
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards automation of chemical process route selection based on data mining†
P.-M.
Jacob
,
P.
Yamin
,
C.
Perez-Storey
,
M.
Hopgood
and
A. A.
Lapkin
 *
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge CB2 3RA, UK. E-mail: aal35@cam.ac.uk
First published on 19th October 2016
Abstract
A methodology for chemical routes development and evaluation on the basis of data-mining is presented. A section of the Reaxys database was converted into a network, which was used to plan hypothetical synthesis routes to convert a bio-waste feedstock, limonene, to a bulk intermediate, benzoic acid. The route evaluation considered process conditions and used multiple indicators, including exergy, E-factor, solvent score, reaction reliability and route redox efficiency, in a multi-criteria environmental sustainability evaluation. The proposed methodology is the first route evaluation based on data mining, explicitly using reaction conditions, and is amenable to full automation.
Introduction
In the field of process and synthetic chemistry ‘clean synthesis’ has become one of the standard criteria for good, commercially viable synthesis routes. As a result synthetic and process chemists must be equipped with adequate methodologies for quantification of ‘cleanness’ or ‘greenness’ of alternative routes at the early phases of the development cycle. These new criteria, and the traditional criteria of cost, security of supply, health and safety (H&S), and risk, provide a balanced picture of sustainability of a future technology. Thus, there are two separate aspects to process chemistry: developing the chemistry and the process, and evaluating the overall process, which must occur in parallel. Evaluation of the proposed routes requires data. As data science rapidly evolves, chemistry will inevitably use more of the new tools of data mining and data analysis to automate the routine tasks, such as evaluation of process metrics. In this paper we show some initial results in automation of process evaluation based on deep data mining of process chemistry and multi-criteria decision making.The evaluation of greenness is a mature field, with a large number of published and standardised approaches, of which many are adopted by industry.1 However, all published methods are highly case-specific and rather labour-intensive. In the field of synthetic routes development one of the most exciting new areas is the potential for automation of synthesis planning using data mining.2 What has never been attempted before is to automate route generation and evaluation in a coherent methodology, which would aid process development at the early, data-lean, stages. For this we show how to automatically generate process options using a network representation of a section of Reaxys database,3 followed by their screening using multi-criteria decision making, see Fig. 1. As the methods mature and become commercially available, such integration and automation will produce significant savings of time, and would deliver a far more detailed view of the competing synthesis route options than is generally possible at the early stages of design.
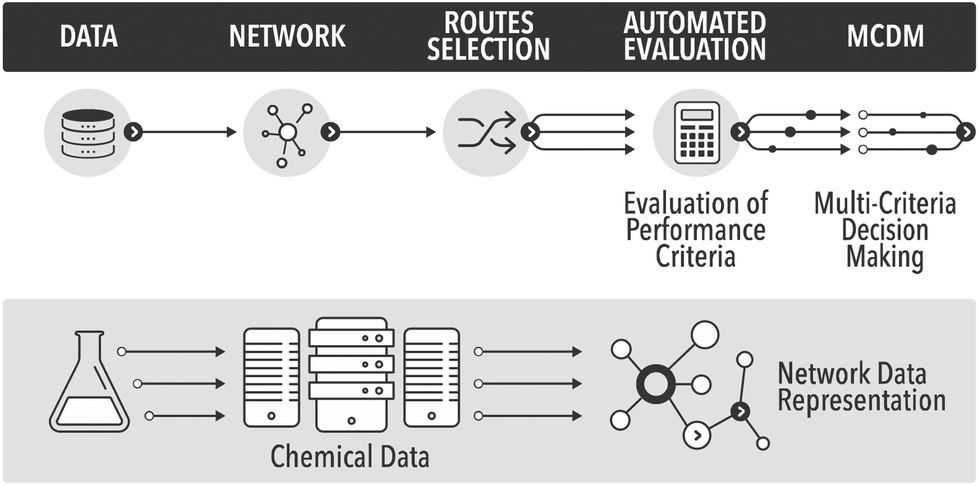 | ||
| Fig. 1 The proposed automated workflow based on deep data mining and multi-criteria decision making. | ||
To date, obtaining the data, assembling the network and finding potential synthesis routes can already be carried out in a fully automated fashion. Due to issues around data availability the connection to the analysis of the routes still has to be initiated manually, involving a data curation step. The subsequent analysis and multi-criteria decision making have been largely automated in this study. To our knowledge this is the first example of the analysis of synthesis routes generated from the network representation of Reaxys obtained through datamining, using reaction conditions and process data.
Process evaluation
Ultimately, life cycle assessment (LCA) should be used for rigorous evaluation of the environmental aspect of sustainability. However, for the early stage of process development mass- and energy proxy measures are frequently used. The mass metrics are favoured by the pharmaceutical industry due to their heavy reliance on solvents, which typically dwarf all material inputs in the environmental impact evaluations.4–6 It is well understood, however, that assessment of a given synthesis route or a process by a single criterion would almost certainly fail to deliver a holistic picture of the process's sustainability. This has been widely recognised in the literature. For example, Andraos over the years has complemented mass-based indicators with energy and health and safety indicators.7–10The indicator-based evaluation methods frequently artificially separate mass and energy streams. Fundamentally, every mass stream carries an associated energy and, thus, also is an energy stream. This separation is avoided by exergy analysis which expresses all streams crossing the system boundaries as streams of energy available to perform useful work, relative to the environment.11 In the chemical industries the use of exergy is particularly attractive, since optimisation for exergy simultaneously considers yield and energy utilisation.
Analysis of the availability of energy (exergy) is gradually finding uptake in the process industry (e.g.ref. 12 and 13) but methodologies combining exergy analysis with other, non-economic metrics are still being developed. Li et al. attempt this by combining exergy analysis with safety and economic assessment. Each criterion was evaluated in turn, eliminating processes if they fail a single criteria, rather than adopting a holistic approach.14 Similarly in ref. 15 a methodology is proposed to expand a set of metrics from simple mass-based indicators to include H&S, critical elements, catalysts, energy and life cycle assessment (LCA). This approach relies on generation of experimental data, which is infeasible for the screening of large datasets, the topic addressed in this paper. Dewulf has applied LCA to exergy analysis such as e.g. in ref. 16 and Fan et al. used a graph theoretical approach to generate paths from a reaction network and then sequentially screened them according to exergy dissipation, profit potential and toxicity indices.17 In addition to mass and exergy efficiency, the H&S and environmental impacts of a process, the redox performance of the reactions and their reliability are also important factors in decision making on selection of novel process routes.
Data mining in chemical route development
In 1990 Lawson and Kallies stated that the “Beilstein data […] forms an explicit network […] being equivalent to a map of practically all known synthetic pathways from almost any starting material to almost any product”.18 The group of Grzybowski explored this idea, termed the Network of Organic Chemistry (NOC), based on the Beilstein database encompassing a total of 7 million reactions and 7 million substances.19 The initial versions of the NOC had simple directed edges; subsequent versions used a bipartite representation, which had separate nodes for species and for reactions. This is important in the planning of syntheses where reactions have several feedstocks or products, thus displaying the true dependency of the various species involved.20 The NOC is a time-evolving scale free network and thus exhibits behaviour very similar to that of the World Wide Web.21 As a consequence, the molecules contained within the network can largely be divided into either those belonging to the “core” or the “periphery”, the two regions varying greatly in size and connectivity. The core is a cluster several thousand times larger than the next largest cluster. Although it only contains 3.5% of all organic substances registered in the NOC, these substances are involved in 35% of all reactions and give rise to 60% of all registered substances.22For illustrative purposes a section of a network of roughly 1 million reactions centred around (3R)-3-isopropyl-6-methyl-7-oxabicyclo[4.1.0]heptane, as downloaded from Reaxys3 during an iterative search, and assembled using Python 2.7 and the toolbox graph-tool23 by us, is shown in Fig. 2.
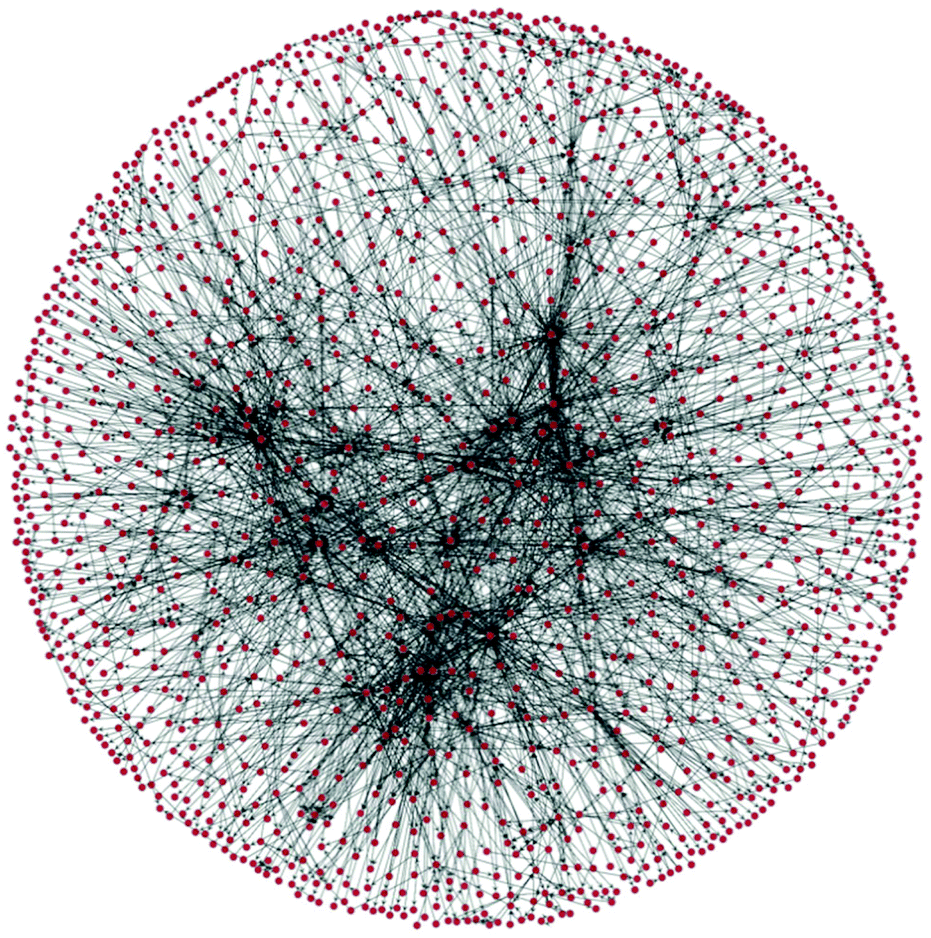 | ||
| Fig. 2 A section of a network of organic chemistry. Dots are species and arrows represent reactions. | ||
Using the network representation of the chemical reactions data allowed the automatic identification of reaction sequences.21 By making explicit use of the existing data it is possible to plan synthesis routes, optimise parallel synthesis routes, identify one-pot conversions or identify purchasing patterns of precursors to controlled substances.19,20,24 In addition to algorithmic connection of molecules, it was possible to deduce from the network structure information about the reactivity of functional groups and how the functional groups present in a molecule influence each other's reactivity with the results matching theory very well.25 The NOC could also be employed to identify “maximally useful” compounds in a manufacturing context.22 More recently the network analysis of the dataset of 10 million reactions was combined with retrosynthetic approaches in Chematica.2
Combination of data mining with retrosynthesis has seen some exciting recent developments.26 Bøgevig et al. report a commercial implementation of this link in a suite from InfoChem by taking a database of 4.4 million reactions and abstracting it into retrosynthetic transforms. During the retrosynthetic analysis it is able to identify papers carrying out similar transformations and use this information in synthesis planning.27 Since September 2015 Wiley offers a commercial platform in the form of ChemPlanner, which includes explicit use of the reported reactions and the retrosynthetic approaches to predict new reactions.28,29 ChemPlanner at present comprises 2 million reactions.30 For a more detailed description of the methodology employed by ARChem, the predecessor of ChemPlanner, the reader is referred to ref. 31.
1.5 million new compounds are claimed to be discovered annually32 and today Reaxys already contains in excess of 74.9 million compounds (27 million compounds from Reaxys itself and the remainder from integrated databases such as PubChem), 40.7 million reactions and 500 million published experimental facts,33 yielding an enormous source of data for analysis. To date, the use of datamining in combination with predictive and heuristic tools in chemistry is at its infancy and very few tools are available. However, such tools are being rapidly developed and advances in data science, automation of experiments, wide adoption of electronic lab-books and development of machine readable formats of chemical data exchange, which are all taking place at present, will undoubtedly make enormous impact on chemical process development.
A missing direction of research is the link of network tools and synthesis planning tools with their evaluation at the early stages of process development. A network search or forward reaction planning will always return a number of possible pathways, raising the question how the paths compare under multi-criteria considerations, including environmental and H&S factors. The approach that we present in this paper attempts to combine the use of chemical data available in databases, such as Reaxys, with automated evaluation of synthesis routes, thus combining the two tasks of industrial process development.
As a case study a hypothetical synthesis route from limonene to benzoic acid will be studied. Terpenes represent a highly versatile and valuable class of natural compounds, attracting significant industrial interest.34–36 Two major sources of natural terpenes are limonene as a byproduct of the citrus industry and crude turpentine from tree resin, which can in turn be converted into limonene, with an annual production of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 350000 tons, respectively,36,37 making it a reasonably abundant feedstock. Though the conversion of limonene to benzoic acid may appear to be destroying economic value due to their relative costs, if limonene is derived from waste and benzoic acid is in turn converted into higher value products, the cost basis would change. The considered route is a hypothetical example of a bio-based route selection and was used to avoid any commercial sensitivity of using current, industrially relevant substances.
000 and 350000 tons, respectively,36,37 making it a reasonably abundant feedstock. Though the conversion of limonene to benzoic acid may appear to be destroying economic value due to their relative costs, if limonene is derived from waste and benzoic acid is in turn converted into higher value products, the cost basis would change. The considered route is a hypothetical example of a bio-based route selection and was used to avoid any commercial sensitivity of using current, industrially relevant substances.
Experimental
Multi-criteria decision making
An implementation of the PROMETHEE methodology (Preference Ranking Organization Method for Enrichment Evaluations) was chosen in the form of the Visual PROMETHEE software suite to perform multi-criteria analysis. Following expert interviews and evaluation of the conflicting criteria, the following weightings were chosen for the specific case of pharmaceutical and speciality chemicals manufacturing: reliability of technology 0.35, exergetic efficiency 0.25, redox economy 0.10, and mass-based environmental indicator 0.15. We should emphasise that weightings will change between different sectors of chemical industries.Evaluation of exergy
As reference environment in this study the annual average temperature at the Teesside refinery complex in Stockton-on-Tees, UK, was taken, which is 9.1 °C,38 and 1 atmosphere of pressure. The kinetic and potential exergy were ignored due to lack of data on the eventual process layout. Thus, the exergy of a species i will be given by their chemical and physical exergies. The chemical exergy of species i, Exi,ch is given by eqn (1).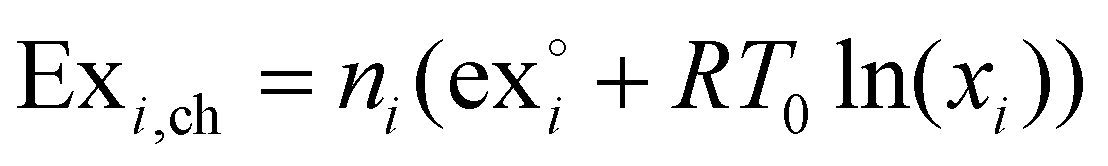 | (1) |
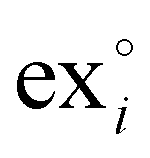 is the standard chemical exergy of species i on a molar basis; subscript 0 represents the reference environment conditions; x stands for a mole fraction, T is temperature and R the universal gas constant.
is the standard chemical exergy of species i on a molar basis; subscript 0 represents the reference environment conditions; x stands for a mole fraction, T is temperature and R the universal gas constant.
The standard chemical exergy of species i is given by the following equation:39,40
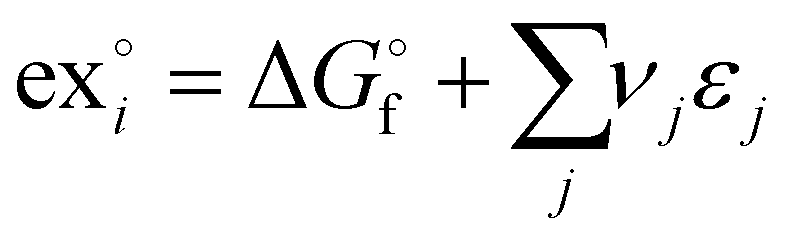 | (2) |
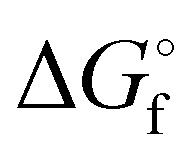 is the Gibb's free energy of formation of a compound i and νj the number of atoms of the constitute element j, each with a standard chemical exergy of εj.
is the Gibb's free energy of formation of a compound i and νj the number of atoms of the constitute element j, each with a standard chemical exergy of εj.
The physical exergy on the other hand is given by the difference in enthalpy and entropy of the stream at its conditions relative to that at the environmental conditions.40
| Exi,ph = [Hi(T,P) − Hi(T0,P0)] − T0[Si(T,P) − Si(T0,P0)] | (3) |
More detailed mathematical descriptions can be found in the ESI.†
Calculation of process heating
We assumed that the reactants for each step are introduced separately at ambient conditions. This assumption is obviously false for a highly integrated chemical plant. However, it is reasonable for the case of batch processes as might be encountered in the pharmaceutical or fine chemicals industries. For simplicity we assumed that any heat is provided to the process via an electric resistance heater and that heat capacities of all mixtures are constant throughout. Though using electricity for heating purposes is less efficient than burning liquid or gaseous fuels directly,41 it is considered an adequate baseline for the purpose of this study. Crucially, changing the fuel source for process heating for the purposes of this model is thus facile. This leads to the following expression for exergy input due to heating (the derivation can be found in the ESI†):| Exelec = 3.57Q | (4) |
Separation energy
Comparing the cost of separation across different processes in monetary terms can be challenging due to the fact that very different processes can be required for different separations. Instead the thermodynamic limit is used as a proxy to quantify the effort due to the required separation. Thus, Gibb's energy change of mixing was considered a proxy to the cost of separation of a reaction mixture. The Gibb's energy change of mixing is the difference in the Gibb's energy of a mixture compared to that of its constituents as pure components (as given by eqn (5)42) and can thus be used as a measure of the absolute minimum energy required to separate a mixture.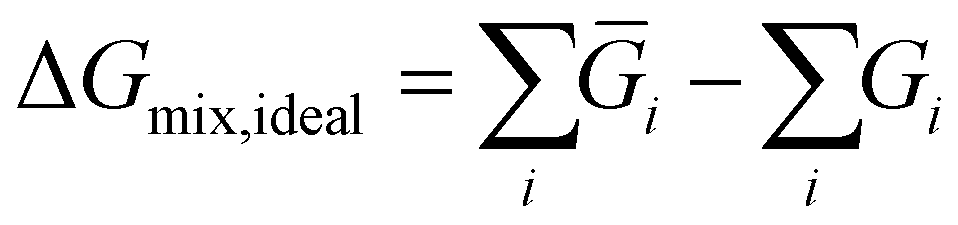 | (5) |
| ΔGmix,ideal = nRT(yalnya + yblnyb) | (6) |
The Gibb's free energy of mixing of the gaseous streams was found using eqn (6) where ya is the mole fraction of gas a in the mixture and yb that of gas b, respectively, n the number of moles of gas present, T is temperature and R the ideal gas constant. For the liquid streams activity coefficients of each species in the mixture (Ḡresi), as well as, as pure compounds (Ḡpurei), were calculated using COSMOthermX Version C30_1401. The Gibb's free energy change of mixing for a given species i was then found using eqn (7) where xi is the mole fraction of species i in the mixture:
| ΔḠmixi = Ḡresi + lnxi − Ḡpurei | (7) |
Exergetic efficiency
Exergetic efficiency of a process is the ratio of useful exergy output to exergy input as illustrated in Fig. 3. When defined as above, solvents and unreacted starting material will appear in the process output without actually having been “produced” by the process, thus artificially inflating the efficiency.43 Instead a more useful approach would be to only consider the produced, utilisable exergy in relation to the consumed exergy, thus excluding the transiting exergy associated to the parts of the mixture not taking part in the reaction. Thus, the transiting exergies were excluded from the efficiency calculation. We also assumed that catalysts do not deactivate and thus could be recycled allowing for their exclusion from the analysis. Further details can be found in ref. 43 and the ESI.†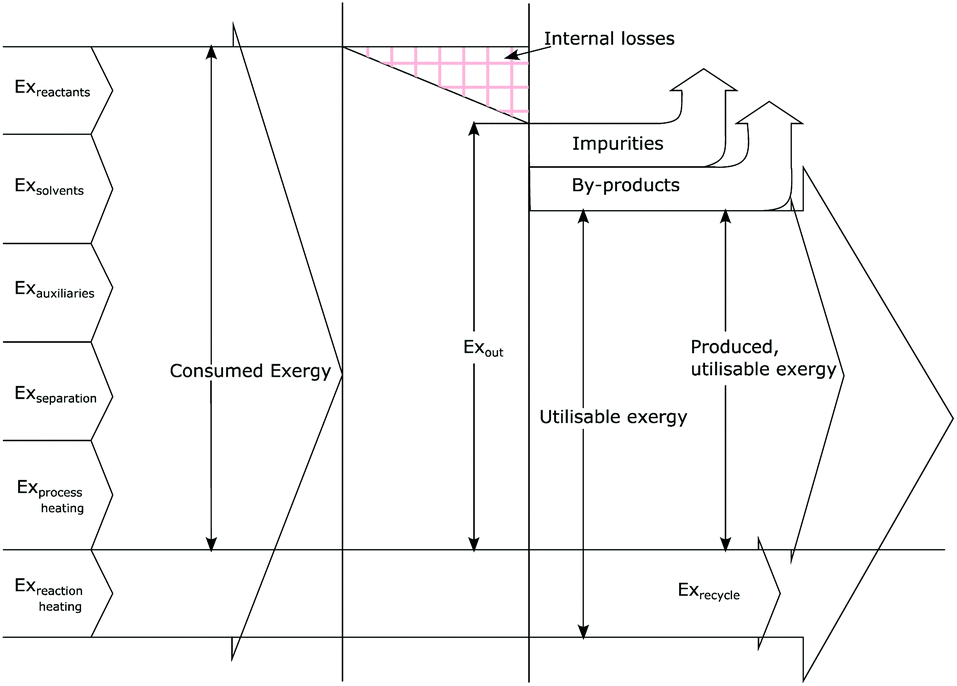 | ||
| Fig. 3 Graphical representation of exergy balance within a chemical process (adapted from ref. 43). | ||
Based on this description, a process step schematic could be represented as shown in Fig. 4. The streams entering the control volume are the reaction mixture input stream, the heat provided by the heat exchanger, the heat required by the reactor (labelled “1” in Fig. 4) in the case of an endothermic reaction and the energy required by the separation unit (labelled “2” in Fig. 4) corresponding to the Gibb's energy of mixing. The streams exiting the control volume are the exergy stream of the product(s), the exergy stream of the unreacted reactants and solvents (which could be recycled) and the exergy stream of the waste species. Then, exergetic efficiency can be described by eqn (8). The overall efficiency of a route, consisting of j steps is the product of the efficiency of each of the individual steps as shown in eqn (9).
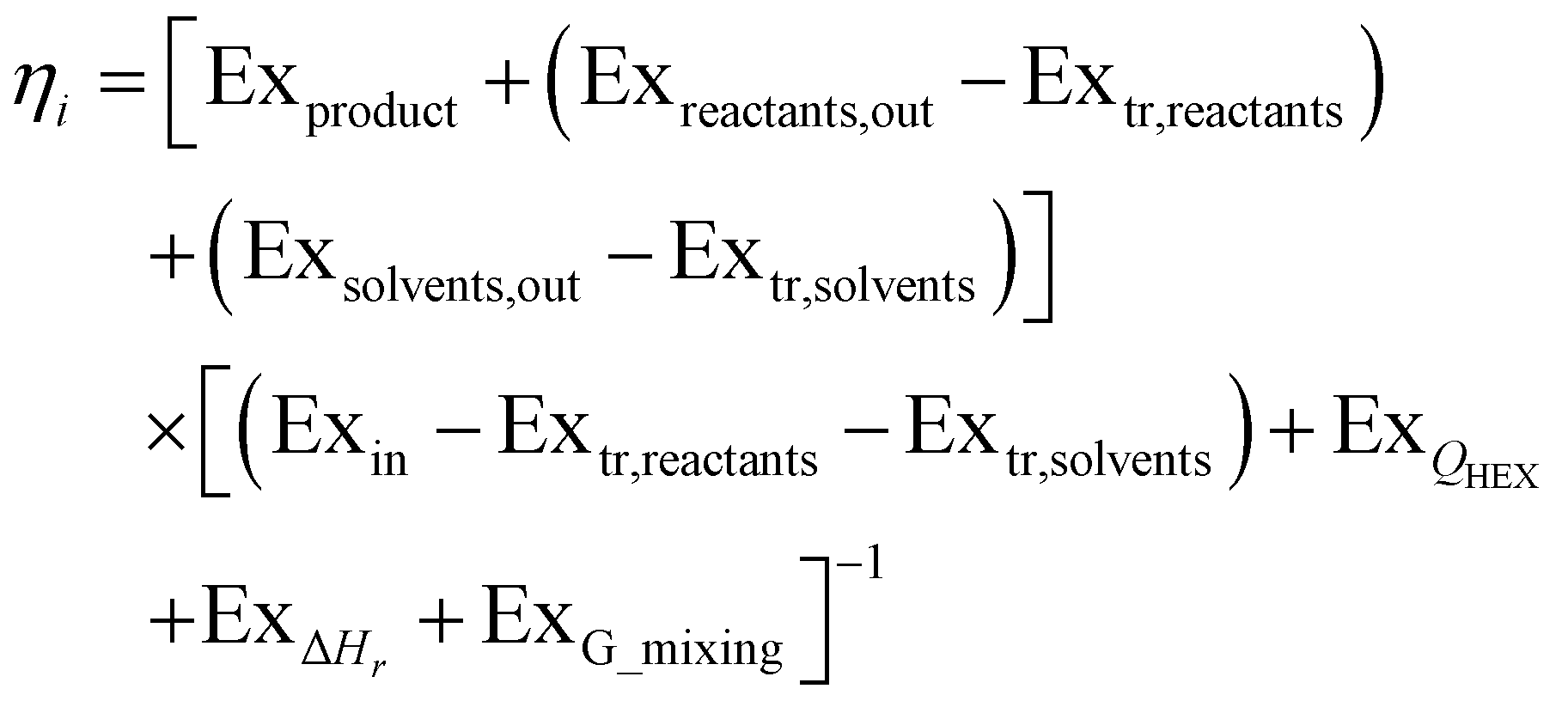 | (8) |
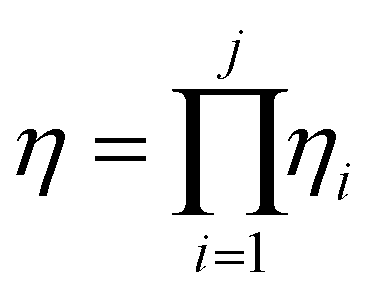 | (9) |
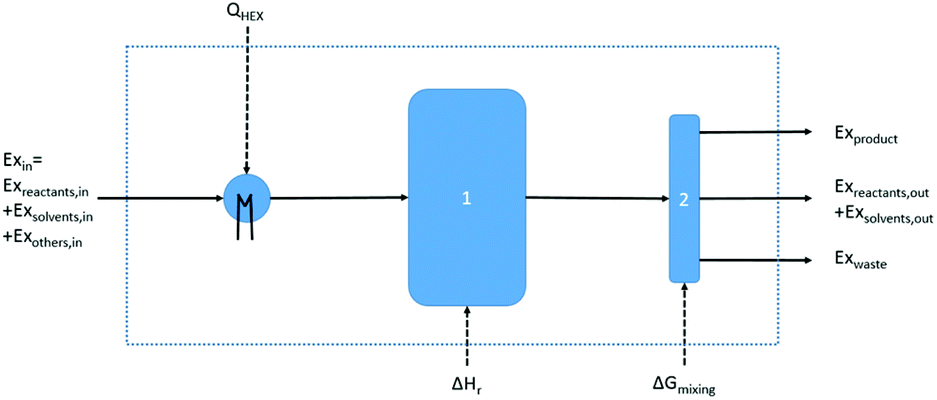 | ||
| Fig. 4 Schematic representation of a control volume drawn around a basic process step. | ||
Mass-based indicators
We use the E-factor as defined by Andraos, but re-derive the expression to account for reaction stoichiometry, thus extending the applicability of the approach. The E-factor was initially proposed by Sheldon in 199244 and reviewed in 2007.45 In contrast to Sheldon's ratio of mass of waste to mass of product, Andraos derived an equivalent methodology to calculate the E-factor, along an entire synthesis route, largely based on yields, atom economy and stoichiometric factors considering all species, including water, and thus making automation and computation more facile as explicit mass balances are not required. The original approach and derivations of Andraos could not be used in the automated route evaluation. In the earlier derivation atom economy, AEj, was defined on the basis of molecular weights. This does not allow inclusion of the actual stoichiometry into the E-factor calculation and was changed in this work. Re-deriving Andraos's equations to account for reaction stoichiometry yields the following expression (details of the derivation can be found in the ESI†):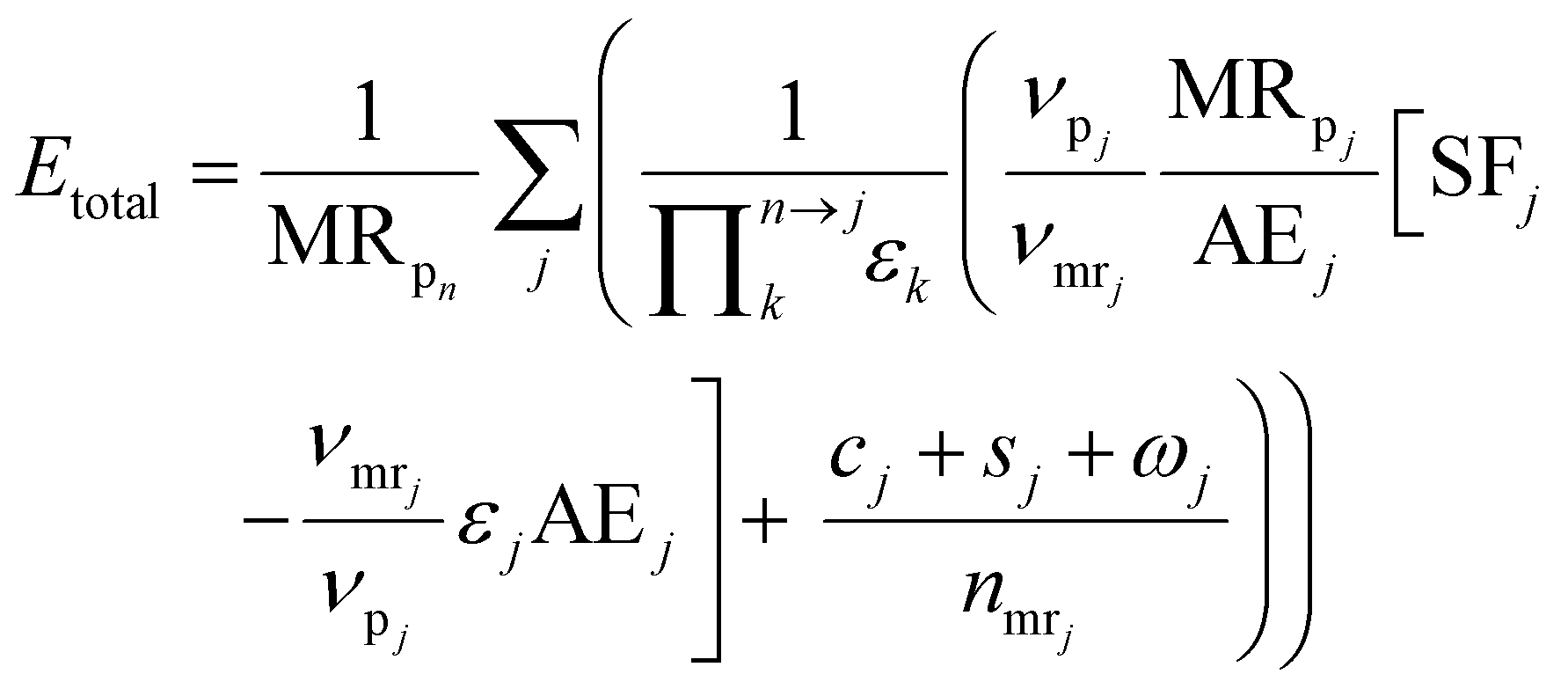 | (10) |
 is the product of reaction yields along the reaction route from the current step to the final step, ignoring any steps carried out prior to the current step, c is the mass of catalyst, s is the mass of solvent, ω is the mass of all other materials used in work-up and purification and nmr is the number of moles of the limiting reagent in step j. SF is the stoichiometric factor, i.e. the ratio of the mass of excess reagent to stoichiometric reagent plus one, νmrj is the stoichiometric coefficient of limiting reagent in step j and νp is the stoichiometric coefficient of the desired product in step j. The atom economy is defined as:
is the product of reaction yields along the reaction route from the current step to the final step, ignoring any steps carried out prior to the current step, c is the mass of catalyst, s is the mass of solvent, ω is the mass of all other materials used in work-up and purification and nmr is the number of moles of the limiting reagent in step j. SF is the stoichiometric factor, i.e. the ratio of the mass of excess reagent to stoichiometric reagent plus one, νmrj is the stoichiometric coefficient of limiting reagent in step j and νp is the stoichiometric coefficient of the desired product in step j. The atom economy is defined as: | (11) |
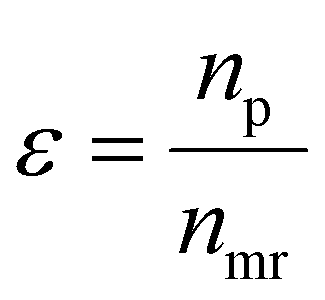 | (12) |
The E-factor as defined here equals to the commonly used Process Mass Efficiency minus 1.6 However, this definition is an easily automatable mass metric, whose evaluation requires the commonly reported data only.
During the network traversal, described subsequently, a list of all routes connecting limonene to benzoic acid was generated. The list of the reactions comprising the synthesis routes for further analysis were written to a file. Similarly, the reaction conditions and data for each reaction in this list were written to another file. The Python script imports these data, computes the required parameters for each individual reaction and then proceeds to combine them for the given synthesis routes in line with eqn (10). Subsequently, Etotal for each route is written to a file (along with further data on each route) to allow comparison.
Heuristics
In addition to calculation of the exergetic efficiency and E-factor, a number of heuristics were extracted from literature and expert knowledge, which were used to calculate a score for different criteria in the route. The performance of a synthesis route under environment, health and safety considerations is a vital factor when assessing the sustainability of a route. Solvents are of crucial importance in much of the pharmaceutical and fine chemical industry having a very large impact on the sustainability balance of a process,4,5 but receive no explicit treatment in the methodology so far. The “CHEM21 selection guide of classical- and less classical-solvents”46 approximates solvents’ performance under health, safety and environmental criteria through use of physical data and, where available, their hazard clauses under the Global Harmonised System and is used in this study to evaluate the solvents used. Seeing as solvents carry some of the greatest toxicological, safety and sustainability concerns as far as pharmaceutical processes are concerned4,5 this is a good proxy, stopping short of a full LCA.Some treatment of how reliable a published reaction is, is desirable. This was achieved by using the number of publications reporting a given reaction as a proxy, following the argument that a reaction reported more frequently is better established and more reliable. This is introduced specifically for process development, rather than discovery.
Another environmental parameter introduced in the evaluation is redox economy. The term redox economy appears to first have been coined by Richter,47 though some of the thinking can be traced back to the works of Hendrickson in 1971 and 1975,48,49 and is being discussed in more detail by Burns.50 One of the key principles of redox economy is that unnecessary changes of the oxidation state may lead to significant environmental impacts.7,50,51 To track changes in the oxidation state the set of atoms involved in the target bond-forming reactions across a route is determined. The oxidation state of each of those atoms is then calculated at each reaction step as described in ref. 9 and then the oxidation state of all such atoms at a given reaction step is summed (Oxi) and the difference ΔOxi = Oxi − Oxtarget is found. Should both ΔOxi and ΔOxi−1be positive (or both negative) and |ΔOxi| > |ΔOxi−1| a penalty for step i will be applied according to eqn (13) as an oxidation or reduction that needs to subsequently be corrected has taken place. If, however both ΔOxi and ΔOxi−1 are of opposite signs an overshoot has taken place and the penalty will be given by eqn (14). The overall penalty for a given route will be given by eqn (15).
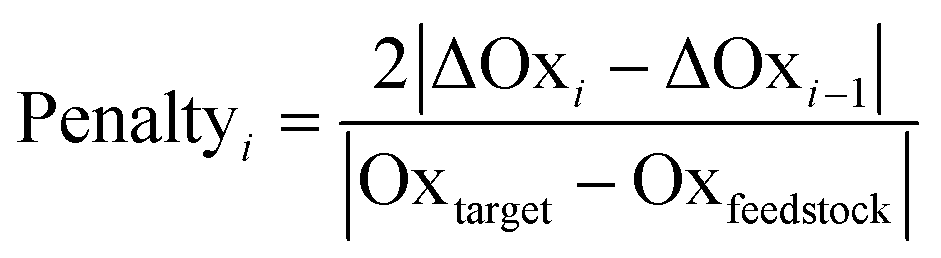 | (13) |
 | (14) |
 | (15) |
Again, every part of this methodology can easily be implemented in a fully automated fashion. Calculation of the oxidation state changes requires some atom mapping, however, tools for this exist.
Network traversal
A network of reactions was constructed based on the reactions contained in Reaxys. To this end all reactions starting from limonene were downloaded. All product species from these reactions were in turn individually queried to obtain all reactions starting from each of these species. This was repeated to obtain data containing three reaction steps as it was known that the desired conversion could be carried out in three steps. This was written to a file, incomplete reactions were deleted and the remaining data was then used to construct a network using “the graph-tool python library”23 in Python 2.7.It was decided to remove acetic acid, formaldehyde, formic acid and isoprene from the network. Due to the architecture of the network any routes via these molecules would mean that limonene would have been decomposed into one of these substances to use them as building blocks to obtain benzoic acid. This destruction of functionality and thus any synthesis routes via these molecules as main reactants was deemed undesirable leading to their exclusion. Using a k-shortest paths algorithm implemented in graph-tool all routes connecting limonene to benzoic acid were found up to a maximum path length of three synthesis steps.
Compound data
The chemical equation, density and heat capacity at constant pressure (at 298 K) were collected from literature where possible.52–63 In cases where no data could be found in literature the values were predicted as described below.Property prediction
 | (16) |
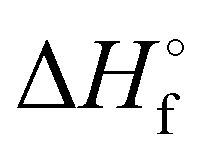 denotes the standard enthalpy of formation of a given compound.
denotes the standard enthalpy of formation of a given compound.
Results and discussion
Network search
The network traversal algorithm returned a total of 228 unique paths. This set contained four two-step syntheses connecting limonene to benzoic acid via 4-ethenylcyclohexene, cumol, maleic anhydride or methyl 4-methylphenyl ketone. At this stage of analysis it is not claimed that these are ideal, or even good, routes, but merely that Reaxys contains a synthesis path involving these molecules. The remaining 224 paths of the result set required three synthesis steps. In order to give the reader an idea of the path taken these paths were classified according to the product of the first step. In total there were 36 different reactions that could be carried out during the first step. The six most frequently encountered options for different products of the first step, together accounting for over 75% of possible routes, and the number of routes (of the 224 remaining) that use this step can be found in Table 1.| Species | Number of occurrences |
|---|---|
| Cumol | 60 |
| Maleic anhydride | 38 |
| p-Cymene | 29 |
| Fumaric acid | 21 |
| Methyl 4-methylphenyl ketone | 19 |
| Thymol | 8 |
Further analysis of this set revealed that many papers were either unavailable online or their reporting of data was insufficient for the desired level of analysis. Thus, two routes for which the required data could be reconstructed, briefly illustrated in Scheme 1 with sources of the reaction routes given, were chosen for proof-of-concept: the first route led via p-cymene and toluene to benzoic acid, while the second route utilised p-cymene and p-toluic acid as intermediates.
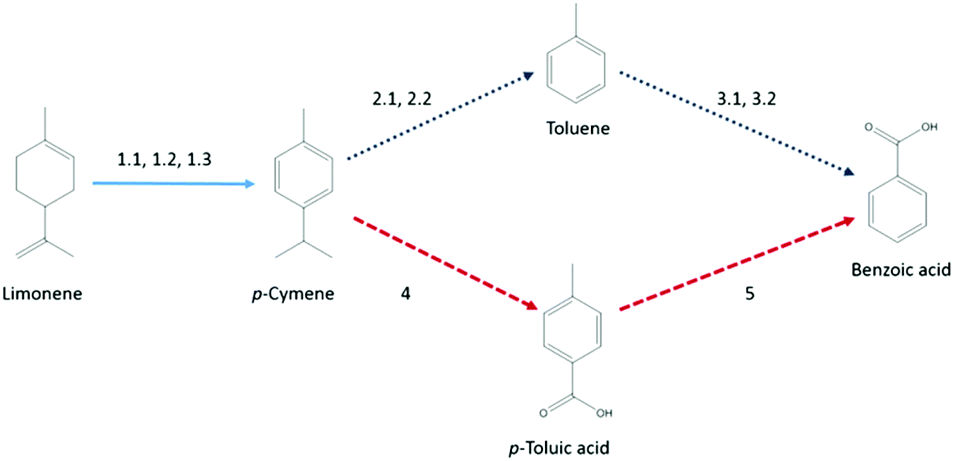 | ||
| Scheme 1 Schematic of the reaction route. Sources: 1.1,75 1.2,75 1.3,76 2.1,77 2.2,78 3.1,79 3.2,80 4,81 5.82 | ||
A further issue that was encountered was the fact that most of the selected papers did not report balanced equations for the reactions, making reconstruction of the reaction systems very difficult when also only the yield of the main product was reported. An attempt was made at balancing the equations purely by using atom balances. It is realised that these may not be the actual equations and are not necessarily based on actual reaction mechanisms but they yield a solvable system, which for this case study is deemed sufficient. The stoichiometric coefficients were then normalised with respect to the main reactant in order to simplify later calculations to give Scheme 2.
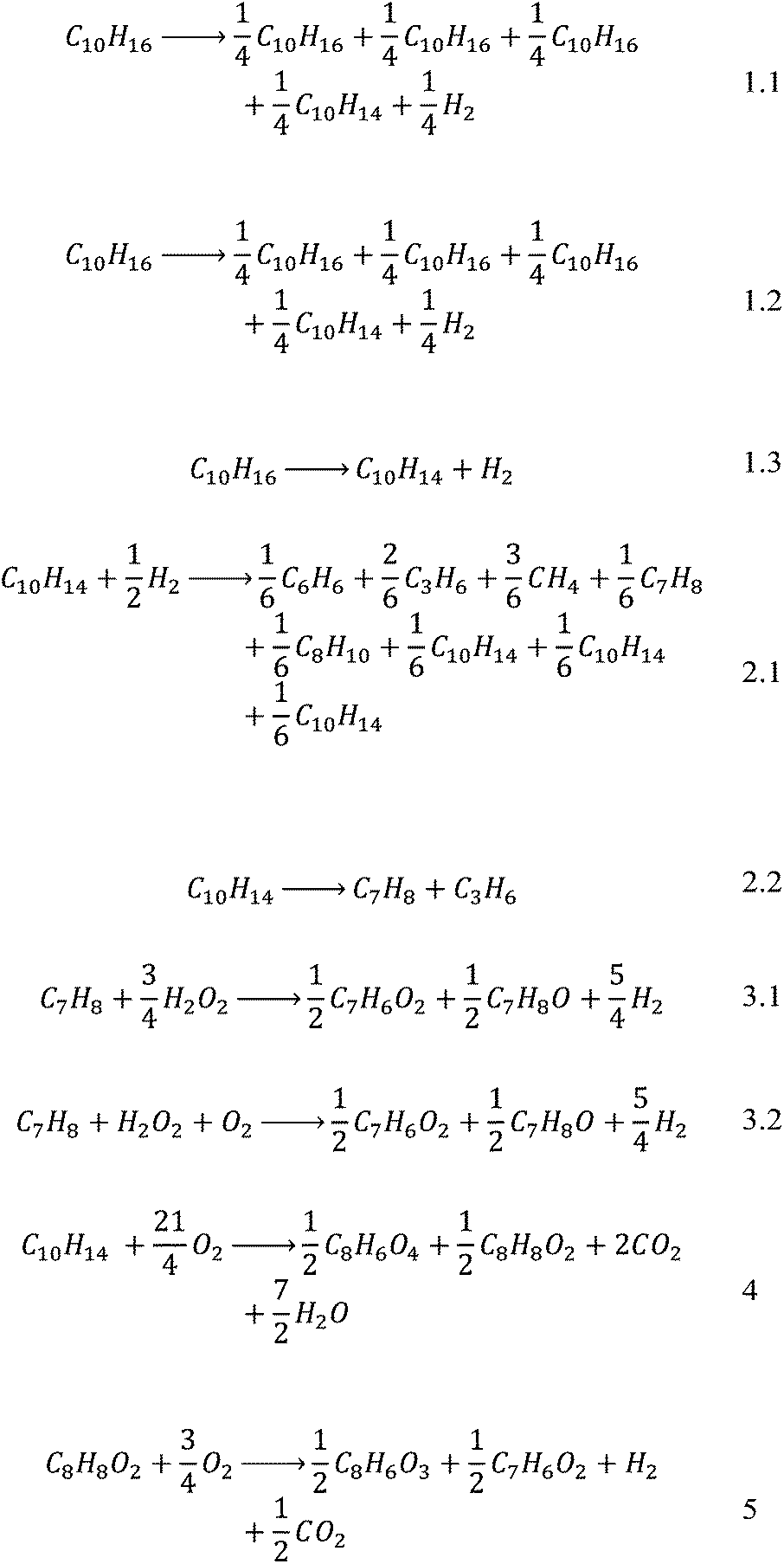 | ||
| Scheme 2 A set of balanced equations used in the case study. | ||
Exergetic efficiency
The in and out flows of each species for each reaction were calculated and, using eqn (1) and (3), it was possible to calculate the exergy associated to each species entering and exiting the system for each reaction. Based on these results the exergetic efficiencies were calculated according to eqn (8). The heating duties for endothermic processes and for stream heating were converted into exergies using eqn (4). The exergetic input required to supply the free energy of mixing was taken to be equivalent to the energy of mixing. The results of the exergy calculations, as well as efficiencies, for each reaction can be found in the ESI in Table S15.† Building on this, the full results for all possible permutations of reaction sequences can be seen in Table 2.| Exergetic efficiency | Yield | ||||||
|---|---|---|---|---|---|---|---|
| 1st step | 2nd step | 3rd step | Πη | 1st step | 2nd step | 3rd step | ΠY |
| 1.3 | 2.2 | 3.2 | 0.1694 | 1.3 | 2.2 | 3.2 | 0.8299 |
| 1.2 | 2.2 | 3.2 | 0.1530 | 1.2 | 2.2 | 3.2 | 0.7303 |
| 1.3 | 2.2 | 3.1 | 0.0918 | 1.3 | 2.1 | 3.2 | 0.6294 |
| 1.2 | 2.2 | 3.1 | 0.0829 | 1.3 | 2.2 | 3.1 | 0.6089 |
| 1.1 | 2.2 | 3.2 | 0.0552 | 1.2 | 2.1 | 3.2 | 0.5538 |
| 1.3 | 2.1 | 3.2 | 0.0411 | 1.2 | 2.2 | 3.1 | 0.5358 |
| 1.2 | 2.1 | 3.2 | 0.0372 | 1.3 | 2.1 | 3.1 | 0.4618 |
| 1.1 | 2.2 | 3.1 | 0.0299 | 1.2 | 2.1 | 3.1 | 0.4063 |
| 1.3 | 2.1 | 3.1 | 0.0223 | 1.1 | 2.2 | 3.2 | 0.0592 |
| 1.2 | 2.1 | 3.1 | 0.0201 | 1.1 | 2.1 | 3.2 | 0.0449 |
| 1.1 | 2.1 | 3.2 | 0.0134 | 1.1 | 2.2 | 3.1 | 0.0435 |
| 1.1 | 2.1 | 3.1 | 0.0073 | 1.1 | 2.1 | 3.1 | 0.0330 |
| 1.3 | 4 | 5 | 0.0003 | 1.3 | 4 | 5 | 0.0139 |
| 1.2 | 4 | 5 | 0.0002 | 1.2 | 4 | 5 | 0.0122 |
| 1.1 | 4 | 5 | 0.0001 | 1.1 | 4 | 5 | 0.0010 |
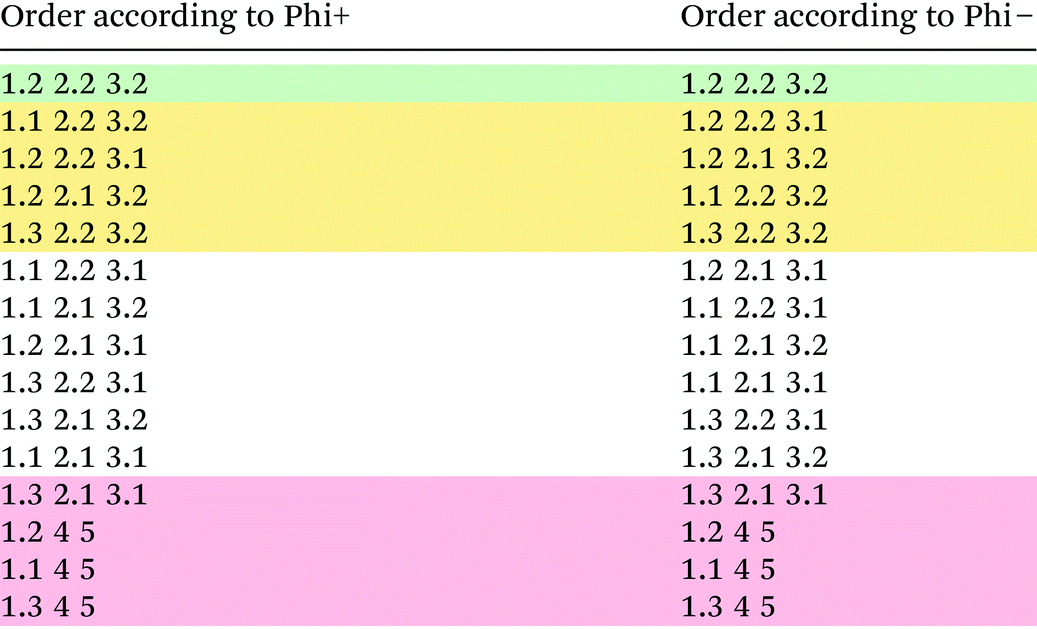
From Table 2 it becomes apparent that the most efficient route would be the sequence of reactions 1.3, 2.2. and 3.2, in that order. In the second place lies the sequence 1.2, 2.2, 3.2 and in the third place 1.3, 2.2, 3.1. This is in contrast to ranking based on the overall yield, where the first two positions are the same but where the third-best option would be 1.3, 2.1, 3.2 as can be seen clearly in the comparison in Table 2. This is caused by the fact that reaction 2.1 both uses a significant amount of hydrogen as carrier gas as well produces a broad range of byproducts, despite a reasonable selectivity towards toluene, which is penalised in the exergy analysis, while reaction 2.2 is carried out without solvent and produces only one byproduct, reducing the separation penalty greatly.
E-Factor
The E-factors for the given reactions were calculated using the developed Python code, based on eqn (10), and ranked in the order of increasing E-factors, i.e. in the order of decreasing environmental efficiency. To ensure the method's accuracy, all calculations were also checked manually. The results can be found in Table 3. Industrial E-factors range between 5 and 50 for the fine chemicals industry across usually 3–4 synthesis steps, while the pharmaceutical industry can reach E-factors exceeding 100 over 6+ steps70 putting most of the computed E-factors into the range.| 1st step | 2nd step | 3rd step | E-Factor |
|---|---|---|---|
| 1.3 | 2.2 | 3.2 | 9.34 |
| 1.2 | 2.2 | 3.2 | 9.55 |
| 1.3 | 2.1 | 3.2 | 15.21 |
| 1.2 | 2.1 | 3.2 | 15.48 |
| 1.1 | 2.2 | 3.2 | 28.92 |
| 1.1 | 2.1 | 3.2 | 41.01 |
| 1.3 | 2.2 | 3.1 | 58.19 |
| 1.2 | 2.2 | 3.1 | 58.47 |
| 1.3 | 2.1 | 3.1 | 66.18 |
| 1.2 | 2.1 | 3.1 | 66.55 |
| 1.1 | 2.2 | 3.1 | 84.87 |
| 1.1 | 2.1 | 3.1 | 101.36 |
| 1.3 | 4 | 5 | 33414.06 |
| 1.2 | 4 | 5 | 33426.34 |
| 1.1 | 4 | 5 | 34585.97 |
Heuristics
Analysing the associated records for each of the reactions stored in Reaxys some variability can be detected but, crucially, the variation in records for the alternative reactions for a given step is very low. Given the fact that a very limited set of reactions was considered carrying out very similar chemistry this was to be expected. The number of records for each reaction can be found in Table 4.| Reaction | Number of records |
|---|---|
| 1.1 | 5 |
| 1.2 | 5 |
| 1.3 | 1* |
| 2.1 | 1 |
| 2.2 | 1* |
| 3.1 | 49 |
| 3.2 | 86 |
| 4 | 1 |
| 5 | 1 |
It was decided that a step having 1–4 associated records would be given a score of 3. A reaction having 5–25 associated records would be given a 2, while any reaction having in excess of 25 associated records would be given a 1. Any route involving a reaction with a score of 3 will be assigned a score of 3, increased by 1 for each additional step scoring 3. Any route involving more than two steps with a 2, and no 3s, will be scored with 2, while any route involving only reactions scoring 1 (and at most one 2)) will be assigned a 1 as overall score.
Only four reactions in all the analysed routes use solvents. Their rating was determined according to the “CHEM21 selection guide of classical- and less classical-solvents”. For the purpose of MCDM the rating was converted to a numeric value as follows: “Recommended” = 1, “Problematic” = 2, “Hazardous” = 3, “Highly hazardous” = 4. Additionally, a score of 0 was given to any reaction that did not use a solvent. If a reaction used more than one solvent the worst solvent score was used for the overall reaction. The results are shown in Table 5. Any route involving more than one step with a score greater than or equal to 3 will have its overall score increased by 1 for each additional step exceeding this threshold.
| Reaction | Solvent score |
|---|---|
| 1.1 | 0 |
| 1.2 | 0 |
| 1.3 | 0 |
| 2.1 | 0 |
| 2.2 | 0 |
| 3.1 | 2 |
| 3.2 | 2 |
| 4 | 1 |
| 5 | 3 |
Multi-criteria decision making
The MCDM was carried out using the preference function parameterisation given in Table 6. PROMETHEE generates three rankings. Firstly, there are the partial rankings according to Phi+ (the positive flow) and Phi− (the negative flow). The sum of the two yields the complete ranking. If a given option ranks more preferably under several criteria but another more preferable on the remaining it is possible for the two to have different positions in the Phi− and Phi+ rankings (which is not apparent from the Phi score). According to the results shown in Table 7 there is a clearly preferred route option: 1.2, 2.2, 3.2. It is also possible to unambiguously determine the four worst options. The ranking of the remainder of the field is somewhat complicated by the fact that when ranking according to the Phi+ and Phi− scores the ordering of the options changes. It is however possible to isolate a field of five choices that are clearly preferable over the remainder as is shown in Table 7. The top entry remains top under both rankings. The following three options are tied for the second place and together with the fifth option occupy places 2–5 under both rankings and thus clearly outperform the non-highlighted block. The bottom four options represent the worst options; their relative position remains unchanged irrespective of the ranking method. It can therefore be concluded that the presented methodology allows differentiation of the different route options investigated and yields a clear favourite followed by a number of equally good alternatives as far as this proof-of-concept study is concerned.| Criterion | Weighting | Preference function | Preference threshold | s | Absolute/percentage |
|---|---|---|---|---|---|
| Exergetic efficiency | 0.25 | Gaussian | 0.0075 | Absolute | |
| E-Factor | 0.15 | Gaussian | 0.13 | Percentage | |
| Solvent score | 0.15 | V-shape | 1 | Absolute | |
| Number of records | 0.35 | V-shape | 1 | Absolute | |
| Oxidation length | 0.1 | V-shape | 1.37 | Absolute |
|
The redox economy penalty of the routes via toluene is 1/3, while that of those via p-toluic acid is 2.
It is possible to carry out a sensitivity analysis and determine the range of values for each of the criterion weightings for which the order of a given number of the top ranked choices remains unchanged according to their complete ranking score. PROMETHEE calls this ‘stability interval’. In this case stability intervals according to the top five choices were calculated. Considering the top five ranks the stability interval with respect to exergetic efficiency is 0.2254–0.3142 and with respect to the E-factor is 0.1138–0.1681. For the solvent score the stability interval lies between 0 and 1 and that for the number of records between 0.2808 and 0.4109 while that of the oxidation length spans from 0 to 1. The results are thus independent of the solvent score and the oxidation length. This was to be expected in this case as all but the worst three choices have the same score and thus the inter-route variation is very low rendering these two criteria potentially redundant in terms of their impact on the final result, though it must be emphasised that they do offer further insights into the underlying problems with some of the routes. This problem is highly specific to this case study. Within a typical process chemistry setting a large number of solvents would be encountered across a number of routes and thus it is expected that the differentiability would be greater in a network with more reactions and a greater number of different solvents, resulting in a greater impact of the solvent score on the final ranking. As expected the results are most sensitive to the weighting of the exergetic efficiency and E-factor, though the stability interval is deemed large enough not to be of concern. This is caused by the fact that due to the different assumptions within the two assessment criteria with regards to solvents, catalysts and separation costs they prefer different options, creating at times conflicting rankings. As a consequence, it is very important to pay close attention to the choice of weightings for the different parameters as their impact on the final outcome is pronounced and the sensitivity of the ultimate result to the relative difference in weightings can be non-negligible.
Uncertainty of the scores and its impact on rankings must be considered. Uncertainty is associated with measurement errors reported or not in the original papers as well as with the accuracy of the estimated or literature properties data. Where experimental errors have not been reported the comparison of different process route options is complicated by the uncertainty in the evaluated metrics. In the ESI† a plot of the exergetic efficiency and E-factors for each route option can be seen in Fig. S1 and S2,† showing uncertainties derived from the property prediction methods and estimates of the uncertainty on experimental data. The mean values were taken forward for MCDM, whereas the overlap of uncertainty intervals provides further information for the final decision making.
Comparing the results obtained using the MCDM methodology to the performance of individual criteria taken in isolation some differences can be observed. One commonly used, much easier to calculate, criterion to assess the performance of a route is the overall yield. Taking this metric compared to the ranking based on exergetic efficiencies the top two performing routes are identical, however, thereafter deviations between the two criteria can be seen. This is caused by the fact that the exergetic efficiency penalises impure product mixtures and the changing of temperature and pressure of the reaction mixture. Comparing performance of yield and E-factor as key metrics the deviations are less well pronounced. However, in this case too, the impact of the yield is soon overridden by differences in the amounts of waste produced. Comparing the top-scoring route using solely the E-factor or solely the exergetic efficiency, 1.3 2.2 3.2 (c.f. Tables 2 and 3), to the MCDM results one notes that due to the inclusion of further decision criteria the route 1.3 2.2 3.2 now only appears in the fifth place. Combining the metrics in the MCDM methodology thus provides a more balanced picture and, as expected, the preferred route under the MCDM approach is different to the preferred route using the more conventional individual indicator metrics, both when comparing it to yield, E-factor or exergetic efficiency taken in isolation. The very high weighting of the industrial reliability plays an important factor in the specific case evaluated. The methodology can be easily adapted to the needs of different scenarios to an extent that the use of a single metric would not be able to, making it an approach yielding not only greater insight but also being more versatile than conventional evaluation methodologies.
A necessary fact that needs to be born in mind when carrying out any assessment of the sustainability of a process is that of system boundaries71 as any metric only focused on the process at hand can be skewed, and outsourcing seemingly encouraged, if sustainability of materials purchased is not considered.11,70 Due to the nature of the database used this, at present, is impossible and thus the system boundaries start at the factory gate, not the cradle. Seeing as the routes compared in this paper necessarily start from the same feedstock the impact of this might be slightly less pronounced but it is a factor to be born in mind when applying the methodology in other circumstances and future development of the methodology will be directed towards life cycle assessment approach.
Performance of algorithm
The automated methodology can be split into several distinct algorithms/tools, mapped onto Fig. 1. The step of data retrieval depends on how access to data is arranged. In the present study a network access to a server was used to download reaction data. Due to the very large size of the network considered, downloading the data and assembly of the network benefits from parallel computing. Running in the order of low tens of parallel processes, the time for this step ranges in the order of a few days. Once the network has been obtained all further analyses take significantly less time and depending on how broadly the network has been defined it can be used for multiple case studies. The network search takes a few minutes to a few hours on a normal desktop machine (albeit requiring a few GBs of RAM). Considering the computational intensity of the analysis carried out, it can be observed that once automated, each of the analysis steps takes in the order of seconds to carry out on a normal desktop machine for the entire set of route options. Setting up the MCDM parametrisation as well as carrying out data curation where still required. These steps add a manual overhead. In addition to the knowledge of the amounts of all substances being fed, yields of all the species and conversion of main reactants, balanced stoichiometric equations need to be available. Calculation of the chemical exergies requires knowledge of the standard chemical exergy of the species or their Gibb's free energy of formation, while calculation of the physical exergy requires temperature and pressure at which the reaction was carried out in addition to the heat capacities for all states encountered in the experiment of all species involved, along with their phase transition enthalpies, if phase transitions are observed. If the analysis is to consider heating then, in addition, knowledge of reaction heat, or alternatively heat of formation of all reaction species, is necessary. Thus, final full automation of the tools described in this paper requires close integration with thermodynamic databases and computational tools.Data scarcity
Reviewing the calculations carried out, a list of properties required for the calculations can be drawn up. First and foremost it is necessary for the reaction stoichiometry to be reported along with at least two out of the list of conversion (of the main reactant), selectivity (for all products) and yield (for all products). Furthermore, it is required that all reactant and solvent species as well as their molar amounts are reported.A brief analysis of the Reaxys database illustrates a crucial problem. 33526757 reactions were downloaded from Reaxys along with some associated data for each reaction entry. This amounts to roughly 80% of all reactions contained in Reaxys33 and should yield a reasonably reliable sample. Due to the way Reaxys reports data any multistep reactions (17686694 reactions) were removed from the analysis set for this section. Additionally, any reactions that were incomplete, i.e. contained either no reactants or products, were pruned from the set. 2.6% of the single step reactions were incomplete. The remaining 15414520 reactions were analysed to determine how scarce the entries associated to these reactions were. As can be seen from Table 8 this data was incredibly scarce. On the one hand this is due to ambiguity of the reported data: ‘ambient temperature’ cannot be easily associated with any specific temperature value and, hence, is not translated into a database entry. On the other hand, this is due to lack of reporting of critically important values, e.g., the yield of all product species is not always clearly reported. These issues could be addressed by the use of clear and accepted data reporting standards.
414520 reactions downloaded from Reaxys, that do not have a value stored in the Reaxys database for the relevant property
| Property | Percent of reactions without value for property |
|---|---|
| Yield | 54.0 |
| Temperature | 53.9 |
| Pressure | 98.4 |
| pH-Value | 99.0 |
| Reaction time | 51.6 |
| Solvent ID | 29.1 |
| Reagent ID | 32.2 |
| Catalyst ID | 95.7 |
The list of properties required for the presented analysis is reasonably long and in some cases quite specialised as far as the experimentalist might be concerned, and perhaps perceived as adding significant additional effort to publishing without an immediately visible pay-off. This however overlooks two important points: (i) publishing such “incomplete” papers prevents their maximum use, and (ii) much of the required data is in fact already available, just spread across different sources.
Regarding the first observation: facilitated by ever greater computational resources and growing online repositories, more and more data is being used primarily by algorithms, and not humans, which needs to be born in mind during the publishing process.72,73 Though a chemist is able to make judgement calls and interpret the data presented in a paper a computer is, in most cases, unable to do so. “Big Data” is a buzzword that is penetrating ever more disciplines and even though some of the hopes placed into it may ultimately prove unfounded it does bear great potential in allowing the unlocking of insights previously impossible purely by leveraging computational resources and available data. Though the possibility of applying automated evaluation routines, such as exergetic and E-factor efficiency evaluations of a synthesis route, are potentially highly useful tools to the process engineer, this is only possible where complete datasets are available. This is a necessary conclusion borne out of the fact that teaching algorithms how to deal with “missing data” is a highly complicated operation. As a consequence, papers not making all required data available would end up being excluded from the result set as a matter of necessity, reducing the practical importance of the initially reported work and results in everyday practice. The responsibility here is necessarily born jointly by author, publisher and database provider. This discussion is being picked up in a forthcoming paper developing an extension to RInChI standard74 to include some of the required data and make their publication and future algorithmic use more straightforward.
Conclusions
In this paper we presented the concept of automated multi-criteria evaluation of new process routes combined with the route development on the basis of data mining. We are convinced that chemical process development will soon be largely based on automated experiments and will make significant use of data sciences and algorithmic decision support tools. This work highlights the possibilities and the necessary components of the methodology, which provides multiple numeric criteria for a balanced decision on the suitability of a novel synthesis route; we also highlight current limitations. Here we have shown the first, to our knowledge, example of a combined development and evaluation of synthesis routes on the basis of datamining of existing chemical knowledge. It is a proof-of-concept demonstration, making use of a hypothetical reaction scenario. At present the actual numerical results are of little significance, whereas the elements of the methodology and the pathway to full automation are important to stimulate further work in this emerging area of chemistry. The emphasis is made on algorithmic tools and multi-criteria decision making, which is particularly important if future chemical processes are considered from the point of view of sustainability. The developed methodology was able to rank the different route options by employing a multi-criteria decision making approach and to identify a preferred option as well as several alternatives. Due to the modularity of the method it is easy to extend the number of criteria considered, to replace some or to readjust the weighting of individual criteria, to match different scenarios or aims, making the methodology highly versatile. The method can be adapted to include results of life cycle assessment instead of individual gate-to-gate indicators. In order to allow wide implementation of the suggested development and evaluation methodology, discussions about the method of publishing reaction data currently being held in the chemoinformatics community need to be picked up by the wider chemistry and chemical engineering community and enforced by publishers to ensure that critical data is electronically available.Acknowledgements
We gratefully acknowledge collaboration with RELX Intellectual Properties AS and their technical support, which enabled us to mine REAXYS. PMJ is grateful to Peterhouse and the Cambridge Trust for scholarships. We are grateful to Dr John Hayler (GSK) for suggestions of reaction criteria and to Dr Martyn Jones (University of Manchester) for his advice on multi-criteria decision making tools. This work was in part funded by EPSRC project “Terpene-based manufacturing for sustainable chemical feedstocks” EP/K014889.Notes and references
- D. J. C. Constable, C. Jimenez-Gonzalez and A. Lapkin, in Green Chemistry Metrics, John Wiley & Sons, Ltd, Chichester, UK, 2009, pp. 228–247 Search PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- Reed Elsevier Properties SA, Login – Reaxys Login Page [Internet], 2014 [accessed 2014 Jun 8]. Available from: https://www.reaxys.com/. Reaxys is a trademark, copyright owned by Relex Intellectual properties SA and used under licence.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- J. Andraos, Org. Process Res. Dev., 2009, 13, 161–185 CrossRef CAS.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS.
- J. Andraos, in Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, ed. A. Lapkin and D. J. C. Constable, John Wiley & Sons Ltd, Chichester, 1st edn, 2008, pp. 69–199 Search PubMed.
- J. Andraos, ACS Sustainable Chem. Eng., 2016, 4, 312–323 CrossRef CAS.
- J. Dewulf, G. Van der Vorst, W. Aelterman, B. De Witte, H. Vanbaelen and H. Van Langenhove, Green Chem., 2007, 9, 785–791 RSC.
- J. Dewulf, H. van Langenhove and B. Van De Velde, Environ. Sci. Technol., 2005, 39, 3878–3882 CrossRef CAS PubMed.
- A. C. Caetano de Souza, J. Luz-Silveira and M. I. Sosa, J. Fuel Cell Sci. Technol., 2006, 3, 346 CrossRef CAS.
- X. Li, A. Zanwar, A. Jayswal, H. H. Lou and Y. Huang, Ind. Eng. Chem. Res., 2011, 50, 2981–2993 CrossRef CAS.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- G. Van der Vorst, H. Van Langenhove, F. De Paep, W. Aelterman, J. Dingenen and J. Dewulf, Green Chem., 2009, 11, 1007 RSC.
- L. T. Fan, T. Zhang, J. Liu, J. R. Schlup, P. A. Seib, F. Friedler and B. Bertok, Ind. Eng. Chem. Res., 2007, 46, 4506–4516 CrossRef CAS.
- A. J. Lawson and H. Kallies, J. Chem. Inf. Model., 1990, 30, 426–430 CrossRef CAS.
- C. M. Gothard, S. Soh, N. A. Gothard, B. Kowalczyk, Y. Wei, B. Baytekin and B. A. Grzybowski, Angew. Chem., 2012, 124, 8046–8051 CrossRef.
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chem., Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed.
- M. Fialkowski, K. J. M. Bishop, V. A. Chubukov, C. J. Campbell and B. A. Grzybowski, Angew. Chem., Int. Ed., 2005, 44, 7263–7269 CrossRef CAS PubMed.
- K. J. M. Bishop, R. Klajn and B. A. Grzybowski, Angew. Chem., Int. Ed., 2006, 45, 5348–5354 CrossRef CAS PubMed.
- T. P. Peixoto, The graph-tool python library, figshare., 2015 Search PubMed.
- P. E. Fuller, C. M. Gothard, N. A. Gothard, A. Weckiewicz and B. A. Grzybowski, Angew. Chem., 2012, 124, 8057–8061 CrossRef.
- S. Soh, Y. Wei, B. Kowalczyk, C. M. Gothard, B. Baytekin, N. Gothard and B. A. Grzybowski, Chem. Sci., 2012, 3, 1497 RSC.
- O. Ravitz, Drug Discovery Today: Technol., 2013, 10, e443–e449 CrossRef PubMed.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Org. Process Res. Dev., 2015, 19, 357–368 CrossRef.
- M. Reubold, Route Planner for Research Chemists [Internet], 2016 [accessed 2016 May 20]. Available from: http://www.chemanager-online.com/en/topics/research-laboratory/route-planner-research-chemists.
- O. Ravitz, Wiley ChemPlanner – Technical Notes [Internet], 2015 [accessed 2016 May 20]. Available from: http://images.news.wiley.com/Web/WileyEnterprise/{84e34101-2105-40fd-8d3a-df6120ebf89e}_Info-RC-CHE-W2627_ChemPlanner_Technical_Notes.pdf.
- Wiley Information Services GmbH, ChemInform – RxnFinder [Internet], 2015 [accessed 2016 May 20]. Available from: https://www.rxnfinder.com/.
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 79–107 CrossRef CAS.
- S. J. Coles, N. E. Day, P. Murray-Rust, H. S. Rzepa and Y. Zhang, Org. Biomol. Chem., 2005, 3, 1832–1834 CAS.
- Elsevier R&D Solutions, Reaxys Fact sheet [Internet], 2015 [accessed 2015 Aug 20]. Available from: http://www.elsevier.com/__data/assets/pdf_file/0005/91616/R_D-Solutions_RX_Fact-Sheet_DIGITAL1.pdf.
- J. L. F. Monteiro and C. O. Veloso, Top. Catal., 2004, 27, 169–180 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, in Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, pp. 329–387 Search PubMed.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- Met Office, Middlesbrough climate [Internet], 2014 [accessed 2015 Nov 29]. Available from: http://www.metoffice.gov.uk/public/weather/climate/gcxn7yvv5.
- G. Song, J. Xiao, H. Zhao and L. Shen, Energy, 2012, 40, 164–173 CrossRef CAS.
- E. Querol, B. Gonzalez-Regueral and J. L. Perez-Benedito, in Practical Approach to Exergy and Thermoeconomic Analyses of Industrial Processes, Springer London, London, 2013, pp. 9–28 Search PubMed.
- G. P. Hammond and A. J. Stapleton, Proc. Inst. Mech. Eng., Part A, 2001, 215, 141–162 CrossRef.
- J. Gmehling, B. Kolbe, M. Kleiber and J. Rarey, in Chemical Thermodynamics for Process Simulations, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 1st edn, 2012, pp. 154–158 Search PubMed.
- M. Sorin, J. Lambert and J. Paris, Chem. Eng. Res. Des., 1998, 76, 389–395 CrossRef CAS.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS PubMed.
- J. B. Hendrickson, J. Am. Chem. Soc., 1971, 93, 6847–6854 CrossRef CAS.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- J. Andraos, ACS Sustainable Chem. Eng., 2013, 1, 496–512 CrossRef CAS.
- E. E. Bolton, Y. Wang, P. A. Thiessen and S. H. Bryant, in Annual Reports in Computational Chemistry, ed. R. Wheeler and D. Spellmeyer, Elsevier, Oxford, 2008, pp. 217–241 Search PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD Search PubMed.
- InfoTherm, release 3.2 [Internet], 2014 [accessed 2015 Oct 31]. Available from: http://www.infotherm.com/.
- Air Liquide, Gas Encyclopedia – Air Liquide [Internet], 2013 [accessed 2015 Nov 11]. Available from: http://encyclopedia.airliquide.com/encyclopedia.asp?LanguageID=11.
- Sigma-Aldrich, α-Terpinene – Sigma Aldrich [Internet], 2015 [accessed 2015 Nov 2]. Available from: http://www.sigmaaldrich.com/catalog/product/aldrich/86473?lang=en®ion=GB.
- Sigma-Aldrich, γ-Terpinene – Sigma-Aldrich [Internet], 2015 [accessed 2015 Nov 2]. Available from: http://www.sigmaaldrich.com/catalog/product/sial/86478?lang=en®ion=GB.
- AlfaAesar, L02120 4-Ethyltoluene, 97% – AlfaAesar [Internet], 2015 [accessed 2015 Oct 31]. Available from: https://www.alfa.com/en/catalog/L02120/.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, T. J. Bruno and D. R. Lide, CRC Press/Taylor and Francis, Boca Raton, FL., 96th edn, 2015, vol. 13, pp. 5-4–5-42 Search PubMed.
- AlfaAesar, AlfaAesar – p-Toluic acid, 98% [Internet], [accessed 2016 Jan 13]. Available from: https://www.alfa.com/en/catalog/A11506/.
- «alpha»-Terpinolene – chemeo [Internet], 2015 [accessed 2015 Nov 3]. Available from: https://www.chemeo.com/cid/75-618-3/«alpha»-Terpinolene.pdf.
- Wired Chemist, Standard Heats and Free Energies of Formation and Absolute Entropies of Organic Compounds [Internet], [accessed 2015 Oct 31]. Available from: http://www.wiredchemist.com/chemistry/data/entropies-organic.
- J. Szargut, A. Valero, W. Stanek and A. Valero, in Proceedings of ECOS 2005, 2005, pp. 409–420 Search PubMed.
- C. F. Chueh and A. C. Swanson, Can. J. Chem. Eng., 1973, 51, 596–600 CrossRef CAS.
- R. C. Reid, J. M. Prausnitz and B. E. Poling, The Properties of Gases and Liquids, McGraw-Hill Book Company, New York, 4th edn, 1987 Search PubMed.
- R. K. Sinnott, Coulson and Richardson's Chemical Engineering Volume 6 - Chemical Engineering Design, Elsevier Butterworth-Heinemann, Oxford, 4th edn, 2005 Search PubMed.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 1987, 57, 233–243 CrossRef CAS.
- B. Rice, S. V. Pai and J. Hare, Combust. Flame, 1999, 118, 445–458 CrossRef CAS.
- D. R. Morris and J. Szargut, Energy, 1986, 11, 733–755 CrossRef CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- C. Jiménez-González, D. J. C. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
- A. M. Clark, B. A. Bunin, N. K. Litterman, S. C. Schürer and U. Visser, PeerJ, 2014, 2, e524 Search PubMed.
- A. M. Clark, A. J. Williams and S. Ekins, J. Cheminf., 2015, 7, 9 Search PubMed.
- G. Grethe, J. M. Goodman and C. H. Allen, J. Cheminf., 2013, 5, 45 Search PubMed.
- M. A. Martin-Luengo, M. Yates, E. S. Rojo, D. Huerta Arribas, D. Aguilar and E. Ruiz Hitzky, Appl. Catal., A, 2010, 387, 141–146 CrossRef CAS.
- M. A. Martin-Luengo, M. Yates, M. J. Martínez Domingo, B. Casal, M. Iglesias, M. Esteban and E. Ruiz-Hitzky, Appl. Catal., B, 2008, 81, 218–224 CrossRef CAS.
- S. Kamiguchi, J. Catal., 2004, 223, 54–63 CrossRef CAS.
- European Patent Office, EP0231569, 1986 Search PubMed.
- M. Debnath, A. Dutta, S. Biswas, K. K. Das, H. M. Lee, J. Vícha, R. Marek, J. Marek and M. Ali, Polyhedron, 2013, 63, 189–198 CrossRef CAS.
- R. Bandyopadhyay, S. Biswas, R. Bhattacharyya, S. Guha and A. K. Mukherjee, Chem. Commun., 1999, 37, 1627–1628 RSC.
- World Intellectual Property Organization, WO2014133433A1, 2014 Search PubMed.
- D. I. Makhon'kov, A. V. Cheprakov, M. A. Rodkin, A. Y. Mil'chenko and I. P. Beletskaya, Russ. J. Org. Chem., 1986, 22, 30–39 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02482c |
| This journal is © The Royal Society of Chemistry 2017 |