
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid, metal-free and aqueous synthesis of imidazo[1,2-a]pyridine under ambient conditions†
Michael R.
Chapman
,
Maria H. T.
Kwan
,
Georgina E.
King
,
Benjamin A.
Kyffin
,
A. John
Blacker
,
Charlotte E.
Willans
* and
Bao N.
Nguyen
*
Institute of Process Research and Development, School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: b.nguyen@leeds.ac.uk
First published on 14th July 2016
Abstract
A novel, rapid and efficient route to imidazo[1,2-a]pyridines under ambient, aqueous and metal-free conditions is reported. The NaOH-promoted cycloisomerisations of N-propargylpyridiniums give quantitative yield in a few minutes (10 g scale). A comparison of common green metrics to current routes showed clear improvements, with at least a one order of magnitude increase in space-time-yield.
The imidazo[1,2-a]pyridine scaffold, found at the core of many pharmaceutical drugs such as zolimidine (peptic ulcer),1 zolpidem (insomnia and brain disorders)2 and rifaximin (hepatic encephalopathy),3,4 is one of the most privileged nitrogen-containing heterocycles in drug design.5–8 Industrial routes to imidazo[1,2-a]pyridines have often been via condensations between 2-aminopyridines and α-bromoketones/α-chloroketones (Scheme 1),9,10 with an aryl or a carboxylate group at the β position for subsequent reduction to an OH group and derivatisation.11–13 Recent synthetic methods have focused on functionalisation of the core scaffold through metal catalysed coupling reactions (e.g. Cu,13–15 Fe,16 Au,7 Ru,18 and Pd19,20),14,15 with attendant problems of product separation, and catalytic routes to assemble the scaffold.16,17 Furthermore, most of these processes employ undesirable solvents such as N,N-dimethylformamide,18–21 1,2-dichloroethane,22 1,4-dioxane,23,24 or acetonitrile,25,26 relatively high temperature, high catalyst loading and long reaction time.16
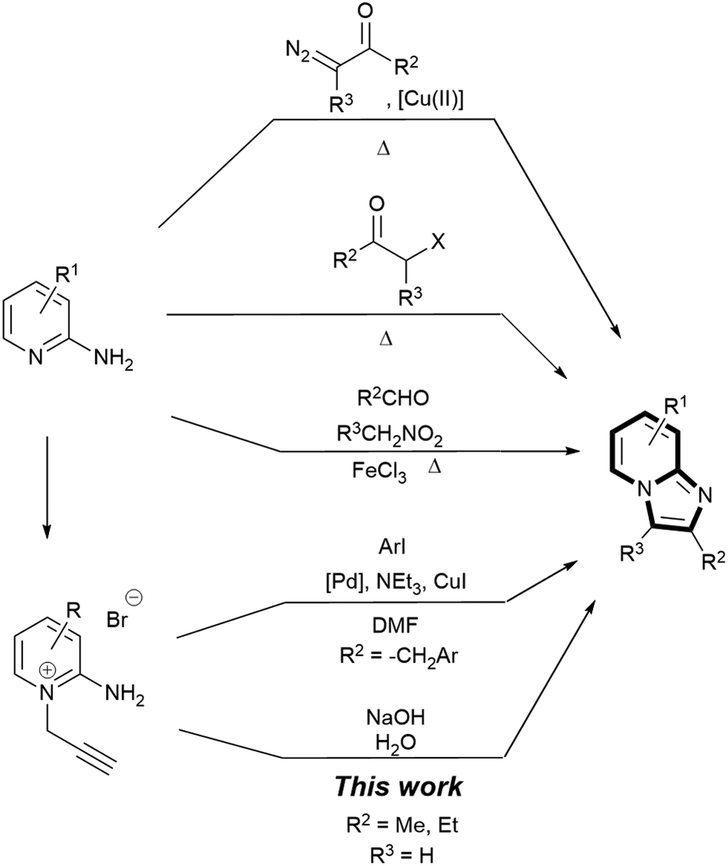 | ||
| Scheme 1 Synthetic methods to assemble imidazo[1,2-a]pyridines. | ||
Bakherad and co-workers reported a Sonogashira-cyclisation cascade reaction catalysed by Pd catalysts.27,28 This reaction has been applied by Marugan et al. to generate a wide range of imidazopyridinium analogues as antagonists of neuropeptide S receptor.29 Further functionalisations at the methyl position (R2 in Scheme 1) are also well established.30–34 We were particularly interested in the cyclisation step, which was proposed to be catalysed by the Pd catalyst. We hypothesised that the same cyclisation could be achieved through a base-catalysed rearrangement of the propargyl fragment to an allenic group35–41 and subsequent Nazarov cyclisation.42–44 This led to a remarkably rapid, mild, general and high yielding NaOH-promoted heterocyclisation approach to imidazo[1,2-a]pyridines via pyridinium bromide precursors under ambient and aqueous conditions, which we are reporting here (Scheme 1).
N-Propargylic pyridinium bromide 1a, which was prepared by reaction of the corresponding 2-aminopyridine with propargyl bromide, was added to an aqueous solution of NaOH (1 equivalent) with vigorous stirring under ambient conditions. A suspension of a pale yellow oil was immediately evident which was subsequently extracted into ethyl acetate, concentrated and identified as spectroscopically pure imidazo[1,2-a]pyridine, 2a, isolated in quantitative yield (Table 1, entry 1). Methanol and ethanol performed almost equally well as solvent compared to water (entries 3 and 4), giving the desired product in high yield. The reaction in water, however, enabled a very simple workup either with an ethyl acetate extraction or simple phase separation at larger scales (vide infra). Employing KOH or DABCO instead of NaOH in water provided the product in quantitative yield (entries 5 and 6). Interestingly, using DABCO as the base in anhydrous DMSO gave no conversion (entry 13). Addition of catalytic amount of water led to some conversion (entry 14), but at much lower rate than that of the same reaction in water. A proton source is therefore essential to the reaction. Weaker bases such as Na2CO3, Cs2CO3 and K2CO3 proved less soluble, requiring slightly elevated temperatures and longer reaction times to achieve comparable yields (entries 7–9). Other organic bases, i.e. NEt3 and (iPr)2EtN, led to biphasic reaction mixtures and low yield of the desired product (entries 10 and 11, respectively), despite similar basicity to DABCO in DMSO.45
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | T (°C) | Time (min) | Yielda,b (%) |
| a Reaction conditions: pyridinium bromide (0.5 mmol), base (0.5 mmol), solvent (5 mL). b Isolated yield. c Extra pure NaOH assay: ≥99.9995% metals basis. d Only 0.2 equivalent of NaOH was employed. e Using anhydrous d6-DMSO. f Using anhydrous d6-DMSO and 20 mol% of H2O, giving 66% conversion after 24 hours. | |||||
| 1 | NaOH | H2O | r.t. | 2 | >99 |
| 2c | NaOH | H2O | r.t. | 2 | >99 |
| 3 | NaOH | MeOH | r.t. | 5 | 95 |
| 4 | NaOH | EtOH | r.t. | 5 | 95 |
| 5 | KOH | H2O | r.t. | 2 | >99 |
| 6 | DABCO | H2O | r.t. | 2 | >99 |
| 7 | Na2CO3 | H2O | 40 | 20 | 94 |
| 8 | K2CO3 | H2O | 40 | 5 | 90 |
| 9 | Cs2CO3 | H2O | r.t. | 5 | 93 |
| 10 | NEt3 | H2O | r.t. | 30 | 48 |
| 11 | iPr2EtN | H2O | r.t. | 30 | 30 |
| 12d | NaOH | H2O | r.t. | 10 | 18 |
| 13e | DABCO | DMSO | r.t. | 2 | 0 |
| 14f | DABCO | DMSO | r.t | 2 | 15 |
To rule out the possibility of a reaction catalysed by trace amount of metal, ultra-high purity NaOH was employed as base (entry 2). The same quantitative yield and reaction time was obtained as with NaOH of standard purity, confirming the metal-free nature of the reaction. Reducing the amount of NaOH to 0.2 eq., however, resulted in only fractional conversion of the starting material (entry 12). Reaction conditions using NaOH as the least expensive and most readily available base, were taken forward for the rest of the study.
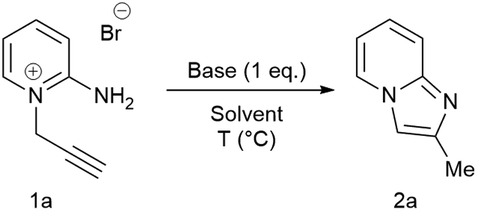
The substrate scope of the reaction was subsequently explored (Scheme 2, 2b–i). Near-quantitative isolated yields were obtained from substrates 1b–i containing electron donating and electron withdrawing substituents. Analytically pure (NMR and microanalysis) imidazopyridines 2b–i were obtained in all cases after a simple extraction. Importantly, the method is tolerant of methyl, chloro, bromo, iodo, trifluoromethyl, nitro and amino-substituents. X-ray crystallographic analysis of 2e, 2g and 2i confirmed their solid-state structures and the positions of substituents on the imidazopyridines (see ESI†). This synthetic method represent a significant advantage over current metal-catalysed routes to imidazo[1,2-a]pyridines as halogens, which are required for subsequent functionalisation, and are well tolerated without compromise in yield and selectivity.
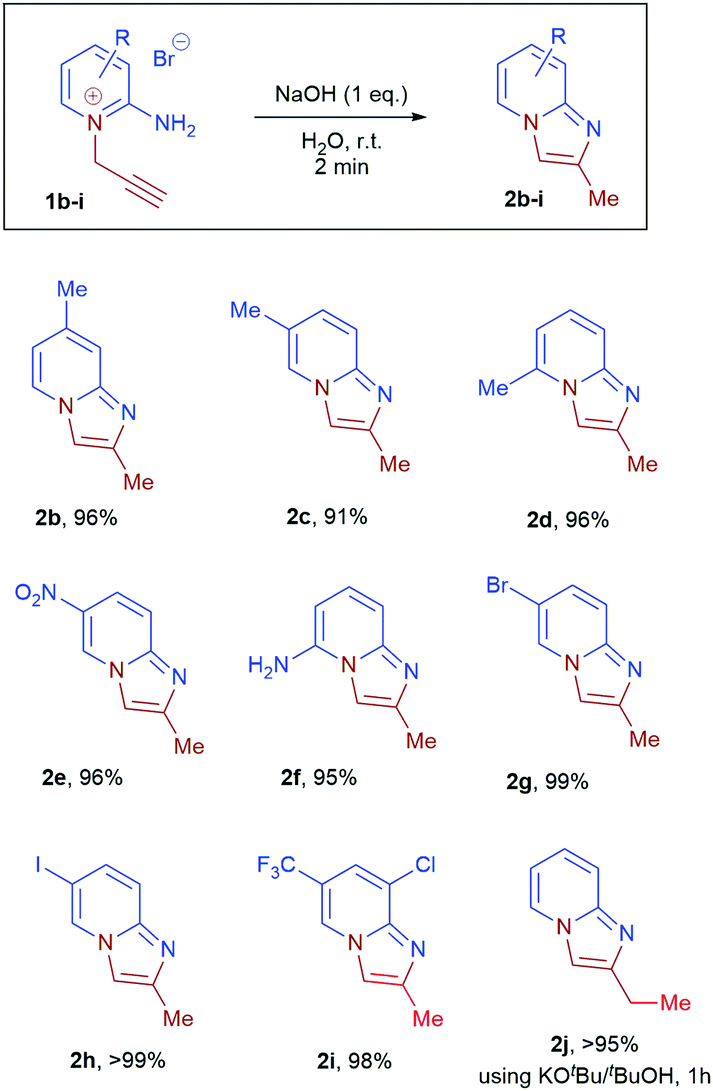 | ||
| Scheme 2 Scope of substrates for aqueous cyclisation of 2-amino-1-propargylpyridiniums 1b–j to product 2b–j. Reaction performed on 1.0 mmol scale in 10 mL of water. | ||
Disubstituted internal alkyne 2-amino-1-(2-butynyl)pyridinium bromide 1j, however, gave no conversion under standard conditions. This was attributed to a decrease in the acidity of the propargyl CH2, which is crucial for isomerisation to the corresponding allene. When tBuOK in tBuOH was employed, nearly quantitative conversion of 1j to product 2j was observed. Further functionalisation of the resulting imidazopyridines 2a, 2b, 2e, 2g and 2i by treatment with benzyl bromide, methyl iodide and allyl bromide led to isolation of imidazopyridiniums 3ak, 3bl, 3gl, 3im, and 3ik in good to excellent yield (Scheme 4).
The mechanism of the reaction was probed in a deuterium labelling experiment. Cyclisation of 4 methyl substituted pyridinium bromide 1b to 2b upon treatment with 1 eq. of NaOH was performed in D2O and the deuterium incorporation evaluated by 1H NMR spectroscopy. Only one methyl signal was observed in the 1H NMR spectrum of the product. The new methyl group, formed at the terminal alkyne position of the substrate, was fully deuterated. An exchange experiment of the non-deuterated product 2b with D2O in the presence of NaOH showed no exchange with deuterium at the same methyl group.
Based on the experimental evidence above, a plausible mechanism is proposed in Scheme 3. It starts with a base-induced alkyne–allene isomerisation.35–41 This process normally requires a stronger base, e.g. KOtBu, BuLi, but the presence of the pyridinium next to the propargyl CH2 makes it significantly more acidic and enables the use of NaOH. Subsequent cyclisation in a Nazarov-type mechanism and tautomerisation leads to the product with a fully deuterated methyl group in D2O. The base plays a dual role as catalyst for the isomerisation to allene and as a reagent to remove the byproduct DBr.
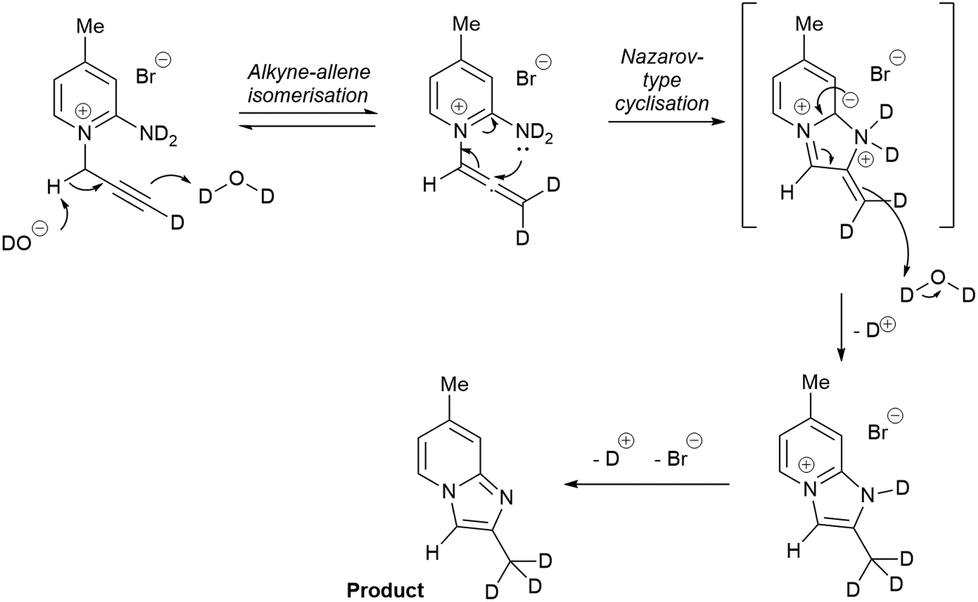 | ||
| Scheme 3 Proposed mechanism for the cyclisation based on deuterium labelling results. | ||
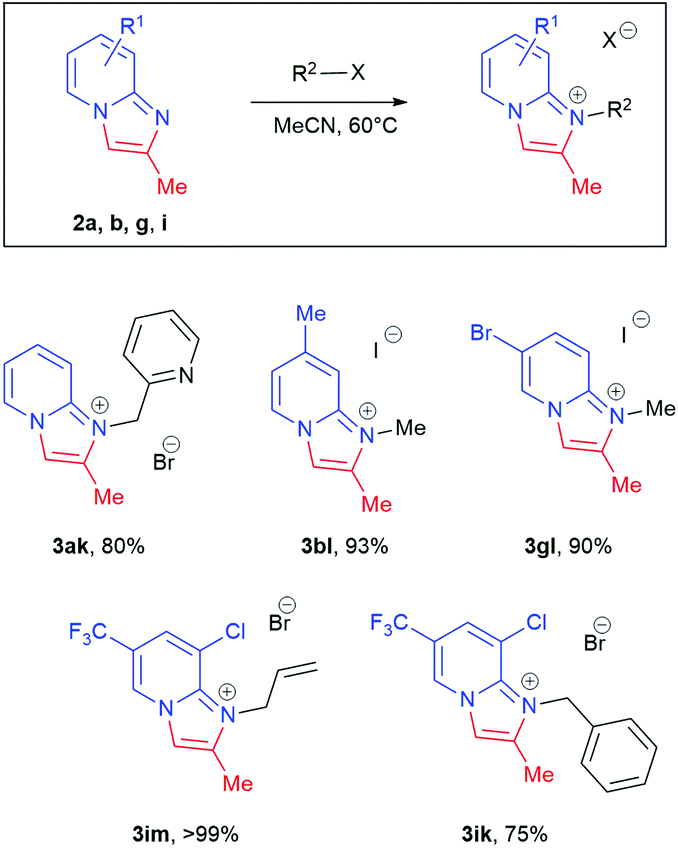 | ||
| Scheme 4 Scope of substrates for functionalisation of imidazo[1,2-a]pyridines. Reaction conditions: imidazo[1,2-a]pyridine (1.5 mmol), R2-X 2.0–2.6 eq., MeCN (30 mL). | ||
The remarkably facile and very rapid nature of the reaction led us to explore the scalability of this novel route to imidazopyridines. Substrate 1a was added over 5 minutes to a vigorously stirred aqueous solution of NaOH using a powder addition funnel on a 47 mmol (10.0 g) scale (Fig. 1). The product was easily separated at the end of the addition, giving the product 2a in the same quantitative yield and high purity.
 | ||
| Fig. 1 A scaled up reaction setup. (a) before reaction; (b) during addition of 1a (zoomed in); (c) phase separation at the end of the reaction (zoomed in). | ||
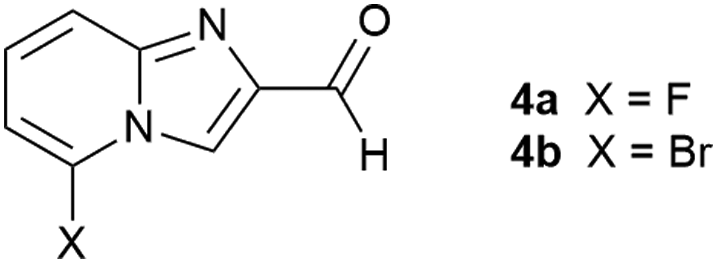
The green metrics for this process were calculated and are summarised in Table 2. These are compared with similar metrics from two industrial syntheses, one on lab scale and one optimised on kilogram scale.9,46 Atom Economy (AE),50 reaction mass efficiency (RME)49 and process mass intensity (PMI)49,51 are three of the currently preferred measures for evaluation of process sustainability.52 These metrics for our reported process are consistently superior to those of the established route using α-halogencarbonyl reagents for 5-fluoro and 5-bromo imidazo[1,2-a]pyridine-2-carbaldehyde, albeit without one functional group in the product.9,46 It is worth noting that the latter has been operated at kilogram scale with optimised work-up to improve E-factor and PMI through reduction of solvent use. The effective mass yield (EMY)53 shows higher efficiency for the new route, even compared to the optimised current process, although this metric requires definition of benign substances which can be contentious. Importantly the space-time-yield (STY) in the alkylation step is 7–14 times higher, and the STY in the cyclisation step is 185–363 times higher, than the comparative processes. In addition, the solvents in our process, i.e.iPrOH, EtOAc and water, are all ‘recommended’ solvents, while the main solvent in the current processes, 1,2-dimethoxyethane, is considered ‘hazardous’.52 Finally, the much faster reactions reported here mean that a telescoped process can potentially be carried out with acceptable solvents, heating and cooling.
|
|
|||
|---|---|---|---|
| Metrics | This work (2a, 10 g scale)a | Current route 1 (4a, 2.8 g scale)47 | Current route 2 (4b, 3 kg scale)9 |
| a Calculated as combined alkylation and cyclisation stages or shown separately. b NaOH, NaHCO3 and CaCO3 were estimated based on assumed stoichiometries. c Curzons’ method of calculating reaction mass efficiency was used, ignoring water.49 d This measure includes solvents but not water. e Space-time-yields exclude work-up time which are not optimised and can be variable. | |||
| Atom economy (%) | 52 | 36 | 51 |
| E-factor without water (g waste/g product)47,48 | 40.1 | 41.9 | 12.0 |
| E-factor with water (g waste/g product) | 51.5 | 77.6 | 22.8 |
| Effective mass yieldb (%) | 39 | 7.2 | 27 |
| Reaction mass efficiencyc (%) | 72 and 51 | 7.2 | 27 |
| Process mass intensity (material/product)d | 42.7 | 45.1 | 19.4 |
| Space-time-yielde (g L−1 min−1) | 0.42 and 10.9 | 0.03 | 0.059 |
In conclusion, we report a novel and green route to imidazo[1,2-a]pyridines which involves no metal catalyst and is extremely fast when performed in water. This route benefits from highly competitive green metrics and 1–2 magnitude higher STY compared to current synthetic processes. Investigation to expand the substrate scope and to understand the effect of water in this reaction is in progress and will be disseminated in the near future.
Acknowledgements
BAK thanks AstraZeneca for an industrial CASE studentship. MRC thanks the University of Leeds and AstraZeneca for a studentship. MHTK thanks the EPSRC, AstraZeneca and YProTech for an Industrial CASE studentship.Notes and references
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305–312 CrossRef CAS PubMed.
- S. Z. Langer, S. Arbilla, J. Benavides and B. Scatton, Adv. Biochem. Psychopharmacol., 1990, 46, 61–72 CAS.
- K. R. Lawrence and J. A. Klee, Pharmacotherapy, 2008, 28, 1019–1032 CrossRef CAS PubMed.
- H. L. DuPont, Clin. Infect. Dis., 2007, 45, S78–S84 CrossRef PubMed.
- Q. Li, M. Zhou, L. Han, Q. Cao, X. Wang, L. Zhao, J. Zhou and H. Zhang, Chem. Biol. Drug Des., 2015, 86, 849–856 CAS.
- S. Kang, R. Y. Kim, M. J. Seo, S. Lee, Y. M. Kim, M. Seo, J. J. Seo, Y. Ko, I. Choi, J. Jang, J. Nam, S. Park, H. Kang, H. J. Kim, J. Kim, S. Ahn, K. Pethe, K. Nam, Z. No and J. Kim, J. Med. Chem., 2014, 57, 5293–5305 CrossRef CAS PubMed.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466–470 CrossRef CAS PubMed.
- E. J. Barreiro, in Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation, ed. S. Bräse, 2015, ch. 1, pp. 1–15 Search PubMed.
- S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781–785 CrossRef CAS.
- R. Bellingham, A. M. Buswell, B. M. Choudary, A. H. Gordon, S. O. Moore, M. Peterson, M. Sasse, A. Shamji and M. W. J. Urquhart, Org. Process Res. Dev., 2010, 14, 1254–1263 CrossRef CAS.
- S. Feng, D. Hong, B. Wang, X. Zheng, K. Miao, L. Wang, H. Yun, L. Gao, S. Zhao and H. C. Shen, ACS Med. Chem. Lett., 2015, 6, 359–362 CrossRef CAS PubMed.
- H. Kishino, M. Moriya, S. Sakuraba, T. Sakamoto, H. Takahashi, T. Suzuki, R. Moriya, M. Ito, H. Iwaasa, N. Takenaga, A. Ishihara, A. Kanatani, N. Sato and T. Fukami, Bioorg. Med. Chem. Lett., 2009, 19, 4589–4593 CrossRef CAS PubMed.
- M. Shiozaki, K. Maeda, T. Miura, M. Kotoku, T. Yamasaki, I. Matsuda, K. Aoki, K. Yasue, H. Imai, M. Ubukata, A. Suma, M. Yokota, T. Hotta, M. Tanaka, Y. Hase, J. Haas, A. M. Fryer, E. R. Laird, N. M. Littmann, S. W. Andrews, J. A. Josey, T. Mimura, Y. Shinozaki, H. Yoshiuchi and T. Inaba, J. Med. Chem., 2011, 54, 2839–2863 CrossRef CAS PubMed.
- J. Koubachi, S. El Kazzouli, M. Bousmina and G. Guillaumet, Eur. J. Org. Chem., 2014, 2014, 5119–5138 CrossRef CAS.
- S. El Kazzouli, J. Koubachi, N. El Brahmi and G. Guillaumet, RSC Adv., 2015, 5, 15292–15327 RSC.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2014, 51, 1555–1575 RSC.
- Y. Tachikawa, Y. Nagasawa, S. Furuhashi, L. Cui, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, RSC Adv., 2015, 5, 9591–9593 RSC.
- R.-L. Yan, H. Yan, C. Ma, Z.-Y. Ren, X.-A. Gao, G.-S. Huang and Y.-M. Liang, J. Org. Chem., 2012, 77, 2024–2028 CrossRef CAS PubMed.
- Z. Wu, Y. Pan and X. Zhou, Synthesis, 2011, 2255–2260 Search PubMed.
- H. Yan, S. Yang, X. Gao, K. Zhou, C. Ma, R. Yan and G. Huang, Synlett, 2012, 2961–2964 CrossRef CAS.
- S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1065–1070 CrossRef CAS.
- J. S. Yadav, B. V. Subba Reddy, Y. Gopal Rao, M. Srinivas and A. V. Narsaiah, Tetrahedron Lett., 2007, 48, 7717–7720 CrossRef CAS.
- N. Shao, G.-X. Pang, C.-X. Yan, G.-F. Shi and Y. Cheng, J. Org. Chem., 2011, 76, 7458–7465 CrossRef CAS PubMed.
- C. He, J. Hao, H. Xu, Y. Mo, H. Liu, J. Han and A. Lei, Chem. Commun., 2012, 48, 11073–11075 RSC.
- M. Chioua, E. Soriano, L. Infantes, M. L. Jimeno, J. Marco-Contelles and A. Samadi, Eur. J. Org. Chem., 2013, 2013, 35–39 CrossRef CAS.
- D. C. Mohan, S. N. Rao and S. Adimurthy, J. Org. Chem., 2013, 78, 1266–1272 CrossRef PubMed.
- M. Bakherad, H. Nasr-Isfahani, A. Keivanloo and N. Doostmohammadi, Tetrahedron Lett., 2008, 49, 3819–3822 CrossRef CAS.
- M. Bakherad, B. Bahramian, H. Nasr-Isfahani, A. Keivanloo and N. Doostmohammadi, J. Heterocycl. Chem., 2009, 46, 100–104 CrossRef CAS.
- S. Patnaik, J. J. Marugan, K. Liu, W. Zheng, N. Southall, S. J. Dehdashti, A. Thorsell, M. Heilig, L. Bell, M. Zook, B. Eskay, K. R. Brimacombe and C. P. Austin, J. Med. Chem., 2013, 56, 9045–9056 CrossRef CAS PubMed.
- R. P. Alexander, J. M. Bentley, G. N. Brace, D. C. Brookings, P. T. Chovatia, H. J. C. Deboves, C. Johnstone; E. P. Jones, B. Kroeplien, F. C. Lecomte, J. Madden, C. A. Miller, J. R. Porter, M. D. Selby, M. A. Shaw, D. G. Vaidya and I. A. Yule, WO2015086506A1, 2015 Search PubMed.
- T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y.-L. Deng, D. Dinh, L. D. Engstrom, M. He, J. Hoffman, R. L. Hoffman, Q. Huang, R. S. Kania, J. C. Kath, H. Lam, J. L. Lam, P. T. Le, L. Lingardo, W. Liu, M. McTigue, C. L. Palmer, N. W. Sach, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed.
- S. Bailey, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, R. L. Hoffman, Q. Huang, T. W. Johnson, R. S. Kania, J. C. Kath, P. T. Q. Le, M. A. McTigue, C. L. Palmer, P. F. Richardson and N. W. Sach, WO2013132376A1, 2013 Search PubMed.
- K. Gudmundsson and E. M. Turner, WO2007087548A2, 2007 Search PubMed.
- K. R. Reddy, G. R. Scarlato, Q. Dang, M. D. Erion, S. R. Kasibhatla and M. R. Reddy, WO, 9839342A1, 1998 Search PubMed.
- A. Navarro-Vázque, Beilstein J. Org. Chem., 2015, 11, 1441–1446 CrossRef PubMed.
- T. W. Bousfield and M. C. Kimber, Tetrahedron Lett., 2015, 56, 350–352 CrossRef CAS.
- W. B. Dickinson and P. C. Lang, Tetrahedron Lett., 1967, 8, 3035–3040 CrossRef.
- J. Huang, H. Xiong, R. P. Hsung, C. Rameshkumar, J. A. Mulder and T. P. Grebe, Org. Lett., 2002, 4, 2417–2420 CrossRef CAS PubMed.
- A. G. Lohse and R. P. Hsung, Org. Lett., 2009, 11, 3430–3433 CrossRef CAS PubMed.
- Á. González-Gómez, L. Añorbe, A. Poblador, G. Domínguez and J. Pérez-Castells, Eur. J. Org. Chem., 2008, 1370–1377 CrossRef.
- H. Fuwa and M. Sasaki, Org. Biomol. Chem., 2007, 5, 2214–2218 CAS.
- W. T. Spencer, M. D. Levin and A. J. Frontier, Org. Lett., 2011, 13, 414–417 CrossRef CAS PubMed.
- V. M. Marx and D. J. Burnell, Org. Lett., 2009, 11, 1229–1231 CrossRef CAS PubMed.
- M. A. Tius, Chem. Soc. Rev., 2014, 43, 2979–3002 RSC.
- M. R. Crampton and I. A. Robotham, J. Chem. Res., Synop., 1997, 22–23 RSC.
- S. D. Boggs and K. Gudmundsson, PTC Int. Appl. Pat., WO2006026703, 2006 Search PubMed.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
- R. A. Sheldon, Chem. Ind., 1997, 12–15 CAS.
- A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- T. Hudlicky, D. A. Frey, L. Koroniak, C. D. Claeboe and L. E. Brammer Jr., Green Chem., 1999, 1, 57–59 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1471645, 1471647–1471650 and 1471653–1471656. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc01601d |
| This journal is © The Royal Society of Chemistry 2016 |