenated to give 4-nonanone. On a Pd/C catalyst, hydrogenation was performed to give >99% conversion with 83% product selectivity at 373 K.
In order to introduce a valorization route for high volume market opportunity of 6PP, nonanone may be reacted with furanics (furfural and HMF) for C–C coupling to yield C14 or C15 range products. C–C coupling of the biomass-derived furanic intermediates with suitable carbonyl compounds is generally performed via aldol condensation or hydroxyalkylation/alkylation pathways.20,21 These pathways for C–C coupling have been reported in our previous publications and by other researchers as well.22,23 For example, an organocatalyst, 1,8-diaza-bicycloundec-7-ene, showed significant improvement in the C–C coupling reactivity of furan aldehyde with ketone and hydroxyl ketone.24–26 Studies on the aldol reactions of biomass-derived furanics have utilized acetone as the reactant, wherein two or more consecutive crossed aldol reactions are necessary to yield C12 or larger fraction drop-in biofuels.20 Handling of acetone is also a problem because of the high volatility and low flash point. Comparatively, nonanone is easier to handle and directly yield C14/C15 fractions on reacting with furanics in a single aldol reaction, showing considerable merits. In addition, as pointed out by Chen et al., the condensation products of nonanones are likely to yield branched alkanes on hydrodeoxygenation (HDO) with higher octane numbers.27 Furthermore, the aldol reaction with nonanone is carried out under neat conditions without the requirement of an organic solvent, thus reducing the cost of the process compared to the furanic-acetone reaction.27 All of the aforementioned reasons make the proposed process an attractive route.
To demonstrate the C–C coupling route, 5-nonanone was allowed to condense with furanic aldehydes in the presence of a Brønsted base to obtain adducts F5N and H5N as shown in Scheme 2. Dumesic, Huber and co-workers have shown the activity of mixed oxide catalysts to achieve a higher yield of the crossed aldol products on reacting furanics with acetone.28 Inspired by their work, Chen et al. have recently experimented with the aldol reaction of 3-pentanone with furfural under solvent free conditions to produce branched alkanes.27 The authors reported a maximum 60% yield of the aldol product of 3-pentanone with furfural on the CaO catalyst at 443 K. Interestingly, the catalytic activity of the CaO catalyst for the aldol reaction was measured to be higher than that of the mixed oxides of MgO–ZrO2, MgO–La2O3 and hydrotalcite of Mg–Al.27 Encouraged by this, an aldol reaction of 5-nonanone with furfural was experimented with the CaO catalyst, under the same reaction conditions.27 However, compared to 3-pentanone, the aldol product yield was significantly reduced to only 30% at 443 K in 8 hours.27
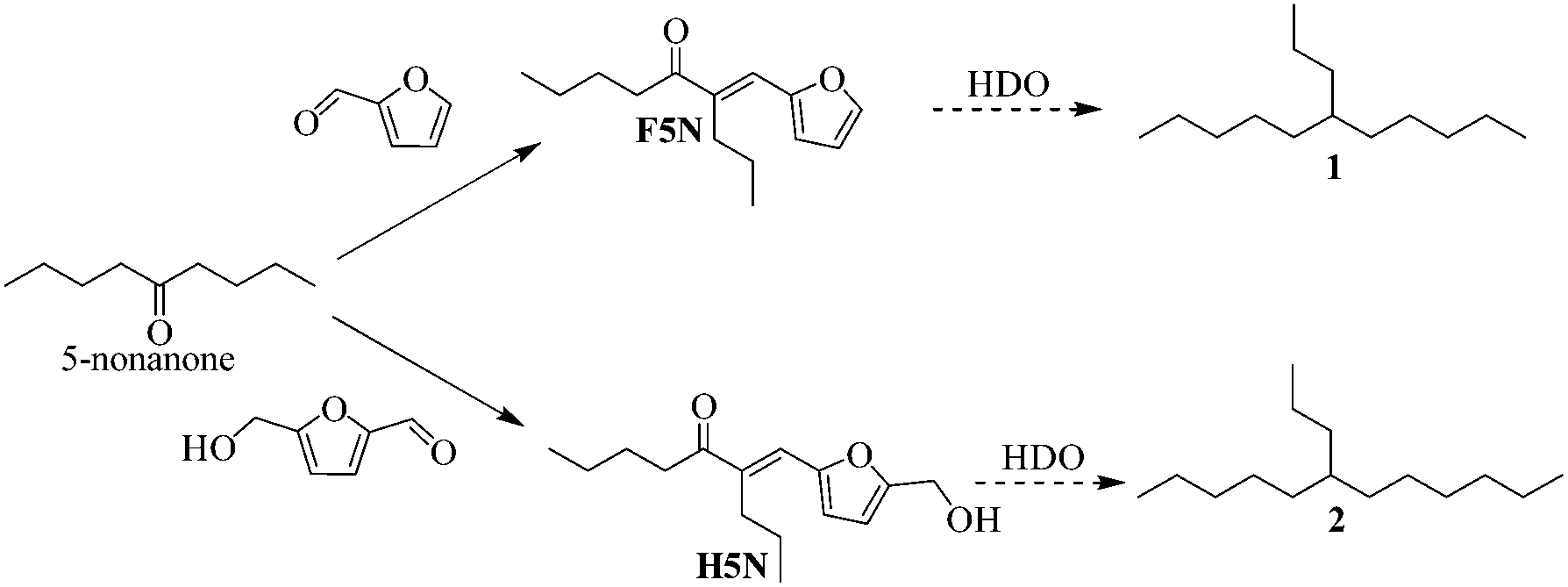 | ||
| Scheme 2 Reaction scheme showing base catalyzed aldol condensation products of 5-nonanone with furfural (F5N) and HMF (H5N). Routes for HDO to yield branched alkanes are shown by the dashed lines. | ||
In order to achieve a higher aldol product yield, mixed oxides of Ca and Mg were synthesized as the base catalyst for the aldol reaction of 5-nonanone with furanics. The XRD pattern showing the peaks corresponding to pure cubic phase CaO and periclase MgO is given in Fig. S6(a).† In the XRD spectra, traces of some CaCO3 and Ca(OH)2 were observed, suggesting rapid carbonation and hydration of the mixed oxide in air and at room temperature which was further confirmed with the FT-IR spectrum (Fig. S6(b)†) of the synthesized CaO–MgO catalyst.
Fig. 2a and b show the obtained yields of aldol products (F5N and H5N) at different temperatures and times of reaction over the CaO–MgO catalyst. The F5N yield measuring up to 40% was observed for a reaction between furfural and 5-nonanone at 443 K for 12 h, which was followed by a plateau on extending the reaction time as shown in Fig. 2a. In the case of HMF condensation with 5-nonanone, 40% yield of H5N was obtained at 393 K for 8 h. A 20% improvement in the yield was observed on continuing the reaction for 12 h with a total yield of 60%. The reaction pattern suggested a faster rate of formation of F5N and H5N in the beginning of the reaction, followed by a slow rate. No improvement in the product yield was observed when the reaction was conducted for 18 hours (Fig. 2a). A higher yield of the aldol product was obtained on reacting 5-nonanone with HMF (compared to furfural), likely due to the mesomeric effect of the electron withdrawing hydroxymethyl group in HMF which increases the positive charge density at the carbonyl carbon (C2) and thus facilitates a favourable C–C coupling with the carbanion.
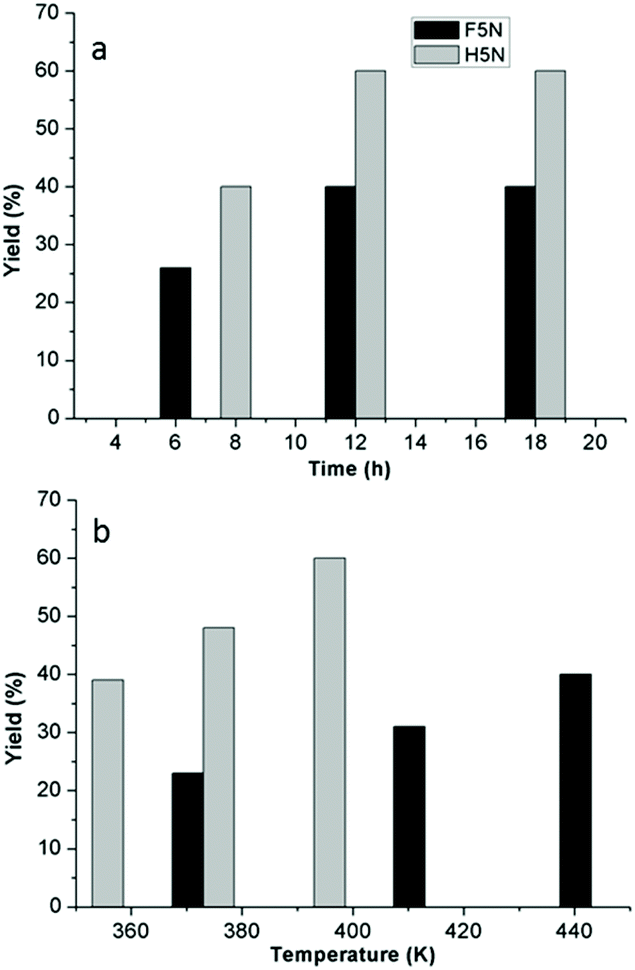 | ||
| Fig. 2 Yields of aldol condensation products (F5N and H5N) of 5-nonanone reaction with furfural and HMF, measured over (a) time and (b) temperature. | ||
The activity of the CaO–MgO catalyst could be attributed to the synergistic effect between the active phase of Ca2+ and Mg2+ in the binary system of the mixed oxide. Both CaO and MgO are known for strong basic sites.28 High basic site density of the catalyst is likely to facilitate the formation of the enolate intermediate by the abstraction of the α-proton, accelerating the C–C coupling.20 The branched aldol products (F5N and H5N) were obtained in the liquid state at room temperature, which may directly undergo a HDO reaction to form C14 and C15 branched alkanes. The HDO route shown in Scheme 2 has been experimented previously to give a high yield of the alkane product.23,27,29 Chen et al. have performed experiments on the HDO of the aldol product of 3-pentanone and HMF using M/SiO2 (M = Fe, Cu, Co, Ni, Pt) catalysts and obtained up to 95% carbon yield of the branched alkane product, with negligible fragmentation.27 Our earlier study on the HDO of the aldol product of HMF with acetone had shown similar results, yielding 96% alkane with the Pd/zeolite-β catalyst.23 The recyclability of the catalyst was studied for the reaction between HMF and 5-nonanone for 12 h at 393 K. The results (Fig. S9†) showed a slight decrease in the yield of H5N from 60% to 48% corresponding to the 1st and 4th cycle, possibly due to a slight loss in the measured basicity of the recovered CaO–MgO catalyst (Table S1†).
Ketones, in general, may show different aldol reactivities based upon the position of the keto functionality resulting in a varying α-H reactivity and steric effect.27 In order to study the effect of the position of the carbonyl group, aldol reactions of HMF with 2-nonanone and 4-nonanone were performed at 393 K for 12 h using the CaO–MgO catalyst (Fig. S7 and S8†). Compared to 5-nonanone, the obtained aldol product yield of HMF with 4-nonanone (57%) was negligibly reduced. However, the aldol reaction of HMF with 2-nonanone showed a slight increase (67%) in the product yield likely due to the less sterically hindered methyl group.
Conclusions and outlook
6PP obtained from the fermentation of lingocellulosic biomass shows considerable potential to be developed as a platform molecule. 6PP underwent ring-opening and decarboxylation in the presence of water, without the requirement of a catalyst. Compared to the non-catalytic reaction, the acid catalyzed ring-opening reactions were accelerated with relatively lower activation barriers. Experimentally measured apparent activation energy for the ring-opening and decarboxylation reaction in water was comparable to the DFT calculated intrinsic barrier for ring-opening, which indicated ring-opening to be rate limiting. Similarly, for the acid-catalyzed reaction in water and in the presence of γ-alumina, the measured apparent activation energy was comparable to the DFT calculated intrinsic endothermicity of the proton transfer step, which was essential for obtaining the ring-opened intermediate. Interestingly, in contrast to the reactivity trend reported for saturated lactones such as GVL, 6PP as an unsaturated lactone showed a different trend in reactivity with a negligible change in conversion and product selectivity on the Brønsted and Lewis acid catalysts. A valorization route for 6PP was envisaged by performing the aldol condensation reaction of nonanones with furfural and HMF. The reaction was performed under neat conditions without the requirement of an organic solvent. A mixed oxide catalyst of Ca and Mg was utilized for obtaining higher yields of the aldol products. The liquid aldol products may directly undergo catalytic upgrading via HDO to yield branched C14 or C15 alkanes to be used as diesel and jet range fuels.Compared to TAL, 6PP as a biomass-derived 2-pyrone shows significant merits for development into a platform molecule. TAL is obtained from the fermentation of glucose.30 In contrast, 6PP is obtained directly from the fermentation of lignocellulosic biomass. So far, the reported maximum yield for TAL by applying metabolic engineering is 5.2 g l−1, which is lower than the reported maximum yield of 6PP.31,32 Interestingly, improvement in the 6PP yield has not been tried with the same rigor using any genetic engineering technique. Once produced in high yield, the fermentation process may be integrated with the catalytic process proposed in this work to produce biorenewable fuels. Similarly, other valorization routes for 6PP may be explored. Structurally, 6PP is a 6-membered unsaturated, heterocyclic compound with a cyclic ester functionality. Numerous possibilities arise on exploring the reactivity of 6PP to produce fuels and chemicals. George Kraus and co-workers have shown the potential for converting the 2-pyrone (TAL) into 2-pyridones, to be used as pharmaceutical building blocks.33 Furthermore, selective partial and complete hydrogenation of 6PP followed by ring-opening and decarboxylation will lead to a myriad of interesting products to be used as fuels, chemicals, polymers and pharmaceuticals.
Acknowledgements
Financial support from the Department of Biotechnology (BT/COE/34/SP15097/2015) and the Science and Engineering Research Board (PDF/2015/001040), Government of India to MAH and MIA respectively is appreciated. Computational resources of High Performance Computing Facility at IIT Delhi are acknowledged. AB would like to thank the University Grant Commission, Government of India, for a D. S. Kothari Post-doctoral Research Fellowship. BS would like to acknowledge the financial support from the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award number DE-SC0001004 for the later stages of this manuscript.Notes and references
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- T. J. Farmer and M. Mascal, in Introduction to Chemicals from Biomass: Second Edition, ed. J. Clark and F. Deswarte, John Wiley & Sons, Ltd., 2nd edn, 2015, pp. 89–155 Search PubMed.
- S. Gupta, M. I. Alam, T. S. Khan, N. Sinha and M. A. Haider, RSC Adv., 2016, 6, 60433–60445 RSC.
- E. Ahmad, M. I. Alam, K. K. Pant and M. A. Haider, Green Chem., 2016, 18, 4804–4823 RSC.
- M. I. Alam, S. Gupta, E. Ahmad and M. A. Haider, in Sustainable Catalytic Process, ed. B. Saha and M. Fan, Elsevier, 2015, pp. 157–177 Search PubMed.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992–999 RSC.
- A. D. S. Ramos, S. B. Fiaux and S. G. F. Leite, Braz. J. Microbiol., 2008, 39, 712–717 CrossRef.
- H. Kono, S. Kawano, K. Tajima, T. Erata and M. Takai, J. Agric. Food Chem., 1972, 20, 415–423 Search PubMed.
- S. Oda, K. Isshiki and S. Ohashi, Process Biochem., 2009, 44, 625–630 CrossRef CAS.
- M. Chia, T. J. Schwartz, B. H. Shanks and J. A. Dumesic, Green Chem., 2012, 14, 1850 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- M. Chia, M. A. Haider, G. Pollock, G. A. Kraus, M. Neurock and J. A. Dumesic, J. Am. Chem. Soc., 2013, 135, 5699–5708 CrossRef CAS PubMed.
- J. A. Dumesic and M. Chia, US Patent, US20120283477, 2013 Search PubMed.
- S. Gupta, R. Arora, N. Sinha, M. I. Alam and M. A. Haider, RSC Adv., 2016, 6, 12932–12946 RSC.
- D. Wang, S. H. Hakim, D. M. Alonso and J. A. Dumesic, Chem. Commun., 2013, 49, 7040–7042 RSC.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- M. Balakrishnan, E. R. Sacia, S. Sreekumar, A. A. Gokhale, C. D. Scown, F. D. Toste, M. Balakrishnan, E. R. Sacia, S. Sreekumar, G. Gunbas and A. A. Gokhale, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1–6 CrossRef PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 2, 4044–4098 CrossRef PubMed.
- A. Bohre, B. Saha and M. M. Abu-Omar, ChemSusChem, 2015, 8, 4022–4029 CrossRef CAS PubMed.
- A. V. Subrahmanyam, S. Thayumanavan and G. W. Huber, ChemSusChem, 2010, 3, 1158–1161 CrossRef CAS PubMed.
- C. Zhu, T. Shen, D. Liu, J. Wu, Y. Chen, L. Wang, K. Guo, H. Ying and P. Ouyang, RSC Adv., 2016, 6, 62974–62980 RSC.
- C. Zhu, T. Shen, D. Liu, J. Wu, Y. Chen, L. Wang, K. Guo, H. Ying and P. Ouyang, Green Chem., 2016, 18, 2165–2174 RSC.
- F. Chen, N. Li, S. Li, J. Yang, F. Liu, W. Wang, A. Wang, Y. Cong, X. Wang and T. Zhang, Catal. Commun., 2015, 59, 229–232 CrossRef CAS.
- C. J. Barrett, J. N. Chheda, G. W. Huber and J. A. Dumesic, Appl. Catal., B, 2006, 66, 111–118 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 823, 1446–1450 CrossRef PubMed.
- W. Zha, Z. Shao, J. W. Frost and H. Zhao, J. Am. Chem. Soc., 2004, 126, 4534–4535 CrossRef CAS PubMed.
- L. P. Saunders, M. J. Bowman, J. A. Mertens, N. A. Da Silva and R. E. Hector, J. Ind. Microbiol. Biotechnol., 2015, 711–721 CAS.
- J. Cardenas and N. A. Da Silva, Metab. Eng., 2014, 25, 194–203 CrossRef CAS PubMed.
- G. A. Kraus, U. K. Wanninayake and J. Bottoms, Tetrahedron Lett., 2016, 57, 1293–1295 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational methods. See DOI: 10.1039/c6gc02528e |
| ‡ Equal first author contribution. |
| This journal is © The Royal Society of Chemistry 2016 |