
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wood biorefinery based on γ-valerolactone/water fractionation†
Huy Quang
Lê
 ,
Yibo
Ma
,
Marc
Borrega
and
Herbert
Sixta
*
,
Yibo
Ma
,
Marc
Borrega
and
Herbert
Sixta
*
Department of Forest Products Technology, School of Chemical Technology, Aalto University, PO Box 16300, 00076 Aalto, Finland. E-mail: herbert.sixta@aalto.fi
First published on 8th August 2016
Abstract
A novel biorefinery concept based on the fractionation of woody biomass in a γ-valerolactone (GVL)/water binary mixture is introduced. Under optimal GVL/water ratio, Eucalyptus globulus wood was effectively fractionated in a single step into its principal components. The pulp fraction, characterized by high yield, high cellulose purity and high bleachability, was directly spun to produce regenerated cellulosic fibers with mechanical properties comparable to the best man-made fibers currently available in the market. Most of the hemicelluloses and lignin in wood were extracted and dissolved into the spent liquor. The dissolved hemicellulose-based fraction may be upgraded to furanic platform chemicals in subsequent catalytic conversion processes. About 50–60% of the extracted lignin was precipitated by the addition of water, an anti-solvent. The precipitated lignin was characterized by low carbohydrate and ash contamination, high phenolic content, relatively low polydispersity and low molecular mass. The lignin extracted by GVL/water fractionation may thus be suitable for a wide range of energy, material or chemical applications.
Introduction
A biorefinery is defined as a facility that integrates biomass conversion processes, which offers the full utilization of biomass components for the production of energy, materials and chemicals. Cellulose is the most abundant biopolymer, constituting 40–50 wt% of dry biomass, and thus cellulose-based products are the most important in the spectrum of biorefinery products. Generally, cellulose is converted to paper-grade pulp, with a global annual production of about 170 million tons in 2013.1 However, the competitiveness of pulp and paper industries is decreasing due to the establishment of new producers in tropical and sub-tropical regions, with higher tree growing rates and cheaper labor costs, which places serious economic pressure on the producers in temperate-climate regions such as Finland. This situation is one of the driving forces for their transformation from the mass production of paper-grade pulp towards other niche products with lower production but higher gross margin. Dissolving pulp is one example. Dissolving pulp, characterized by high cellulose content (>90%), high brightness and low macromolecular polydispersity, is used for the production of regenerated fibers and cellulose derivatives, comprising a much higher value than conventional paper. Global dissolving pulp production is currently small (about 6.4 million tons in 2013,2i.e. less than 4% of that of paper-making pulp), however, the demand for dissolving pulp is significantly increasing due to a persistent growth of the cellulosic textile fiber consumption during the coming years.3 To meet this increasing demand, global production of dissolving pulp is expected to double in the next two decades.4Currently, dissolving pulp is commercially produced from wood, by either the acid sulfite or the prehydrolysis kraft (PHK) pulping process, or from cotton linters by refining, with a production share of 50, 35 and 15%, respectively.5 Despite being the most significant natural source of pure cellulose, pulp production from cotton linters is expected to stagnate in the near future, due to unavailability of cotton growing land and high production costs.6 Additionally, the cultivation and refining of cotton raise various environmental issues such as intensive irrigation and excessive application of pesticides and fertilizers.7 Therefore, cotton linters pulps are probably more suitable for applications requiring high purity, for example, the production of cellulose derivatives,8 while lower-grade dissolving wood pulps can be used for commodity products like textile fibers. Being the oldest commercial pulping technology, acid sulfite pulping has serious disadvantages, such as limited flexibility in term of raw material and inefficient chemical recovery.9 Furthermore, hemicelluloses are oxidized by bisulfite anions to aldonic acids, which are difficult to recover10 and, moreover, hamper the efficient recovery of aldoses such as xylose after the separation of the lignocellulose fraction.6 Kraft pulping is by far the dominant and the most efficient commercial process for the production of paper-grade pulp. Production of dissolving pulp by kraft process can be enabled by the installation of an extra prehydrolysis stage. The PHK process, however, suffers from low pulp yield11–13 and the underutilization of the hemicellulosic stream. A significant part of the hemicelluloses in wood are extracted during prehydrolysis, but their recovery is hindered by the formation of sticky lignin precipitates.14 Above all, both acid sulfite and PHK are sulfur-containing processes, thus raising environmental concerns.
Intensive research efforts have been made on finding alternative fractionation methods to produce dissolving pulp as well as to recover the hemicellulose and lignin fractions for their utilization as value-added products, following biorefinery principles. Ionic liquids have recently emerged as novel solvents in biomass fractionation processes. Ionosolv fractionation can be performed homogenously or heterogeneously. In the homogenous approach, all biomass constituents are dissolved, then different fractions are separately regenerated by the addition of anti-solvents. In the heterogeneous approach, certain biopolymer is selectively dissolved from the biomass. However, the use of ionic liquids reveals several drawbacks for both approaches. Firstly, the thermal instability of ionic liquids prevents the process to be conducted at temperatures higher than 130 °C, resulting in long fractionation time, typically more than 8 hours for milled wood and longer for wood chips.15–17 Secondly, low consistency (<5%) is required for the effective dissolution of biomass constituents, making the process unpractical in large scale.18–20 And thirdly, dissolution power of ionic liquids is usually sensitive to the presence of water,21 which requires intensive drying of the raw material before processing. Especially, the research on heterogeneous fractionation is still at initial stage due to limited dissolution selectivity. Delignification in ionic liquid is accompanied by significant carbohydrate loss.22–24 Therefore, ionic liquids extraction are more useful as pulp purification method than biomass fractionation. For example, by the recently introduced IONCELL-P process, where xylan is selectively dissolved, bleached kraft pulp can be converted to high purity dissolving pulp.25–27
Besides, the use of organic solvents offers a promising solution for the effective isolation and full utilization of wood components.28–30 Organosolv pulping are mostly sulfur- and inorganic-free processes to fractionate biomass, but the corrosive organic solvents and the typically high pressure during the cooking process demand costly equipment investment. ALCELL®, Acetocell, Formacell and Milox are representative examples of conventional organosolv fractionation processes which employ ethanol, acetic acid, formic acid and peroxyformic acid as cooking chemicals. During the last decade, novel concepts on organosolv pulping have been developed; the two other representatives are the SEW,31 a sulfur-containing organosolv process and the Clean Fractionation,32 which employ ternary solvents such as SO2–ethanol–water or methyl isobutyl ketone–ethanol–water, respectively. These processes have shown the potential to produce high quality dissolving pulp from lignocellulosic biomass, and the SEW process has already reached the pilot scale, operated under the trademark AVAP™ by American Process Inc. Despite the initial success of those two novel fractionation concepts, we believe that the forest industry would need a wider range of biomass processing methods in order to be able to match and compete with the vast product spectra offered by the oil-refining industry. Therefore, it is still worth investigating in other alternative fractionation processes.
Recently, gamma-valerolactone (GVL) has been identified as a promising organic solvent for biomass conversion.33,34 GVL is a green, non-toxic and non-volatile (vapor pressure of 0.44 mbar at 25 °C) solvent, soluble in water but does not form azeotrope, and has a low melting point (−31 °C) and a high boiling point (207 °C).35,36 Furthermore, the recognizable smell of GVL enables easy detection of leakage or spilling and more importantly, it is a stable chemical unsusceptible to degradation and oxidation at standard conditions, making it a safe substance for large-scale storage, transportation and application.33 For biomass fractionation, GVL is coupled with water as a binary mixture in which water provides hydrolytic power towards hemicelluloses while GVL dissolves the lignin fraction. GVL/water brings a clear advantage in terms of reaction pressure over the most renowned organosolv process, ALCELL®, where a binary mixture of ethanol/water is employed.37 For example, at the fractionation temperature of 180 °C, 50/50 wt% ethanol/water mixture delivers a pressure of 18 bars,38 while that of GVL/H2O is about 9.9 bars, which would potentially reduce the cost for pressure-resistant vessels.
The conversion of lignocellulosic biomass into monomeric carbohydrates in GVL/water medium without the need for an additional enzymatic hydrolysis stage has been investigated by Luterbacher et al.39 Their results showed that carbohydrates from corn stover could be converted to their monomeric and oligomeric forms at high yield, which could then be further processed to platform biochemicals or biofuels. Nevertheless, this approach did not include separate value chains for the cellulose and hemicellulose streams (both were combined and hydrolyzed to sugars), thus underutilizing cellulose, which may be employed in its polymeric form in the manufacture of high value-added biomaterials. More recently, Fang et al. tested the fractionation of birch sawdust in GVL/water mixtures34 and concluded that GVL-based fractionation of wood may be employed to produce dissolving pulp. However, sawdust fractionation is not feasible in industrial scale due to the high energy input required to mill the raw material into fine particle size. Therefore, we hereby suggest a biorefinery concept based on GVL/water fractionation of eucalyptus wood chips to fully utilize the biomass components. Fractionation trials under optimum GVL/water ratios for wood delignification are performed and the effects on the extraction of wood components are reported. The cellulose fraction is converted, with and without bleaching, to textile fibers, and their mechanical properties are compared to those of commercial fibers. The hemicelluloses fraction in the spent liquor is then characterized and discussed in relation to its valorization pathways to furanic platform chemicals and to GVL. Finally, the lignin fraction is precipitated and characterized, and based on the lignin properties possible applications are evaluated.
Materials and methods
Chemicals and material
Eucalyptus globulus wood chips were delivered by ENCE, Spain. The chips were screened according to the SCAN-CM 40:01 standard and stored at −20 °C. Some chips were air-drided and then ground to sawdust in a Wiley mill (Arthur H. Thomas Co., model No. 2 with screen opening 0.5 mm) while others were used directly for cooking trials. Only those milled wood particles with size smaller than 125 microns were collected. The identified chemical composition of the wood was: 44.1% glucose, 15.2% xylose, 3.1% other sugars, 27.7% lignin and 1.3% extractive. Wood chips and sawdust were used for the GVL/water fractionation experiments. The GVL was supplied by Sigma Aldrich with ≥98 wt% purity. Pure water was obtained from Millipore Synergy® UV purification system (water resistivity of 18.2 MΩ cm).The ionic liquid 1,5-diazabicyclo[4.3.0]non-5-ene acetate, [DBNH][OAc], was employed as cellulose solvent in the production of regenerated fibers from GVL/water pulp. [DBNH][OAc] was synthesized from 1,5-diazabicyclo[4.3.0]non-5-ene (DBN, supplied by Fluorochem, UK, 99% purity) and acetic acid (glacial, supplied by Merck, Germany, 100% purity). An equimolar amount of acetic acid was slowly added to DBN. The temperature was controlled and allowed to rise to 70 °C in order to avoid solidification of the ionic liquid.
GVL/water fractionation
To identify the optimum GVL concentration for delignification, small scale fractionation trials with eucalyptus sawdust were conducted in 30 mL vials heated in a microwave reactor (Anton Paar Monowave 300). For each trial, 1.5 g of oven-dried sawdust were used, with a liquid-to-wood ratio (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W) of 10 L kg−1. The GVL content in the fractionation liquor ranged from 0–98 wt%. The reaction mixture was rapidly heated (less than 2 min) to 180 °C, held at that temperature for 120 min, and then cooled down by compressed air to 55 °C. The pulp and the spent liquor were separated by filtration with a Robu® glass crucible (porosity 4). The pulp was washed with 200 mL of hot water (ca. 80 °C) and then oven-dried at 105 °C. The pulp yield was determined gravimetrically. The spent liquor and washing water were collected for subsequent analyses.
:W) of 10 L kg−1. The GVL content in the fractionation liquor ranged from 0–98 wt%. The reaction mixture was rapidly heated (less than 2 min) to 180 °C, held at that temperature for 120 min, and then cooled down by compressed air to 55 °C. The pulp and the spent liquor were separated by filtration with a Robu® glass crucible (porosity 4). The pulp was washed with 200 mL of hot water (ca. 80 °C) and then oven-dried at 105 °C. The pulp yield was determined gravimetrically. The spent liquor and washing water were collected for subsequent analyses.
The fractionation trials with wood chips were conducted in 225 mL bombs heated in a silicon oil-bath reactor (Haato-tuote, model 43427). An impregnation stage at 120 °C for 60 minutes was used to facilitate the penetration of the cooking liquor into the wood cellular structure. A reaction temperature of 180 °C and a L:W ratio of 10 L kg−1 were used. The GVL content in the liquor was 50 and 60 wt%, and the fractionation time (retention time at 180 °C) ranged from 60 to 180 minutes. The reaction was quenched by submerging the bombs in cold water, and the pulp and spent liquor were separated using a nylon filtration bag. In comparison with the sawdust fractionation experiments, an extra washing stage with 60 wt% ethanol (with ratio of about 10 L kg−1) was added to prevent lignin deposition upon water addition. The use of ethanol instead of the more expensive GVL was here adopted solely as an economical lab-scale approach, while in industrial scale, pulp washing will be executed with a GVL/water mixture. The ethanol-washed pulp was subjected to a final wash with hot (ca. 80 °C) water. Spent liquor and washing liquids were collected for subsequent analyses. After washing, the pulp was screened in a table-top screener (G.A. Serlachius A.B., model 16140-567, with 0.35 mm mesh-opening) to determine the rejects amount. Pulp yield was determined gravimetrically.
A selected pulp sample from a GVL/water fractionation was bleached using an ECF (Elemental-Chlorine-Free) sequence of D0–Ep–P. The bleaching was done in plastic bags, heated by steam in a water bath. The conditions for each bleaching stage were: D0: 50 °C, 60 minutes, 10% consistency, active chlorine charge according to a Kappa factor of 0.25; Ep: 70 °C, 60 minutes, 10% consistency, 1.5% NaOH, 0.5% H2O2; P: 70 °C, 120 minutes, 10% consistency, 0.6% NaOH, 0.5% H2O2.
Analytical characterization of pulp and liquor
The carbohydrate and lignin content in the pulps were analyzed in accordance to the 2-step hydrolysis method described in the NREL/TP-510-42618 standard. The pulp was firstly hydrolyzed in 72% H2SO4, with an acid-to-material ratio of 10 mL g−1, at 30 ± 3 °C, for 60 ± 5 minutes. The hydrolyzed suspension was subjected to the second hydrolysis in 4% H2SO4, with an acid-to-material ratio of 300 mL g−1, at 121 ± 1 °C, for 60 minutes. The monosaccharides were analyzed by high performance anion exchange chromatography (HPAEC-PAD) in a Dionex ICS-3000 system, equipped with a CarboPac PA20 column. From the amount of neutral monosaccharides, the cellulose and hemicelluloses content in wood and pulp samples was estimated with the Janson formula.40 Acid insoluble (Klason) lignin was determined gravimetrically while acid soluble lignin (ASL) was determined by measuring the absorbance at the wavelength of 205 nm (spectrophotometer Shimadzu UV-2550). An extinction coefficient of 148 L (g cm)−1 was used for quantification of ASL. This extinction coefficient was calibrated for the lignin extracted by GVL/water fractionation (ESI 1†). The hexenuronic acid (HexA) content in pulp was quantified by UV Resonance Raman spectroscopy with the method developed by Jääskeläinen et al. (2005).41 Selected pulps were analyzed for viscosity and kappa number in accordance to the SCAN-CM 15:88 and SCAN-C 1:00 standards, respectively. Additionally, pads from the bleached pulps were prepared in accordance to the SCAN-C 11:95 standard, and their ISO-brightness was measured according to the SCAN-P 3:93 standard.The molar mass distribution (MMD) of selected pulps was determined by gel permeation liquid chromatography (GPC). Prior to the analyses, the samples were activated by a water–acetone-N,N-dimethylacetamide (DMAc) sequence. Activated samples were dissolved in 90 g L−1 lithium chloride (LiCl) containing DMAc at room temperature and under gentle stirring. The samples were then diluted to 9 g L−1 LiCl/DMAc, filtered with 0.2 μm syringe filters, and fed to a Dionex Ultimate 3000 system equipped with four PLgel MIXED-A 7.5 × 300 mm columns, refractive index detector Shodex RI-101and with LiCl/DMAc as the eluent. Pullulan standards (343 Da–708 kDa, Polymer Standard Service GmbH, Mainz, Germany, and 1600 kDa, Fluka GmbH, Germany) were used to calibrate the system. The molar masses of pullulan standards were converted to correspond to those of cellulose, using the equation MMcellulose = q × (MMPullulan)p, developed by Berggren et al.42
The carbohydrate and lignin content in the spent liquors and washing liquids were analyzed in accordance to the method described in the NREL/TP-510-42623 standard. The monomeric sugars were determined by direct injection in the HPAEC-PAD, while the oligomeric ones were quantified by the difference after hydrolysis at 121 ± 1 °C for 60 minutes, with sulfuric acid concentration of 4%. The lignin content in the spent and washing liquors was determined by UV-Vis spectrophotometry (Shimadzu UV-2550 spectrometer) after diluting with ethanol solution of 35 wt% and measuring the absorption at the wavelength of 205 nm, using the extinction coefficient of 148 L (g cm)−1.
The content of furanic compounds (furfural and HMF) and organic acids (formic acid, acetic and levulinic acid) in the spent liquor was determined by high performance liquid chromatography in a Dionex UltiMate 3000 (Dionex, Sunnyvale, CA, USA) device equipped with Refractive Index (RI) and UV diode array detectors and HyperREZ XP Carbohydrate Ca+ column 7.7 × 300 mm (Thermo Scientific, Waltham, MA, USA). The eluent was 0.005 mol L−1 sulfuric acid. The column temperature and the RI detector temperature were set to 70 °C and 55 °C, respectively.
Production and characterization of regenerated fibers
Selected bleached and unbleached pulps were spun to regenerated cellulosic fibers according to the IONCELL-F spinning process.43,44 Prior to pulp dissolution, [DBNH][OAc] was liquefied at 70 °C. The spinning dopes were then prepared in a vertical kneader by mixing air-dried pulps with the IL with a concentration of 13 wt%, and kneaded for 1.5 hours at 80 °C and 10 rpm at reduced pressure (50–200 mbar). The resulting solution was filtered by a hydraulic press filter device (metal filter mesh with 5 μm absolute fineness, Gebr. Kufferath AG, Germany) at 2 MPa and 80 °C to remove undissolved substrate which would lead to unstable spinning. The dope was then shaped into the dimension of the spinning cylinder and solidified upon cooling.Filament spinning was carried out by a customized laboratory piston spinning system (Fourné Polymertechnik, Germany) as described by Hummel et al. (2015).44 The solidified dope was loaded in the cylinder and then heated to 70 °C to form a highly viscous, air-bubble-free spinning dope. The molten solution was extruded through a 36-hole spinneret (with capillary diameter of 100 μm and a length-to-diameter ratio (L/D) of 0.2) to a 1 cm air gap and subsequently the filaments were coagulated in a water bath (15 °C), where they were guided by Teflon rollers to the godet couple. The extrusion velocity (Ve) was set to 1.6 mL min−1 (11.4 m min−1) while the take-up velocity (Vt) of the godet varied from 10 to 90 m min−1 depending on different samples. The draw ratio (DR) is calculated as DR = Vt/Ve. The spun fibers were washed off-line in hot water (ca. 60 °C) and air-dried.
The linear density (titer) and tenacity of fibers in dry and wet conditions were determined by using a Vibroskop 400 and a Vibrodyn 400 (Lenzing Instruments GmbH & Co KG, Austria) at 23 °C and 50% relative humidity (RH). The gauge length in the Vibrodyn 400 was set to 20 mm, the strain rate was 20 mm min−1 and the pretension was 5.9 ± 1.2 mN per tex. 10 fibers of each sample were measured. The elastic modulus of the spun fibers was calculated from the slope of the entire elastic region of the stress–strain curves with a Matlab script according to ASTM standard D2256/D2256 M.45
Conversion of xylose to furfural
The spent liquor produced by fractionation of wood chips in 50 wt% GVL and after 150 min reaction time was selected for production of furfural. The spent liquor was first concentrated with pure GVL to reach a 90 wt% GVL solution, and then heated at 200 °C in the presence of 0.1 M sulfuric acid. The experiments were conducted in 30 mL vials in the microwave reactor (Anton Paar Monowave 300), with holding times ranging from 5 to 120 min. The resulting solutions were analyzed for xylose, furanic compounds and organic acids by the same analytical procedure as described above for the analysis of the crude spent liquors.Lignin isolation and characterization
Lignin in the spent liquor after GVL/water fractionation was precipitated by addition of water (water-to-spent-liquor ratio from 1 to 6). The suspensions were centrifuged at a relative centrifugal force of 3000g for 30 minutes. The precipitated lignin was collected and washed 3 times by addition of water (with the same amount as the original spent liquor), followed by ultrasonic treatment for 15 minutes. The washed lignin was air-dried and manually ground to finer particles. Precipitated lignin sample from the experiment with water-to-spent-liquor = 3 was selected for further analyses.Lignin elemental analysis (nitrogen, carbon, hydrogen, oxygen and sulfur content determination) was performed with a FlashEA 1112 elemental analyzer series CHNS/O with MAS200R Auto-sampler (Thermo Fisher Scientific). Lignin methoxyl groups were quantified in accordance with the Zeisel–Vieböck–Schwappach method, as described in Zakis (1994).46 The carbohydrate and lignin content of the lignin sample was analyzed in accordance to the 2-step hydrolysis method described above for the characterization of pulp.
The molecular mass distribution (MMD) of lignin was obtained by GPC using the UV detector (UV-vis Detector 2487). Dimethyl sulfoxide (DMSO) containing 0.1% LiBr was used as a column eluent (1 mL min−1 flow rate). The GPC system consisted of two analytical columns (Suprema 1000 and Suprema 100, 20 μm, 8 mm I.D. × 300 mm) and one pre-column (Suprema 20 μm). The columns, injector and UV detector were maintained at 80 °C during the analysis.
The content of structural groups (hydroxyl, β-O-4, etc.) in the precipitated lignin was analyzed using a nuclear magnetic resonance (NMR) spectrometer (Varian Unity Inova 500, 5 mm broadband probe head at 27 °C, 600 MHz 1H-frequency). Lignin samples were acetylated in pyridine/acetic anhydride (1:1 by volume), washed with ethanol and subsequently freeze-dried.47 The sample for NMR analysis was dissolved in deuterated chloroform (CDCl3) containing 0.03% tetramethylsilane with concentration of 150 mg mL−1. For quantitative 13C experiments, inverse gated 1H-decoupling and 30-degree excitation pulse flip angle were utilized. Spectral width was 36182.7 Hz, relaxation delay was 5 seconds, and acquisition time was 0.2 second. Number of transients varied between 41215 and 49683. Free induction decays were apodized using exponential multiplication with 10 Hz line broadening and zero filled up to 16384 complex points prior to Fourier transformation. For 13C experiments, the samples were doped with relaxation agent chromium(III) acetylacetonate Cr(acac)3 to 10 mM concentration.
Results and discussion
Eucalyptus wood fractionation
Small scale fractionation trials with sawdust were first conducted to determine the optimum GVL/H2O ratio for wood delignification. The behavior of the main components of wood, i.e. cellulose, hemicelluloses and lignin, along with varying GVL/H2O ratios is shown in Fig. 1. The wood was not extracted before the fractionation trials, and thus a small amount of extractives (about 1.3%) in the starting material appears as lignin in the ASL analysis by UV-spectrometry. | ||
| Fig. 1 Effect of GVL content in the fractionation liquor on the removal of wood components from eucalyptus sawdust. Fractionation trials were conducted at 180 °C for 120 minutes, with a L:W = 10 L kg−1 (odw: oven-dried wood). | ||
The results in Fig. 1 indicate that the cellulose fraction in wood was recovered almost quantitatively at any GVL/H2O content, due to the low extent of hydrolysis under the relatively low reaction temperature. On the other hand, the removal of hemicelluloses increased with increasing water content in the fractionation liquor, due to enhanced hydrolytic degradation. Delignification reached a maximum when the fractionation liquor contained about 50–60 wt% GVL. These results are in agreement with those reported by Fang and Sixta for the fractionation of birch sawdust.34Table 1 summarizes important results obtained from eucalyptus sawdust fractionation, with emphasis on pulp chemical composition and properties. The GVL/H2O fractionation appears to be a mild acid-driven process; the pH of the spent liquor measured at room conditions ranged between 2.8 and 3.7, predominantly increasing with increasing GVL content. However, the fundamental mechanisms behind delignification reactions are not yet understood and will be the subject of future investigations.
:W = 10 L kg−1, for 120 minutes
| GVL in liquor (wt%) | Solid fraction (pulp) | Spent liquor pH | |||||
|---|---|---|---|---|---|---|---|
| Yieldc (% odw) | Cellulosed (% odp) | C5a (% odp) | C6b (% odp) | Lignin (% odp) | Viscosity (mL g−1) | ||
| a C5 hemicelluloses (xylan and arabinan). b C6 hemicelluloses (galactan, mannan and rhamnan). c Percent on oven-dried wood. d Percent on oven-dried pulp. | |||||||
| 35 | 54.5 | 79.9 | 3.2 | 1.0 | 15.9 | 177 | 3.03 |
| 50 | 49.9 | 85.0 | 5.3 | 1.8 | 7.9 | 245 | 3.20 |
| 60 | 50.5 | 85.1 | 6.3 | 1.5 | 7.1 | 305 | 3.53 |
| 75 | 59.1 | 76.0 | 10.0 | 2.6 | 11.4 | 402 | 3.74 |
The high cellulose content in the pulps produced in 50% and 60% GVL, coupled with the relatively low hemicellulose and lignin content (Table 1) suggests the possibility to convert GVL/water pulp to dissolving pulp of viscose grade after bleaching. However, the use of sawdust for pulp production cannot be realized at industrial scale. Therefore, the potential of GVL/water fractionation in dissolving pulp production was investigated by fractionating wood chips in GVL/water mixtures containing 50 and 60 wt% GVL (Table 2). The results indicate that after 60 min fractionation time, about 75% of the hemicelluloses and over 90% of the lignin from wood were removed, while cellulose was quantitatively preserved. Extending reaction time beyond 60 min slightly increased the removal of wood components, but considerably decreased the intrinsic viscosity. Using 50 wt% instead of 60 wt% GVL in the liquor gave a slight advantage on delignification and hemicellulose removal, but on the other hand, resulted in lower pulp viscosity. Compared to the sawdust fractionation trials, the inclusion of an additional washing stage with GVL/water further improved the removal of spent liquor entrapped within the fibers, resulting in pulps with lower lignin content.
:W = 10 L kg−1 (odw: oven-dried wood)
| Samplea | Pulp yield (% odw) | Intrinsic viscosity (mL g−1) | Wood components (% odw) | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pulp | Spent liquorb | |||||||||||
| Cellulose | Hemicellulose | Ligninc | Cellulose | Hemicellulose | Ligninc | Furfural | HMF | Acidsd | ||||
| a The sample is named as: GVL content in fractionation liquor (in wt%) − fractionation time (in minutes). b Component content in SL is the sum of that in free SL and washing liquids. c As the raw material is unextracted wood, extractive are shown as lignin in analysis. d Organic acids: formic acid, acetic acid and levulinic acid. e Uronic acid which is not bound to xylan (about 2.6% odw)40 is not taken into account. | ||||||||||||
| Wood | 100 | — | 46.4 | 22.1 | 29.0 | — | — | — | — | — | — | 97.8 |
| 50–60 | 52.7 | 833 | 45.8 | 4.4 | 2.6 | 0.8 | 9.2 | 26.8 | 1.5 | 0.1 | 8.5 | 99.7 |
| 50–90 | 47.8 | 818 | 42.5 | 3.6 | 1.9 | 0.4 | 7.8 | 26.9 | 2.5 | 0.2 | 9.5 | 95.3 |
| 50–120 | 46.8 | 630 | 42.7 | 3.0 | 1.2 | 0.5 | 6.5 | 27.8 | 3.7 | 0.3 | 8.5 | 94.2 |
| 50–150 | 45.0 | 551 | 41.4 | 2.5 | 1.2 | 0.5 | 5.1 | 28.0 | 4.8 | 0.4 | 8.1 | 92.0 |
| 50–180 | 44.6 | 456 | 41.3 | 2.3 | 1.0 | 0.6 | 4.1 | 28.1 | 5.6 | 0.5 | 7.8 | 91.3 |
| 60–60 | 51.9 | 936 | 44.9 | 4.6 | 2.7 | 0.4 | 8.1 | 24.7 | 1.0 | 0.1 | 8.4 | 94.9 |
| 60–90 | 48.6 | 862 | 42.4 | 4.1 | 2.2 | 0.4 | 7.7 | 26.3 | 1.9 | 0.1 | 10.1 | 95.2 |
| 60–120 | 47.3 | 737 | 42.7 | 3.4 | 1.3 | 0.3 | 5.7 | 28.2 | 3.0 | 0.2 | 10.3 | 95.1 |
| 60–150 | 45.8 | 710 | 41.0 | 3.3 | 1.5 | 0.4 | 4.9 | 27.6 | 3.2 | 0.3 | 9.0 | 91.2 |
| 60–180 | 44.8 | 592 | 40.7 | 2.9 | 1.3 | 0.5 | 4.3 | 28.1 | 4.5 | 0.4 | 9.9 | 92.6 |
The material mass balances are shown in Table 2. Over 91% of the starting wood material could be identified in the pulp and in the spent liquor. Lignin was quantitatively recovered, while over 90% of the cellulose was identified. About 30–65% of the hemicelluloses were recovered as sugar fractions in both the pulp and the spent liquor, but this amount increased to over 90% when the furans and organic acids found in the spent liquor were accounted for. The formation of furanic compounds and organic acids from carbohydrates and their presence in the spent liquor will be discussed in Hemicellulose-based compounds section.
Valorization of pulp fraction
According to the results in Table 2, GVL/water fractionation may be tailored to yield either paper-grade pulp (with higher GVL content in the liquor and shorter reaction time) or dissolving pulp (with lower GVL content and longer reaction time). In this work, we have focused on the production of viscose-grade dissolving pulp and its conversion into regenerated cellulosic fibers by the IONCELL-F spinning process.44 Therefore, the pulps produced after 150 and 180 minutes in 50 wt% GVL/water, having the lowest hemicellulose and lignin content, were selected for spinning. The detailed composition and properties of all pulps is shown in ESI 2.†The pulp produced after 180 minutes of fractionation time was spun directly without bleaching, while the pulp produced after 150 min fractionation time was bleached with the short ECF sequence D0–Ep–P. The viscosity of the bleached pulp dropped to 470 mL g−1 during bleaching, which was within the range of the 400–500 mL g−1 preferably required for spinning. Since GVL/water fractionation is a mildly acidic process, no HexA were detected in the pulps, resulting in high pulp bleachability. The chemical composition and macromolecular properties of the pulps used in the spinning trials are shown in Table 3. A commercial bleached acid sulfite pulp from hardwood, with similar molecular mass distribution as the GVL pulps (see Fig. 2), was selected as reference and spun to regenerated cellulose fibers with the same procedure.
 | ||
| Fig. 2 Molecular mass distribution of unbleached and bleached GVL/water pulps, in comparison with a commercial bleached acid sulfite hardwood dissolving pulp used as reference (dw/dlog(MW): differential mass fraction). | ||
| Sample | Chemical composition [% odp] | ISO brightness | Viscosity (mL g−1) |
M
wa (kDa) |
PDIb | DP > 2000c (wt%) | ||
|---|---|---|---|---|---|---|---|---|
| Cellulose | Hemicellulose | Lignin | ||||||
| a Weight-average molecular mass. b Polydispersity index. c Fraction with degree of polymerization higher than 2000. d U-GVL and B-GVL: unbleached and bleached GVL/water pulps, respectively. | ||||||||
| U-GVLd | 92.7 | 5.2 | 2.1 | — | 456 | 352 | 8.2 | 0.27 |
| B-GVLd | 93.6 | 5.8 | 0.6 | 86% | 470 | 309 | 8.0 | 0.24 |
| Reference | 94.9 | 4.2 | 0.9 | 89% | 524 | 334 | 8.9 | 0.26 |
Prior to spinning, the GVL and reference pulps were readily dissolved in [DBNH][OAc] with no solid retained on the filter. In a previous study, the optimum visco-elastic properties for stable spinning of [DBNH][OAc]-based dopes (13 wt% eucalyptus pre-hydrolyzed kraft pulp, viscosity 468 mL g−1) were observed at 70 °C, where a zero shear viscosity of 25000–35000 Pa s was obtained.43 Due to the similarity of the pulp viscosity and chemical compositions, 13 wt% dopes for the three pulps used in this study were prepared and spun at temperature of 70–80 °C. The spun fibers from GVL/water pulps are shown in Fig. 3.
 | ||
| Fig. 3 Regenerated cellulosic fibers spun from unbleached (left) and bleached (right) pulps produced by GVL/water fractionation of eucalyptus wood. | ||
The mechanical properties of both unbleached and bleached regenerated fibers are shown in Table 4. Despite a slightly lower wet tenacity at maximum draw ratio, the fibers produced from the bleached GVL/water pulp exhibited higher strength and stretchability (elongation) than those produced from the unbleached GVL/water pulp. The tenacity of the fibers produced from the bleached GVL pulp was similar to that of the fibers produced from the reference pulp. The lower mechanical performance of the fibers from unbleached GVL pulp might be attributed to the higher content of lignin present in the pulp. The mechanical properties of the spun fibers were further compared to those of commercial viscose and TENCEL® fibers. As shown in Fig. 4, the reference fiber (produced by IONCELL-F process) demonstrated the strongest tenacity among the commercial fibers. However, the tensile properties of the GVL fibers were comparable to those of the reference fiber and clearly higher than those of commercial TENCEL® and viscose fibers. The elastic moduli of the GVL fibers were determined from the stress–strain curves and compared to those of commercial fibers in Fig. 4. The elastic moduli were similar for both GVL fibers and higher than those of viscose and modal fibers, but slightly lower than those of the reference and TENCEL® fibers. These results demonstrate that dissolving pulps produced by GVL/water fractionation can be successfully spun into regenerated cellulosic fibers with mechanical properties comparable to those of the best man-made fibers existing in the market.
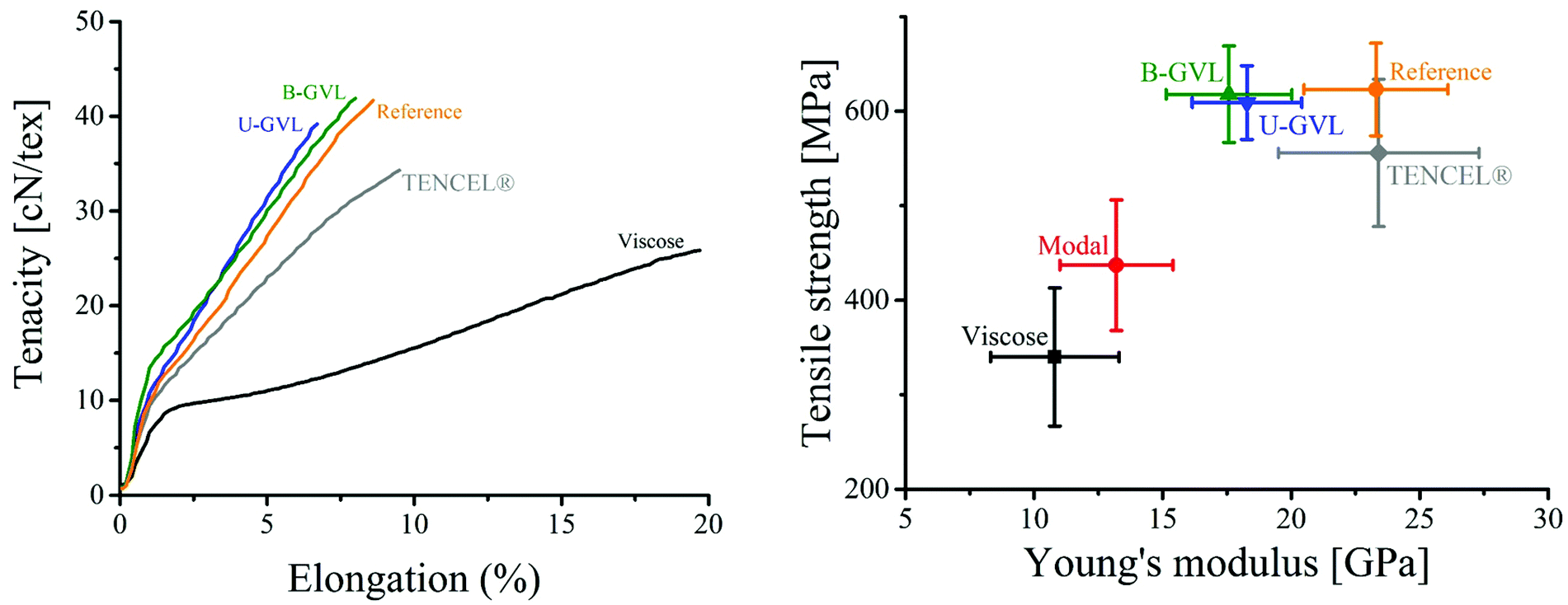 | ||
| Fig. 4 (Left) Stress–strain curves of GVL fibers in comparison to commercial textile fibers. (Right) Young's modulus of different composite and commercial fibers. Values for viscose, modal and lyocell fibers are adopted from Adusumali et al. (2006).48 | ||
| Sample | Max DR | Titer (dtex) | Tenacity (cN per tex) | Wet tenacity (cN per tex) | Elongation (%) | Wet elongation (%) |
|---|---|---|---|---|---|---|
| a U-GVL and B-GVL: unbleached and bleached GVL/water pulps, respectively. | ||||||
| U-GVLa | 14.1 | 1.30 ± 0.1 | 40.6 ± 2.6 | 37.1 ± 2.8 | 8.2 ± 0.8 | 9.5 ± 0.7 |
| B-GVLa | 15.9 | 1.30 ± 0.2 | 41.2 ± 3.4 | 34.7 ± 2.4 | 10.0 ± 1.2 | 11.0 ± 0.6 |
| Reference | 15 | 1.45 ± 0.2 | 41.5 ± 3.3 | — | 8.2 ± 1.2 | — |
Valorization of spent liquor
The production of furanic compounds is a promising pathway for the valorization of GVL/water spent liquor. As shown in Table 2, the formation of furans increased with increasing fractionation time at both 50 wt% and 60 wt% GVL/water ratios. After 180 minutes of reaction time, the amounts of furfural and HMF were about 5–6% odw and 0.5% odw, respectively. The furfural formation corresponds to a conversion of 37–43 mol% of the extracted xylose (ESI 4†). Moreover, the amount of dissolved sugars in the spent liquor was about 4% odw, which may increase the maximum potential yield of furans to 11% odw. Aqueous systems are known to facilitate degradation reactions of furanic compounds, via fragmentation (production of smaller molecules like acids, aldehydes), resinification (coupling of two furfural molecules) and condensation (coupling of furfural with pentoses or intermediates),51 thus limiting the yield of furans to less than 60 mol%.52 However, Gürbüz et al. have reported that GVL/water systems largely suppress these degradation reactions, resulting in higher furfural yields.28 In order to maximize the conversion of sugars to furfural, the presence of a strong acid catalyst is typically required; the use of conventional sulfuric acid yields >80 mol% of furanic compounds.53 In this study, the production of furfural was evaluated in a selected spent liquor (50 wt% GVL and 150 min), after concentrating to 90% GVL and adding 0.1 M sulfuric acid. The results showed that the treatment of spent liquor at 200 °C for 5 min converted the xylose into furfural at a yield of 84 mol%, raising the overall conversion to 51 mol% of the extracted xylose. Nevertheless, extending reaction time decreased the furfural yield due to its degradation (ESI 5†). However, and despite the effective conversion of xylose into furfural by addition of sulfuric acid, the introduction of a homogenous acid catalyst results in equipment corrosion, poses environmental issues, and complicates the recovery process. An innovative approach is the use of solid acid catalysts, as introduced by Dumesic and co-workers,53 who obtained comparable furfural yields to H2SO4-catalyzed conversion when xylose was dehydrated in 90 wt% GVL solution in the presence of H-mordenite. Moreover, they also showed a good recyclability of the catalyst. Nonetheless, it is worth noting that the production of furans in the spent liquor by using solid acid catalysts would likely be more complicated than from pure xylose due to the complex chemical nature of the spent liquor, particularly in the presence of lignin, which may deposit onto the catalyst and deactivate it. Therefore, the use of a solid acid catalyst in our spent liquor may require the previous development of an efficient fractionation and recycling scheme, which is the subject of current investigations. An alternative to the production of furans for the valorization of the hemicellulosic fraction in the spent liquor may be the production of GVL via the hydrogenation of levulinic acid intermediate,54–59 which in turn could be used to account for possible solvent losses during the fractionation.
In addition to furanic compounds, three organic acids were also identified in the spent liquors: formic, acetic and levulinic acid (ESI 3†). These acids are known to be degradation products of the carbohydrates. Acetic acid originates from the cleavage of acetyl groups in glucuronoxylan, which is the most abundant hemicellulose in hardwoods.60 Formic and levulinic acid are formed simultaneously from the degradation of hexoses via HMF intermediate.61 Additionally, formic acid can also originate from the degradation of pentoses.62,63 The total amount of organic acids ranged between 8–10% odw, with formic acid being the most abundant, while the presence of levulinic acid was only minor (<0.5% odw). These acids may be isolated by reactive distillation of the spent liquor, for example, with methanol,64 thus increasing the valorization of the sugar-based side-streams.
 | ||
| Fig. 5 Relative lignin precipitation from GVL/water fractionation spent liquor by addition of water. | ||
| Property | Value |
|---|---|
|
a Number of methoxyl group per phenylpropane unit (C9, lignin elementary unit).
b Lignin moieties relative amount p-hydroxyphenyl (H):guaiacyl (G):syringyl (S).
c Number of hydroxyl groups per phenylpropane unit.
|
|
| Empirical formula | C9H6.42O2.23(OCH3)1.39 |
| Weight-average molar mass (g mol−1) | 2745 |
| Polydispersity | 3.5 |
| Carbon (wt%) | 64.24 |
| Hydrogen (wt%) | 5.46 |
| Nitrogen (wt%) | 0.20 |
| Oxygen (wt%) | 30.11 |
| Ash (wt%) | 0.16 |
| Hexose carbohydrate (wt%) | 0.26 |
| Pentose carbohydrate (wt%) | 0.21 |
| Acid soluble lignin (wt%) | 5.04 |
| Acid insoluble lignin (wt%) | 87.93 |
| Methoxyl group (OCH3) (wt%) | 22.29 |
| OCH3/C9a |
1.39 |
| H:G:Sb |
19.1:22.8:58.1 |
| Primary aliphatic OH/C9c |
0.30 |
| Secondary aliphatic OH/C9c |
0.25 |
| Phenolic OH/C9c |
0.58 |
| β-O-4/C9 | 0.12 |
The combustion of the lignin to produce energy to sustain the fractionation process might be less complicated than in the case of sulfite and kraft lignin, due to the absence of sulfur and ash, which would lower the investment for combustion equipment and exhaust gas treatment. As lignin is the only high-volume renewable feedstock comprised of aromatics,65 the relative ease to isolate the GVL lignin and its purity may enable the production of aromatic compounds by depolymerization. High content of syringyl (S) units in the lignin (58.1%, Table 5) would also favor thermal or chemical depolymerization. This is because methoxyl groups at position 3 and 5 on the phenyl units diminish relatively unreactive C–C interunit linkages, resulting in a more reactive lignin.66 Furthermore, the lignin isolated from our process may be a precursor for the manufacturing of cost-competitive carbon fibers, because minimization of lignin interunit C–O bonds, and especially the β-O-4 linkage, is reported to favor the production of structural carbon fibers.67 Low β-O-4 content (12% per C9, Table 5) of GVL lignin is a good starting point to further lower the C–O bond content via chemical modification. However, carbon fibers produced from lignin are currently inferior to those produced from poly-acrylonitrile, because the amorphous nature of lignin limits the graphitic stacking, making the fiber more brittle and with significantly lower mechanical properties.68,69
Conclusions
GVL/water mixtures enable a quantitative and selective fractionation of all lignocellulosic components in just one single step, with a solid cellulose fraction that can be directly spun to high-quality cellulose regenerated fibers, and a liquid fraction that contains the extracted sugars and lignin which can be further processed to valuable chemicals and materials. Via our proposed biorefinery concept, about 80% of the starting material is potentially converted to final products. The cellulosic fraction, under the form of unbleached textile fibers, is the main product with a biorefinery yield of 47.5%. Additionally, 32.5% of the raw material can be processed to side products, which are: 15.4% as sulfur-free lignin, 8.3% as organic acids and 8.6% as furanic compounds (including the conversion of dissolved hemicelluloses under optimum conditions).Furthermore, the process is sulfur-free and the high pulp bleachability offers the possibility for a TCF (Total Chlorine Free) bleaching sequence, thus making the process more environmentally friendly. Altogether, our process fulfills the requirements of a modern biorefinery. The possibility to lower the L:W ratio for industrial commercialization, as well as the development of efficient solvent recovery schemes, will be the subject of future investigations.
Acknowledgements
Funding from Aalto University, School of Chemical Technology and Finnish Bioeconomy Cluster Oy (FIBIC) via the Advanced Cellulose to Novel Products (ACel) research program is gratefully acknowledged. The authors would like to thank Ms Olga Ershova for her experimental support with the furfural production and Ms. Rita Hataka for her support with the chromatographic analyses.References
- K. Conley, Annual Review of Global Pulp & Paper Statistics, RISI Inc., PPI, 2014 Search PubMed.
- R. Young, World Dissolving Pulp Monitor, RISI Inc., PPI, 2014 Search PubMed.
- The Fiber Year 2016: World Survey on Textiles & Nonwovens, Issue 16, May 2016, http://www.thefiberyear.com/ Search PubMed.
- F. M. Hämmerle, Lenzinger Ber., 2011, 89, 12 Search PubMed.
- H. Sixta, Handbook of Pulp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, vol. 2, p. 743 Search PubMed.
- H. Sixta, M. Iakovlev, L. Testova, A. Roselli, M. Hummel, M. Borrega, A. Heiningen, C. Froschauer and H. Schottenberger, Cellulose, 2013, 20, 1547 CrossRef CAS.
- J. Carpenter, A. Felsot, T. Goode, M. Hammig, D. Onstad and S. Sankula, Comparative environmental impacts of biotechnology-derived and traditional soybean, corn, and cotton crops, Council for Agricultural Science and Technology, Ames (IA), 2002 Search PubMed.
- M. Iakovlev, X. You, A. van Heiningen and H. Sixta, Cellulose, 2014, 21, 1419 CAS.
- H. Sixta, Handbook of Pulp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, vol. 1, p. 608 Search PubMed.
- S. A. Rydholm, Pulping Chemistry, R.E. Krieger Publisher Company, Malabar (FL), 1985 Search PubMed.
- H. Sixta and G. Schild, Lenzinger Ber., 2009, 87, 26 CAS.
- C. V. T. Mendes, M. G. V. S. Carvalho, C. M. S. G. Baptista, J. M. S. Rocha, B. I. G. Soares and G. D. A. Sousa, Food Bioprod. Process., 2009, 87, 197 CrossRef CAS.
- H. Håkansson, U. Germgård and D. Sens, Cellulose, 2005, 12, 621 CrossRef.
- M. Leschinsky, H. Sixta and R. Patt, BioResources, 2009, 4, 687 CAS.
- X. Wang, H. Li, Y. Cao and Q. Tang, Bioresour. Technol., 2011, 102, 7959 CrossRef CAS PubMed.
- J. Viell and W. Marquardt, Holzforschung, 2011, 65, 519 CrossRef CAS.
- L. K. J. Hauru, Y. Ma, M. Hummel, M. Alekhina, A. W. T. King, I. Kilpeläinen, P. A. Penttilä, R. Serimaa and H. Sixta, RSC Adv., 2013, 3, 16365 RSC.
- W. Lan, C. Liu and R. Sun, J. Agric. Food Chem., 2011, 59, 8691 CrossRef CAS PubMed.
- W. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038 RSC.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodriguez and R. D. Rogers, Green Chem., 2009, 11, 646 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS PubMed.
- S. S. Y. Tan, D. R. MacFarlane, J. Upfal, L. A. Edye, W. O. S. Doherty, A. F. Patti, J. M. Pringle and J. L. Scott, Green Chem., 2009, 11, 339 RSC.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368 CrossRef CAS PubMed.
- A. Pinkert, D. F. Goeke, K. N. Marsh and S. Pang, Green Chem., 2011, 13, 3124 RSC.
- C. Froschauer, M. Hummel, G. Laus, H. Schottenberger, H. Sixta, H. K. Weber and G. Zuckerstätter, Biomacromolecules, 2012, 13, 1973 CrossRef CAS PubMed.
- A. Roselli, M. Hummel, A. Monshizadeh, T. Maloney and H. Sixta, Cellulose, 2014, 21, 3655 CrossRef CAS.
- A. Roselli, S. Asikainen, A. Stepan, A. Monshizadeh, N. von Weymarn, K. Kovasin, Y. Wang, H. Xiong, O. Turunen, M. Hummel and H. Sixta, Holzforschung, 2016, 70, 291 CrossRef CAS.
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270 CrossRef PubMed.
- M. A. Mellmer, D. Martin Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Green Chem., 2014, 16, 4659 RSC.
- S. Dutta and S. Pal, Biomass Bioenergy, 2014, 62, 182 CrossRef CAS.
- M. Iakovlev, T. Pääkkönen and A. van Heiningen, Holzforschung, 2009, 63, 779 CrossRef CAS.
- J. J. Bozell, S. K. Black, M. Myers, D. Cahill, W. P. Miller and S. Park, Biomass Bioenergy, 2011, 35, 4197 CrossRef CAS.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- W. Fang and H. Sixta, ChemSusChem, 2015, 8, 73 CrossRef CAS PubMed.
- I. T. Horvath, Green Chem., 2008, 10, 1024 RSC.
- V. N. Emel'yanenko, S. A. Kozlova, S. P. Verevkin and G. N. Roganov, J. Chem. Thermodyn. Thermochem., 2008, 40, 911 CrossRef.
- E. K. Pye and J. H. Lora, Tappi J., 1991, 74, 113 CAS.
- S. Laure, M. Leschinsky, M. Froehling, F. Schultmann and G. Unkelbach, Cellul. Chem. Technol., 2014, 48, 793 CAS.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277 CrossRef CAS PubMed.
- J. Janson, Pap. Puu, 1970, 52, 323 CAS.
- A. Jääskeläinen, A. Saariaho and T. Vuorinen, J. Wood Chem. Technol., 2005, 25, 51 CrossRef.
- R. Berggren, F. Berthold, E. Sjöholm and M. Lindström, J. Appl. Polym. Sci., 2003, 88, 1170 CrossRef CAS.
- H. Sixta, A. Michud, L. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpelainen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43 CAS.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. K. J. Hauru, A. Parviainen, A. W. T. King, I. Kilpeläinen and H. Sixta, Ionic Liquids for the Production of Man-Made Cellulosic Fibers: Opportunities and Challenges, Springer, Berlin, Heidelberg, 2015 Search PubMed.
- L. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471 CrossRef CAS.
- G. F. Zakis, Functional analysis of lignins and their derivatives, TAPPI PRESS, Atlanta (GA), 1994 Search PubMed.
- S. Y. Lin and C. W. Dence, Methods in lignin chemistry, Springer, 1992, p. 578 Search PubMed.
- R. Adusumali, M. Reifferscheid, H. Weber, T. Roeder, H. Sixta and W. Gindl, Macromol. Symp., 2006, 244, 119 CrossRef.
- G. Marcotullio and W. de Jong, Carbohydr. Res., 2011, 346, 1291 CrossRef CAS PubMed.
- K. J. Zeitsch, The chemistry and technology of furfural and its many by-products, Elsevier Science B. V., Amsterdam, the Netherlands, 2000, vol. 13 Search PubMed.
- A. P. Dunlop, Ind. Eng. Chem., 1948, 40, 204 CrossRef CAS.
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2013, 6, 76 CAS.
- A. P. Dunlop and J. W. Madden, US Patent2786862, 1957 Search PubMed.
- R. S. Assary and L. A. Curtiss, Chem. Phys. Lett., 2012, 541, 21 CrossRef CAS.
- P. P. Upare, J. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J. Chang, J. Ind. Eng. Chem., 2011, 17, 287 CrossRef CAS.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755 RSC.
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321 CrossRef CAS.
- D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Catal. Sci. Technol., 2013, 3, 927 CAS.
- O. Theander and D. A. Nelson, Advances in carbohydrate chemistry and biochemistry, Harcourt Brace Jovanovich, San Diego (CA), 1988, p. 273 Search PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696 CrossRef CAS.
- T. Ahmad, L. Kenne, K. Olsson and O. Theander, Carbohydr. Res., 1995, 276, 309 CrossRef CAS.
- M. R. Nimlos, X. Qian, M. Davis, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 11824 CrossRef CAS PubMed.
- D. Painer, S. Lux and M. Siebenhofer, Sep. Sci. Technol., 2015, 50, 2930 CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 CrossRef CAS PubMed.
- M. Foston, G. A. Nunnery, X. Meng, Q. Sun, F. S. Baker and A. Ragauskas, Carbon, 2013, 52, 65 CrossRef CAS.
- D. Saha, E. A. Payzant, A. S. Kumbhar and A. K. Naskar, ACS Appl. Mater. Interfaces, 2013, 5, 5868 CAS.
- J. L. Braun, K. M. Holtman and J. F. Kadla, Carbon, 2005, 43, 385 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01692h |
| This journal is © The Royal Society of Chemistry 2016 |