
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmine dehydrogenases: efficient biocatalysts for the reductive amination of carbonyl compounds†
Tanja
Knaus‡
 ,
Wesley
Böhmer‡
and
Francesco G.
Mutti
*
,
Wesley
Böhmer‡
and
Francesco G.
Mutti
*
Van't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, 1098 XH, The Netherlands. E-mail: f.mutti@uva.nl
First published on 6th September 2016
Abstract
Amines constitute the major targets for the production of a plethora of chemical compounds that have applications in the pharmaceutical, agrochemical and bulk chemical industries. However, the asymmetric synthesis of α-chiral amines with elevated catalytic efficiency and atom economy is still a very challenging synthetic problem. Here, we investigated the biocatalytic reductive amination of carbonyl compounds employing a rising class of enzymes for amine synthesis: amine dehydrogenases (AmDHs). The three AmDHs from this study – operating in tandem with a formate dehydrogenase from Candida boidinii (Cb-FDH) for the recycling of the nicotinamide coenzyme – performed the efficient amination of a range of diverse aromatic and aliphatic ketones and aldehydes with up to quantitative conversion and elevated turnover numbers (TONs). Moreover, the reductive amination of prochiral ketones proceeded with perfect stereoselectivity, always affording the (R)-configured amines with more than 99% enantiomeric excess. The most suitable amine dehydrogenase, the optimised catalyst loading and the required reaction time were determined for each substrate. The biocatalytic reductive amination with this dual-enzyme system (AmDH–Cb-FDH) possesses elevated atom efficiency as it utilizes the ammonium formate buffer as the source of both nitrogen and reducing equivalents. Inorganic carbonate is the sole by-product.
Introduction
Amines are the most widely used chemical intermediates for the production of active pharmaceutical ingredients, fine chemicals, agrochemicals and a significant number of bulk chemicals.1,2 Nowadays, the production of amines in the laboratory, as well as on an industrial scale, relies principally on the reductive amination of carbonyl containing compounds.3 Nonetheless, the chemocatalytic reductive amination requires precious, and sometimes toxic, metal catalysts coordinated to sophisticated organic ligands that operate at high pressure of hydrogen gas. Furthermore, the overall process is quite lengthy as various protection and deprotection steps are involved. Finally, a follow-up recrystallization of the amine product is often needed in order to improve the enantiomeric excess, and the traces of heavy metals from the catalyst have to be removed to comply with legislative requirements. On the other hand, tremendous advancements in biocatalytic methods for chiral amines have been achieved during the past two decades.4 An industrially applied method is the kinetic resolution of racemic amines via selective acylation catalysed by a lipase; however, this process is limited by a maximum of 50% theoretical yield.5 Quantitative yield of enantiopure amines can be obtained by dynamic kinetic resolution (e.g. combining a hydrolase with a metal-catalyst)6 or deracemisation and desymmetrisation (e.g. combining an amine oxidase with a chemical reducing reagent or artificial metal-enzymes or Pd nanoparticles).7–16 Besides these earlier established methods, the arsenal of enzymes for asymmetric amine synthesis has been enriched, for instance encompassing wild-type as well as engineered ω-transaminases.17–22 However, the formal reductive amination of carbonyl compounds by ω-transaminases requires supra-stoichiometric amounts of an amine donor (e.g. 5 equivalents of alanine or ca. 10 equivalents of 2-propylamine) and strategies for shifting the unfavorable thermodynamic equilibrium, for example, the use of an additional enzyme for removing the co-product pyruvate19 or special equipment for the selective evaporation of the co-product acetone.21 Whilst the use of alternative amine donors has been demonstrated to provide an improved thermodynamic driving force, these molecules are expensive and require multi-step chemical synthesis or generate co-products that polymerise and therefore complicate the work-up of the reaction.23–25 More recently, imine reductases have gained interest, but the substrate scope of these enzymes is practically limited to cyclic secondary imines.26–30 Finally, other enzymes have also been applied for chiral amine synthesis, for example, ammonia lyases,31–36 Pictet-Spenglerases,37–40 berberine bridge enzymes41–43 and engineered P450 monooxygenases.44–46 However, all these last mentioned classes of enzymes are active on rather specific, yet valuable, families of substrates.In this context, amine dehydrogenases (AmDHs) are a new class of enzymes that possess tremendous potential for the development of the next generation of processes for the synthesis of α-chiral amines.47 The applicability of this class of enzymes in organic synthesis has been demonstrated in our notable biocatalytic dual-enzyme hydrogen-borrowing amination of alcohols.48,49 In general, AmDHs catalyse the reductive amination of ketone and aldehyde substrates using NAD(P)H as the hydride source. The study and exploitation of AmDHs for biocatalytic processes are, however, underdeveloped. In particular, only one report on a natural occurring amine dehydrogenase has been reported about two decades ago but the experiments have not been reproduced again.50 As a consequence, a narrow panel of AmDHs have been created recently through protein engineering starting from wild-type amino acid dehydrogenases as scaffolds51–53 or DNA shuffling of first generation variants.54 Although initial reaction rates have been determined for the amination of a limited number of ketones, a systematic investigation on the substrate acceptance, optimal reaction conditions as well as chemo- and stereoselectivity of the known AmDHs has not been undertaken to date. This study aims at providing this knowledge and at showing the potential of AmDHs for the efficient asymmetric synthesis of α-chiral amines.
Results and discussion
Optimisation of the reaction conditions
In our previous study, the most elevated reaction rates for the reductive amination of (para-fluorophenyl)acetone (1a), catalysed by the AmDH variant originating from wild-type L-phenylalanine dehydrogenase from Bacillus badius (for the sake of clarity, referred to, in this work, as Bb-PhAmDH), were observed in ammonium chloride and ammonium formate at pH between 8.2 and 8.8.48 Besides the elevated activity and stability of the enzymes under the operational reaction conditions, another important and often neglected parameter to be considered for the reductive amination using AmDHs is the stability of the coenzyme NADH/NAD+ in solution at more basic pH. In fact, in practical biocatalytic reactions, the coenzyme has to be applied in catalytic amounts and recycled at the expense of a sacrificial substrate such as, among others, formate in combination with formate dehydrogenase (FDH, Scheme 1) or glucose in combination with glucose dehydrogenases (GDH, not shown).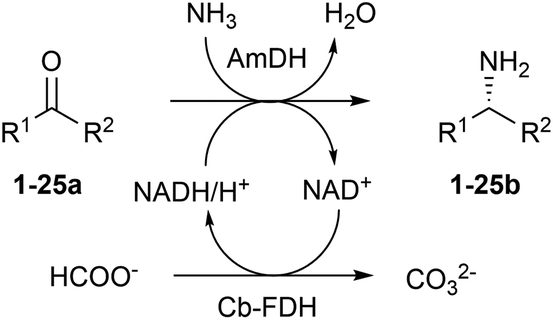 | ||
| Scheme 1 Amine dehydrogenases catalyze the reductive amination of ketones and aldehydes (50 mM) to chiral amines. A catalytic amount of nicotinamide coenzyme (1 mM) is applied. The reducing equivalents as well as the nitrogen source originate from the buffer of the reaction: ammonia/ammonium formate (pH 8.5, 1 M). The only by-products are water and carbonate. | ||
Surprisingly, only a few studies on the stability of nicotinamide coenzymes dissolved in aqueous buffers at different pH values and temperatures are found in the literature. Although NADH is reported to be fairly stable at basic pH, other publications suggest that the decomposition of NADH in solution occurs at ambient temperature in alkali55 or even at nearly neutral pH with certain types of buffers.56–58 Hence, in this study, the structural stability of the reduced form of the coenzyme NADH was monitored spectrophotometrically (λ 325 nm) over time in buffers at different pH values (see ESI S5.1†). No significant decrease of the absorbance of NADH was detected in a buffer at pH 6.5 for 24 h, indicating that the coenzyme is stable under these conditions. At pH 8.8, the absorbance started to diminish smoothly after 5 h, but it was still two thirds of the initial value after 24 h. In contrast, NADH was significantly more unstable at pH 10. In fact, the absorbance decreased linearly and was halved just after 2 h and depleted after 24 h. Finally, the absorbance of NADH in NaOH (0.1 N) was reduced to 20% of the initial value in 3 minutes and fully depleted within 1 h. These data showed that the ideal pH of 8.2–8.8 for the biocatalytic reductive amination might also be a consequence of a diminished stability of the nicotinamide coenzyme at higher values of pH. Hence, ammonium chloride and ammonium formate buffers at pH 8.5–8.7 were used in the continuation of our studies.
In our previous study, we also determined that ca. 700 mM of ammonium cation/ammonia was required to achieve >99% conversion, at 30 °C, for substrate 1a (20 mM). In that case, NAD+ was applied in a catalytic amount (1 mM) and recycled using glucose (60 mM) and a commercial engineered GDH.48 Nevertheless, the reaction with glucose as a cosubstrate generates a stoichiometric amount of gluconic acid, hence reducing the atom economy of the reaction.59 Furthermore, we had to employ a large amount of GDH (300 U mL−1) for sustaining the amination in ammonium buffer (pH 8.7, >700 mM), due to its mediocre stability under the reaction conditions. Consequently, in the present study, we envisaged the recycling system based on formate and FDH (recombinant enzyme from Candida boidinii)60 to be the preferable alternative because formate was already present in the reaction buffer as a counteranion of the ammonium species. Moreover, these new experiments showed that an extremely low amount of FDH (2.0–3.0 U mL−1) was sufficient to obtain a quantitative amination. Therefore, we compared the performance of the reductive amination in the following cases: (i) glucose/GDH (150 U) as a system for the recycling of NADH in ammonium chloride buffer (pH 8.7, 1 M); (ii) glucose/GDH (150 U) in ammonium formate (pH 8.5, 1 M) and (iii) formate/FDH (purified, 14 μM equal to 1.5 U) in ammonium formate (pH 8.5, 1 M) (for details, ESI S5.2 and S5.3†). This investigation was extended to the three amine dehydrogenases that are available in our collection: (i) Bb-PhAmDH, (ii) a variant originating from the L-phenylalanine dehydrogenase from Rhodococcus sp. M4 (in this work, indicated as Rs-PhAmDH)53 and (iii) a previously described chimeric AmDH (in this work, indicated as Ch1-AmDH)48,54 obtained by domain shuffling of Bb-PhAmDH with a variant from the leucine dehydrogenase from Bacillus stearothermophilus. The reductive aminations were carried out with the representative best substrate for each AmDH, according to our own data and other data from the literature ((1a) for Bb-PhAmDH, 4-phenylbutan-2-one (24a) for Rs-PhAmDH and 2-heptanone (10a) for Ch1-AmDH). The initially tested reaction conditions were: substrate concentration (20 mM), NAD+ concentration (1 mM), AmDH concentration (80–130 μM) and ammonium buffer (1 M), at 30 °C, for 24 h. Under these reaction conditions it was not possible to reach quantitative conversion (within 21 h) using the glucose/GDH recycling system in ammonium chloride buffer with any of the three AmDHs, despite the use of 3 equivalents of glucose (Table 1, entries 1, 5 and 9). Switching from ammonium chloride to formate and maintaining the same composition of the reaction mixture resulted in quantitative conversion for the amination of substrates 24a and 10a with Rs-PhAmDH and Ch1-AmDH (Table 1, entries 6 and 10). However, no improvement was observed in the case of Bb-PhAmDH (Table 1, entry 2). When the third, preferred, option with formate as a cosubstrate was tested, all the amination reactions afforded the related product with >99% conversion (Table 1, entries 3, 7 and 11). Hence, we deduced that the employed FDH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 can recycle NADH more efficiently than GDH, likely due to its higher stability in ammonium/ammonia buffer at pH 8.5. It is notable that the stereoselective outcome of the reaction was perfect in all the cases (Table 1, >99% (R)).
60 can recycle NADH more efficiently than GDH, likely due to its higher stability in ammonium/ammonia buffer at pH 8.5. It is notable that the stereoselective outcome of the reaction was perfect in all the cases (Table 1, >99% (R)).
| Entry | Enzyme | Substrate | Substrate concentration [mM] | Enzyme concentration [μM] | Coenzyme/buffer system | Conversion [%] | ee% (R) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: NAD+ (1 mM); recycling enzyme (GDH or Cb-FDH) in ammonium chloride buffer (1 M, pH 8.7) or ammonium formate buffer (1.005 M, pH 8.5); reaction volume 0.5 mL, temperature 30 °C, reaction time 24 h; agitation on an orbital shaker (190 rpm). | |||||||
| 1 | Bb-PhAmDH | 1a | 20 | 115 | GDH/NH4Cl | 79 | >99 |
| 2 | Bb-PhAmDH | 1a | 20 | 115 | GDH/HCOONH4 | 76 | >99 |
| 3 | Bb-PhAmDH | 1a | 20 | 115 | Cb-FDH/HCOONH4 | >99 | >99 |
| 4 | Bb-PhAmDH | 1a | 50 | 115 | Cb-FDH/HCOONH4 | 88 | >99 |
| 5 | Rs-PhAmDH | 24a | 20 | 130 | GDH/NH4Cl | 72 | >99 |
| 6 | Rs-PhAmDH | 24a | 20 | 130 | GDH/HCOONH4 | >99 | >99 |
| 7 | Rs-PhAmDH | 24a | 20 | 130 | Cb-FDH/HCOONH4 | >99 | >99 |
| 8 | Rs-PhAmDH | 24a | 50 | 50 | Cb-FDH/HCOONH4 | >99 | >99 |
| 9 | Ch1-AmDH | 10a | 20 | 80 | GDH/NH4Cl | 61 | >99 |
| 10 | Ch1-AmDH | 10a | 20 | 80 | GDH/HCOONH4 | 99 | >99 |
| 11 | Ch1-AmDH | 10a | 20 | 80 | Cb-FDH/HCOONH4 | >99 | >99 |
| 12 | Ch1-AmDH | 24a | 50 | 32 | Cb-FDH/HCOONH4 | 98 | >99 |
Aiming at understanding the overall catalytic efficiency of the reductive amination under the optimized reaction conditions, we increased gradually the concentration of the substrate up to 50 mM while maintaining the same concentrations of AmDH (80–130 μM), NAD+ (1 mM), FDH (14 μM), and ammonium buffer (1 M). Rs-PhAmDH and Ch1-AmDH converted the related substrates 24a and 10a (50 mM), respectively, with >99% conversion and perfect stereoselectivity (>99% (R)) within 21 h (ESI S5.4†). In contrast, Bb-PhAmDH turned out to be a less efficient catalyst in this regard as the conversion of 1a at 50 mM concentration slightly dropped to 88% within 21 h reaction time (Table 1, entry 4, and a full data set in ESI S5.4†). Finally, the catalyst loading was reduced for the reductive amination employing Rs-PhAmDH and Ch1-AmDH. The amination of 24a and 10a (50 mM) proceeded quantitatively within 21 h using 50 μM of Rs-PhAmDH and 32 μM of Ch1-AmDH, respectively (Table 1, entries 8 and 12). The calculated turnover number (TON)61 was equal to or more than 1000 and therefore comparable, or even superior, to the values previously obtained for the amination of ketones in aqueous buffers with other enzymes such as ω-transaminases.21,62,63 Moreover, compared to the bio-amination with ω-transaminases, AmDHs do not require an enantiopure amine donor (e.g.L- or D-alanine)19 and inhibition phenomena are not observed (i.e. inhibition due to cosubstrate alanine and/or coproduct pyruvate).64–66 As an additional parameter, the concentration of FDH could be lowered to only 9.5 μM, still providing the same conversion.
Influence of the temperature and time studies
Under the selected reaction conditions (ammonium formate buffer pH 8.5, 1 M; substrate concentration 50 mM; NAD+ 1 mM; FDH 14 μM; varied concentrations of AmDHs), we studied the influence of temperature on the progress of the reductive amination. In fact, we postulated that an increase in temperature might accelerate the kinetics of the reaction, whereas excessive temperature may be detrimental for the stability of the enzymes.The progress of the reductive amination of 1a (50 mM) using Bb-PhAmDH (46 μM) showed a consistent increase in the reaction rate when the temperature was raised from 20 °C to 30 °C and finally 40 °C (Fig. 1A). It is interesting to note that, for this enzyme, the conversion increased almost linearly over time for every temperature tested. Furthermore, the final conversion (taken after 24 h) at 40 °C doubled the value observed at 20 °C (83% vs. 37%). Nonetheless, the progress of the reaction at 50 °C was worse than that at 20 °C, leading to a mediocre conversion of 21% after 24 h. A further increase of the temperature up to 60 °C provoked a complete loss of the enzymatic activity (ESI S5.6†). The lack of conversion at 60 °C cannot be attributed to the deactivation of the FDH or the decomposition of the coenzyme NAD since the reductive amination performed well up to 60 °C with Ch1-AmDH as the biocatalyst (Fig. 1C). Furthermore, our data are in agreement with the profile of the activity vs. stability of Bb-PhAmDH that shows a rapid denaturation of the enzyme above 50 °C.52
 | ||
| Fig. 1 Progress of the reaction vs. the time for the reductive amination of: (A) 1a using Bb-PhAmDH (46 μM), (B) 24a using by Rs-PhAmDH (48 μM), and (C) 10a using Ch1-AmDH (33 μM). The study was carried out at different temperatures: 20 °C (black), 30 °C (red), and 40 °C (blue), 50 °C (green), and 60 °C (grey). Reaction conditions: 0.5 mL, 50 mM substrate, ammonium formate buffer (1.00 M, pH 8.5), FDH (14 μM). | ||
The reaction profiles for the reductive amination of substrates 24a and 10a (50 mM) with Rs-PhAmDH (48 μM) and Ch1-AmDH (33 μM), respectively, were significantly different from the previous one. For both Rs-PhAmDH and Ch1-AmDH, the conversions increased hyperbolically over time (Fig. 1B and C). In particular, Rs-PhAmDH is an extremely active enzyme on its preferred substrate 10a. Considering the first hour of the reaction, wherein the conversion correlated linearly with time, the maximum turnover frequency (TOF) was reached already at 20 °C. In particular the increase of the temperature in the range of 20 °C, 30 °C, 40 °C and 50 °C led always to the same conversion after 1 h (varying from 80% to 83%). Quantitative conversion (>98%) of 24a was obtained at 20 °C and 30 °C within 3 h (Fig. 1B and ESI S5.7†). The efficiency at 40 °C was slightly lower as the reaction required 5 h to overcome 99% conversion. In contrast, the kinetics of the reaction was negatively influenced at 50 °C with a drop in the catalytic activity after 1 h. In fact, an additional 29 h was required to increase the conversion from 83% (after 1 h) to 98% (after 30 h) at this temperature. The activity of Rs-PhAmDH was affected at 60 °C as the conversion rose smoothly, reaching a maximum of 93% only after 30 h.
Conversely, the chimeric enzyme Ch1-AmDH performed the amination of 10a almost equally well in the range of temperatures investigated that spans from 30 °C to 60 °C. In fact, after 5 h the conversion was above 90% for the aminations at 30 °C, 40 °C and 50 °C and reached 82% at 60 °C. The rate of reductive amination, instead, was lower at 20 °C.
Nevertheless, only with Ch1-AmDH, quantitative conversion (>98%) was obtained at every temperature from 20 °C to 60 °C at the end of the reaction (30 h, ESI S5.8†). The Ch1-AmDH–Cb-FDH dual enzyme system for reductive amination was instead inapplicable at 70 °C, albeit a mediocre conversion (8%) was observed at this temperature after 18 h. Our observation is in agreement with the previously determined half-life of 40 min for Ch1-AmDH at 70 °C.54
Regardless of the degree of conversion, the AmDH enzyme and the substrate employed, the enantiomeric excess was not affected by the reaction time or the temperature. The stereoselectivity remained always perfect (>99% (R)).
Current substrate scope of the reductive amination using AmDHs
The initial reaction rates for the reductive amination of a limited number of carbonyl compounds and the oxidative deamination of a few amines catalysed by AmDHs have been previously measured.51,52 However, a study describing the substrate scope of these enzymes for the organic synthesis of amines from prochiral ketones and aldehydes has not been published so far. Moreover, information regarding the stereoselectivity for the amination with AmDHs is limited to very few compounds. Therefore, in this research, we tested an extensive library of structurally diverse prochiral ketones such as phenylacetone derivatives and phenylacetaldehyde (1–7a), aliphatic methylketones and aldehydes (8–13a), acetophenone derivatives (14–19a) and, finally, a selection of more sterically demanding ketones (20–25a) (Fig. 2). The substrate concentration was kept at 50 mM, whereas the amount of enzyme and the reaction time were varied in order to achieve the maximum efficiency (i.e. highest ratio of [S]/[E] with highest conversion).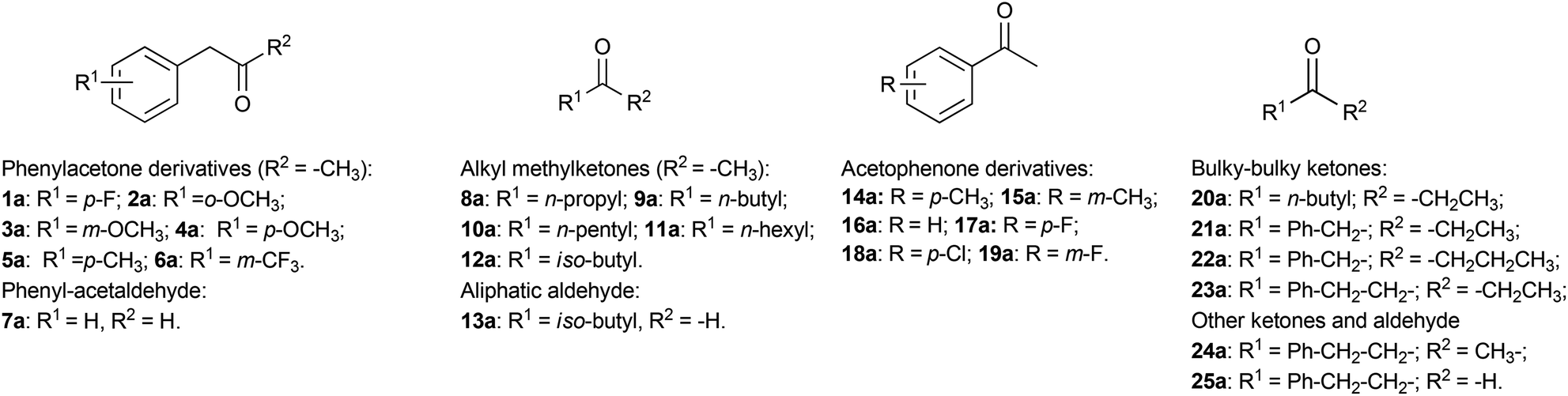 | ||
| Fig. 2 Substrate scope tested using amine dehydrogenases. | ||
First, we examined the family of phenylacetone derivatives (Fig. 2 and Table 2). It was previously shown that Bb-PhAmDH accepts (para-fluorophenyl)acetone (1a) as the best substrate.52 In our independent experiment (Table 2, entry 2), Bb-PhAmDH (50 μM) converted 1a (50 mM) to 93% of the amine product 1b within 48 h and in perfect stereoselectivity (>99% (R)). Hence, it may be logical to assume that Bb-PhAmDH can also be a useful biocatalyst for the amination of other substituted phenylacetones. Indeed, Bb-PhAmDH converted ortho-, meta- and para-methoxy substituted phenylacetone derivatives (2–4a), but the conversion was mediocre (from 3% to 21%, ESI Table S7†) although the reaction time was prolonged up to 48 h. Bb-PhAmDH also accepted (para-methyl)phenylacetone (5a, 7% conversion), whilst (meta-trifluoromethyl)phenylacetone (6a) was not converted at all (ESI Table S7†).
| Entry | No. | Enzyme | Enzyme concentration [μM] | Time [h] | Conversion substrate [%] | ee% (R) |
|---|---|---|---|---|---|---|
| Reaction conditions: substrate 50 mM, AmDH 30–130 μM, Cb-FDH 14 μM, ammonium formate buffer (1.005 M, pH 8.5), T 30 °C, agitation on an orbital shaker (190 rpm). n.a.: not applicable. | ||||||
| 1 | 1a | Ch1-AmDH | 30 | 24 | 93 | >99 |
| 2 | 1a | Bb-PhAmDH | 50 | 48 | 93 | >99 |
| 3 | 2a | Ch1-AmDH | 130 | 48 | >99 | >99 |
| 4 | 3a | Rs-PhAmDH | 50 | 24 | 98 | >99 |
| 5 | 4a | Rs-PhAmDH | 50 | 24 | >99 | >99 |
| 6 | 5a | Rs-PhAmDH | 130 | 48 | 98 | >99 |
| 7 | 6a | Rs-PhAmDH | 130 | 48 | 98 | >99 |
| 8 | 7a | Bb-PhAmDh | 50 | 48 | 34 | n.a. |
Surprisingly the chimeric enzyme Ch1-AmDH, known to be active on aliphatic ketones48 and acetophenone derivatives,54 was a superior catalyst for the amination of 1a. Compared to the amination with Bb-PhAmDH, Ch1-AmDH afforded the product 1b with the same conversion but in half of the reaction time (24 h) and at a significantly lower enzyme loading (30 μM, Table 2, entry 1). Ch1-AmDH was also the best catalyst for the amination of 2a that reached quantitative conversion in 48 h (Table 2, entry 3).
The third AmDH from this study, Rs-PhAmDH, has been developed and tested only for the reductive amination of 4-phenylbutan-2-one as the substrate (24a, Table 5, entry 6).53 In this study we disclosed that Rs-PhAmDH has a much wider substrate scope than expected and reported before. Interestingly Bb-PhAmDH and Rs-PhAmDH were engineered from their respective parent wild-type phenylalanine dehydrogenases by mutating similar positions in the active site (K78 and N277 for Bb-PhAmDH and K66, S149 and N262 for Rs-PhAmDH). In fact, in the wild-type enzymes, the side chains of the K and N residues located in the active site generate hydrogen bond interactions with the oxygens of the carboxylic moiety of the natural substrate. Furthermore, the two parent phenylalanine dehydrogenases have 34% identity. Despite these similarities, we have shown before (Fig. 1) that the two variants have a different thermal stability and reactivity measured on their respective preferred substrate: Rs-PhAmDH can operate efficiently at 60 °C, whereas Bb-PhAmDH is completely deactivated above 50 °C. Table 2 reveals that Rs-PhAmDH is a superior biocatalyst than Bb-PhAmDH also in terms of substrate acceptance. In fact, Rs-PhAmDH was the optimal AmDH for the conversion of 3a, 4a, 5a and 6a, affording the related amines in elevated conversion (more than or equal to 98%) and excellent stereoselectivity (>99% (R)) (Table 2, entries 4–7). On the other hand, Bb-PhAmDH proved to be still a useful catalyst for the amination of phenyl-acetaldehyde (7a) (Table 2, entry 8). In contrast, both Rs-PhAmDH and Ch1-AmDH were inactive on this type of aldehyde.
A similar scenario was revealed also in the case of the reductive amination of aliphatic ketones and aldehydes (Fig. 2 and Table 3). Initially, we investigated the less sterically demanding alkyl methylketones. Ch1-AmDH and Rs-PhAmDH were the most active biocatalysts on this family of substrates. Substrates (50 mM) bearing a medium length linear chain such as 2-hexanone (9a) and 2-heptanone (10a) were efficiently converted by Ch1-AmDH (30 μM) within 24 h (Table 3, entries 2 and 3). Ketones bearing a shorter linear chain (2-pentanone, 8a) or a branched chain (4-methylpentan-2-one, 12a) were converted with lower turnover numbers by Ch1-AmDH and, therefore, an elevated concentration of enzyme was necessary (130 μM, Table 3, entries 1 and 7). A ketone bearing a longer chain such as 2-octanone (11a) was a challenging substrate for Ch1-AmDH (Table 3, entry 5). Fortunately, Rs-PhAmDH seems to be a complementary enzyme in this respect: Rs-PhAmDH (50 μM) aminated 2-octanone (50 mM, 11a) with elevated conversion (93%, Table 3, entry 6). Notably, the enantiomeric excess was perfect for all the reductive aminations (>99% (R)).
| Entry | No. | Enzyme | Enzyme concentration [μM] | Time [h] | Conversion substrate [%] | ee% (R) |
|---|---|---|---|---|---|---|
| Reaction conditions: 0.5 mL, 50 mM substrate, 30–130 μM enzyme, ammonium formate buffer (1.005 M, pH 8.5), 14 μM Cb-FDH. n.a.: not applicable. | ||||||
| 1 | 8a | Ch1-AmDH | 130 | 48 | 75 | >99 |
| 2 | 9a | Ch1-AmDH | 30 | 24 | 92 | >99 |
| 3 | 10a | Ch1-AmDH | 30 | 24 | 98 | >99 |
| 4 | Rs-PhAmDH | 50 | 24 | 99 | >99 | |
| 5 | 11a | Ch1-AmDH | 30 | 48 | 50 | >99 |
| 6 | Rs-PhAmDH | 50 | 48 | 93 | >99 | |
| 7 | 12a | Ch1-AmDH | 130 | 48 | 96 | >99 |
| 8 | Rs-PhAmDH | 130 | 48 | 91 | >99 | |
| 9 | 13a | Ch1-AmDH | 30 | 24 | >99 | n.a |
| 10 | Rs-PhAmDH | 130 | 48 | 99 | n.a | |
Interestingly, Ch1-AmDH and Rs-PhAmDH rapidly converted 3-methylbutanal (13a, Table 3, entries 9 and 10) even though both enzymes were inactive on the arylaliphatic aldehyde 7a. These data demonstrate that all the known AmDHs are capable of converting ketones and aldehydes although with different substrate scopes.
Acetophenone derivatives (Fig. 2) are discussed separately as the type and position of the substituents on the phenyl ring affect considerably the reactivity of these substrates, due to the existence of resonance and field effects.67 This phenomenon is not always considered for enzymatic reactions, for which a low catalytic rate is often solely – and sometimes misleadingly – interpreted as the result of a poor affinity of the substrate to the active site of the enzyme or the intrinsic low enzymatic turnovers (kcat). In contrast, a number of publications on the reactivity of acetophenone derivatives with other oxidoreductases such as alcohol dehydrogenases showed that resonance and field effects can play a major, and sometimes unexpected, role.68–70
In this regard, the chimeric Ch1-AmDH and Rs-PhAmDH turned out to be the most efficient enzymes for the amination of acetophenone derivatives (Table 4 and ESI Table S8†). The reductive amination of these substrates (50 mM) required generally a higher amount of enzyme (up to 130 μM) for obtaining moderate conversions. para-Methylacetophenone (14a) was the least converted substrate (9%, Table 4, entry 1) whereas meta-fluoroacetophenone (19a) afforded the most elevated conversion (43%, Table 4, entry 6). Along the series of acetophenone derivatives reported in Table 4, the para-methyl substituent in 14a is the one possessing the most intense electron donating character, while the meta-fluoro in 19a is the strongest electron withdrawing substitution. Furthermore, para-hydroxy acetophenone, whose hydroxyl substituent has an even higher electron donating impact than a para-methyl,67,68 was not converted at all (ESI Table S6†).
| Entry | No. | Enzyme | Enzyme concentration [μM] | Time [h] | Conversion substrate [%] | ee% (R) |
|---|---|---|---|---|---|---|
| Reaction conditions: 0.5 mL, 50 mM substrate, 30–130 μM enzyme, ammonium formate buffer (1.005 M, pH 8.5), 14 μM Cb-FDH. | ||||||
| 1 | 14a | Ch1-AmDH | 130 | 48 | 9 | >99 |
| 2 | 15a | Ch1-AmDH | 50 | 48 | 39 | >99 |
| 3 | 16a | Ch1-AmDH | 130 | 48 | 34 | >99 |
| 4 | 17a | Ch1-AmDH | 130 | 48 | 22 | >99 |
| 5 | 18a | Rs-PhAmDH | 100 | 48 | 33 | >99 |
| 6 | 19a | Ch1-AmDH | 130 | 48 | 43 | >99 |
Although not based on a more rigorous determination of the initial reaction rates, this initial observation may suggest that the enzymatic reductive amination with AmDHs is favored by the delocalization of a higher partial positive charge on the reactive carbonyl carbon during the reaction. This assumption is corroborated by the fact that the same influence of the substituents was revealed for the reduction of acetophenone to alcohols with alcohol dehydrogenases (ADH). In fact, both ADHs and AmDHs belong to the class of oxidoreductases (EC1) and share the same cofactor (NAD) and a similar reaction mechanism.68–71 Additionally, none of the AmDHs from this study accepted ortho-methyl acetophenone as a substrate, indicating that the steric effect might also play a significant role in enzymatic reductive amination (ESI Table S6†).
Finally, the reactivity of AmDHs was investigated on more sterically demanding substrates. Bulky–bulky ketones (20–23a) (Fig. 2) are challenging substrates for the amination catalysed by other enzymes such as ω-transaminases. Naturally occurring ω-transaminases, which are suitable for application in biotechnology, seem to accept mainly ketones possessing a bulky group on one side and a small methyl group on the other side.19,20,72–77 Alternatively, wild-type ω-transaminases are active on bicyclic ketones.78 To the best of our knowledge, only one scaffold from a wild-type ω-transaminase was engineered for accepting bulky–bulky substrates.21,76
Thus, we were intrigued in understanding whether AmDHs also share the same limitation in relation to the range of ketones that can be converted. Interestingly, all the tested bulky–bulky ketones bearing the carbonyl moiety conjugated with the phenyl ring such as 1-phenylpropan-1-one, 1-phenylbutan-1-one and 1-phenylpentan-1-one were either not accepted or afforded mediocre conversions (ESI Table S6†). Conversely, when the carbonyl moiety was positioned further from the aromatic ring, the ketones (50 mM) were converted efficiently by Rs-PhAmDH (90–130 μM). For instance, 1-phenylbutan-2-one (21a), 1-phenylpentan-2-one (22a) and 1-phenylpentan-3-one (23a) afforded the amine product with a conversion of up to >99% and perfect stereoselectivity (>99% (R)) (Table 5, entries 3–5). In contrast, Bb-PhAmDH and Ch1-AmDH were poorly active on these substrates or were not active at all (ESI Tables S7 and S8†).
| Entry | No. | Enzyme | Enzyme concentration [μM] | Time [h] | Conversion substrate [%] | ee% (R) |
|---|---|---|---|---|---|---|
| Reaction conditions: 0.5 mL, 50 mM substrate, 30–130 μM enzyme, ammonium formate buffer (1.005 M, pH 8.5), 14 μM Cb-FDH. n.a.: not applicable. | ||||||
| 1 | 20a | Ch1-AmDH | 90 | 48 | 57 | >99 |
| 2 | Rs-PhAmDH | 100 | 48 | 32 | >99 | |
| 3 | 21a | Rs-PhAmDH | 130 | 48 | >99 | >99 |
| 4 | 22a | Rs-PhAmDH | 100 | 48 | 71 | >99 |
| 5 | 23a | Rs-PhAmDH | 100 | 48 | 87 | >99 |
| 6 | 24a | Rs-PhAmDH | 50 | 24 | >99 | >99 |
| 7 | 25a | Rs-PhAmDH | 100 | 48 | 96 | n.a. |
As an example of an aliphatic and more sterically demanding ketone, 3-heptanone (20a) was tested. In this case, Ch1-AmDH was the most active enzyme (Table 5, entry 1), in agreement with the general trend for this enzyme (i.e. elevated conversions for aliphatic ketones, Table 3).
For the sake of completeness, 4-phenyl-butan-2-one (24a) and an aldehyde such as hydrocinnamaldehyde (25a) were assayed for the reductive amination. Rs-PhAmDH (50 μM) was capable of quantitatively aminating 24a at the 50 mM scale within 24 h (Table 5, entry 6) with more than 99% ee. Rs-PhAmDH was also the optimal biocatalyst for the reduction of the aldehyde 25a (96%, Table 5, entry 7). Ch1-AmDH also quantitatively converted 25a. However, surprisingly, besides the desired product 3-phenylpropan-1-amine 25b (70%), hydrocinnamic alcohol was obtained as a side-product (30%). To the best of our knowledge, this is the only documented case wherein an amine dehydrogenase reduces a carbonyl compound leading to the formation of a significant amount of alcohol as a by-product.
Representative biocatalytic reductive amination on a preparative scale
In order to ascertain that our optimized reaction conditions at an analytical scale are applicable to preparative scale biocatalytic reactions, we attempted the asymmetric amination of (para-methoxy)phenylacetone (4a, 208 mg) using Rs-AmDH (Scheme 2).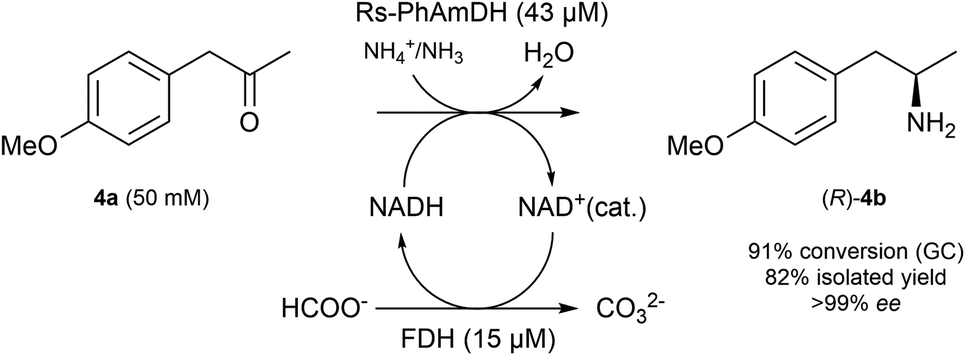 | ||
| Scheme 2 Preparative reductive amination of (para-methoxy phenyl)acetone (4a, 208 mg) using Rs-PhAmDH. Reaction conditions: ammonium formate buffer (1 M, pH 8.5), T 30 °C, agitation on an orbital shaker (190 rpm), reaction time 24 h. | ||
The reaction was successfully performed with ca. 50 mM substrate (208 mg), 43 μM of Rs-AmDH, 15 μM FDH, and 1 mM of NAD+ in ammonium formate buffer (1 M, pH 8.5) at the temperature of 30 °C. The substrate was converted into the optically pure amine (R)-4b with 91% conversion and >99% ee within 24 h reaction time. After work-up, (R)-4b was isolated with 82% yield. The purity and authenticity of the product were confirmed by NMR and GC (ESI S6†). Amine (R)-4b was recently reported as an important building block for the synthesis of tacrine–selegiline hybrids that possess cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer's disease.79
Additionally, amine (R)-4b is the optically active intermediate for the synthesis of the blockbuster pharmaceutical formoterol,80,81 sold under various trade names including Foradile and Oxeze.
Conclusions
In this work, we have shown that amine dehydrogenases hold the basis for the development of the next generation of chemical processes for the synthesis of α-chiral amines.47 The applicability of amine dehydrogenases was demonstrated for the asymmetric amination of a range of structurally diverse prochiral ketones and aldehydes. The most suitable enzyme and the optimal catalyst loading and reaction times were determined for each substrate from this study. The majority of the substrates tested were aminated with elevated conversion and elevated TONs; moreover, all the α-chiral amine products were obtained with perfect optical purity (>99% R). This fact is of particular interest as ω-transaminases capable of giving access to (R)-configured amines are rare in nature. Only very few (R)-selective ω-transaminases have been discovered82,83 and only one enzyme was engineered to convert a specific bulky–bulky ketone with elevated stereoselectivity.21,62 From this study, it became also evident that a single amine dehydrogenase capable of accepting a large variety of substrates is not available. For instance, the chimeric Ch1-AmDH is very active on aliphatic ketones and acetophenone derivatives whereas Rs-PhAmDH is an excellent biocatalyst for the amination of phenylacetone derivatives and more sterically demanding ketones. Additionally, the influence of temperature on the biocatalytic reductive amination with the three AmDH variants was determined. In particular, a pH in the range of 8.2–8.8 is preferable as a consequence of the improved stability of the enzymes (AmDHs and FDH) and, especially, of the coenzyme (NAD). Finally, the optimised reaction parameters were applied to the synthesis of an important drug precursor on a scale of hundreds of milligrams.The reductive amination catalyzed by amine dehydrogenases operating in tandem with formate dehydrogenase possesses an elevated atom efficiency as the ammonium formate buffer is simultaneously the source of nitrogen and reducing equivalents. Stoichiometric inorganic carbonate is the sole by-product. Additionally, the reductive amination catalyzed by AmDH/FDH is performed under atmospheric pressure. In contrast, the amination catalysed by ω-transaminases on an industrial scale, in an aqueous system and using 2-propylamine (ca. 10 equivalents) as an amine donor, requires removal of the co-product acetone by employing reduced pressure and nitrogen sweep. Hence, besides the requirement of a supra-stoichiometric amount of 2-propylamine and the generation of one equivalent of acetone, a significant amount of energy is consumed to operate at low pressure. We expect therefore that the herein described reductive amination will be applied increasingly in the future when new amine dehydrogenase variants possessing expanded substrate scope and complementary stereoselectivity will become available. Co-expression of AmDH/FDH in a single host organism and/or co-immobilisation of both enzymes will enhance the practical applicability of the biocatalytic process.84–86
Experimental
General
The AmDH variants and Cb-FDH were expressed as recombinant enzymes in E. coli BL21 (DE3). Details are reported in the ESI, paragraph S4.†General optimized procedure for the biocatalytic reductive amination on an analytical scale
The reactions were conducted in ammonium formate buffer (1.005 M, pH 8.5, final volume 0.5 mL) containing NAD+ (final concentration 1 mM). The enzymes AmDH (30–130 μM) and Cb-FDH (14.1 μM) and the substrate (50 mM) were added. The reactions were run at 30 °C in an incubator for 21 hours (190 rpm) or longer if required in selected cases. Work-up was performed by the addition of KOH (100 μL, 10 M) followed by extraction with dichloromethane (600 μL). The water layer was removed after centrifugation and the organic layer was dried with MgSO4. Conversion was determined by GC with an Agilent DB-1701 column. The enantiomeric excess of the amine product was determined after derivatization to acetamido. Derivatization of the samples was performed by adding 4-dimethylaminopyridine into acetic anhydride (40 μL of 50 mg mL−1 stock solution). The samples were shaken in an incubator at RT for 30 minutes. Afterwards, water (300 μL) was added and the samples were shaken for an additional 30 minutes. After centrifugation, the organic layer was dried with MgSO4. Enantiomeric excess was determined by GC with a Variant Chiracel DEX-CB column. Details of the GC analysis and methods are reported in the ESI, paragraph S7.†Preparative biocatalytic reductive amination for the synthesis of (R)-4b
NAD+ (final concentration 1 mM) was dissolved in ammonium formate buffer (30 mL, 1.005 M, pH 8.5) in a 50 mL round bottom flask. Ketone 4a (195 μL, 1.27 mmol), FDH (233 μL from 80.7 mg mL−1 stock solution, final concentration 15 μM), and Rs-PhAmDH (1.02 mL from 48.8 mg mL−1 stock solution, final concentration 43 μM) were added and the reaction mixture was shaken in an incubator at 30 °C for 24 hours. The progress of the reaction was monitored by TCL and GC. When quantitative conversion was achieved, the reaction mixture was acidified to pH 2–4 via addition of HCl (1 M). The water layer was washed with methyl tert-butyl ether (15 mL) to remove any possible remaining ketone starting material. The pH of the water phase was increased to basic pH via KOH (10 M) while cooling in an ice bath. The water layer was extracted with methyl tert-butyl ether (2 × 15 mL). The organic fractions containing the amine product were combined and dried over MgSO4. After filtration and evaporation of the solvent, the product was obtained in a pure form. Column chromatography was not required. The authenticity of the product was confirmed by 1H-NMR (ESI Fig. S4†). 1H NMR (400 MHz, CDCl3): δ 7.12 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 3.81 (s, 3H), 3.13 (m, 1H), 2.67 (dd, J = 13.4, 5.3 Hz, 1H), 2.47 (dd, J = 13.4, 8.1 Hz, 1H), 1.12 (d, J = 6.3 Hz, 3H).Competing financial interest
The authors declare to have no competing interests, or other interests that might be perceived to influence the results and/or discussion reported in this article.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 638271, BioSusAmin). Dutch funding from the NWO Sector Plan for Physics and Chemistry is also acknowledged.Notes and references
- H. A. Wittcoff, B. G. Rueben and J. S. Plotkin, in Industrial Organic Chemicals, Wiley-Interscience, New York, 2nd edn, 2004 Search PubMed.
- Chiral amine synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley-VCH, Weinheim, 2010 Search PubMed.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS.
- S. K. Au, J. Groover, B. D. Feske and A. S. Bommarius, in Organic Synthesis Using Biocatalysis, Elsevier, 1st edn, 2015, pp. 187–212 Search PubMed.
- F. Balkenhohl, K. Ditrich, B. Hauer and W. Ladner, J. Prakt. Chem., 1997, 339, 381–384 CrossRef CAS.
- Y. Kim, J. Park and M.-J. Kim, ChemCatChem, 2011, 3, 271–277 CrossRef CAS.
- G. Li, J. Ren, P. Yao, Y. Duan, H. Zhang, Q. Wu, J. Feng, P. C. K. Lau and D. Zhu, ACS Catal., 2014, 4, 903–908 CrossRef CAS.
- N. J. Turner, Chem. Rev., 2011, 111, 4073–4087 CrossRef CAS PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed.
- C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224–2225 CrossRef CAS PubMed.
- R. Carr, M. Alexeeva, A. Enright, T. S. Eve, M. J. Dawson and N. J. Turner, Angew. Chem., Int. Ed., 2003, 42, 4807–4810 CrossRef CAS PubMed.
- M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 114, 3309–3312 CrossRef.
- V. Kohler, Y. M. Wilson, M. Durrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knorr, D. Haussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef CAS PubMed.
- T. Li, J. Liang, A. Ambrogelly, T. Brennan, G. Gloor, G. Huisman, J. Lalonde, A. Lekhal, B. Mijts, S. Muley, L. Newman, M. Tobin, G. Wong, A. Zaks and X. Zhang, J. Am. Chem. Soc., 2012, 134, 6467–6472 CrossRef CAS PubMed.
- J. M. Foulkes, K. J. Malone, V. S. Coker, N. J. Turner and J. R. Lloyd, ACS Catal., 2011, 1, 1589–1594 CrossRef CAS.
- V. Kohler, K. R. Bailey, A. Znabet, J. Raftery, M. Helliwell and N. J. Turner, Angew. Chem., Int. Ed., 2010, 49, 2182–2184 CrossRef PubMed.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 CrossRef CAS PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed.
- D. Koszelewski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozzell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 9337–9340 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- A. Cuetos, M. Garcia-Ramos, E. M. Fischereder, A. Diaz-Rodriguez, G. Grogan, V. Gotor, W. Kroutil and I. Lavandera, Angew. Chem., Int. Ed., 2016, 55, 3144–3147 CrossRef CAS PubMed.
- B. Wang, H. Land and P. Berglund, Chem. Commun., 2013, 49, 161–163 RSC.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2016, 358, 1618–1624 CrossRef.
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Muller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- H. Li, Z.-J. Luan, G.-W. Zheng and J.-H. Xu, Adv. Synth. Catal., 2015, 357, 1692–1696 CrossRef CAS.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 CrossRef CAS PubMed.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- F. Parmeggiani, S. L. Lovelock, N. J. Weise, S. T. Ahmed and N. J. Turner, Angew. Chem., Int. Ed., 2015, 54, 4608–4611 CrossRef CAS PubMed.
- N. J. Weise, F. Parmeggiani, S. T. Ahmed and N. J. Turner, J. Am. Chem. Soc., 2015, 137, 12977–12983 CrossRef CAS PubMed.
- S. L. Lovelock, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 4652–4656 CrossRef CAS PubMed.
- B. Wu, W. Szymanski, G. G. Wybenga, M. M. Heberling, S. Bartsch, S. de Wildeman, G. J. Poelarends, B. L. Feringa, B. W. Dijkstra and D. B. Janssen, Angew. Chem., Int. Ed., 2012, 51, 482–486 CrossRef CAS PubMed.
- N. J. Turner, Curr. Opin. Chem. Biol., 2011, 15, 234–240 CrossRef CAS PubMed.
- B. J. Verkuijl, W. Szymanski, B. Wu, A. J. Minnaard, D. B. Janssen, J. G. de Vries and B. L. Feringa, Chem. Commun., 2010, 46, 901–903 RSC.
- E.-M. Fischereder, D. Pressnitz and W. Kroutil, ACS Catal., 2016, 6, 23–30 CrossRef CAS.
- F. Wu, H. Zhu, L. Sun, C. Rajendran, M. Wang, X. Ren, S. Panjikar, A. Cherkasov, H. Zou and J. Stockigt, J. Am. Chem. Soc., 2012, 134, 1498–1500 CrossRef CAS PubMed.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS.
- A. Bonamore, I. Rovardi, F. Gasparrini, P. Baiocco, M. Barba, C. Molinaro, B. Botta, A. Boffi and A. Macone, Green Chem., 2010, 12, 1623 RSC.
- J. H. Schrittwieser, B. Groenendaal, V. Resch, D. Ghislieri, S. Wallner, E. M. Fischereder, E. Fuchs, B. Grischek, J. H. Sattler, P. Macheroux, N. J. Turner and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 3731–3734 CrossRef CAS PubMed.
- J. H. Schrittwieser, V. Resch, J. H. Sattler, W. D. Lienhart, K. Durchschein, A. Winkler, K. Gruber, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2011, 50, 1068–1071 CrossRef CAS PubMed.
- A. Winkler, A. Lyskowski, S. Riedl, M. Puhl, T. M. Kutchan, P. Macheroux and K. Gruber, Nat. Chem. Biol., 2008, 4, 739–741 CrossRef CAS PubMed.
- T. K. Hyster, C. C. Farwell, A. R. Buller, J. A. McIntosh and F. H. Arnold, J. Am. Chem. Soc., 2014, 136, 15505–15508 CrossRef CAS PubMed.
- J. A. McIntosh, P. S. Coelho, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed.
- E. M. Brustad, Nat. Chem. Biol., 2014, 10, 170–171 CrossRef CAS PubMed.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- J. B. Wang and M. T. Reetz, Nat. Chem., 2015, 7, 948–949 CrossRef CAS PubMed.
- N. Itoh, C. Yachi and T. Kudome, J. Mol. Catal. B: Enzym., 2000, 10, 281–290 CrossRef CAS.
- M. J. Abrahamson, E. Vazquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 CrossRef CAS PubMed.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS.
- B. R. Bommarius, M. Schürmann and A. S. Bommarius, Chem. Commun., 2014, 50, 14953–14955 RSC.
- O. H. Lowry, J. V. Passonneau and M. K. Rock, J. Biol. Chem., 1961, 236, 2756–2759 CAS.
- S. G. A. Alivisatos, F. Ungar and G. Abraham, Nature, 1964, 203, 973–975 CrossRef CAS PubMed.
- S. G. A. Alivisatos, F. Ungar and G. J. Abraham, Biochemistry, 1965, 4, 2616–2630 CrossRef CAS PubMed.
- L. Rover, J. C. B. Fernandes, G. D. O. Neto, L. T. Kubota, E. Katekawa and S. L. H. P. Serrano, Anal. Biochem., 1998, 260, 50–55 CrossRef CAS.
- R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- H. Schutte, J. Flossdorf, H. Sahm and M.-R. Kula, Eur. J. Biochem., 1976, 62, 151–160 CrossRef.
- TON is here defined as the number of molecules of substrate converted per molecule of enzyme in the given reaction time.
- F. G. Mutti, J. Sattler, K. Tauber and W. Kroutil, ChemCatChem, 2011, 3, 109–111 CrossRef CAS.
- F. G. Mutti and W. Kroutil, Adv. Synth. Catal., 2012, 354, 3409–3413 CrossRef CAS.
- J. S. Shin and B. G. Kim, Biotechnol. Bioeng., 2002, 77, 832–837 CrossRef CAS PubMed.
- J.-S. Shin and B.-G. Kim, Biosci., Biotechnol., Biochem., 2001, 65, 1782–1788 CrossRef CAS PubMed.
- J.-S. Shin and B.-G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS PubMed.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Inc., 6th edn, 2007 Search PubMed.
- M. Vogl, R. Kratzer, B. Nidetzky and L. Brecker, Org. Biomol. Chem., 2011, 9, 5863–5870 CAS.
- M. L. Contente, I. Serra, L. Palazzolo, C. Parravicini, E. Gianazza, I. Eberini, A. Pinto, B. Guidi, F. Molinari and D. Romano, Org. Biomol. Chem., 2016, 14, 3404–3408 CAS.
- D. Zhu, B. E. Rios, J. D. Rozzell and L. Hua, Tetrahedron: Asymmetry, 2005, 16, 1541–1546 CrossRef CAS.
- C. Rodriguez, W. Borzecka, J. H. Sattler, W. Kroutil, I. Lavandera and V. Gotor, Org. Biomol. Chem., 2014, 12, 673–681 CAS.
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS PubMed.
- E. O'Reilly, C. Iglesias, D. Ghislieri, J. Hopwood, J. L. Galman, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 2447–2450 CrossRef PubMed.
- D. Koszelewski, M. Göritzer, D. Clay, B. Seisser and W. Kroutil, ChemCatChem, 2010, 2, 73–77 CrossRef CAS.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, N. G. Turrini, J. H. Sattler, A. Lerchner, A. Skerra and W. Kroutil, Eur. J. Org. Chem., 2012, 1003–1007 CrossRef CAS.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef.
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS.
- C. Lu, Q. Zhou, J. Yan, Z. Du, L. Huang and X. Li, Eur. J. Med. Chem., 2013, 62, 745–753 CrossRef CAS PubMed.
- F. Campos, M. P. Bosch and A. Guerrero, Tetrahedron: Asymmetry, 2000, 11, 2705–2717 CrossRef CAS.
- G. P. Anderson, Life Sci., 1993, 52, 2145–2160 CrossRef CAS PubMed.
- A. Iwasaki, Y. Yamada, N. Kizaki, Y. Ikenaka and J. Hasegawa, Appl. Microbiol. Biotechnol., 2006, 69, 499–505 CrossRef CAS PubMed.
- M. Hohne, S. Schatzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: A full list of the substrates with related structures, detailed descriptions of the expression and purification of the enzymes, full data sets and explanation of the biocatalytic reactions, NMR spectra, GC analytical methods and data as well as GC traces. See DOI: 10.1039/c6gc01987k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |