
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompositional and structural feedstock requirements of a liquid phase cellulose-to-naphtha process in a carbon- and hydrogen-neutral biorefinery context†
A.
Deneyer
a,
T.
Ennaert
a,
G.
Cavents
a,
J.
Dijkmans
a,
J.
Vanneste
ab,
C. M.
Courtin
c,
M.
Dusselier
*a and
B. F.
Sels
*a
aCenter for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200f, 3001 Heverlee, Belgium. E-mail: michiel.dusselier@kuleuven.be; bert.sels@kuleuven.be
bMaterials Department, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400 Mol, Belgium
cCenter for Food and Microbial Technology, KU Leuven, Kasteelpark Arenberg 22, 3001 Heverlee, Belgium
First published on 27th July 2016
Abstract
Processing raw (ligno)cellulosic feedstock into renewable light naphtha alkanes could lead to a gradual replacement of fossil feedstock for the production of chemicals, materials and fuels. The production of drop-in alkanes is a preferable short term strategy because of its practical implementation and integration in existing infrastructure and processes. A handful of promising cellulose-to-alkane biorefinery initiatives were recently reported, both processing in gas and liquid phase. This contribution presents a detailed study of the two-liquid phase hydrodeoxygenation of cellulose to n-hexane under relatively mild circumstances, proceeding through the recently communicated HMF route, in presence of a soluble acid and Ru/C metal catalyst. Two main points were addressed here: (i) the importance (or not) of the lignocellulose pretreatment and purification to the alkane yield, and (ii) the renewability of the consumed hydrogen in the process. A systematic study of the effect of cellulose purity, crystallinity, degree of polymerization and particle size (surface area) on the light naphtha yield was performed to tackle the first part. As fibrous cellulose with large particles was the most favourable feedstock with regard to alkane yield and as the presence of hemicellulose and lignin impurities had no effect on the cellulose-to-naphtha conversion, costly mechanical and purification steps are redundant to the process, in contrast to their notable importance in other cellulose valorisation processes (e.g. to glucose, sorbitol, isosorbide and acids). The second point regarding sustainable hydrogen supply is discussed in detail by calculating hydrogen and carbon mass and energy balances of the chemical conversions, assuming selected scenarios among others to recuperate the hydrogen by steam-reforming of waste streams (like gaseous C<6 hydrocarbons and aqueous polyol fractions) and (partial) aromatization of the C6 fraction into benzene. The study shows potential to integrate the liquid phase cellulose-to-naptha (LPCtoN) technology into a self-sufficient biorefinery, in which the chemical processes may run without consumption of external (non-renewable) hydrogen, carbon and energy, except for solar light.
Introduction
Current climate problems, geo-political frictions and the exhausting long term availability of fossil resources require at least a partial transition to renewable alternatives in the chemical industry. Biomass and more specifically lignocellulose may become an important feedstock for the production of chemicals, materials and in some cases (additives for) fuels.1–14 The carbon skeleton and the high functionality per C-atom offer beautiful possibilities to synthesize novel components and platform molecules.15–25 Lignocellulose is for instance also a great alternative resource for the production of fossil-based alkanes.1,6,13 The light naphtha fraction is nowadays an important group of hydrocarbons with a myriad of applications in the chemical industry. Light naphtha reforming for instance leads to the production of high-octane gasoline, which is typically composed of short and branched alkanes,26 cracking gives ethylene, propylene and butadiene building blocks,27,28 while selective catalytic cyclization/dehydrogenation produces aromatics.29–31 One important light naphtha compound cyclohexane is oxidized to adipic acid, a nylon precursor.32,33 Today, a handful of cellulose-to-alkane technologies are presented. Recent thermochemical approaches such as (hydro)pyrolysis are robust technologies producing complex and sometimes unstable product mixtures.34–43 Mild catalytic liquid phase approaches offer the advantage of better preserving the original C skeleton, for instance by forming n-hexane from cellulose. Much work has been reported on the depolymerisation of cellulose into its building blocks and their subsequent conversion to different platform molecules. Hydrodeoxygenation (HDO) of these molecules, with or without C–C coupling, leads to the formation of alkanes.6,44–52 Recently described one-pot liquid phase cellulose-to-naphtha (LPCtoN) conversion strategies, as depicted in Fig. 1, though promising, are in its infancy and less studied so far.53–56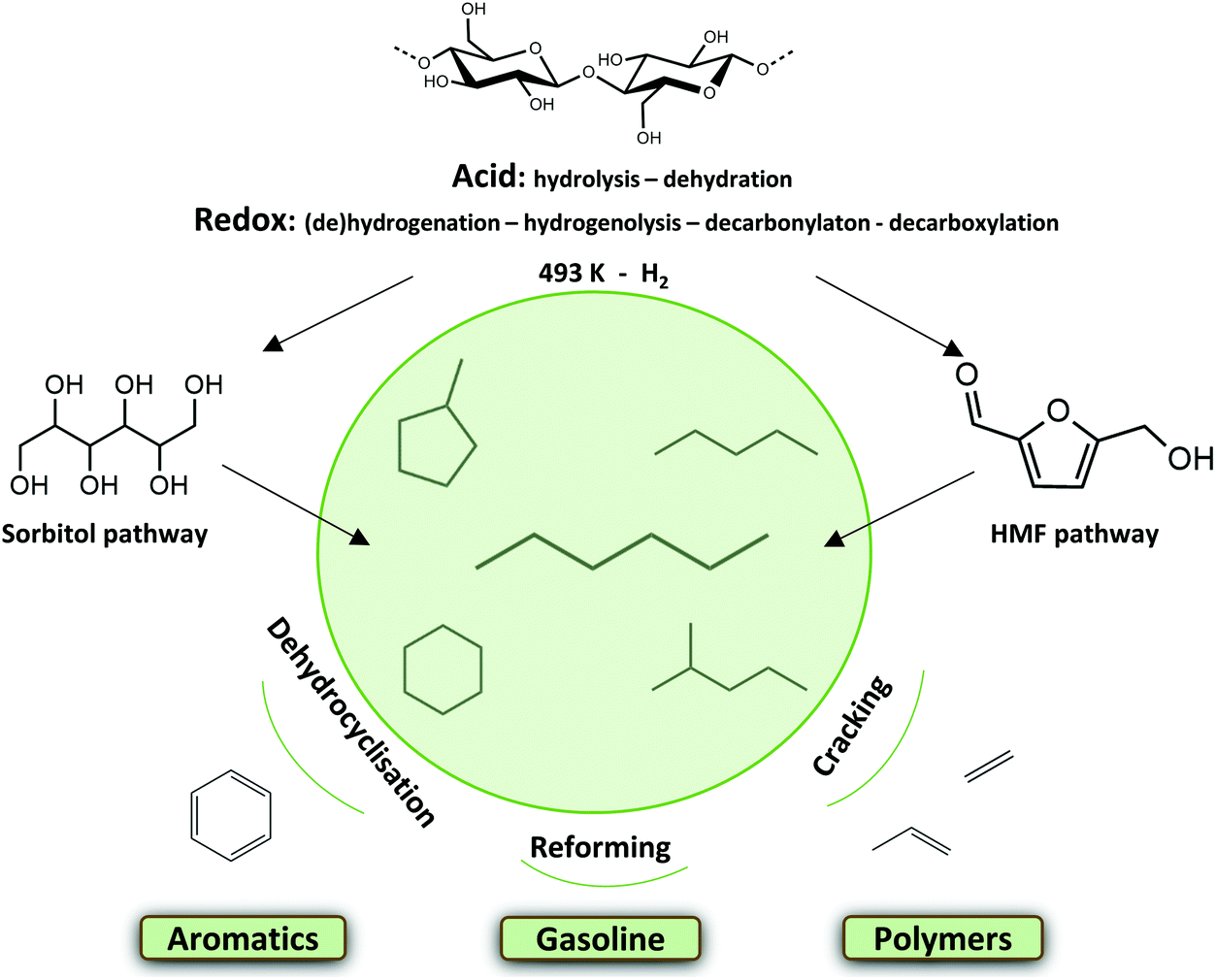 | ||
| Fig. 1 Mild catalytic conversion of cellulose to light naphtha via two reaction pathways, through sorbitol or HMF, and their subsequent valorisation. | ||
Tomishige et al. developed such a one-pot process producing n-hexane from cellulose in a biphasic solvent system of water and n-dodecane. Hydrolysis of cellulose to glucose takes place in the aqueous phase in presence of HZSM-5 zeolite. The metal catalyst Ir-ReOx/SiO2 hydrogenates glucose to sorbitol in the aqueous phase, and subsequent C–O hydrogenolysis cycles forms n-hexane. After 24 h, a yield of 78 mol% C hexane (in total of 90% light naphtha alkanes) from microcrystalline cellulose was reached at 483 K.53 This sorbitol pathway was also investigated using a Ru/C catalyst combined with LiNbMoO6 in aqueous phosphoric acid yielding 72 mol% C of C6 alkanes after 24 h at 503 K. The layered structure of the catalysts is said to inhibit the formation of (too stable) isosorbide.54
Alternatively to the sorbitol pathway, there is the 5-hydroxymethylfurfural (HMF) pathway, as recently introduced by Op de Beeck et al.55 The combination of a soluble tungstate acid and a modified Ru/C in a biphasic water–alkane environment stimulates glucose dehydration to HMF and its further hydrogenation, while retarding direct sugar hydrogenation to sorbitol. The authors assumed that Ru sintering, altered through modification, favors the HMF pathway. To corroborate this hypothesis, characterization and differences in activity between a typical Ru/C and the modified Ru/C (htTSA(2)Ru/C) were reported in the manuscript of Op de Beeck et al.55 Consecutive dehydration and hydrogenation cycles from HMF ultimately lead to the production of alkanes from microcrystalline cellulose (Avicel PH-101), yielding 42 mol% C of hexane (of a total of 60 mol% C light naphtha alkanes) within 3 h at 493 K. Other components in the organic phase are mostly oxygenates, such as (di)methyl-tetrahydrofuran, oxepane, (methyl-)tetrahydropuran and hexanols. The aqueous phase consists of isosorbide and sorbitan isomers.
Though promising, these cellulose-to-alkane technologies require more intense research. The atom and energy efficiency of the conversion of preferentially raw cellulose is essential herein.57 To bridge the gap between model and real cellulosic feedstock, this paper attempts to map the influence of the cellulose characteristics like purity, particle size (surface area), degree of polymerization and crystallinity on its conversion to light naphtha for the HMF route technology. A second part attempts the development of reaction (process) schemes to realize closed energy and mass balances for the LPCtoN biorefinery, suggesting several strategies to recuperate hydrogen from product and waste streams. While the latter goal is essential in light of devising an overall carbon and hydrogen neutral process, the first objective will assess the requirement (or not) of (costly) feedstock pretreatment.
Experimental
For a list of all used chemicals and materials as well as a more additional description of the experimental procedures, the reader is referred to the ESI.†For the pretreatment, a Ru/C (5 wt%) metal catalyst is modified in a 100 ml batch reactor (Parr Instrument Co.). The reactor is typically loaded with Ru/C (1 g), TSA hydrate (0.25 g) and water (40 ml). After flushing with N2, the reactor was pressurized with a H2 pressure of 50 bar at room temperature. Subsequently, a stirring rate of 700 rpm was installed. A temperature program, starting at room temperature to 483 K (±10 K min−1) and staying for 1 h at 483 K, is followed. Finally, the synthesized catalyst (htTSA(2)Ru/C) was filtered, washed with water until neutral pH of the effluent and dried to constant weight.
Catalytic conversion of various cellulose substrates (2 g) was carried out in a 100 ml batch reactor (Parr Instrument Co.). In addition, a biphasic system water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-decane (30:10 ml), TSA hydrate (4.8 g) and htTSA(2)Ru/C (0.25 g) was loaded into the reactor. After flushing with N2, the reactor was pressurized at 50 bar of hydrogen and room temperature. Subsequently, a stirring rate of 700 rpm was installed. A temperature program, going from room temperature to 423 K (±12 K min−1) and further to 493 K (±0.5 K min−1), is followed. After a reaction of 5 h, the reactor is cooled, depressurized and opened for sampling and analysis. Products yields are expressed as mol% C and mostly presented with just % in the results section.
:n-decane (30:10 ml), TSA hydrate (4.8 g) and htTSA(2)Ru/C (0.25 g) was loaded into the reactor. After flushing with N2, the reactor was pressurized at 50 bar of hydrogen and room temperature. Subsequently, a stirring rate of 700 rpm was installed. A temperature program, going from room temperature to 423 K (±12 K min−1) and further to 493 K (±0.5 K min−1), is followed. After a reaction of 5 h, the reactor is cooled, depressurized and opened for sampling and analysis. Products yields are expressed as mol% C and mostly presented with just % in the results section.
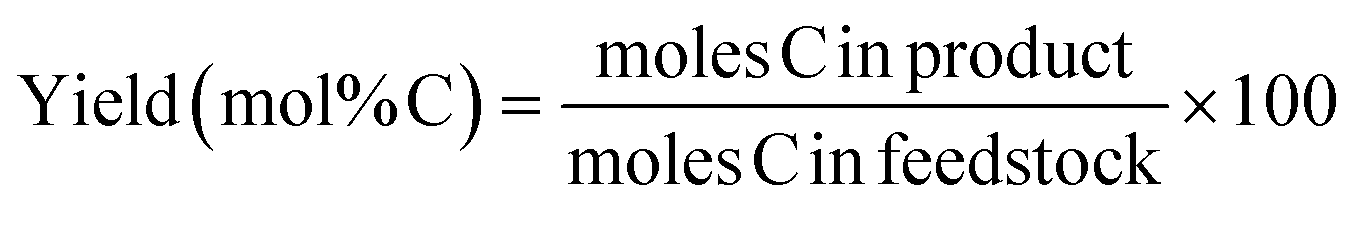
Cellulose powders were characterized by different techniques: FTIR, PXRD, laser diffraction, SEM and viscosimetry. FTIR spectra were recorded by using a Bruker IFS 66v/S instrument (KBr method). Crystallinity (CrI%) was recorded with powder X-ray diffractions (PXRD) patterns at room temperature on a STOE STADI P Combi diffractometer. The diffracted intensity of the CuKα radiation (λ = 0.154 nm) was measured in a 2θ range between 0° and 62.5°. Crystallinity based on PXRD was determined with the peak height method.58 Particle sizes (μm) were determined by laser diffraction with a Microtrac S 3500. To remove all particles from earlier measurements, the machine was first flushed with air. After measuring some blanks, powders were systematically sucked for 10 s towards the cell where particles were measured with a TRI-LASER multi-detection system. Data handling was done with Microtrac flex 11.0.0.3 software. SEM pictures were recorded by using a JEOL JSM-6010 JV microscope after coating the samples with gold with a JEOL JSC-1300 sputter. Viscosimetry based on the NF G 06-037 norm was used to measure the degree of polymerization (DP). Typically, 0.125 g (or 0.015 g when a high DP was expected) cellulose was dissolved in 50 ml of a 0.5 M cuprietethylenediamine solution and stirred for 2 hours at room temperature. Viscosity data were determined in a UBBELOHDE thermostated capillary tube viscosimeter at 298 K and afterwards calculated according to the NF G 06-037 norm.
The thermodynamics of the proposed processes were calculated under common process conditions (with regard to temperature and pressure) by using Aspen Tech 7.3 software. The process streams were based on stoichiometric relations after equalizing the amount of H2 in the entire process. The mass and energy balances are calculated taking into account a stand-alone biorefinery for n-hexane and benzene production, which is self-sufficient in hydrogen and energy.
Results and discussion
Influence of cellulose properties
A systematic study on the LPCtoN conversion was initially performed with common pure cellulose powders, viz. commercial Avicel PH-101 and various Sigmacell celluloses as well as with unusual substrates such as Vivapur, Whatman filters and cotton. The different cellulose feedstocks with their physical characteristics and the corresponding alkane yield after the cellulose-to-naphtha catalytic processing are summarized in Table 1. Vivapur cellulose proved to be structurally similar to Avicel, but with bigger particle sizes (Table 1, entry 8). Whatman filters (Fig. S1 in ESI,† FTIR spectra), which are manufactured from cotton, and cotton itself are characterized by a high degree of polymerization in comparison with the other cellulose substrates (Table 1, entries 11–14, viscosimetry). SEM pictures show a fibrous, highly dense structure for the filter material, whereas the classic cellulose powders consist of aggregated particles/spheres (Fig. S2 in ESI†).| Entry | Type cellulose | Pretreatment | Propertiesc | Yield (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Particle size (μm) | Crystallinity (CrI%) | DP (# units) | n-Pen | n-Hex | Other alkanesb | Light naphtha | |||
|
a Conditions: 2 g cellulose; 4.8 g TSA; 0.25 g htTSA(2)Ru/C; biphasic system: water/n-decane (30:10); 50 bar H2 at RT; temperature programme: RT to 423 K (±12 K min−1) and from 423 K to 493 K (±0.5 K min−1); total reaction time of 5 h. Blank reactions with n-decane (and squalane) as organic solvent did not give any cracking products.
b Other alkanes: n-butane; 2-methylpentane; 3-methylpentane; methylcyclopentane and cyclohexane.
c Ennaert et al. recently published the properties of certain cellulose substrates (entries 1–7).23 Characterization data is also available in section B of the ESI. Particle size measurements indicate diameters. n.d.r.: could not be determined reliably (fibrous).
|
|||||||||
| 1 | Avicel PH-101 | None | 68 | 79 | 161 | 7 | 46 | 9 | 62 |
| 2 | Ball-milling 0.5 h | 56 | 53 | 149 | 8 | 43 | 7 | 58 | |
| 3 | Ball-milling 2 h | 56 | <5 | 137 | 9 | 43 | 8 | 60 | |
| 4 | Ball-milling 6 h | 57 | <5 | 116 | 8 | 46 | 8 | 62 | |
| 5 | Sigmacell Type 20 | None | 29 | 76 | 168 | 8 | 41 | 7 | 56 |
| 6 | Sigmacell Type 50 | None | 97 | 80 | 165 | 8 | 48 | 10 | 66 |
| 7 | Sigmacell Type 101 | None | 16 | 48 | 442 | 5 | 38 | 7 | 50 |
| 8 | Vivapur | None | 204 | 78 | 170 | 10 | 48 | 9 | 67 |
| 9 | Sieving: <125 μm | 93 | 74 | 179 | 8 | 45 | 9 | 62 | |
| 10 | Sieving: >125 μm | 291 | 80 | 170 | 10 | 51 | 9 | 70 | |
| 11 | Whatman filter 1 | None | n.d.r. | 69 | 1125 | 10 | 46 | 9 | 65 |
| 12 | Whatman filter 4 | None | n.d.r. | 65 | 685 | 10 | 49 | 10 | 69 |
| 13 | Whatman filter 5 | None | n.d.r. | 60 | 729 | 10 | 47 | 9 | 66 |
| 14 | Cotton | None | n.d.r. | 64 | 1141 | 15 | 48 | 9 | 72 |
The influence of crystallinity is systematically investigated first by prior ball-milling of microcrystalline Avicel at various contact times.59 After an initial drop, the particle sizes stay more or less constant with the milling time (Table 1, entries 1–4). This treatment creates amorphous regions especially when the milling time is high.60 Surprisingly, cellulose amorphization has no influence on the cellulose-to-naphtha efficiency. The second property of this series, i.e. the degree of polymerization (DP, expressed in # glucose units per chain), which drops by milling, is thus also an unimportant parameter to the alkane yield.
In contrast to crystallinity and degree of polymerization, the effect of particle size (diameter in μm, laser diffractometry) seems more pronounced: indeed, larger particles result in substantially higher light naphtha yields, as seen slightly for the Avicel samples (e.g. compare entry 1 with 2), but more clear in the Sigmacell series (compare entries 5 and 6, with similar DP and crystallinity). To confirm the hypothesis, Vivapur cellulose, which comprises of large particles, is tested before and after sieving it into two size fractions (entries 8–10). The Vivapur fraction with particles above 125 μm has a three-times average particle size compared to the small-particle fraction according to the particle distribution, as ascertained by laser diffraction (Fig. 2A). Processing the large-particle fraction gives a substantial 10% higher yield of light naphtha in comparison to reaction with the small particle fraction (<125 μm), corresponding to a 13% relative yield increase. Reactions with the other celluloses, of which the results are displayed in Fig. 2E, substantiate the relationship between light naphtha yield and particle size. The figure plots the naphtha yield in function of cellulose particles radius, together with the calculated external surface areas, while assuming spherical particles and constant cellulose densities of 1.5 g cm−3 (section C of the ESI†). It is important to notice the rapid decay of surface area, indicating that a small increase in radius for small particles (<30 μm) results in a strong decrease of external surface. Clearly, the naphtha yield increases in a reverse trend with the surface area. This evolution, going from small (Sigmacell Type 101), over medium (Vivapur <125 μm), to very large particles (Vivapur >125 μm) is visualized with the corresponding SEM pictures and laser diffraction data (Fig. 2A–D). Taken together, our observations prove that the particle size, and linked with that, the external surface area of the cellulose particles is the most important substrate parameter, impacting the cellulose-to-naphtha yield. This can be rationalized as follows: as the soluble acid catalyst needs to hydrolyze cellulose at its external surface,61,62 larger particles infer a more gradual release of the monomeric sugar units to the solution because of its low surface area. In our strongly acidic environment at 493 K, soluble sugars or other reaction intermediates, notably HMF, can lead to side-products such as humins through polymerization63,64 and this is usually more pronounced at higher sugar concentrations.65,66 In line with these findings, a higher cellulose loading (here: Sigmacell Type 50, from 5 wt% to 10 wt%) gives a decrease in light naphtha alkanes (from 66% to 42%). Likely, this is due to the higher concentration of glucose and reactive intermediates such as HMF, at a certain moment, causing more degradation products to form. A slower sugar release by hydrolysis (resembling a glucose fed-batch reactor), giving lower concentrations of intermediates, could thus be at the basis of the more efficient conversion of large-sized cellulose to light naphtha. This observation is in stark contrast to most other catalytic cellulose conversion systems, as they preferentially require a more accessible and broken down cellulose structure. This is for instance the case in the LPCtoN process, following the sorbitol pathway. As a heterogeneous acid catalyst was used here, cellulose accessibility is essential, while the sugars are stabilized into hexitols via fast hydrogenation.53 Recently, also Ennaert et al. confirmed the need for small and amorphous cellulose particles for the conversion of cellulose into sorbitol.23 Interestingly, the LPCtoN process following the HMF route has no need to such usually costly (mechanical) feedstock pretreatment.67–69 The catalytic results of the Whatman filter cellulose and cotton (Table 1, entries 11–14) are in line with the above conclusions: use of dense, fibrous celluloses with high DP show the highest naphtha yields, up to 72% in case of cotton.
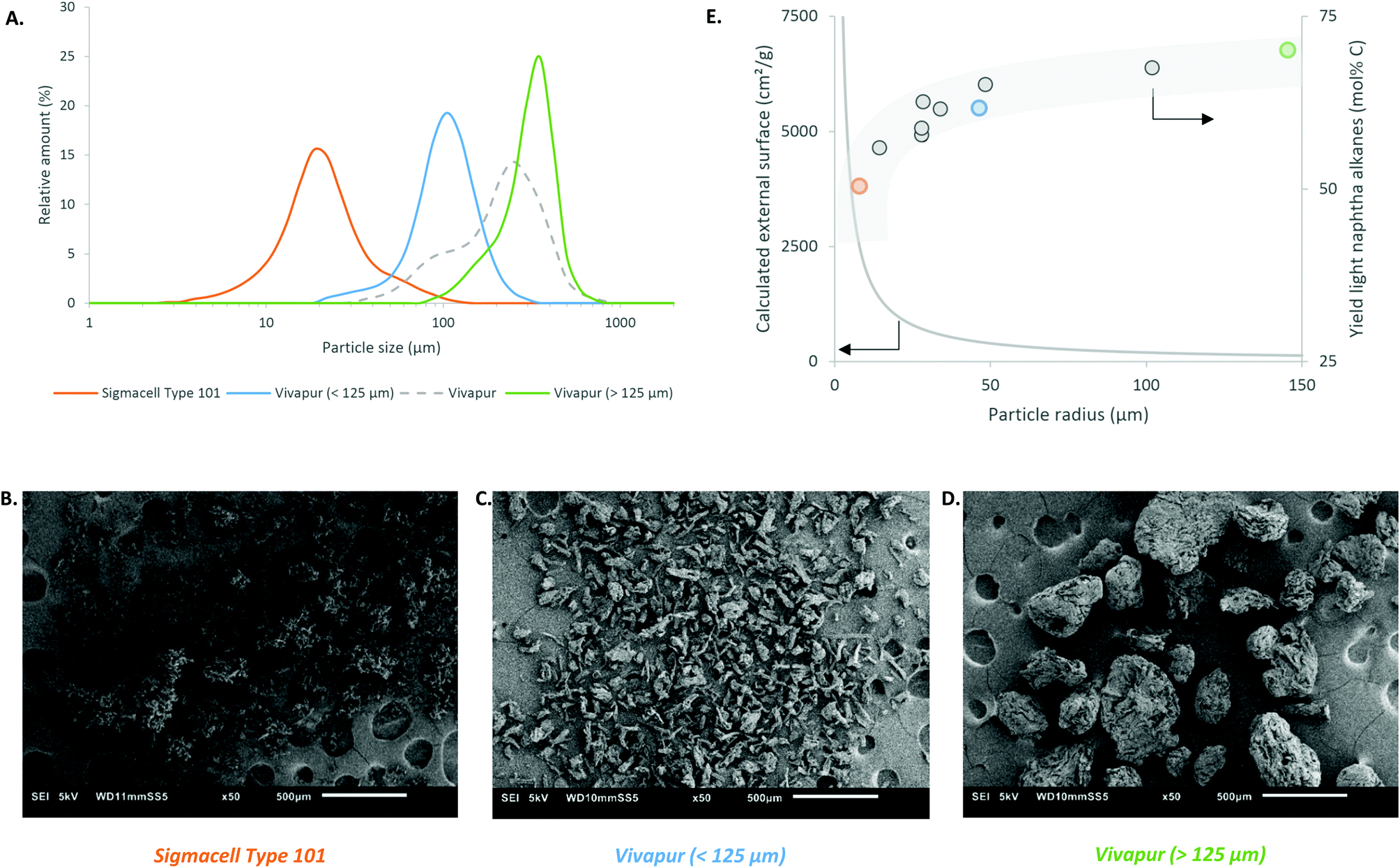 | ||
| Fig. 2 Particle size distribution of the three cellulose substrates, measured with laser diffraction (A). SEM pictures of Sigmacell Type 101 (B); Vivapur (<125 μm) (C) and Vivapur (>125 μm) (D). Yield of light naphtha alkanes (E), starting from different cellulose substrates with Sigmacell Type 101, Vivapur (<125 μm) and Vivapur (>125 μm) highlighted. Improvement of the yield can be realized by using cellulose (2 g) with a bigger particle size. The highest improvements in yield (bullets) are seen, if the decrease in accessibility, external surface, is the highest. The colours mark the same substrates in all figures A–E. | ||
Towards real (ligno)cellulosic feedstocks
The pure cellulose model substrates used above, underwent several process cleaning steps, starting from wood and other plant-based products. Usually, raw woody materials consists of 40–50 wt% cellulose, 20–35 wt% hemicellulose and 15–20 wt% lignin.15 After Kraft pulping, lignin and hemicellulose are (partially) degraded and dissolved, leaving insoluble α-cellulose which is filtered off.70–72 The α-cellulose consists of cellulose fibers (Fig. S2 in ESI†), that contains small amounts of hemicellulose (17%). Organosolv is another technology to isolate purified cellulose fractions containing residual hemicellulose and lignin parts.23,73–77Three cellulose pulps with a small amount of impurities were tested to address the impact of cellulose purity: α-cellulose, still containing some hemicellulose; wheat straw organosolv pulp containing both hemicellulose and lignin; and the bleached version of the latter. Their composition and the corresponding light naphtha yield are found in Table 2. To evaluate the efficiency of the catalytic system in the presence of impurities, the yield of C6 alkanes (mainly methylcyclopentane and n-hexane) should be considered, as they originate from C6 sugars, and thus mainly from cellulose. In addition, Whatman filter 4, which is from a physical point of view similar to authentic highly dense cellulose fibers in a lignocellulosic matrix, is chosen as benchmark. The similar C6 alkane yields (in Table 2) for the different celluloses suggest that the presence of impurities has no influence on the efficiency of cellulose-to-naphtha reactions (Table 2, entries 1–4). Moreover, the mild pretreatment procedures, which partly removed hemicellulose and lignin fractions, keep intact the original and dense cellulose structure, which is very interesting for the catalytic system under study here considering our findings regarding the impact of the cellulose external surface. While the efficiency of cellulose conversion is almost constant, the total yield of light naphtha is lower compared to that from pure cellulose substrates. This decrease is caused by the inefficient conversion of hemicellulose (mainly C5 sugars) and lignin under the applied reaction circumstances. Though these impurities do not affect the cellulose-to-naphtha reaction, their conversion to light naphtha alkanes is less efficient and therefore their presence in the cellulosic feedstock should be minimized (Table 2, entries 1–4). If their valorisation is pursued for instance for economic reasons, their fractionation will thus be a prerequisite.
| Entry | Substratef | Compositionb (wt%) | Yield from C6 sugars (%) | Total yieldd (%) | ||||
|---|---|---|---|---|---|---|---|---|
| C6 sugars | C5 sugars | Lignin | Othersc | n-Hex | Other C6 alkanese | Light naphtha | ||
|
a Conditions: 2 g substrate; 4.8 g TSA; 0.25 g htTSA(2)Ru/C; biphasic system: water/n-decane (30:10); 50 bar H2 at RT; temperature programme: RT to 423 K (±12 K min−1) and from 423 K to 493 K (±0.5 K min−1); total reaction time of 5 h.
b Ennaert et al. recently published the composition of the respective substrates.23
c Others: sum of ash; proteins; acetyl; uronic acids and extractives.
d The total yield is based on the amount of carbon coming from C5 sugars, C6 sugars and lignin.
e Other C6 alkanes: 2-methylpentane; 3-methylpentane; methylcyclopentane and cyclohexane.
f Characterization data is available in section B of the ESI.
|
||||||||
| 1 | Whatman filter 4 | 100 | — | — | — | 49 | 9 | 68 |
| 2 | α-Cellulose | 83 | 17 | — | — | 48 | 8 | 54 |
| 3 | Wheat straw organosolv | 70 | 8 | 16 | 6 | 45 | 12 | 45 |
| 4 | Wheat straw organosolv bleached | 74 | 3 | 6 | 16 | 42 | 8 | 51 |
For the hemicellulose fraction, the current conditions are likely too harsh to convert these amorphous and branched sugar polymers selectively into alkanes. Tomishige et al. noticed the same issue and reported the formation of humins as major drawback.78 Recent work by Ennaert et al. also warned for the use of unbalanced bifunctional (metal-acidic) catalytic systems for hemicellulose conversion to pentitols.79 Moreover, the acidic condition, required to convert the crystalline rigid celluloses, chemically transforms lignin through acid-catalysed condensation into (usually larger and) stable lignin derivatives, which may foul the catalyst sites.76,80,81 Therefore, in order to efficiently valorise the complete lignocellulosic biomass, a multistep process, which involves practical fractionation of the individual lignocellulose constituents, viz. cellulose, hemicellulose and lignin, followed by their individual chemical conversion, is highly advised. This strategy will lead to the most effective biorefinery, since the processing conditions can be optimised to the needs of each fraction. Whereas most fractionation processes focus on the isolation of one purified fraction, usually cellulose, more recent initiatives attempts to refine the biomass with regard to produce two or three separate fractions. Selective removal of lignin with formation of a fairly pure and easily processable sugar pulp was recently introduced in the lignin-first processing of lignocellulose,82–89 but also other initiatives are under development.90 Lignin-first technology in presence of small quantities of H3PO4 is perhaps the most notable one. This technology generates a purified cellulose pulp from woody biomass, besides a solution containing a soluble lignin oil and methyl sugars from the hemicellulose fraction.91
Closed energy and mass balances
There are many ways to assess the viability and sustainability of a biorefinery as multiple factors such as feedstock cultivation, biomass transportation, process efficiency and product added-value are involved. Moreover, these parameters are influenced by specific boundary conditions (e.g. biomass density in the surrounding area and infrastructure for transportation).92,93 By consequence, concepts such as net atmospheric carbon ration (NACR) and indirect land use change emissions (ILUC) are important parameters to evaluate the entire life cycle of each process from a sustainable point of view.94–98 The most important stumbling blocks associated with biorefineries are the following: feedstock price, food competition, loss of biodiversity, additional emissions through the replacement of wood and grassland in new cropland, new infrastructure needs, etc. The present LPCtoN technology may be considered as a potential second generation biomass-to-liquid process replacing crude oil to reach the government-imposed renewable end-objectives.99,100 It creates a drop-in light naphtha from non-edible lignocellulose feedstock, ideally from waste or fast-growing trees (e.g. poplar and willow) from sustainably managed forests. However, implementation of such processes is only realistic within a certain framework, e.g. for making additives for the large fuel markets in expectation of electrification or, more likely, as valuable alternative in the smaller markets of chemicals and materials.1 Moreover, large quantities of hydrogen are consumed and therefore the self-sustainability (with regard to carbon, hydrogen and energy neutrality) may be argued.Beside use of renewable (ligno)cellulose, one should also be able to realize an integrated biorefinery with a closed-loop of mass and energy. Recent biofactories, in which chemicals are produced from renewable resources and solar energy is used to co-valorise CO2, have been discussed, but also integration of petrochemistry and solar has been suggested. Centi et al. recently introduced the concept of the biofactory, taking into account the whole production process by integration of renewable energy (e.g. solar) and valorisation of CO2.101–103 Also, Jacobs et al. proposed a conceptual biorefinery, merging petroleum and solar refineries.104 The generation of CO2, e.g. to be considered as the end-of-life of most hydrocarbon chemicals, but also the result of H2 generation from carbon containing resources, can be seen as looped by photosynthesis during lignocellulosic plant growth, and may be largely regarded as carbon neutral as long as renewable resources were used. Natural or artificial techniques, requiring a limited input of energy (e.g. solar), could be used in the long term here to convert such spent carbon into working carbon.105,106
Sustainable production of hydrogen in the biorefinery is an important challenge. Today, most H2 is produced through steam-reforming of (shale) natural gas, while in light of a 100% renewability criterion, a non-fossil resource is required. Though economically sound on a short term to initiate biorefinery activities, the search for and the integration of other techniques such as biological hydrogen production,107 photocatalytic splitting of water,108–111 gasification of biomass,112 aqueous-phase-reforming (APR)113–117 and steam-reforming118,119 of biomass derived gaseous products should be considered to offer more sustainable opportunities on the long term, but they are still in their infancy at the commercial scale. In addition to APR and steam-reforming, there might be a third and fourth option to recuperate additional hydrogen, which is particularly valid for the presented LPCtoN technology: steam or catalytic cracking of n-hexane to short olefins27,28 and dehydrocyclisation of n-hexane (and the other cyclic C6 alkanes) to benzene.29–31 The latter is in particular interesting since the technology could lead to the on-purpose production of bio-benzene. The cyclisation to benzene, producing hydrogen gas, for instance described in presence of Pt–Ba loaded L zeolite at 773 K,29–31,120 may pave the way to the production of bio-based ethylbenzene and styrene. Other aromatics such as phenols and anilines will more likely be targeted starting from the lignin (oil) fraction.121–124 In section D of the ESI† a short case study, with respect to the typical capacity of a biorefinery, illustrates the feasibility of synthesizing such renewable chemicals and materials.
Though highly relevant in general, evaluating the above issues is very complex. Therefore, rather than focussing already on the complete life cycle analysis of the proposed biorefinery, this contributions wants to discuss the sustainability at the technical level, with regard to carbon and hydrogen neutrality of a potential stand-alone process.
In what follows, we will conceptually close mass and energy balances for an integrated LPCtoN process, and give suggestions for the carbon and hydrogen neutral production of the renewable alkanes and thereof derived bio-benzene. First, this exercise is done taking into account two scenarios, one which includes only reforming for hydrogen recuperation and a second one, which includes both reforming and dehydrocyclisation. The scenarios are illustrated in Fig. 3. Before evaluating the two scenarios for the real situation (see later), the different process streams were calculated in thermodynamic equilibrium using Aspen Tech 7.3 software assuming no net production/consumption of hydrogen in the process. Rather than directly using the precise (waste) products available in the LPCtoN process, obvious model molecules to produce hydrogen were selected. The calculations used therefore n-hexane (as a model of potential waste alkanes) for steam-reforming, ethylene glycol (as a model of polyol waste) for APR and n-hexane for the dehydrocyclisation to benzene. Note that the water gas shift reaction is taken into account in the steam-reforming process as to maximize the potential hydrogen production. Details of the calculations are presented in the ESI (section E in the ESI†). The thermodynamic data and the calculated streams for the specific case of the combination of reforming of n-hexane (here by steam) and dehydrocyclisation are illustrated in Table 3 and Fig. 4.
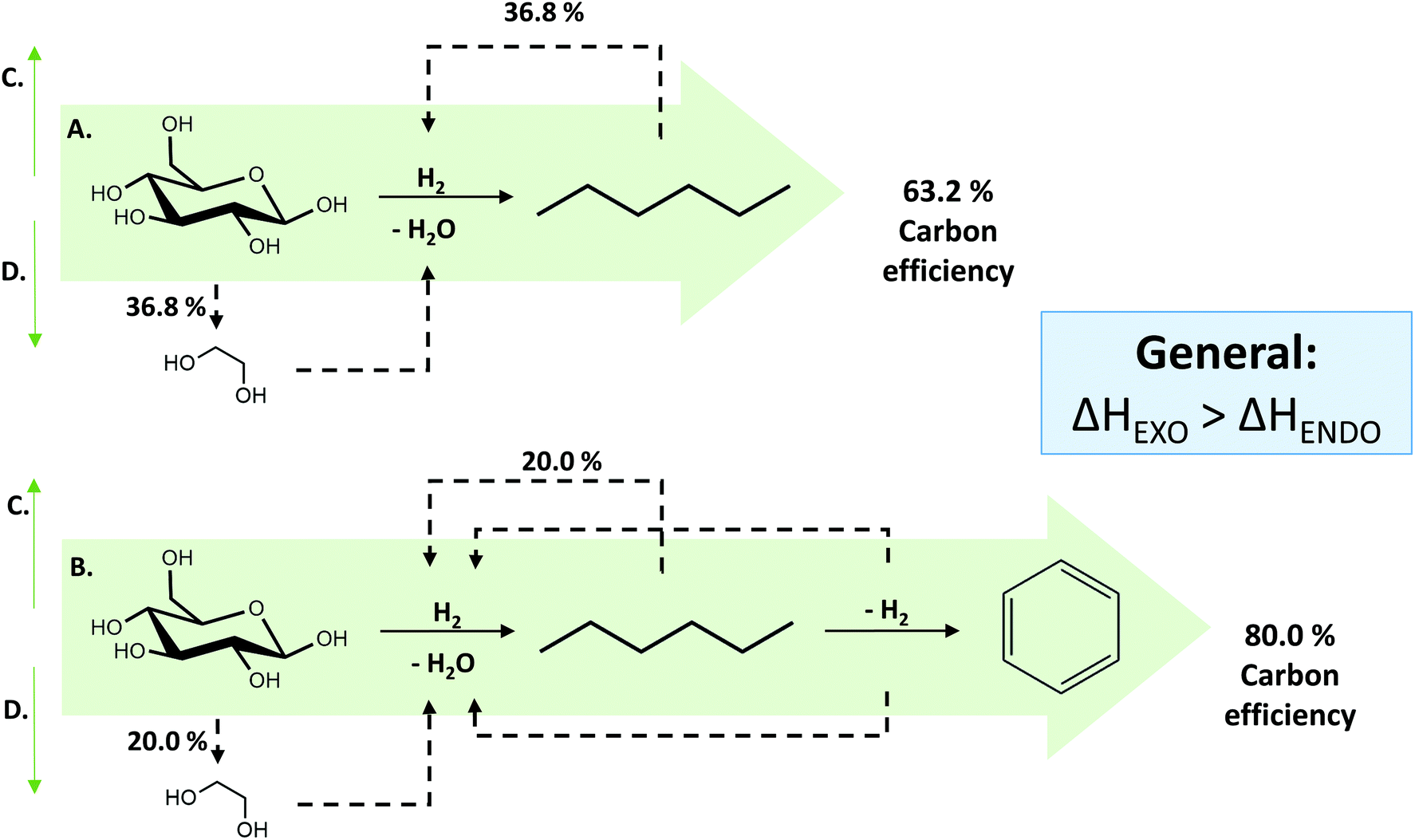 | ||
| Fig. 3 Overview of the 2 scenarios to generate end products (n-hexane or benzene) with 100% renewable carbon and hydrogen. The amount of carbon used for H2 recovery is based on equal consumption and production of H2 for 100% efficient reactions. The thermodynamics are calculated at the most common reactions conditions by using AspenTech 7.3 software. Different reactions are HDO of glucose to n-hexane (A); HDO of glucose to n-hexane and subsequently dehydrocyclisation to benzene (B); steam-reforming of n-hexane (C) and production of ethylene glycol form sugars and subsequently APR (D). | ||
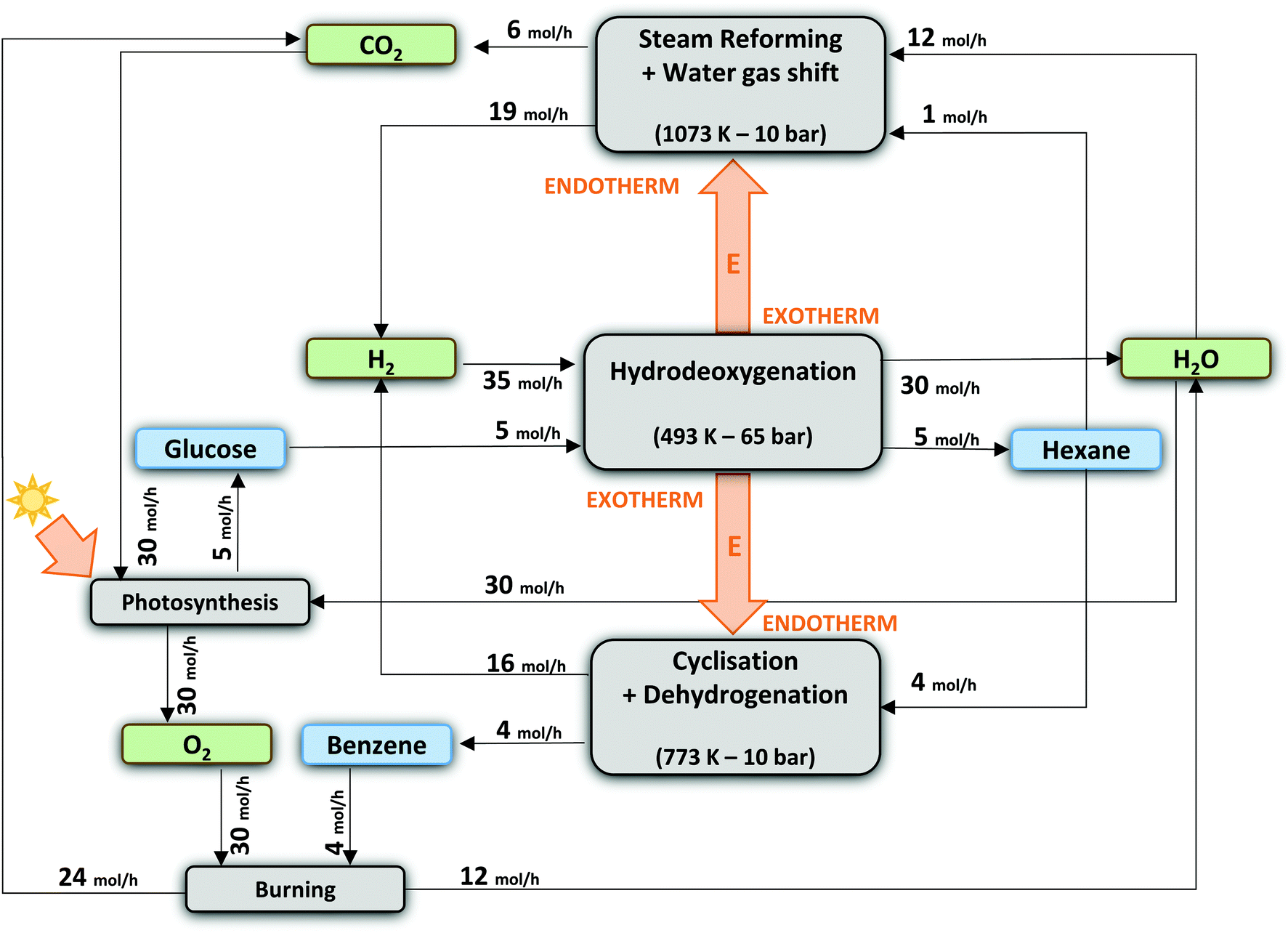 | ||
| Fig. 4 Theoretical process scheme assuming 100 mol% C of n-hexane yield (of which 1/5 is steam-reformed for H2-recuperation) and 80 mol% C of benzene yield. | ||
| Component | HDO of glucosea | Dehydrocyclisation of n-hexaneb | Steam-reforming of n-hexanec | ||||
|---|---|---|---|---|---|---|---|
| Mixedd | Vapore | Liquide | Vapord | Vapore | Vapord | Vapore | |
| Conditions:a T = 493 K; p = 65 bar.b T = 773 K; p = 10 bar.c T = 1073 K; p = 10 bar.d Starting mixture and input in AspenTech software.e End streams. | |||||||
| C6H12O6 (mol h−1) | 5.0 | 1.2 × 10−8 | 1.2 × 10−3 | 0 | 0 | 0 | 0 |
| H2 (mol h−1) | 35.0 | 8.1 × 10−3 | 1.3 × 10−4 | 0 | 12.4 | 0 | 18.2 |
| C6H14 (mol h−1) | 0 | 5.0 | 1.9 × 10−2 | 4.0 | 1.0 | 1.0 | 4.1 × 10−2 |
| H2O (mol h−1) | 0 | 2.8 | 27.2 | 0 | 0 | 12.0 | 4.9 × 10−1 |
| C6H6 (mol h−1) | 0 | 0 | 0 | 0 | 3.1 | 0 | 0 |
| CO2 (mol h−1) | 0 | 0 | 0 | 0 | 0 | 0 | 5.8 |
| Total (mol h−1) | 40.0 | 7.7 | 27.3 | 4.0 | 16.5 | 13.0 | 24.5 |
| Molar H (kJ mol−1) | −1.4 × 102 | −1.7 × 102 | −2.7 × 102 | −6.0 × 101 | 3.5 × 101 | −1.9 × 102 | −7.1 × 101 |
| Molar S (kJ mol−1 K−1) | −1.3 × 10−1 | −3.4 × 10−1 | −1.2 × 10−1 | −3.8 × 10−1 | −2.0 × 10−2 | −3.4 × 10−2 | 2.9 × 10−2 |
| ΔH (kJ h−1) | −2948.0 | 815.6 | 782.6 | ||||
Three main conclusions emerge from these calculations. Firstly, the carbon efficiency of the overall process depends strongly on the envisioned end product, showing a value of 63.2% and 80.0% for n-hexane and benzene, respectively. The latter number indicates that the overall production of two benzene molecules is accompanied with the formation of three bio-derived CO2 molecules, which will enter the photosynthesis cycle to reform cellulose using sun light input. Secondly, there is no additional energy requirement to obtain the above mentioned reaction outcomes, because the energy release during cellulose-to-hexane processing overcompensates the endothermicity of the reforming and dehydrocyclisation reaction. Our examples show an excess of 300 kJ mol−1 (per mol glucose converted), which may well be used to support other unit operations such as separation. Thirdly, considering the end-of-life of the products, burning of the products ultimately delivers a CO2 to water molar ratio of unity, which enters the photosynthesis cycle regenerating cellulose. Though not yet mature, also artificial techniques can (partially) be used to valorise the amount of released CO2 in the long term, e.g. using the excess of released energy or sun light.105,106
Similar calculations were performed using the real product outcome of an experimental cellulose-to-naphtha reaction. We have selected the data of Vivapur cellulose processing (Table 1, entry 10), as this feedstock produces the highest C6 alkane yield, approaching 60 mol% C, consisting of 86% n-hexane, 2% methylpentanes, 3% cyclohexane and 9% methylcyclopentane. The total content of light hydrocarbons, almost exclusively straight, shorter alkanes (C<6) amounts to 15 mol% C, consisting of 74% C5, 7% C4 and 19% C1, while the aqueous phase contains about 15 mol% C oxygenates such as hexitols and other polyols. The deficiency in mass balance, about 10 mol% C, is because of unknowns. The product distribution is seen in Fig. 5 (right). These data show that, in contrast to a large production of lights such as in fast-hydropyrolysis, a more selective production of C6 alkanes is realized. While pyrolysis is useful to obtain a mixture of gaseous hydrocarbons, the two-phase liquid catalytic strategy with Ru/C and tungstate heteropolyacids is more appropriate for chemical applications. The hydrogen consumption of both processes is very similar and amounts to roughly 0.05 kg H2 per kg of dry cellulose feed.
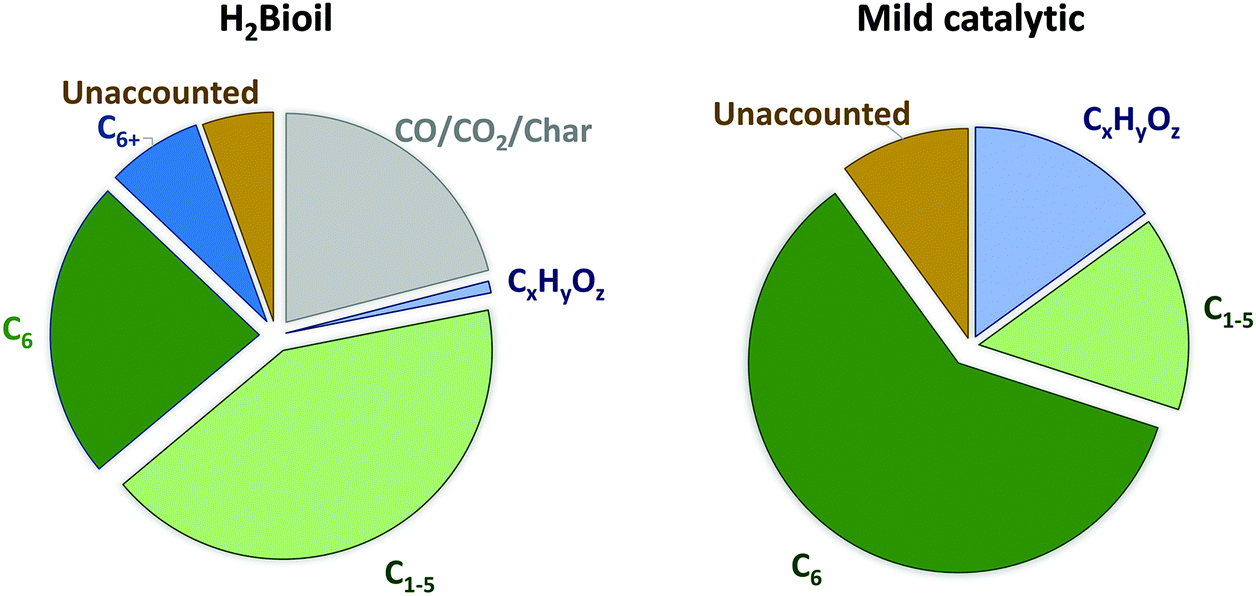 | ||
| Fig. 5 Product distributions (mol% C) for the H2Bioil process (consecutive two-step process of fast-hydropyrolyis and downstream vapor-phase catalytic hydrodeoxygenation)41 and the here described mild catalytic one-pot process (hydrodeoxygenation) from the experiments with cellulose. | ||
Hydrogen can be recovered from both light gas C<6 mixture as well as from the organic waste in the aqueous phase. Steam-reforming of the first waste stream foresees about 57% of the consumed hydrogen. An additional reforming of the aqueous waste, either through aqueous or steam-reforming reactions, led to almost 80% of the required hydrogen. This scenario indicates a hydrogen shortness, pointing out that an additional dehydrocyclisation of the C6 fraction is a nice option to fill the hydrogen gap. A 50% conversion of the C6 fraction to benzene is sufficient to balance the hydrogen in the overall process. Full details of the calculations can be found in section F of the ESI.†
If benzene is the desired product in the biorefinery, 43% of hydrogen may be recuperated through dehydrocyclisation. Steam-reforming of the light gaseous fraction provides the remaining hydrogen. This scenario fulfils a 100% renewable hydrogen LPCtoN biorefinery (section F in the ESI†). Ideally, two out of ten carbons of cellulose should end up in the light gas waste to foresee the hydrogen, the remaining of the carbons ultimately ending in benzene. A stand-alone LPCtoN biorefinery thus should not necessary operate exclusively towards one specific product, as the remaining waste is usefully recuperated into renewable hydrogen streams. A possible process scheme for such a closed-loop (stand-alone) biorefinery is illustrated in Fig. 6, showing the central cellulose-to-naphtha unit, combined with benzene formation, and the hydrogen recuperation units.
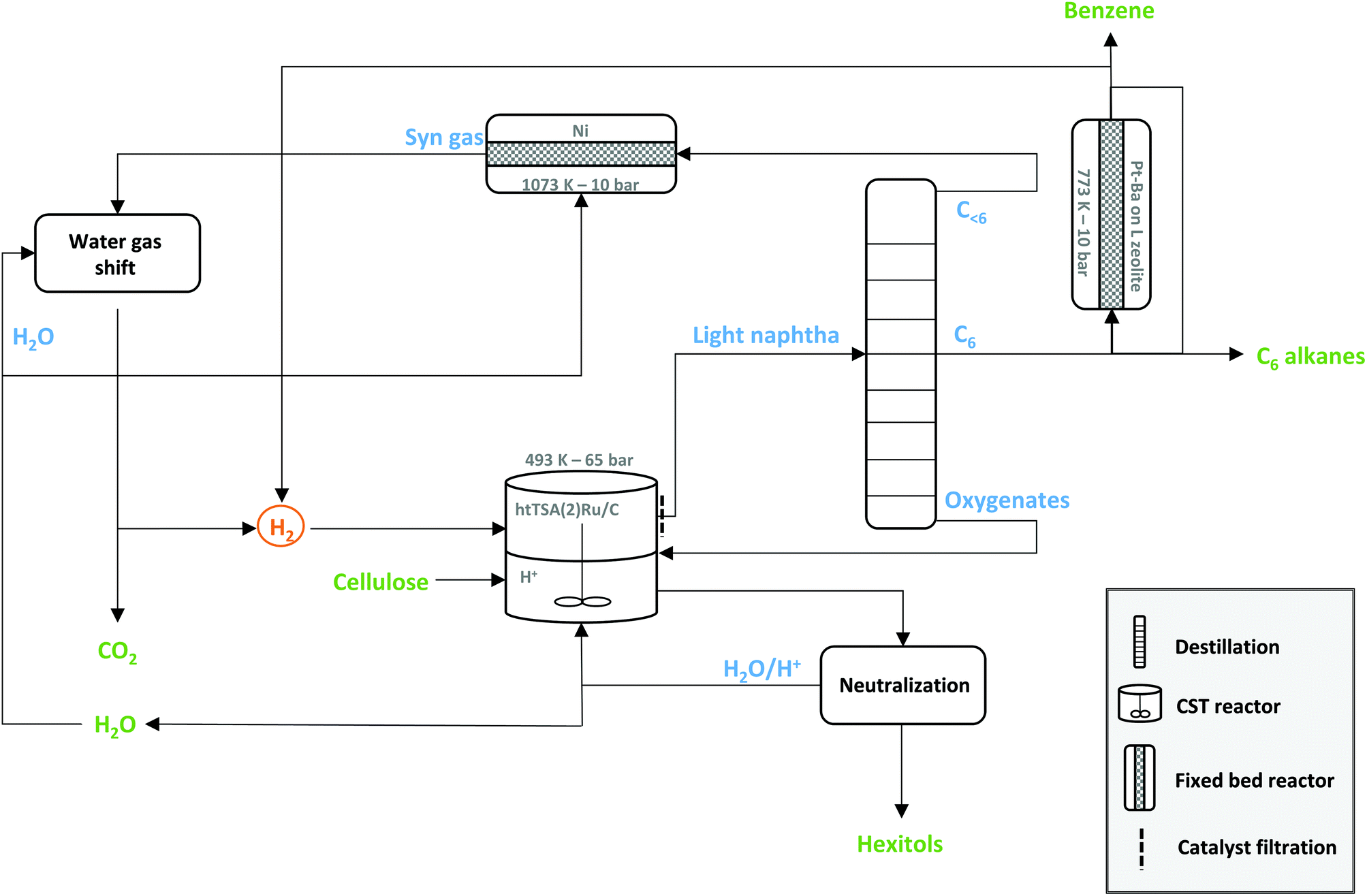 | ||
| Fig. 6 Biorefinery scenario for the conversion of cellulose into n-hexane, benzene and hextiol-derived products, with additional strategies toward closed mass and energy streams. Compounds presented in green and blue are end-products and intermediate products, respectively. | ||
Conclusions
The presented LPCtoN biorefinery can be an interesting opportunity to selectively produce bio-derived light naphtha alkanes, and more specifically C6 alkanes, and as such it may gradually fuel the existing naphtha facilities with bio-derived carbon. Two key points of the biorefinery were addressed here: (i) structural and compositional aspects of the cellulose biomass, and (ii) the renewable origin (or not) of hydrogen. Perhaps somewhat surprising, this study reveals the highest alkane yields from large-sized fibrous crystalline cellulose. The rationale is to keep cellulose hydrolysis the slowest step as to protect the reactive sugar molecules in the cellulose structure. A too fast release of reactive sugars in solution otherwise results in side-reactions producing for instance humins. Therefore costly mechanical or other treatments to lower cellulose crystallinity are not necessary. Also interesting to notice, presence of hemicellulose and lignin impurities also preserves the catalytic outcome of the LPCtoN process. As they are hardly converted to alkanes, but rather end-up in an oligomeric waste stream, a rough cellulose purification step is advised. The separated hemicellulose and lignin fractions may be better used in other valorisation processes. From economic point of view, the ability of directly processing large and impure cellulose fibres obviates the need of a series of costly pretreatment steps in the overall process.Sustainable biorefinery schemes should attempt not only to use a renewable feedstock such as lignocellulose, but also other reagents such as hydrogen should preferably be renewable. The issue of renewable hydrogen is countered here by integrating hydrogen producing technologies like reforming and dehydrocyclisation. Both mass and energy balances prove the viability of a stand-alone biorefinery for making n-hexane and benzene. In the first case, reforming of different side streams is required, while in the benzene case, reforming of only the light hydrocarbon gases (C<6 fraction) is already sufficient to foresee hydrogen. Considering a hydrogen neutral process, 59% of renewable benzene, e.g. for styrene and aniline production, can be technically produced with excess of process energy (300 kJ per mol glucose converted), available to the other unit operations. In the end, a completely self-supporting biorefinery process is put forward using solar energy in form of biomass production as the only input to produce useful drop-in chemicals. Research is ongoing to make a more complete life cycle analysis of the integrated process taking into account net atmospheric carbon ratios and indirect land use change emissions.
Acknowledgements
This work was performed in the framework of IWT-SBO project ARBOREF. A. D. and T. E. acknowledge the Agency for Innovation by Science and Technology (IWT) for a Ph.D. grant. J. V. thanks the Flemish Institute for Technological Research (VITO) for a doctoral fellowship. M. D. acknowledge Research Foundation-Flanders (FWO) for financial support. The authors kindly thank Arjan T. Smit, Wouter J. J. Huijgen and Annick Vanhulsel for respectively providing the organosolv samples and use of DLS equipment.Notes and references
- A. Deneyer, T. Renders, J. Van Aelst, S. Van den Bosch, D. Gabriëls and B. F. Sels, Curr. Opin. Chem. Biol., 2015, 29, 40–48 CrossRef CAS PubMed
.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC
- J. C. Serrano-Ruiz, R. M. West and J. A. Dumesic, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 79–100 CrossRef CAS PubMed
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC
-
M. Dusselier, M. Mascal and B. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, 2014, ch. 544, vol. 353, pp. 1–40 Search PubMed
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed
- R. De Clercq, M. Dusselier, C. Christiaens, J. Dijkmans, R. I. Iacobescu, Y. Pontikes and B. F. Sels, ACS Catal., 2015, 5, 5803–5811 CrossRef CAS
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC
-
J. A. Dumesic, Y. J. Pagán-Torres, T. Wang and B. H. Shanks, US8642791B2, 2014 Search PubMed
-
J. A. Dumesic, S. G. Wettstein, D. M. Alonso and E. I. Gurbuz, US8962856B2, 2015 Search PubMed
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 CAS
- R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 CAS
- T. Ennaert, B. Op de Beeck, J. Vanneste, A. T. Smit, W. J. J. Huijgen, A. Vanhulsel, P. A. Jacobs and B. F. Sels, Green Chem., 2016, 18, 2095–2105 RSC
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS
- M. R. Rahimpour, M. Jafari and D. Iranshahi, Appl. Energy, 2013, 109, 79–93 CrossRef CAS
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS
- K. M. Van Geem, M.-F. Reyniers, G. B. Marin, J. Song, W. H. Green and D. M. Matheu, AIChE J., 2006, 52, 718–730 CrossRef CAS
-
W. C. Buss and T. R. Hughes, US4435283A, 1984 Search PubMed
- P. Mériaudeau and C. Naccache, Catal. Rev., 1997, 39, 5–48 Search PubMed
-
C. T. O'Connor, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, 2008, vol. 1, pp. 3123–3133 Search PubMed
- M. L. Campbell, Ullmann's Encycl. Ind. Chem., 2012, 673–710 Search PubMed
- M. Dugal, G. Sankar, R. Raja and J. M. Thomas, Angew. Chem., Int. Ed., 2000, 39, 2310–2313 CrossRef CAS
- R. C. Baliban, J. A. Elia and C. A. Floudas, Ind. Eng. Chem. Res., 2013, 52, 3381–3406 CrossRef CAS
-
S. D. Phillips, J. K. Tarud, M. J. Biddy and A. Dutta, Gasoline from Wood via Integrated Gasification, Synthesis, and Methanol-to- Gasoline Technologies, 2011 Search PubMed
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed
- H. Jahangiri, J. Bennett, P. Mahjoubi, K. Wilson and S. Gu, Catal. Sci. Technol., 2014, 4, 2210–2229 CAS
- S. Wang, Q. Yin, J. Guo, B. Ru and L. Zhu, Fuel, 2013, 108, 597–603 CrossRef CAS
- M. Dalil, M. Sohrabi and S. J. Royaee, J. Ind. Eng. Chem., 2012, 18, 690–696 CrossRef CAS
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS
- V. K. Venkatakrishnan, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Green Chem., 2015, 17, 178–183 RSC
- N. R. Singh, D. S. Mallapragada, R. Agrawal and W. E. Tyner, Biomass Convers. Biorefin., 2012, 2, 141–148 CrossRef
- N. R. Singh, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Environ. Sci. Technol., 2010, 44, 5298–5305 CrossRef CAS PubMed
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 CAS
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC
- H. Olcay, A. V. Subrahmanyam, R. Xing, J. Lajoie, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2013, 6, 205–216 CAS
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 CAS
- J. Xin, S. Zhang, D. Yan, O. Ayodele, X. Lu and J. Wang, Green Chem., 2014, 16, 3589–3595 RSC
- D. Liu and E. Y. X. Chen, ACS Catal., 2014, 4, 1302–1310 CrossRef CAS
- J. Yang, N. Li, G. Li, W. Wang, A. Wang, X. Wang, Y. Cong and T. Zhang, Chem. Commun., 2014, 50, 2572–2574 RSC
- Y. Nakagawa, S. Liu, M. Tamura and K. Tomishige, ChemSusChem, 2015, 8, 1114–1132 CrossRef CAS PubMed
- E. R. Sacia, M. H. Deaner, Y. Louie and A. T. Bell, Green Chem., 2015, 17, 2393–2397 RSC
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS
- Y. Liu, L. Chen, T. Wang, X. Zhang, J. Long, Q. Zhang and L. Ma, RSC Adv., 2015, 5, 11649–11657 RSC
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 CAS
- Y. Liu, L. Chen, T. Wang, Q. Zhang, C. Wang, J. Yan and L. Ma, ACS Sustainable Chem. Eng., 2015, 3, 1745–1755 CrossRef CAS
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS
- M. Benoit, A. Rodrigues, K. De Oliveira Vigier, E. Fourré, J. Barrault, J.-M. Tatibouët and F. Jérôme, Green Chem., 2012, 14, 2212–2215 RSC
- F. Jérôme, G. Chatel and K. De Oliveira Vigier, Green Chem., 2016, 18, 3903–3913 RSC
- M. Sasaki, B. Kabyemela, R. Malaluan, S. Hirose, N. Takeda, T. Adschiri and K. Arai, J. Supercrit. Fluids, 1998, 13, 261–268 CrossRef CAS
- S. A. Maurer, C. N. Bedbrook and C. J. Radke, Ind. Eng. Chem. Res., 2012, 51, 11389–11400 CrossRef CAS
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS PubMed
- R. Ooms, M. Dusselier, J. A. Geboers, B. Op de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC
- G. Zhao, M. Zheng, J. Zhang, A. Wang and T. Zhang, Ind. Eng. Chem. Res., 2013, 52, 9566–9572 CrossRef CAS
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed
- F. Boissou, N. Sayoud, K. De Oliveira Vigier, A. Barakat, S. Marinkovic, B. Estrine and F. Jérôme, ChemSusChem, 2015, 8, 3263–3269 CrossRef CAS PubMed
- H. Krässig, J. Schurz, R. G. Steadman, K. Schliefer, W. Albrecht, M. Mohring and H. Schlosser, Ullmann's Encycl. Ind. Chem., 2012, 6, 279–332 Search PubMed
- M. Ragnar, G. Henriksson, M. E. Lindström, M. Wimby, J. Blechschmidt and S. Heinemann, Ullmann's Encycl. Ind. Chem., 2014, 6, 3–92 Search PubMed
-
G. E. Reier, Avicel PH Microcrystalline Cellulose, NF, Ph Eur., JP, BP, 2000, pp. 1–27 Search PubMed
- W. J. J. Huijgen, A. T. Smit, J. H. Reith and H. Den Uil, J. Chem. Technol. Biotechnol., 2011, 86, 1428–1438 CrossRef CAS
- W. J. J. Huijgen, A. T. Smit, P. J. de Wild and H. den Uil, Bioresour. Technol., 2012, 114, 389–398 CrossRef CAS PubMed
- W. J. J. Huijgen, J. H. Reith and H. Den Uil, Ind. Eng. Chem. Res., 2010, 49, 10132–10140 CrossRef CAS
- J. Wildschut, A. T. Smit, J. H. Reith and W. J. J. Huijgen, Bioresour. Technol., 2013, 135, 58–66 CrossRef CAS PubMed
- X. Pan, C. Arato, N. Gilkes, D. Gregg, W. Mabee, K. Pye, Z. Xiao, X. Zhang and J. Saddler, Biotechnol. Bioeng., 2005, 90, 473–481 CrossRef CAS PubMed
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC
- T. Ennaert, S. Feys, D. Hendrikx, P. A. Jacobs and B. F. Sels, Green Chem., 2016 10.1039/C6GC01439A
- R. El Hage, N. Brosse, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2010, 95, 997–1003 CrossRef CAS
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS
- S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Energy Environ. Sci., 2015, 8, 2657–2663 CAS
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS
- V. Balan, Int. Scholarly Res. Not., 2014, 2014, 1–31 Search PubMed
- A. W. Bhutto, K. Qureshi, R. Abro, K. Harijan, Z. Zhao, A. A. Bazmi, T. Abbas and G. Yu, RSC Adv., 2016, 6, 32140–32170 RSC
- D. J. Garcia and F. You, ACS Sustainable Chem. Eng., 2015, 3, 1732–1744 CrossRef CAS
- P. Havlík, U. A. Schneider, E. Schmid, H. Böttcher, S. Fritz, R. Skalský, K. Aoki, S. De Cara, G. Kindermann, F. Kraxner, S. Leduc, I. McCallum, A. Mosnier, T. Sauer and M. Obersteiner, Energy Policy, 2011, 39, 5690–5702 CrossRef
- H. H. Khoo, W. L. Ee and V. Isoni, Green Chem., 2016, 18, 1912–1922 RSC
- R. J. Plevin, J. Beckman, A. A. Golub, J. Witcover and M. O'Hare, Environ. Sci. Technol., 2015, 49, 2656–2664 CrossRef CAS PubMed
- T. Searchinger, R. Heimlich, R. A. Houghton, F. Dong, A. Elobeid, J. Fabiosa, S. Tokgoz, D. Hayes and T. Yu, Science, 2008, 319, 1238–1240 CrossRef CAS PubMed
- J. Murray and D. King, Nature, 2012, 481, 433–435 CrossRef CAS PubMed
- R. Mukhopadhyay, Chem. Eng. News, 2011, 10–15 Search PubMed
- S. Abate, P. Lanzafame, S. Perathoner and G. Centi, ChemSusChem, 2015, 8, 2854–2866 CrossRef CAS PubMed
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC
- S. Bensaid, G. Centi, E. Garrone, S. Perathoner and G. Saracco, ChemSusChem, 2012, 5, 500–521 CrossRef CAS PubMed
- P. A. Jacobs, M. Dusselier and B. F. Sels, Angew. Chem., Int. Ed., 2014, 53, 8621–8626 CrossRef CAS PubMed
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS
- P. C. Hallenbeck, M. Abo-Hashesh and D. Ghosh, Bioresour. Technol., 2012, 110, 1–9 CrossRef CAS PubMed
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295–295 CrossRef CAS PubMed
- K. Shimura and H. Yoshida, Energy Environ. Sci., 2011, 4, 2467–2481 CAS
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed
- J. Dai, J. Saayman, J. R. Grace and N. Ellis, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 77–99 CrossRef CAS PubMed
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS PubMed
- T. P. Vispute and G. W. Huber, Green Chem., 2009, 11, 1433–1445 RSC
- H. Jeong and M. Kang, Appl. Catal., B, 2010, 95, 446–455 CrossRef CAS
- B. T. Schädel, M. Duisberg and O. Deutschmann, Catal. Today, 2009, 142, 42–51 CrossRef
- R. J. Davis and E. G. Derouane, Nature, 1991, 349, 313–315 CrossRef CAS
- D. Verboekend, Y. Liao, W. Schutyser and B. F. Sels, Green Chem., 2015, 18, 297–306 RSC
- J. S. Kruger, N. S. Cleveland, S. Zhang, R. Katahira, B. A. Black, G. M. Chupka, T. Lammens, P. G. Hamilton, M. J. Biddy and G. T. Beckham, ACS Catal., 2016, 6, 1316–1328 CrossRef CAS
- Y. Ono and H. Ishida, J. Catal., 1981, 72, 121–128 CrossRef CAS
- J. Ma, H. Wang, M. Sun, F. Yang, Z. Wu, D. Wang and L. Chen, Bull. Korean Chem. Soc., 2012, 33, 387–392 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains SEM pictures, particle size distributions and PXRD patterns of celluloses, as well as detailed mass and energy balance schemes and calculations. See DOI: 10.1039/c6gc01644h |
| This journal is © The Royal Society of Chemistry 2016 |